Au@Pt dendrimer encapsulated nanoparticles as model electrocatalysts for comparison of experiment and theory†
David F.
Yancey
abc,
Liang
Zhang
acd,
Richard M.
Crooks
*abc and
Graeme
Henkelman
*acd
aDepartment of Chemistry and Biochemistry, The University of Texas at Austin, 1 University Station, A5300, Austin, TX 78712-0165, USA. E-mail: crooks@cm.utexas.edu; henkelman@mail.utexas.edu; Tel: 512-471-4179; Tel: 512-475-8674; Fax: 512-471-4179; Fax: 512-475-8674
bCenter for Electrochemistry, The University of Texas at Austin, 1 University Station, A5300, Austin, TX 78712-0165, USA
cTexas Materials Institute, The University of Texas at Austin, 1 University Station, A5300, Austin, TX 78712-0165, USA
dInstitute for Computational and Engineering Sciences, The University of Texas at Austin, 1 University Station, A5300, Austin, TX 78712-0165, USA
First published on 30th January 2012
Abstract
In this paper we report the electrochemical synthesis of core@shell dendrimer-encapsulated nanoparticles (DENs) consisting of cores containing 147 Au atoms (Au147) and Pt shells having ∼54 or ∼102 atoms (Au147@Ptn (n = 54 or 102)). The significance of this work arises from the correlation of the experimentally determined structural and electrocatalytic properties of these particles with density functional theory (DFT) calculations. Specifically, we describe an experimental and theoretical study of Pb underpotential deposition (UPD) on Au147 DENs, the structure of both Au147@Pbn and Au147@Ptn DENs, and the activity of these DENs for the oxygen reduction reaction (ORR). DFT calculations show that Pb binding is stronger on the (100) facets of Au as compared to (111), and the calculated deposition and stripping potentials are consistent with those measured experimentally. Galvanic exchange is used to replace the surface Pb atoms with Pt, and a surface distortion is found for Au147@Ptn particles using molecular dynamics simulations in which the Pt-covered (100) facets shear into (111) diamond structures. DFT calculations of oxygen binding show that the distorted surfaces are the most active for the ORR, and that their activity is similar regardless of the Pt coverage. These calculations are consistent with rotating ring-disk voltammetry measurements.
Introduction
In this paper we report the electrochemical synthesis of core@shell dendrimer-encapsulated nanoparticles (DENs) consisting of cores containing 147 Au atoms (Au147) and Pt shells having ∼54 or ∼102 atoms (Au147@Ptn (n = 54 or 102)). The significance of this work arises from the correlation of the experimentally determined structural and electrocatalytic properties of these particles with density functional theory (DFT) calculations. Specifically, we describe an experimental and theoretical study of Pb underpotential deposition (UPD) on Au147 DENs, the structure of both Au147@Pbn and Au147@Ptn DENs, and the activity of these DENs for the oxygen reduction reaction (ORR). Our findings illustrate how the coupling of DENs with DFT calculations serves as a good model for understanding electrocatalysis at a fundamental level.DENs are synthesized by sequestering metal ions within the interior of poly(amidoamine) (PAMAM) dendrimers, followed by chemical reduction of the metal-ion/dendrimer complex with a reducing agent such as borohydride.1–4 Variations of this homogeneous synthetic method have yielded bimetallic DENs having different configurations, including random alloy and core@shell structures.5 We have also developed electrochemical methods for synthesizing core@shell DENs.6,7 DENs have been used to study electrocatalysis since 2005.8 They are valuable for such studies for the following reasons. First, their size, composition, and structure can be precisely controlled.9,10 Second, the dendrimers stabilize the encapsulated nanoparticles and provide a handle for linking them to an electrode surface, but they do not interfere significantly with reactions on the particle surface.8 Third, DENs are in a size range (1–2 nm diameter) that makes it possible to directly compare theoretical and experimental results from structural and catalytic studies.
We have recently published several studies that are relevant to the results reported here. First, we described the synthesis and properties of Au147@Pt DENs and demonstrated that they are electrocatalytically active for the ORR.6 These core@shell DENs were prepared by the UPD of Cu onto the surface of a Au core containing 147 atoms, followed by galvanic exchange of the Cu UPD layer for Pt. Second, we published a pair of comprehensive experimental and theoretical structural studies of Cu UPD onto Pt DENs consisting of ∼55 to ∼225 atoms.7,11 Finally, we reported on cluster-based DFT calculations of the structure and ORR activity of bimetallic nanoparticles in the same size range used for the experimental DEN studies.12 In contrast to these earlier studies, we now report a direct correlation between the experimentally determined structural and catalytic properties of DENs and first-principles calculations of materials having the same nominal structures and compositions.
Core@shell nanostructures are important because they exhibit tunable properties arising from the ability of the core metal to influence the properties of the shell.12–16 This phenomenon has been applied to electrocatalytic14,15,17 and spectroscopic applications.16 Early examples demonstrating the importance of Pt-shell nanoparticles as active electrocatalysts for the ORR were reported by Adzic and coworkers.14,15,18 The relationship between bimetallic nanoparticle structure and catalytic activity has been correlated to DFT calculations by a number of groups.17,19–21 Notably, Nørskov and coworkers have demonstrated trends in oxygen reduction electrocatalyst activity based on the binding energy of oxygen and other reaction intermediates to the metal surface.22,23
Pb UPD onto bulk Au surfaces, as well as onto Au nanostructures, has been studied extensively by Feliu and coworkers.24–26 They have shown that Pb UPD onto Au is a surface-selective reaction and that the morphology of the Au surface can be determined by analyzing voltammetric UPD peaks. With regard to nanoparticles, Feliu and his group have shown that the location of voltammetric Pb UPD peaks for well-defined single-crystal surfaces can be related to peak positions derived from nanoparticles, and that this correspondence can be used to help elucidate details of nanoparticle surface structure.26 One fact to emerge from these studies is that Pb UPD occurs on the low-index faces of Au in the following order: first Au(110), then Au(100), and finally Au(111).26
In the present paper, we show that Au DENs containing an average of ∼147 atoms are active for UPD and that particular facets on these materials can be selectively decorated with Pb. Specifically, a single monolayer of Pb is deposited first on the Au(100) facet, and then, at more negative potentials, the Au(111) facet. A principal outcome of this study is that the measured electrochemical potentials at which UPD occur are in good agreement with those determined by DFT modeling. We also found that UPD Pb deposited onto only the (100) facet, or both the (100) and (111) facets of Au147 DENs, could be exchanged for Pt. This galvanic exchange reaction results in Au147@Ptn DENs, where n = 54 or 102, respectively. Interestingly, the experimentally determined ORR activities for both Au147@Pt54 and Au147@Pt102 are very similar. This finding is interpreted in terms of DFT oxygen binding energy calculations on models of the corresponding particles. While these particles may not necessarily be practical ORR catalysts, they offer the ability to study the ORR on a fundamental level. Taken together, the results presented here indicate that DENs are good nanoparticle models for correlating DFT calculations with experimental measurements.
Experimental section
Chemicals
Amine-terminated, sixth-generation PAMAM dendrimers (G6-NH2) in methanol were purchased from Dendritech, Inc. (Midland, MI). The methanol was evaporated under vacuum and the dendrimer was reconstituted in H2O at a concentration of 100 μM. The following chemicals were purchased from Sigma-Aldrich: HAuCl4, K2PtCl4, Pb(NO3)2, NaBH4, NaOH, and ultrapure HClO4. Solutions were prepared using high-purity water obtained from a Millipore Milli-Q water system (18.2 MΩ cm). Ultra-high-purity N2 and O2 (99.999%) were purchased from Praxair. All chemicals were used as received.Synthesis of Au DENs
Au DENs were synthesized using a method similar to one reported previously.27 Typically, 20.0 mL of 2.0 μM G6-NH2(Au147) were prepared by adding 147 equiv. of HAuCl4 to a stirred solution containing 2.0 μM G6-NH2. The Au3+ was allowed to complex with the dendrimer for 10 min, and then a 10-fold excess of NaBH4 in 0.30 M NaOH was added to reduce the G6-NH2(Au3+)147 precursor to G6-NH2(Au147) DENs. This solution was stirred in air for 12 h to deactivate excess BH4−, and the resulting DENs were used without further purification.Characterization
UV-vis spectra were obtained using a Hewlett-Packard 8453 spectrometer. The cuvette path length was 0.200 cm and the spectra were background corrected using an aqueous solution of 2.0 μM G6-NH2 dendrimer. TEM images of G6-NH2(Au147) DENs were obtained using a JEOL 2010F TEM. Carbon-coated copper TEM grids (400 mesh) were purchased from Electron Microscopy Sciences. TEM grids were prepared by dropping 4.0 μL of a 2.0 μM G6-NH2(Au147) DENs solution onto the grid and drying in a desiccator. TEM images of the of G6-NH2(Au147@Pt102) DENs were obtained using both the JEOL 2010F TEM and a Hitachi HD2700C aberration-corrected STEM. The TEM grid preparation for the G6-NH2(Au147@Pt102) DENs is discussed later.Electrochemistry
Electrochemical measurements were performed using a CH Instruments 1202B potentiostat (Austin, TX, USA). All potentials were recorded and are reported versus a Hg/Hg2SO4 reference electrode (CH Instruments) unless otherwise noted. This electrode was calibrated against an eDAQ Hydroflex hydrogen reference electrode each day to ensure accurate potential control. Rotating ring-disk voltammetry (RRDV) was carried out using a AFASR rotator (Pine Instruments) and a Pine Instruments E7R9 series working electrode (5.61 mm-diameter glassy carbon disk and 0.84 mm wide Pt ring).The Au DENs were immobilized on the electrode surface as follows. The G6-NH2(Au147) DENs were dissolved in an aqueous solution containing 20% isopropyl alcohol and then sonicated for 30 min with 2 mg mL−1 Vulcan EC-72R carbon. Next, 6.0 μL of this catalyst ink was pipetted onto the glassy carbon disk of a freshly polished rotating ring-disk electrode (RRDE). Finally, the catalyst was dried in air at room temperature (23 ± 3 C).
DFT calculations
DFT was used to calculate the deposition process of Pb UPD on Au147 as well as the structures and oxygen binding energies to the Au147@Ptn particles. All calculations were performed with the VASP code28,29 where electron correlation was evaluated within the generalized gradient approximation using the PW91 functional.30 Core electrons were described with the projector augmented-wave method.31,32 Kohn–Sham wave functions for the valence electrons were expanded in a plane wave basis set with an energy cutoff of 250 eV. The energy cutoff was increased to 400 eV to test for convergence, and oxygen binding energies were found to vary by less than 0.01 eV. Spin polarization was tested and used as required.The Au147 particles were modeled as face-centered cubic (FCC) crystallites in the shape of a cuboctahedron having 6 (100) facets and 8 (111) facets. The structures of Au147@Pb and Au147@Pt were built by attaching atoms on the facets of the Au nanoparticles. In all calculations, the nanoparticles were isolated in a cubic box having edge lengths of 28 Å. The vacuum gap in all directions was large enough to avoid artificial interactions between periodic images.
Results and discussion
Characterization of G6-NH2(Au147) DENs
The synthesis of Au147 DEN cores is described in the Experimental Section. These DENs were characterized using UV-vis spectroscopy and TEM to ensure their size and degree of monodispersity were consistent with previous reports (ESI, Figure S1†). The TEM data indicate a diameter of 1.5 ± 0.2 nm, which is very close to the calculated value for a 147-atom cuboctahedron of 1.6 nm.10 The very weak plasmon peak in the UV-vis spectrum also indicates a Au particle size of <2 nm.27Electrochemical properties of Au147 DENs
As described in the Experimental Section, electrochemical experiments were carried out using glassy carbon electrodes modified with Au DENs. The electroactive surface area of the Au DENs was determined by scanning the electrode potential 5 times between −0.67 V and +0.87 V in N2-saturated 0.10 M HClO4. The positive scan limit was set to a potential just before bulk Au oxidation, and the negative limit was defined by the onset of H2 evolution on platinum. The final cycle of these scans is shown as the black cyclic voltammogram (CV) in Fig. 1. The onset of Au surface oxidation is at +0.50 V, and upon scan reversal the surface oxide is reduced between +0.50 V and +0.10 V. The charge under the reduction peak corresponds to a total Au surface area of 0.29 cm2. This value assumes a charge-per-unit-area of 390 μC cm−2, which has been established for bulk, polycrystalline Au33 and which we have used previously to estimate the surface area of DENs.6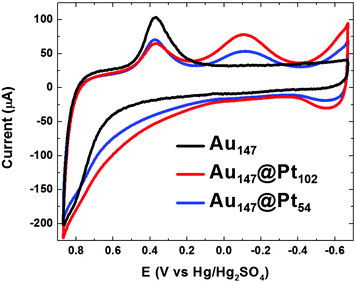 | ||
| Fig. 1 CVs of the indicated DEN-modified glassy carbon electrodes (geometrical area = 0.247 cm2). The scan rate was 100 mV s−1 and the aqueous electrolyte contained N2-saturated 0.10 M HClO4. | ||
Pb UPD onto G6-NH2(Au147) DENs
Pb UPD onto the electrode-immobilized Au147 DENs was carried out in a N2-saturated aqueous solution containing 1.0 mM Pb(NO3)2 and 0.10 M HClO4. Pb UPD and stripping (oxidation) were accomplished by scanning the electrode potential between 0.00 and −0.90 V, which is just before the onset of bulk Pb2+ reduction (Fig. 2, black trace). With one key exception, the major features of the Pb UPD voltammetry obtained using the Au147 DENs are similar to those observed on bulk Au (ESI, Figure S2†) or larger Au nanoparticle surfaces.24 For example, the potentials of the two most prominent Pb stripping peaks obtained using the Au147-modified electrode, are at −0.54 and −0.69 V. These values compare well those associated with Pb stripping from the (100) and (111) facets of bulk, polycrystalline Au: −0.60 and −0.67 V, respectively (Figure S2†).26 Importantly, however, the peak observed for Pb stripping from the Au(110) facets of the polycrystalline electrode (−0.40 V) is absent on the Au147 DENs. All of these observations are reassuringly consistent with our contention that Au147 DENs have a cuboctahedral structure, because in this case the (100) and (111) facets are present while (110) facets are absent.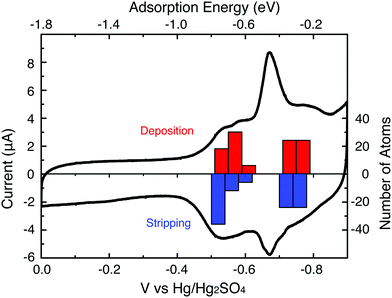 | ||
| Fig. 2 A CV showing the Pb UPD process at a Au147 DEN-modified glassy carbon electrode. The scan rate was 5 mV s−1 and the aqueous electrolyte solution contained 1.0 mM Pb(NO3)2 in 0.10 M HClO4. The DFT-calculated potentials for Pb deposition (red bars) and stripping (blue bars) are also shown. | ||
The coverage of UPD Pb on Au147 DENs was determined by measuring the charge required to strip UPD Pb from the Au surface (Fig. 2), and then converting this to surface area using a calculated conversion factor of 413 μC cm−2 (see ESI†). The resulting Pb surface area, 0.33 cm2, divided by the initial Au surface area (vide supra, 0.29 cm2) is defined as θPb.7 The value of θPb is equivalent to the number of Pb atoms in the UPD layer divided by the number of underlying Au atoms, because Pb forms a commensurate layer on the small Au domains. The experimentally determined value of θPb (1.14) is very close to that calculated (θPb = 1.11) using a model that places 102 UPD Pb atoms (48 Pb atoms on the 8 Au (111) facets and 54 Pb atoms on the 6 Au (100) facets) onto the 92 surface atoms of Au147 (Fig. 3b).
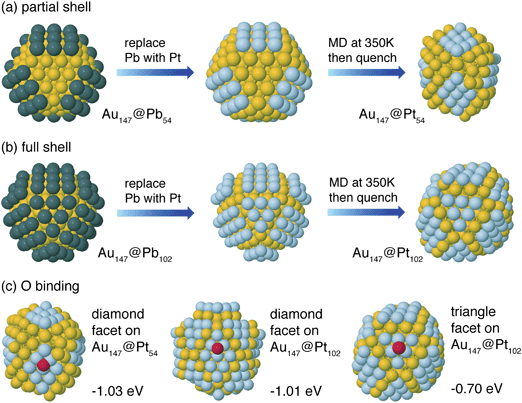 | ||
| Fig. 3 DFT-calculated structures for (a) Au147@Pb54 and Au147@Pt54 DENs and (b) Au147@Pb102 and Au147@Pt102 DENs. (c) Oxygen binding sites and energies for Au147@Pt54 and Au147@Pt102 DENs. | ||
Pb UPD DFT calculations
Pb UPD was modeled on Au147 particles using DFT calculations. First, the stability of Pb monolayers on the (100) and (111) facets was investigated by comparing two models. In the first model, each of the 6 Au(100) facets were covered with 9 Pb atoms. In the second model, each of the 8 Au(111) facets were covered with 6 Pb atoms. The average adsorption energies of the above two models were calculated using eqn (1).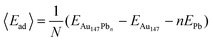 | (1) |
To model the Pb UPD process in detail, an atom-by-atom deposition calculation was done. Pb atoms were successively added to the lowest energy site on the Au particle, and the particle was relaxed before the addition of the next atom. The adsorption energy of the nth Pb atom, Ead(n), was calculated using eqn (2).
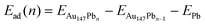 | (2) |
The calculated Ead values of each successive Pb atom binding site on (100) and (111) facets are plotted in Figure S3.† The first (or lowest energy) binding sites on the Au147 particle are on the (100) facets. Once the (100) facet sites are covered, the next Pb atoms deposit on the (111) sites. This means that the order of facet decoration on Au147 is consistent with bulk Au and larger Au nanoparticles.24
The values of Ead were converted to potentials, VDFTPb, for comparison with the experimental voltammograms. For this conversion, we assume that the binding energy per atom in bulk Pb corresponds to the onset potential of bulk Pb deposition. The DFT-calculated potential is given by eqn (3).
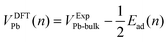 | (3) |
Fig. 2 compares a histogram of VDFTPb values for deposition and stripping of Pb atoms on Au147 with the UPD voltammogram. Based upon the order of deposition, and the separation of the peaks corresponding to UPD on the (100) and (111) facets, we constructed partially and fully covered models for the Au147@Pbn particles (left side of frames a and b, Fig. 3). In the partially covered Au147@Pb54 model, the 6 (100) facets of the Au147 core are covered by 9 Pb atoms. In the fully covered Au147@Pb102 model, the Au147 core has an additional 6 Pb atoms on each of the 8 (111) facets.
Synthesis of Au@Ptn DENs
A full or partial Pt shell can be added to the surface of Au147 particles via galvanic exchange of UPD Pb. Monolayer Pt deposition on bulk Au surfaces is thought to be conformal, although thicker layers result in island growth.34,35 It follows that displacement of a Pb monolayer on Au DENs will result in a conformal Pt layer too. The data presented in this section is consistent with this expectation.To avoid oxidation of the intermediate UPD Pb layer prior to galvanic exchange, the entire electrosynthesis was completed without removing the DEN-modified electrode from the N2-purged electrolyte solution. In addition, the electrode was rotated at 1600 RPM to ensure a high flux of the electrolyte solution to the electrode surface throughout the following electrosynthetic procedure. First, the disk of a RRDE was functionalized with Au147 DENs immobilized on Vulcan carbon as discussed earlier. Second, the disk electrode was held at the open circuit potential (OCP) for 5 min in a solution containing 100 mL of 0.10 M HClO4 and 1.0 mM Pb(NO3)2, and then its potential was stepped to −0.85 V for 3 s to facilitate the UPD of a complete monolayer of Pb on the Au147 DENs. Third, the electrode was returned to the OCP, and then 10 mL of a 1.0 mM K2PtCl4 solution was added to the solution to initiate galvanic exchange of UPD Pb for Pt. The OCP was monitored for 5 min following the addition of Pt2+ to the solution to confirm the galvanic exchange reaction (Figure S4†). Next, the electrode was removed from solution and rinsed thoroughly with H2O. The electrode was then rotated at 1600 RPM in ultrapure H2O for 5 min, followed by 0.10 M HClO4 for 5 min, to remove any free metal ions from the dendrimer. We refer to the resulting DENs as Au147@Pt102. To prepare a partial Pt monolayer on Au147 DENs, the same UPD and galvanic exchange processes were used, except the electrode potential for preparing the Pb UPD layer was set to −0.63 V. This potential corresponds to Pb deposition on just the Au(100) facets. These partial-monolayer DENs are denoted Au147@Pt54.
The Au147@Pt102 DENs were imaged by TEM to verify their size and degree of monodispersity following addition of the Pt shell. Somewhat surprisingly, very good micrographs could be obtained by simply wiping a TEM grid across the electrode surface, which apparently dislodges flakes of Vulcan carbon onto which the DENs are adsorbed. A representative image of Au147@Pt102 is provided in Fig. 4a. The average measured particle size is 2.3 ± 0.4 nm, reflecting the addition of a Pt monolayer to the original Au147 cores (1.5 ± 0.2 nm). The calculated size of Au147@Pt102 DENs is ∼2.1 nm. It is important to note that the particles remain relatively monodisperse after Pb UPD and galvanic exchange, which suggests they remain encapsulated within the protective dendrimer throughout electrosynthesis.
 | ||
| Fig. 4 TEM micrographs of Au147@Pt102 DENs (a) before and (b and c) after being used for the ORR. The particle diameters before and after the ORR were 2.3 ± 0.4 and 2.4 ± 0.4 nm, respectively. | ||
Information about the distribution and identity of atoms on the surfaces of the Au147@Pt102 and Au147@Pt54 DENs can be obtained using CV. The results of this analysis are provided in Fig. 1 alongside a CV for the original Au147 DEN cores. The CVs of the Au147@Ptn DENs have several features in common. For example, the Pt shell begins to oxidize well negative of the onset potential for surface oxidation of the Au147 DENs (+0.1 V vs. +0.5 V, respectively). Upon scan reversal, reduction of the Au surface oxide occurs at about the same potential observed for Au147 DENs, but the peak height is attenuated because Pt masks some of the surface Au atoms. At more negative potentials, a peak is observed for reduction of Pt oxide, but note that this peak is absent for the Au147 DENs. Likewise, H atom adsorption and desorption features appear negative of −0.4 V for the Au147@Ptn DENs but are absent for the Au147 DENs. The features here are not as pronounced as they are on bulk Pt, but they are consistent with previous reports of H adsorption and desorption on monometallic Pt DENs in the same size range.7,10 The important results of this analysis are that Pb UPD followed by galvanic exchange results in features characteristic of a Pt surface and impedes formation of Au oxide, thereby confirming formation of the Au147@Ptn DENs.6
The surface coverage of Pt on Au147@Ptn DENs can be estimated by integrating the area under the hydrogen adsorption peaks in Fig. 1. This requires correction of the current for double-layer charging and an assumption about the relationship between charge and surface area. For the latter, we used the commonly accepted conversion factor of 210 μC cm−2,36 but this value has only been confirmed for bulk surfaces and therefore is just a best estimate for DENs. As discussed earlier, the surface coverage of Au can also be estimated using the Au oxide reduction peak, which we integrated from the onset of the Au reduction to the onset of Pt oxide reduction. Using this analysis, the surface areas of Pt on the Au147@Pt102 and Au147@Pt54 DENs were determined to be 0.17 and 0.11 cm2, respectively, and the Au surface areas were 0.13 and 0.17 cm2, respectively. When the Au and Pt coverages are added together, the total surface areas are 0.30 and 0.28 cm2 for the Au147@Pt102 and Au147@Pt54 DENs, respectively. These areas are slightly larger than those of the original Au147 DENs (0.29 and 0.27 cm2, respectively), which is consistent with addition of a Pt shell. Table 1 provides a summary of the electrochemically derived surface coverages of Au and Pt on the Au147, Au147@Pt102, and Au147@Pt54 DENs.
| Au147@Pt102 | Au147@Pt54 | |
|---|---|---|
| a Percent surface Pt is calculated as the Pt electrochemical surface area divided by the total Au and Pt electrochemical surface area. b Total Au@Pt surface area is the Au@Pt: Au surface area plus the Au@Pt: Pt surface area. c Percent area increase is calculated as the total Au@Pt surface area divided by the starting Au147 surface area. | ||
| Initial Au147 surface area | 0.29 cm2 | 0.27 cm2 |
| Final Au surface area | 0.13 cm2 | 0.17 cm2 |
| Final Pt surface area | 0.17 cm2 | 0.11 cm2 |
| Percent Surface Pta | 57% | 39% |
| Total Au@Pt surface areab | 0.30 cm2 | 0.28 cm2 |
| Percent surface area increasec | 3% | 4% |
Oxygen reduction reaction on Au147@Ptn DENs
The activity of Au147@Pt102, Au147@Pt54, and Au147 DENs for the ORR was determined using a RRDV method that we10 and others36,37 have reported previously. Briefly, the DEN surfaces were cleaned electrochemically by cycling the electrode potential 20 times between 1.00 and 0.05 V vs. RHE (unless otherwise noted, potentials in this section are reported vs. RHE to be consistent with previous reports). Next, the DEN-modified glassy carbon disk electrode was scanned from 0.05 to 1.00 V in freshly prepared, O2-saturated 0.10 M HClO4, while holding the ring at +1.30 V to collect (oxidize) any peroxide generated at the disk. The measured disk and ring currents were background corrected by performing the same scan program in N2-saturated 0.10 M HClO4. The resulting polarization curves for the disk electrode are given in Fig. 5a and the corresponding ring currents are provided in Fig. 5b.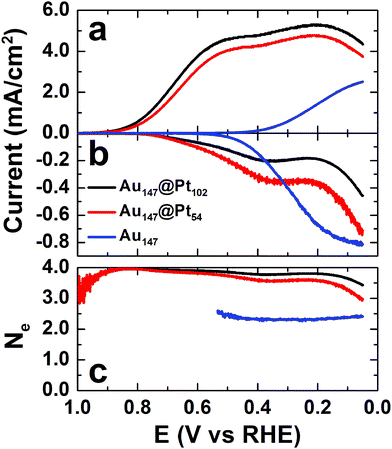 | ||
| Fig. 5 (a) ORR polarization curves for the indicated DEN-modified glassy carbon electrodes. (b) Ring currents arising from the oxidation of peroxide generated at the disk electrode during the ORR. (c) The apparent number of electrons calculated from the disk and ring currents. The electrolyte solution was O2-saturated 0.10 M HClO4, the scan rate was 20 mV s−1, and the rotation rate was 1600 RPM. | ||
The onset potential for the ORR for both Au147@Pt102 and Au147@Pt54 DENs is at ∼0.9 V, and the current attains limiting behavior at ∼0.5 V. However, at slightly more negative potentials, a second current plateau is observed. Importantly, the onset of current for the ORR at the Au147-modified electrode (blue line, Fig. 5a) is coincident with the appearance of the second plateau observed for the Au147@Ptn DENs. We conclude that the first current plateau for the Au147@Ptn DENs arises from the ORR on the fraction of the DEN surface covered with Pt atoms, and the second plateau arises from the fraction covered with Au atoms. This observation strongly suggests that the ORR proceeds independently on the surface of the full or partial Pt shell and the exposed Au core (structural models are shown in Fig. 3).
The number of electrons (Ne) involved in the ORR was calculated using eqn (4), where IR and ID are the ring and disk currents, respectively. The ring collection efficiency was experimentally determined to be 0.37.
| Ne = 4 − (2 × IR)/(0.37 × ID) | (4) |
The value of Ne is plotted as a function of disk electrode potential in Fig. 5c. In the potential region corresponding to the limiting current, Ne = 3.8 and 3.6 for the Au147@Pt102- and Au147@Pt54-modified electrodes, respectively. The slight decrease in Ne observed for the partial Pt shell can be attributed to peroxide generated on the exposed Au facets.38
Theoretical limiting currents for the ORR at Au147@Ptn-modified electrodes can be calculated using the Levich equation (eqn (5)).10
| Ilim = 0.20 × Ne × F × Ageo × D2/3O × ω1/2 × ν−1/6 × CO | (5) |
The kinetic currents (Ik) for the Au147@Pt102- and Au147@Pt54-modified electrodes at E = 0.80 V was calculated using eqn (6).36
| Ik = (Ilim × I0.8V)/(Ilim − I0.8V) | (6) |
The resulting values of Ik were then normalized to the Pt surface areas of the electrocatalysts, determined using the hydrogen adsorption charge (vide supra), to yield specific activities of 0.92 and 0.98 mA cm−2 and for the Au147@Pt102 and Au147@Pt54 electrocatalysts, respectively.
In the next section, we will correlate the experimentally determined ORR measurements to DFT calculations. For such a comparison to be valid, it is necessary to ensure that no gross changes in catalyst structure occurred during the electrochemical measurements. Accordingly, a TEM grid was wiped across the electrode surface to harvest Au147@Pt102 DENs after the RRDV data in Fig. 5 were collected. Representative TEM micrographs of these post-catalysis Au147@Pt102 DENs are shown in Fig. 4b and 4c. The average DEN diameter was found to be 2.4 ± 0.4 nm, which compares well with the DEN diameter measured before catalysis (2.3 ± 0.4 nm). The absence of agglomeration suggests that the DENs are stable and remain within their dendrimer hosts throughout the electrocatalytic experiments.
DFT calculations of Au147@Ptn structure and ORR activity
For simplicity, Pb atoms were assumed to be directly replaced by Pt in the galvanic exchange process. MD simulations were carried out on the Au147@Pt102 and Au147@Pt54 particles for 10 ps at 350 K to allow them to escape from any shallow local minima. Forces from DFT were used for these dynamics, as well as in a subsequent minimization for comparison with the initial (Au147@Pbn) structures. Fig. 3a shows the considerable deformation for the partial-shell structure induced by the short-time MD simulations, indicating the instability of the ordered structures upon substitution of the Pb shell atoms with Pt. The deformation has two distinct features: First, Au atoms in the sublayer rise to the surface edge sites. Second, the original square (100) Pt facets deform to diamond-shaped (111) facets. The relaxed Au147@Pt54 particle is 0.2 eV per Pt atom lower in energy than the ordered structure. A similar distortion occurs on the full shell Au147@Pt102 particle. The (100) ordered facets of the Au147@Pt102 structure, shown in Fig. 3b, spontaneously deform to diamond-shaped (111) facets.To better understand these deformations, we calculated the (100) and (111) surface energies of Pt, Au, and a Pt monolayer on Au using 4-layer slab models containing 9 atoms per layer. The results indicate that the surface energy of Au(111) is 0.11 eV atom−1 lower (more stable) than that of Au(100), and Pt(111) is 0.25 eV atom−1 lower than Pt(100). Moreover, the cohesive energy of a Pt monolayer on Au(111) is 0.17 eV atom−1 lower than on Au(100). These two results account for the deformation of Pt-covered Au(100) facets to a (111) diamond structure by lowering of the Pt surface energy and increasing the stability of the particle.
To determine the catalytic activity of the Au147@Ptn particles, we rely on the binding energy of O as a descriptor for ORR activity.22,23 A volcano plot analysis of the ORR shows that the O binding energy on Pt(111) is near optimal.22Fig. 3c shows O binding to hollow sites on different Pt facets of Au147@Pt54 and Au147@Pt102. Oxygen binding to the deformed diamond (111) facets of the two nanoparticle structures have similar values of −1.03 and −1.01 eV, respectively. The triangular (111) facet of the Au147@Pt102 particle has an O binding energy that is 0.3 eV weaker. All of these O binding energies are weaker than for Pt(111), which was calculated with the slab model to be −1.55 eV. This is in contrast to other reported cases where a Pt monolayer on a Au surface leads to a lattice expansion of the Pt, and stronger binding of oxygen to the surface.39–41 In the present case, Au atoms that rise to the surface during the structural reorganization actually compress the small Pt(111) domains. The Pt–Pt bond length in the diamond-shaped domains, as calculated from the DFT structures of Au147@Pt54 and Au147@Pt102, is 2.65 Å, which is shorter than that of bulk Pt (2.77 Å). This compression of the Pt–Pt bonds leads to a weaker binding of O on the nanoparticle Pt surface as compared to bulk Pt(111).39,40 Because O binding on the nanoparticle facets is weaker than on Pt(111), the particle facets are too noble to give optimal activity, and the ORR mechanism is limited by O–O dissociation. However, the diamond facets (with stronger O-binding) are expected to have a higher activity than the triangular facets, so it is the diamond facets that are active for the ORR on both the Au147@Pt102 and Au147@Pt54 particles. Note also that the nearly identical calculated O binding energy for these two structures is consistent with the experimentally determined specific ORR activities measured for the partial and full shell Au147@Ptn DENs (vide supra).
Summary and conclusions
Here we have reported the electrochemical synthesis and electrocatalytic properties of Au147@Ptn DENs (n = 0, 54, and 102), and correlated the experimental measurements to theoretical calculations. The synthesis of the Au147@Ptn DENs is carried out by first reducing Pb2+via UPD, and then exchanging the resulting surface Pb atoms for Pt atoms. We model the Au147 DEN cores as cuboctahedra and show that the UPD/galvanic exchange approach can be used to selectively decorate particular facets with Pt. DFT calculations accurately predict the potentials for Pb deposition onto the Au(100) and Au(111) facets of the Au147 DENs. Moreover, the structures of Au147@Pbn and Au147@Ptn DENs having full and partial coverage were also predicted by theory. Ordered structures are observed for Au147@Pbn DENs, while Au147@Ptn particles undergo a deformation such that all Pt surface atoms reorganize to (111) facet orientations. The experimentally measured electrocatalytic activities of Au147@Ptn DENs having full or partial shells (n = 102 and 54, respectively) were found to be similar. The calculations, which are based on the binding energy between O and Pt, are in accord with this finding.The most important result of this study is that core@shell DENs are good experimental models for testing the predictive power of first-principles calculations. This is because DENs are sufficiently small and structurally well-defined that direct correlations between theory and experiment can be made. In the present report we have reconciled the theory to experimental measurements, but our findings have been sufficiently encouraging that theory will lead the experiments as we move forward. The results of those studies will be reported in due course.
Acknowledgements
We gratefully acknowledge support from the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U. S. Department of Energy (Contract: DE-FG02-09ER16090). We thank Dr Dong Su (Brookhaven National Laboratory) for help with collection of the STEM micrographs and Dr J. P. Zhou (UT-Austin) for assistance with the other TEM measurements. RMC thanks the Robert A. Welch Foundation (Grant F-0032) for sustained support. GH would like to acknowledge support from the Robert A. Welch Foundation (Grant F-1601) and the W. A. “Tex” Moncrief, Jr. Endowment in Simulation-Based Engineering Sciences through Grand Challenge Faculty Fellowships from the Institute of Computational and Engineering Sciences at UT-Austin. The computational work was done primarily at the National Energy Research Scientific Computing Center and the Texas Advanced Computing Center.References
- R. W. J. Scott, O. M. Wilson and R. M. Crooks, J. Phys. Chem. B, 2005, 109, 692 CrossRef CAS.
- D. Yamamoto, S. Watanabe and M. T. Miyahara, Langmuir, 2010, 26, 2339 CrossRef CAS.
- H. Lang, R. A. May, B. L. Iversen and B. D. Chandler, J. Am. Chem. Soc., 2003, 125, 14832 CrossRef CAS.
- M. Bernechea, S. García-Rodríguez, P. Terreros, E. de Jesús, J. L. G. Fierro and S. Rojas, J. Phys. Chem. C, 2011, 115, 1287 CrossRef CAS.
- V. S. Myers, M. G. Weir, E. V. Carino, D. F. Yancey, S. Pande and R. M. Crooks, Chem. Sci., 2011, 2, 1632 RSC.
- D. F. Yancey, E. V. Carino and R. M. Crooks, J. Am. Chem. Soc., 2010, 132, 10988 CrossRef CAS.
- E. V. Carino and R. M. Crooks, Langmuir, 2011, 27, 4227 CrossRef CAS.
- H. Ye and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 4930 CrossRef CAS.
- H. Ye and R. M. Crooks, J. Am. Chem. Soc., 2007, 129, 3627 CrossRef CAS.
- H. Ye, J. A. Crooks and R. M. Crooks, Langmuir, 2007, 23, 11901 CrossRef CAS.
- E. V. Carino, H. Y. Kim, G. Henkelman and R. M. Crooks, J. Am. Chem. Soc., 2011 Search PubMed , in press.
- W. Tang and G. Henkelman, J. Chem. Phys., 2009, 130, 194504 CrossRef.
- N. S. Froemming and G. Henkelman, J. Chem. Phys., 2009, 131, 234103 CrossRef.
- J. X. Wang, H. Inada, L. Wu, Y. Zhu, Y. Choi, P. Liu, W.-P. Zhou and R. R. Adzic, J. Am. Chem. Soc., 2009, 131, 17298 CrossRef CAS.
- K. Gong, D. Su and R. R. Adzic, J. Am. Chem. Soc., 2010, 132, 14364 CrossRef CAS.
- S. Pande, S. K. Ghosh, S. Praharaj, S. Panigrahi, S. Basu, S. Jana, A. Pal, T. Tsukuda and T. Pal, J. Phys. Chem. C, 2007, 111, 10806 CrossRef CAS.
- P.-P. Fang, S. Duan, X.-D. Lin, J. R. Anema, J.-F. Li, O. Buriez, Y. Ding, F.-R. Fan, D.-Y. Wu, B. Ren, Z. L. Wang, C. Amatore and Z.-Q. Tian, Chem. Sci., 2011, 2, 531 RSC.
- S. Ball, S. L. Burton, J. Fisher, R. O'Malley, B. Tessier, B. R. C. Theobald, D. Thompsett, W. P. Zhou, D. Su, Y. Zhu and R. Adzic, ECS Trans., 2009, 25, 1023 Search PubMed.
- A. U. Nilekar, Y. Xu, J. Zhang, M. B. Vukmirovic, K. Sasaki, R. R. Adzic and M. Mavrikakis, Top. Catal., 2007, 46, 276 CrossRef CAS.
- G. Barcaro, A. Fortunelli, M. Polak and L. Rubinovich, Nano Lett., 2011, 11, 1766 CrossRef CAS.
- F.-J. Lai, H.-L. Chou, L. S. Sarma, D.-Y. Wang, Y.-C. Lin, J.-F. Lee, B.-J. Hwang and C.-C. Chen, Nanoscale, 2010, 2, 573 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef CAS.
- J. K. Norskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937 CrossRef CAS.
- J. Hernández, J. Solla-Gullón, E. Herrero, A. Aldaz and J. M. Feliu, J. Phys. Chem. B Lett., 2005, 109, 12651 Search PubMed.
- J. Hernández, J. Solla-Gullón, E. Herrero, A. Aldaz and J. M. Feliu, J. Phys. Chem. C, 2007, 111, 14078 CrossRef CAS.
- J. Hernández, J. Solla-Gullón, E. Herrero, J. M. Feliu and A. Aldaz, J. Nanosci. Nanotechnol., 2009, 9, 2256 CrossRef.
- Y.-G. Kim, S.-K. Oh and R. M. Crooks, Chem. Mater., 2004, 16, 167 CrossRef CAS.
- G. Kresse, Phys. Rev. B: Condens. Matter, 2000, 62, 8295 CrossRef CAS.
- G. Kresse and J. Hafner, Surf. Sci., 2000, 459, 287 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B, 1992, 45, 13244 CrossRef.
- P. E. Blochl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758 CrossRef CAS.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711 CrossRef CAS.
- Y. Ding, M. Chen and J. Erlebacher, J. Am. Chem. Soc., 2004, 126, 6876 CrossRef CAS.
- A. Mathur and J. Erlebacher, Surf. Sci., 2008, 602, 2863 CrossRef CAS.
- Y. Garsany, O. A. Baturina, K. E. Swider-Lyons and S. S. Kocha, Anal. Chem., 2010, 82, 6321 CrossRef CAS.
- K. J. J. Mayrhofer, D. Strmcnik, B. B. Blizanac, V. Stamenkovic, M. Arenz and N. M. Markovic, Electrochim. Acta, 2008, 53, 3181 CrossRef CAS.
- T. Inasaki and S. Kobayashi, Electrochim. Acta, 2009, 54, 4893 CrossRef CAS.
- F. H. B. Lima, J. Zhang, M. H. Shao, K. Sasaki, M. B. Vukmirovic, E. A. Ticianelli and R. R. Adzic, J. Phys. Chem. C, 2007, 111, 404 CrossRef CAS.
- J. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., Int. Ed., 2005, 44, 2132 CrossRef CAS.
- R. R. Adzic, J. Zhang, K. Sasaki, M. B. Vukmirovic, M. Shao, J. X. Wang, A. U. Nilekar, M. Mavrikakis, J. A. Valerio and F. Uribe, Top. Catal., 2007, 46, 249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2sc00971d |
| This journal is © The Royal Society of Chemistry 2012 |