Condensation of malononitrile with salicylaldehydes and o-aminobenzaldehydes revisited: solvent and catalyst free synthesis of 4H-chromenes and quinolines†
Subrahmanya Ishwar
Bhat
a,
Angshuman Roy
Choudhury
b and
Darshak R.
Trivedi
*a
aSupramolecular Chemistry Laboratory, Department of Chemistry, National Institute of Technology Karnataka (NITK), Srinivasnagar, Surathkal, Mangalore 575025 Karnataka, India. E-mail: darshak_rtrivedi@yahoo.co.in; Fax: +91-824-2474033; Tel: +91-824-2474000 Ext. No:3205
bIndian Institute of Science Education and Research, Mohali, Sector 81, S. A. S. Nagar, Manauli PO, Punjab, India 140306
First published on 5th September 2012
Abstract
The reaction of malononitrile with salicylaldehyde under solvent and catalyst free conditions was re-investigated using mechanochemical mixing, thermal heating and a direct crystallization process. The resulting condensation product by all three types of molecular activation, was found to be (2-amino-3-cyano-4H-chromene-4-yl)malononitrile, which is not the previously reported benzofuran-2-carbonitrile. The structure of the resulting chromene derivative was confirmed by FT-IR, MS, 1H, 13C NMR and single crystal and powder X-ray diffraction. The reaction pathway under neat conditions (mechanochemical mixing) at ambient temperature was monitored by IR spectral measurements. The versatility of the current green protocol was examined through the reaction of eleven derivatives of o-hydroxybenzaldehyde with malononitrile to obtain 2-amino-3-cyano-4H-chromene derivatives. In addition, malononitrile was further reacted with o-aminobenzaldehydes under neat conditions to yield quinoline derivatives.
Introduction
Organic reactions under solid state and solvent free conditions1 have become more attractive as a result of the growing awareness for green and sustainable chemical transformations. Solvent free organic reactions are preferred over conventional synthesis because of their numerous advantages, e.g., increased selectivity, lower energy consumption, minimized waste, hazards, toxicity and cost.2The Knoevenagel condensation of aldehydes with an active methylene compound is one of the classical methods for olefin synthesis. Although the Knoevenagel condensation is well known since 1898,3 several advantages like reaction simplicity, mild conditions, application in synthesizing biologically privileged compounds have made it an incessant field of research among organic chemists. Owing to the importance of this reaction, several green approaches have been developed under solvent free conditions,4 on solid supports,5 and through promotion by infrared,6 ultrasonic,7 microwave,8 electrochemical9 and thermal heating10 conditions. Kaupp et al. conducted quantitative Knoevenagel condensation reactions of aldehydes with various active methylene components under solid state and melt conditions.11 Inspired by their work, Ondruschka et al. studied the Knoevenagel condensation by reacting a series of aldehydes with malononitrile under solvent and waste free conditions,12 and at ambient temperature.13 The resulting product of the reaction malononitrile with salicylaldehyde was reported as benzofuran-2-carbonitrile, achieved through intramolecular cyclization followed by elimination of hydrocyanic acid. However, the structure of the product was assigned with inadequate spectroscopic data. A detailed literature survey reveals that it could be plausible to synthesize chromene derivatives (Fig. 1)14–16 by the delicate control of the experimental conditions of the conventional reaction of salicylaldehyde with malononitrile.
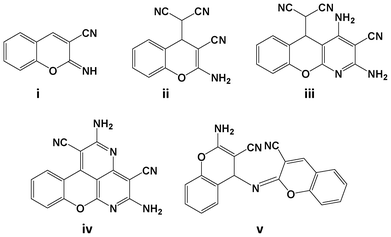 | ||
| Fig. 1 Various products observed in the reaction of salicylaldehyde and malononitrile under conventional methods. | ||
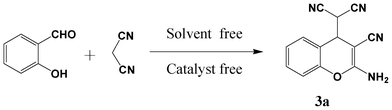
With the aim to elucidate the exact structure of the product and to determine the reaction pathway, herein, we re-investigated the neat reaction of salicylaldehyde and malononitrile under mechanochemical mixing, thermal heating and a direct crystallization process. The structure of the product was fully characterized by spectroscopic measurements (mp, FT-IR, 1H and 13C NMR, MS) and it was further confirmed with the reported literature.14,16 Surprisingly, the resulting condensation product was found to be (2-amino-3-cyano-4H-chromene-4-yl)malononitrile, not the previously reported benzofuran-2-carbonitrile by all three types of molecular activation. Further, the versatility of the developed methodology was examined by reacting eleven derivatives of o-hydroxybenzaldehyde and three o-aminobenzaldehydes with malononitrile to obtain 4H-chromene derivatives and quinoline derivatives respectively.
Results and discussion
Initially the reaction of salicylaldehyde with malononitrile was performed by mechanochemical activation, using a mortar and pestle. Equimolar amounts of the reactants were taken in a pre-weighed Agate mortar and mixed thoroughly with a pestle. The reaction mixture slowly converted to a viscous liquid after 5 min of mixing, and then it was allowed to stand for 3 h at room temperature (Table 1, entry 1). During this period the reaction mixture slowly converted to a wet white solid. The white solid thus obtained was partially soluble in ethyl acetate (EtOAc) and completely dissolved when 1 drop of DMSO was added. The TLC analysis [30% EtOAc in petroleum ether (60–80 °C fraction)] showed incomplete conversion of salicylaldehyde. The extended standing time did not allow the complete conversion of salicylaldehyde. Then, the wet solid was washed with ethanol (EtOH) to obtain a pure white solid, and was characterized by mp (151–153 °C), FT-IR (neat) and 1H NMR (in DMSO-d6 solvent). The structure assignment of the product was achieved based on the spectral analysis and was confirmed with the reported literature.15,16 Surprisingly, the obtained product was neither benzylidene-malononitrile nor benzofuran-2-carbonitrile, as reported earlier,12,13 but it was found to be (2-amino-3-cyano-4H-chromene-4-yl)malononitrile as shown in Fig. 1(ii). The resultant product was formed from a condensation reaction of malononitrile with aldehyde in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. Further, no by-product or the 1:1 condensation product 2-imino-2H-chromene-3-carbonitrile [Fig. 1(i)] was observed. This may be attributed to the relatively unstable nature of 2-imino-2H-chromene-3-carbonitrile [Fig. 1(i)].15
:1 molar ratio. Further, no by-product or the 1:1 condensation product 2-imino-2H-chromene-3-carbonitrile [Fig. 1(i)] was observed. This may be attributed to the relatively unstable nature of 2-imino-2H-chromene-3-carbonitrile [Fig. 1(i)].15
|
|
||||
|---|---|---|---|---|
| Entry | Reaction conditions | Reactant ratio used | Reaction timea (h) | Yieldb (%) |
| a Reactions were monitored by TLC. b Yield of pure product. c Reaction carried out at 60 °C | ||||
| 1 | Mechanochemical grinding | 1:1 |
3 | 49 |
| 2 | Thermal heatingc | 1:1 |
0.5 | 42 |
| 3 | Direct crystallization | 1:1 |
3.5 | 48 |
| 4 | Mechanochemical grinding | 1:2 |
3 | 98 |
Inspired by the interesting condensation product achieved by mechanical activation, the stoichiometric reaction of malononitrile with salicylaldehyde was further carried out under thermal conditions, using a conventional pre-heated oil bath at 150 °C. A vigorous reaction was observed as reported.12 The reaction mixture was cooled to room temperature after 10 min to obtain a brown coloured solid. The pure product was yielded after washing with ethanol, and was found to be identical to the chromene that was obtained from mechanochemical activation. A parallel reaction, with elongated heating, resulted in the complete decomposition of the product. Further, by optimization of the reaction temperature, we found that the reaction was carried out at 60 °C, resulting in the desired product after 30 min, without decomposition (Table 1, entry 2).
Furthermore, the reaction of salicylaldehyde with malononitrile via the direct crystallization process17 was performed at ambient temperature. Equimolar amounts of the reactants were mixed by brief swirling of the reactants in a 10 ml round bottom flask (RB), and was kept at room temperature under nitrogen atmosphere. A white crystalline solid contaminated with a light yellowish liquid was observed after 3.5 h (Table 1, entry 3). The crystals were filtered and washed with a small amount of EtOH. The structure of the resultant product was assigned by FT-IR and 1H NMR spectral analysis, and confirmed unequivocally by single crystal X-ray diffraction analysis (Fig. 2). Further, the X-ray powder diffraction data of the solids obtained by mechanochemical grinding, thermal heating and the bulk solid obtained by direct crystallization process were compared as shown in Fig. 3. The virtually superimposable patterns indicate that the crystal phase of the solid obtained by all three methods and that of the single crystal are identical.
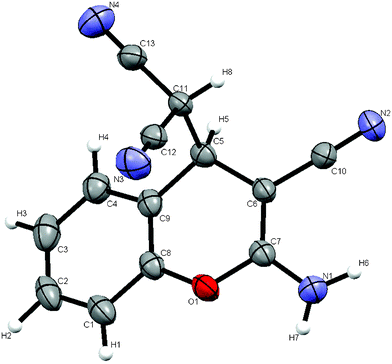 | ||
| Fig. 2 ORTEP (50% probability) diagram of the crystal structure of 3a obtained from the direct crystallization process. | ||
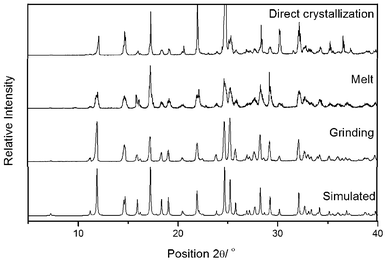 | ||
| Fig. 3 Powder X-ray diffraction pattern of 3a obtained by the reaction of a 1:1 molar ratio of salicylaldehyde and malononitrile under various conditions followed by an ethanol wash. | ||
The structural elucidation, based on spectral analysis, revealed that the product obtained from mechanical activation, thermal conditions and by the direct crystallization process was identical. These results are dissimilar to earlier reports,13 in which, no product was observed by the direct crystallization process in the absence of solvent and at ambient temperature.
To achieve the maximum conversion of the starting materials to the final product, the reaction was then performed with a 1:2 molar equivalent of salicylaldehyde and malononitrile. Complete conversion of the reactants to product 3a took place when the reaction mixture was mixed thoroughly for 5 min and then allowed to stand for 3 h at room temperature in presence of atmospheric air (Table 1, entry 4). The white solid product thus formed was characterized without further purification and the structure was confirmed by comparing the data with that obtained in the earlier methods. Thus, the present protocol is an example for the liquid state reaction in which the product crystallizes during the reaction18 to obtain near quantitative yields.
The conventional methods used for the synthesis of (2-amino-3-cyano-4H-chromene-4-yl)malononitrile derivatives suffer from many disadvantages such as long reaction times, usage of solvent and catalyst etc. Also, the vast biological importance of 2-aminochromenes19 inspired us to extend the scope of the present protocol. Hence, to evaluate the generality of the present protocol and to confirm similar chromene formation, eleven salicylaldehyde derivatives were reacted with malononitrile at room temperature as shown in Scheme 1 while the experimental information is listed in Table 2. All the reactions proceeded with liquifaction of the reaction mixture during grinding or thorough mixing of the reactants in an Agate mortar with a pestle followed by solidification of the final product, as explained by the Scott group.20 The purity of the final products obtained was good enough for spectroscopic analysis, however, all compounds were further washed with ethanol.
 | ||
| Scheme 1 Reaction of salicylaldehydes with malononitrile under solvent and catalyst free conditions. | ||
| Entry | Aldehyde | Reaction timeab (h) | Chromene derivative | Yieldc (%) | ||
|---|---|---|---|---|---|---|
|
a All the reactions were carried out by grinding/mixing 1:2 molar ratio of reactants with Agate mortar and pestle for 5 min followed by allowing the mixture to standing.
b Reactions were monitored by checking TLC every 30 min after mixing.
c Yield of pure product.
|
||||||
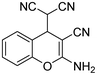
| 1 | 1a |
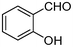
|
3 | 3a |
|
98 |
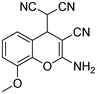
| 2 | 1b |
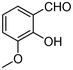
|
4.5 | 3b |
|
97 |
| 3 | 1c |
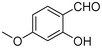
|
6 | 3c |

|
89 |
| 4 | 1d |

|
5.5 | 3d |
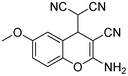
|
95 |
| 5 | 1e |

|
4 | 3e |

|
98 |
| 6 | 1f |

|
3 | 3f |

|
97 |
| 7 | 1g |
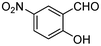
|
0.5 | 3g |

|
97 |
| 8 | 1h |

|
0.5 | 3h |

|
98 |
| 9 | 1i |
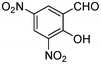
|
0.5 | 3i |
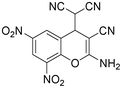
|
96 |
| 10 | 1j |

|
1 | 3j |

|
96 |
| 11 | 1k |

|
2 | 3k |

|
94 |
In order to elucidate the reaction pathway, the neat reaction of salicylaldehyde (1a) and malononitrile in a 1:2 molar ratio was monitored by IR spectral measurements. The IR spectra were recorded at 3 min intervals until the completion of the reaction (Fig. S1, ESI†). The absorption peaks were assigned by comparison with the literature.21 The IR spectrum obtained just after mixing of both the reactants displayed a superimposed spectrum of both the starting materials (Fig. 4). At the beginning of the measurement, two absorption peaks appeared at 2960 cm−1 and 2927 cm−1, which have been assigned to the asymmetrical and symmetrical methylene C–H stretching of malononitrile.22 These peaks became merged towards the completion of the reaction and appeared as a single peak at 2932 cm−1, corresponding to the methine C–H stretching absorption of the product. Also, the absorption bands of the aldehyde group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1654 cm−1, C–H stretching Fermi doublet at 2857 cm−1 and 2758 cm−1) gradually disappeared as the condensation reaction proceeds, and concomitantly a new absorption band, corresponding to the vinyl nitrile CC stretching, appeared at 1636 cm−1.23 After 6 min of reaction, a new absorption peak appeared at νmax = 2195 cm−1 and as the reaction proceeds, the peak intensity was rapidly enhanced with a small shift towards a lower frequency. This has been assigned to the conjugated C
O stretching at 1654 cm−1, C–H stretching Fermi doublet at 2857 cm−1 and 2758 cm−1) gradually disappeared as the condensation reaction proceeds, and concomitantly a new absorption band, corresponding to the vinyl nitrile CC stretching, appeared at 1636 cm−1.23 After 6 min of reaction, a new absorption peak appeared at νmax = 2195 cm−1 and as the reaction proceeds, the peak intensity was rapidly enhanced with a small shift towards a lower frequency. This has been assigned to the conjugated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching absorption.24 After 15 min, new asymmetrical and symmetrical νNH absorptions and a νC–N absorption of product 3a appeared at 3450 cm−1, 3329 cm−1 and 1223 cm−1 respectively (Fig. 4).
N stretching absorption.24 After 15 min, new asymmetrical and symmetrical νNH absorptions and a νC–N absorption of product 3a appeared at 3450 cm−1, 3329 cm−1 and 1223 cm−1 respectively (Fig. 4).
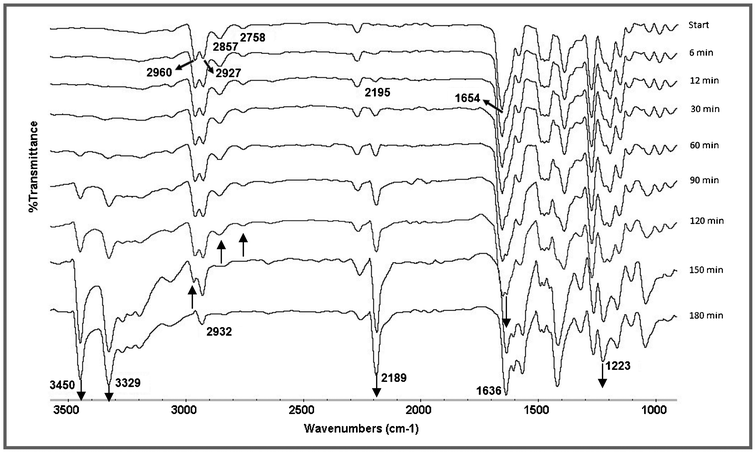 | ||
| Fig. 4 IR monitoring by the ATR method of the solvent and catalyst free reaction between salicylaldehyde and malononitrile under grinding conditions at room temperature. | ||
Taking into consideration the above facts and the final product characterization data, a possible mechanistic pathway is proposed, as shown in Scheme 2, which uses 1a as an example. Initially, the condensation reaction of salicylaldehyde and malononitrile, with the elimination of a water molecule, for the formation of salicylidene-malononitrile (A) occurs (known to take place under catalyst free grinding conditions).4,25 Since, malononitrile is the most reactive methylene component12 and the intervention of a liquid phase allows a large number of productive molecular collisions,20 the proper mixing and energy generated by the friction of the mortar and pestle may drive the reaction. The Knoevenagel adduct thus formed undergoes simultaneous Michael addition with malononitrile and intramolecular cyclization to obtain the stable product 3a. This was further supported by the IR spectrum, due to the appearance of an absorption peak corresponding to the conjugated cyano group (2193 cm−1), well before the appearance of absorption bands corresponding to the amine group present in the bicyclic product (as shown in Fig. S2, ESI†).
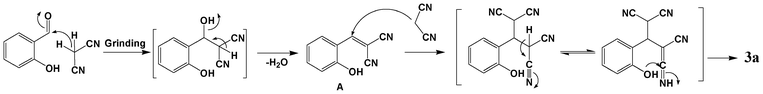 | ||
| Scheme 2 Proposed mechanism for the reaction between salicylaldehyde and malononitrile to obtain chromene derivative 3a. | ||
It was also found that the nature of the substitution on the aromatic ring of salicylaldehyde had a great influence on the reaction. The presence of electron-withdrawing groups on the aromatic ring leads the very fast reaction, and in contrast, electron-donating groups lead to a slower reaction . For instance, the reaction with 5-nitrosalicylaldehyde (1g) was very fast, which was complete faster than with 5-methoxy salicylaldehyde (1d).
Some of the solvent free reactions suffer from the possibility of runaway reactions at larger scale.1c Thus, to explore the present protocol at the gram scale, the reaction of salicylaldehyde (1a, 20 mmol) with malononitrile (40 mmol) was carried out under neat conditions. The reactants were mixed thoroughly using a pre-weighed mortar and pestle and the homogeneous liquid thus formed was left standing at room temperature. Complete conversion of the reactants to product 3a took place after standing for 5 h. Also, we did not notice any highly exothermic reaction or thermal runaway during the course of the reaction.
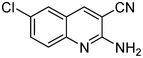
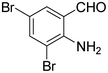
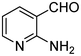
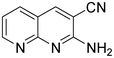
The next series of experiments concerns the Knoevenagel product formed from the reaction of malononitrile with aldehydes containing an amine group in the ortho-position under neat conditions. The reaction with 5-chloro-2-aminobenzaldehyde resulted in very poor conversion under solvent and catalyst free grinding conditions at room temperature. As a main reason for the decreased conversion, we assume the poorer reactivity of aldehydes is due to the presence of a more nucleophilic amine group in the ortho-position. The reaction was then attempted under melt conditions. The neat reactants (1:2 ratio) were taken in a 10 ml RB and then heated to 150 °C using a conventional preheated-oil bath. The solid thus obtained was recrystallized with 60% EtOAc in pet. ether and was then characterized. As expected, the bicyclic product thus obtained was 2-amino-3-cyano-quinoline derivative (Scheme 3). The stability caused by aromaticity26 may be the reason for avoiding the Michael addition product, as in the case of o-hydroxybenzaldehydes (Scheme 2). A similar product was reported for the solution synthesis using a base catalyst under reflux conditions.27 Further, different ratios of reactants were tested to derive the optimized conditions. The ratio 1:1.2 of aldehydes and malononitrile was found to be the more suitable system to obtain the product with good yield. Under similar conditions, the reaction of malononitrile with 3,5-dibromo-2-aminobenzaldehyde resulted in the corresponding quinoline derivative. Further, the reaction proceeded successfully with heteroaromatic 2-amino-pyridine-3-carboxaldehyde to obtain the corresponding quinoline derivative. Two possible mechanisms for the product formation are shown in Scheme 4. The pure products were obtained in good yield by recrystallization from 60% EtOAc in pet. ether (Table 3).
 | ||
| Scheme 3 Reaction of o-aminobenzaldehydes with malononitrile under solvent and catalyst free conditions. | ||
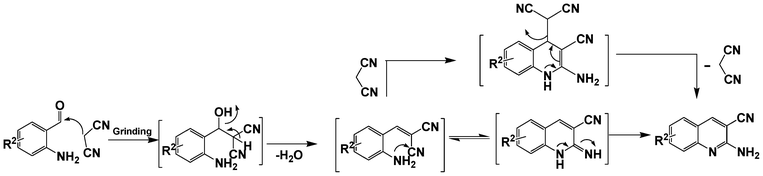 | ||
| Scheme 4 Possible mechanism for the reaction between o-aminobenzaldehyde and malononitrile. | ||


Conclusion
We have demonstrated that the solvent and catalyst free reaction of salicylaldehyde with malononitrile leads to the formation of (2-amino-3-cyano-4H-chromene-4-yl)malononitrile, instead of benzofuran-2-carbonitrile as reported previously. The reaction, monitored by IR measurements, shows that the Knoevenagel adduct undergoes simultaneous Michael addition with malononitrile and intramolecular cyclization to yield the bicyclic chromene derivative. Further, eleven salicylaldehyde derivatives were successfully reacted with malononitrile to generate the corresponding chromene derivatives. The products were characterized by spectroscopic techniques and were further confirmed by single crystal X-ray diffraction (3a). Neat conditions, short reaction time, high product yield without tedious work up, and the applicability to gram scale are the green relevance and notable advantages of the present methodology over conventional syntheses.Additionally, it was observed that the reactions of 2-aminobenzaldehydes with malononitrile yield 2-amino-3-cyanoquinolines under melt conditions. The Knoevenagel adduct undergoes simultaneous cyclization and aromatization to form the product in good yield.
The overall findings of these studies show that the Knoevenagel condensation of benzaldehydes containing a hydroxyl or amine group in the ortho-position leads to bicyclic heterocycles via six membered cyclization. It is conceivable that the present study offers a facile and expedient way to synthesize various medicinally privileged chromene and quinoline derivatives.
Experimental
General
All chemicals were purchased from commercial suppliers and used without further purification. Melting points were determined with Stuart SMP3 melting point apparatus and are uncorrected. The IR spectra were recorded on a Thermo scientific Nicolet Avatar 330 FT-IR spectrometer as a neat sample. The absorption is given in wavelength ν (cm−1). NMR spectra were recorded as a solution in DMSO-d6 500 MHz and 400 MHz instrument. Chemical shifts (δ) are reported in parts per million (ppm) with tetramethylsilane (TMS) as an internal standard. J-Values are given in Hz. NMR raw data was analysed with the program MestReNova 7.0.0-8331. Mass spectra were performed on Waters Micromass Q-Tof Micro spectrometer with an ESI source. The X-ray single-crystal diffraction was performed on the Bruker AXS KAPPA APEX II system. X-ray powder diffraction patterns were recorded on an XPERT Philips (Cu-Kα radiation) diffractometer. Elemental analysis was performed using Perkin Elmer, Series II, 2400 analyzer. Thin layer chromatography was performed on silica gel 60 F254 (Merck).General procedure for the preparation of 3
General procedure for the preparation of 4
:1.2 molar ratios of aldehyde (2 mmol) and malononitrile (2.4 mmol) were taken in a 10 ml round bottom flask and subjected to melt conditions using a conventional oil bath at 150 °C and was stirred for 1 h. The reaction mixture was cooled to room temperature and recrystallized with 60% EtOAc in pet. ether (boiling range: 60–80 °C), filtered and dried under vacuum to obtain the pure solid product in good yield (as shown in Table 3).
Acknowledgements
DRT and SIB acknowledge the Director and the HOD (Department of Chemistry), NITK Surathkal for providing the research infrastructure. SIB is thankful to NITK for the research fellowship. DRT and SIB are also thankful to CSMCRI Bhavnagar, IISc Bangalore, and SAIF Panjab University, for providing analytical support. Further, X-ray facility of IISER Mohali is acknowledged for the single crystal X-ray diffraction data collection.References
- (a) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS; (b) G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701–8708 CrossRef CAS; (c) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, L. Buriol and P. Machado, Chem. Rev., 2009, 109, 4140–4182 CrossRef CAS; (d) G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159–2169 RSC; (e) G. Kaupp, CrystEngComm, 2003, 5, 117–133 RSC; (f) K. Tanaka, Solvent-free Organic Synthesis, Wiley-VCH, Weinheim, 2003 Search PubMed.
- (a) M. Lancaster, Green Chemistry: An Introductory Text, Royal Society of Chemistry, Cambridge, UK, 2002 Search PubMed; (b) F. Toda, Top Curr. Chem., 2005, 254, 1–40 CAS; (c) G. Kaupp, J. Schmeyers and J. Boy, Chemosphere, 2001, 43, 55–61 CrossRef CAS; (d) F. Schneider, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Green Chem., 2009, 11, 1894–1899 RSC; (e) F. Toda, Acc. Chem. Res., 1995, 28, 480 CrossRef CAS; (f) G. Choudhary and R. K. Peddinti, Green Chem., 2011, 13, 276–282 RSC.
- E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1898, 31, 2585–2595 CrossRef CAS.
- Z. Ren, W. Cao and W. Tong, Synth. Commun., 2002, 32, 3475–3479 CrossRef CAS.
- S. Wada and H. Suzuki, Tetrahedron Lett., 2003, 44, 399–401 CrossRef CAS.
- (a) F. Delgado, J. Tamariz, G. Zepeda, M. Landa, R. Miranda and J. Garcia, Synth. Commun., 1995, 25, 753–759 CrossRef CAS; (b) E. Obrador, M. Castro, J. Tamariz, G. Zepeda, R. Miranda and J. Garcia, Synth. Commun., 1998, 28, 4649–4663 CrossRef CAS.
- S. S. Shindalkar, B. R. Madje and M. S. Shingare, Indian J. Chem., Sect. B, 2005, 44, 1519–1521 Search PubMed.
- (a) J. S. Biradar and B. S. Sasidhar, Eur. J. Med. Chem., 2011, 46, 6112–6118 CrossRef CAS; (b) S. Balalaie and N. Nemati, Synth. Commun., 2000, 30, 869–875 CrossRef CAS; (c) S. Balalaie and N. Nemati, Heterocycl. Commun., 2001, 7, 67 CrossRef CAS; (d) A. K. Mitra, A. De and N. Karchaudhuri, Synth. Commun., 1999, 29, 2731–2739 CrossRef CAS; (e) B. P. Bandgar, L. S. Uppalla and D. S. Kurule, Green Chem., 1999, 1, 243–245 RSC; (f) G. Sabitha, B. V. S. Reddy, R. S. Babu and J. S. Yadav, Chem. Lett., 1998, 773–774 CrossRef CAS; (g) D. Villemin and B. Labiad, Synth. Commun., 1990, 20, 3333–3337 CrossRef CAS.
- M. Feroci, M. Orsini, L. Palombib and A. Inesi, Green Chem., 2007, 9, 323–325 RSC.
- (a) P. S. Rao and R.V. Venkataratnam, Tetrahedron Lett., 1991, 32, 5821–5822 CrossRef CAS; (b) D. S. Bose and A.V. Narsaiah, J. Chem. Res., 2001, 2001, 36 CrossRef.
- G. Kaupp, M. R. Naimi-Jamal and J. Schmeyers, Tetrahedron, 2003, 59, 3753–3760 CrossRef CAS.
- R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 767–772 RSC.
- R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 873–878 RSC.
- (a) J. S. Yadav, B. V. S. Reddy, M. K. Gupta, I. Prathap and S. K. Pandey, Catal. Commun., 2007, 8, 2208–2211 CrossRef CAS; (b) R. G. Vaghei, Z. T. Semiromi and R. K. Nami, J. Braz. Chem. Soc., 2011, 22, 905–909 Search PubMed; (c) A. Sakurai, Y. Motomura and H. Midorikawa, J. Org. Chem., 1972, 37, 1523–1526 CrossRef CAS; (d) F. Fringuelli, O. Piermatti and F. Pizzo, Synthesis, 2003, 15, 2331–2334 Search PubMed; (e) M. N. Elinson, A. S. Dorofeev, S. K. Feducovich, R. F. Nasybullin, S. V. Gorbunov and G. I. Nikishin, Electrochem. Commun., 2006, 8, 1567–1571 CrossRef CAS.
- C. N. O'Callaghan, T. B. H. McMurry and J. E. O'Brien, J. Chem. Soc., Perkin Trans. 1, 1995, 417–420 RSC.
- M. Costa, F. Areias, L. Abrunhosa, A. Venancio and F. Proencüa, J. Org. Chem., 2008, 73(5), 1954–1962 CrossRef CAS.
- G. Kaupp, CrystEngComm, 2006, 8, 794–804 RSC.
- G. Kaupp, Angew. Chem., Int. Ed., 2001, 40, 4506–4508 CrossRef CAS.
- (a) For recent examples of biologically active 2-aminochromene derivatives see: S. G. Das, B. Srinivasan, D. L. Hermanson, N. P. Bleeker, J. M. Doshi, R. Tang, W. T. Beck and C. Xing, J. Med. Chem., 2011, 54, 5937–5948 CrossRef CAS; (b) R. R. Kumar, S. Perumal, J. C. Menendez, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2011, 19, 3444–3450 CrossRef CAS; (c) T. R. Reddy, L. S. Reddy, G. R. Reddy, V. S. Nuthalapati, Y. Lingappa, S. Sandra, R. Kapavarapu, P. Misra and M. Pal, Bioorg. Med. Chem. Lett., 2011, 21, 6433–6439 CrossRef; (d) M. N. Erichsen, T. H. V. Huynh, B. Abrahamsen, J. F. Bastlund, C. Bundgaard, O. Monrad, A. B. Jensen, C. W. Nielsen, K. Frydenvang, A. A. Jensen and L. Bunch, J. Med. Chem., 2010, 53, 7180–7191 CrossRef CAS; (e) S. G. Das, J. M. Doshi, D. Tian, S. N. Addo, B. Srinivasan, D. L. Hermanson and C. Xing, J. Med. Chem., 2009, 52, 5937–5949 CrossRef CAS; (f) A. Jensen, M. N. Erichsen, C. W. Nielsen, T. B. Stensbol, J. Kehler and L. Bunch, J. Med. Chem., 2009, 52, 912–915 CrossRef CAS; (g) X. Fan, X. Zhang, L. Zhou, K. A. Keith, M. N. Prichard, E. R. Kern and P. F. Torrence, J. Med. Chem., 2006, 49, 4052–4054 CrossRef CAS; (h) D. R. Anderson, S. Hedge, E. Reinhard, L. Gomez, W. F. Vernier, L. Lee, S. Liu, A. Sambandam, P. A. Snider and L. Masih, Bioorg. Med. Chem. Lett., 2005, 15, 1587 CrossRef CAS.
- G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701–8708 CrossRef CAS.
- (a) R. M. Silverstein, in Spectrometric Identification of Organic Compounds, ed. F. X. Webster, Wiley, New Delhi, 6th edn, 2007, ch. 3, pp. 81–104 Search PubMed; (b) B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, John Wiley & Sons, Ltd, Chichester, 2004, ch. 4, pp. 71–88 Search PubMed.
- Y. I. Binev, J. A. Tsenov, I. N. Juchnovski and G. Binev Ivan, THEOCHEM, 2003, 625, 207–214 CrossRef CAS.
- S. Makarem, A. A. Mohammadi and A. R. Fakhari, Tetrahedron Lett., 2008, 49, 7194–7196 CrossRef CAS.
- C. Sridevi, G. Shanthi and G. Velraj, Spectrochim. Acta, Part A, 2012, 89, 46–54 CrossRef CAS.
- A. Kumar and S. Sharma, Green Chem., 2011, 13, 2017–2020 RSC.
- J. D. Hepworth, in Aromatic Chemistry, ed. D. R. Waring and M. J. Waring, The Royal Society of Chemistry, Cambridge, 2002 Search PubMed.
- B. M. Kiran and K. M. Mahadevan, Heterocycl. Commun., 2006, 12, 481–484 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data, including copies of selected 1H and 13C NMR spectra. CCDC reference number 870509. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21849f |
| This journal is © The Royal Society of Chemistry 2012 |