Investigations on a series of novel ionic liquids containing the [closo-B12Cl12]2− dianion†‡
Na
Zhou
ab,
Guoying
Zhao
*b,
Kun
Dong
b,
Jian
Sun
b and
Huawu
Shao
*a
aNatural Products Research Center, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, 610041, P. R. China. E-mail: shaohw@cib.ac.cn
bBeijing Key Laboratory of Ionic Liquids Clean Process, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: gyzhao@home.ipe.ac.cn
First published on 13th August 2012
Abstract
A series of novel imidazolium, ammonium, phosphonium and pyridinium based salts with the [closo-B12Cl12]2− dianion were synthesized by straight forward metathetic reactions and characterized by physical methods. The melting point of each salt with [closo-B12Cl12]2− dianion was measured to investigate if it can be classified in the field of ionic liquids. Thermodynamic results showed observed higher melting points, which can be illustrated both from the higher symmetry of the dianion and the enhanced electrostatic forces of the salts synthesized. Given the above mentioned reasons, the [closo-B12Cl12]2− dianion containing ionic liquids can be designed either by optimizing the structure of cation, especially of the alkylation pattern which naturally affected the cations' packing efficiency, or by shielding the cation–dianion interactions with appropriate hydrogen bond (H-bond) donors to form ‘quasi-eutectic ILs’. This can be rationalized by the increased interionic separation and the consequent weakening of the electrostatic forces. Additionally, influence of H-bond interactions on the phase transition temperatures of [closo-B12Cl12]2− dianion containing salts was also investigated and related changes were explained by ab initio calculations, illustrating weak H-bond interactions between the hydroxy protons and the chlorine atoms which showed modest effects on the melting points of the salts with [closo-B12Cl12]2− dianion.
Introduction
Ionic liquids1 (ILs) with melting point below 100 °C have attracted great scientific and industrial interests2 owing to their special and useful properties, like the negligible vapor pressure, non-flammability, wide electrochemical windows, high thermal and chemical stability, and above all, enhanced solvent quality. Boron cluster anions, which are of extraordinarily weak nucleophilicity, redox inertness and possess delocalized charges,3 have already been applied to the field of ILs chemistry. Compared with the common weakly coordinating anions (BF4− and PF6−), which are prone to undesirable decomposition and release the highly corrosive toxic acid (HF),4 boron cluster anion based ILs are more moisture-stable, greener and cleaner.Reed and co-workers prepared the first ILs with boron cluster anions, namely the imidazolium salts with inert carborane anions, and reported their synthesis, characterization as well as the factors affecting their melting points.5 Several years later, Larsen and co-workers reported that the salts with [nido-C2B9H12]− anion have lower melting points than the ones with [closo-CB11H12]− anion.6 Gabel and co-workers prepared ILs with [closo-B12H11NR3]− anions (R = alkyl) and found that even the potassium, lithium and unsolvated proton cations could yield lower melting salts.7 In the following years, Viñas et al. have comprehensively studied ILs containing the boron cluster anions.8 Among all the boron cluster anions studied, the derivatives of [closo-B12Cl12]2− dianion, being a class of novel materials, can be applied to stabilize reactive cations7,9,10 and taken as starting materials for the preparation of diprotic superacid H2B12Cl1211 as well as strong methylating agent Me2B12Cl12.12
However, most of the existing [closo-B12Cl12]2− dianion containing derivatives synthesized previously were all ionic salts instead of the liquids, thus greatly limiting their applications as solvents or catalysts in the field of catalysis chemistry. Furthermore, the concrete reasons attributed to their higher melting points were still not completely understood. Thereby, investigations on [closo-B12Cl12]2− dianion containing ILs with lower melting points are urgently required. According to the literature,5,6,8 the melting points could be regulated by optimizing the cations' structures, especially of the alkylation pattern. Moreover, it is well known that the salts with higher melting points can liquify at temperatures below 100 °C by mixing with appropriate H-bonds donors, which are generally named as ‘deep eutectic ILs'.25–27
In our present work, we synthesized a series of novel [closo-B12Cl12]2− dianion containing salts in order to investigate their abilities as ILs and the studied cations included imidazolium, ammonium, phosphonium and pyridinium, shown in Scheme 1. Furthermore, we characterized the salts and analysed the reasons attributed to their higher melting points both from the alkylation pattern on the cation and from the anion. With respect to the influence of H-bond interactions on melting points of the salts synthesized, we introduced the hydroxyl-substituent on cation or the appropriate ILs based H-bonds donors by forming the ‘deep eutectic ILs’, and the related changes were explained by ab initio calculations.
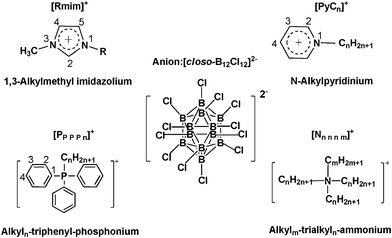 | ||
| Scheme 1 Structures and abbreviations for the cations and the dianion. | ||
Results and discussion
Synthesis
The imidazolium, ammonium, phosphonium and pyridinium based salts with the [closo-B12Cl12]2− dianion were prepared through conventional metathetic reactions of Cs2B12Cl12 with the corresponding bromide in acetonitrile. However, sometimes water was added as co-solvent in particular of the synthesis of [Nn n n m]2[B12Cl12] salt owning to the lower solubility of the precursor [Nn n n m]X (X = Cl/Br) in acetonitrile. The whole process was based on a simple preparation method in which the solid (CsCl/CsBr) formed was removed by filtration and the filtrate was evaporated to dryness under vacuum to yield the desired [closo-B12Cl12]2− dianion containing salts, which were obtained mainly as white crystalline solids at ambient temperature and dried under vacuum at 100 °C before analysis. The 1H nuclear magnetic resonance of the synthesized [closo-B12Cl12]2− dianion containing salts is shown in the ESI.‡ Additionally, densities of the salts prepared were measured at room temperature to further explore their physical properties.It is worth noting that the [closo-B12Cl12]2− dianion containing salts were not especially sensitive to air and moisture, which made them attractive for application purposes. In contrast, the ILs with PF6− and BF4− start to decompose in the presence of traces of water only at the temperature of 60 °C.13 Thereby, in order to better illustrate the stabilities of the salts with [closo-B12Cl12]2− dianion, [N11116]2[B12Cl12] (Td = 158 °C) was selected as a representative due to its lower decomposition temperature, and the relevant experiment was undertaken in our laboratories. Treated with water at about 60 °C for 2 h, the selected sample showed little change in its structure and composition, which was demonstrated by the FT-IR, 1H NMR, ESI and elemental analysis. The related information is shown in the ESI.‡ The above sufficiently proved the higher hydrolysis stability for the [closo-B12Cl12]2− dianion containing salts.
Thermal stability
Thermogravimetric analysis (TGA) curves for representative samples under a dinitrogen atmosphere are shown in Fig. 1. Decomposition temperatures taken from the onset of weight loss are presented in Table 1. Values listed are dynamic data rather than static. Thereby, the existed disparities are undisputed.14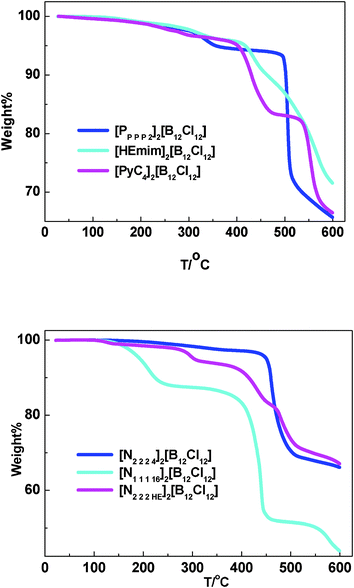 | ||
| Fig. 1 TGA curves for representative samples with different cations. | ||
| Entry | Materials | T m/°Ca | T d/°Cb | Density (g mL−1 25 °C) |
|---|---|---|---|---|
| a T m = Melting point. b T d = Decomposition temperature. c 1,3-Hydrogen-methylimidazolium [B12Cl12]. d Data from Ref. 8. e 1,3-Benzylmethyl-imidazolium [B12Cl12]. f 1,2-Dimethyl-3-butylimidazolium [B12Cl12]. g 1,3-Hydroxyethylmethylimidazolium [B12Cl12]. h Hydroxyethyltriethylammonium [B12Cl12]. i Hydroxyethyltriphenylphosphonium [B12Cl12]. | ||||
| 1c | [Hmim]2[B12Cl12] | 153 | — | 1.6855 |
| 2 | [C2mim]2[B12Cl12] | 265 | 480 | 1.6136 |
| 3 | [C3mim]2[B12Cl12] | 269 | — | 1.5961 |
| 4 | [C4mim]2[B12Cl12] | 131 | — | 1.5534 |
| 5 | [C8mim]2[B12Cl12] | 127 | — | 1.3874 |
| 6 | [C10mim]2[B12Cl12] | 174 | — | 1.4278 |
| 7d | [C16mim]2[B12Cl12] | 105 | — | — |
| 8d | [C18mim]2[B12Cl12] | 110 | — | — |
| 9e | [Bnmim]2[B12Cl12] | 256 | — | 1.7607 |
| 10f | [C4C1mim]2[B12Cl12] | 236 | — | 1.5166 |
| 11g | [HEmim]2[B12Cl12] | 275 | 412 | 1.6712 |
| 12 | [N2 2 2 4]2[B12Cl12] | >300 | 444 | 1.4567 |
| 13 | [N2 2 2 6]2[B12Cl12] | 221 | — | 1.6580 |
| 14 | [N1 1 1 16]2[B12Cl12] | 104 | 158(390) | 1.2155 |
| 15h | [N2 2 2 HE]2[B12Cl12] | 295 | 390 | 1.5629 |
| 16 | [PyC4]2[B12Cl12] | 222 | 398(531) | 1.6827 |
| 17 | [PP P P 2]2[B12Cl12] | 293 | 494 | 1.6559 |
| 18i | [PP P P HE]2[B12Cl12] | 240 | — | 1.5197 |
In general, the [closo-B12Cl12]2− dianion, being a weak nucleophilic reagent to react via SN2 mechanism, usually produces more thermally stable salts with higher recorded decomposition temperatures. Nevertheless, the salts with [closo-B12Cl12]2− dianion showed a progressive weight loss through the whole temperature scan (up to 600 °C) once they started decomposing.
For example, weight loss for the imidazolium, ammonium and phosphonium based salts of [HEmim]2[B12Cl12], [N2 2 2 4]2[B12Cl12] and [PP P P 2]2[B12Cl12] all started above 400 °C, at ∼412 °C, ∼444 °C and ∼494 °C, respectively (Fig. 1).
The pyridinium based salt of [PyC4]2[B12Cl12] decomposed firstly at 398 °C corresponding to a weight loss of 27.64% (229 g mol−1), and then came to the second decomposition at 530 °C. The weight loss value mentioned above is close to the quantity of the cation {[PyC4]2}2+ (272 g mol−1), suggesting the decomposition started from the cation and then turned to the dianion. Further experiments will be performed in our laboratories to gain more precise information about the process of decomposition.
Here it is worth noting that the thermally stability of ammonium based salts decreased with elongation of the alkyl chain on cation (Fig. 1, lower graph). For example, [N1 1 1 16]2[B12Cl12] (Table 1, entry 14), showing a weight loss firstly at 158 °C and then at 390 °C, is less thermally stable compared with [N2 2 2 4]2[B12Cl12] (Table 1, entry 12, Td = 444 °C).
Interestingly, both [HEmim]2[B12Cl12] and [N2 2 2 HE]2[B12Cl12] (Table 1, entries 11 and 15) showed decreased decomposition temperatures when to comparing with the salts of [C2mim]2[B12Cl12] and [N2 2 2 4]2[B12Cl12] (Table 1, entries 2 and 12), indicating that the incorporation of hydroxyl-substituent on the cation would have an effect on the thermal stability of the [closo-B12Cl12]2− dianion containing salts.
Solubility
Aside from the above mentioned physicochemical properties, the solubility of the salts with boron cluster anions in organic solvents is another focus of the chemists. The solubility of the [closo-B12Cl12]2− dianion containing salts in various organic solvents was then investigated at room temperature and the corresponding results are shown in Table 2.| Entry | Materials | H2O | EtOH | MeCN | DMSO | Me2CO | CH2Cl2 | CHCl3 | EtOAc | Et2O |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined at room temperature; m: miscible; nm: non-miscible; pm: partially miscible. | ||||||||||
| 1 | [Hmim]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 2 | [C2mim]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 3 | [C3mim]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 4 | [C4mim]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 5 | [C8mim]2[B12Cl12] | nm | nm | m | m | m | m | pm | nm | nm |
| 6 | [C10mim]2[B12Cl12] | nm | nm | m | m | m | m | m | m | nm |
| 7 | [Bnmim]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 8 | [C4C1mim]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 9 | [HEmim]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 10 | [N2 2 2 4]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 11 | [N2 2 2 6]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 12 | [N1 1 1 16]2[B12Cl12] | nm | nm | m | m | m | m | m | pm | nm |
| 13 | [N2 2 2 HE]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 14 | [PyC4]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 15 | [PP P P 2]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
| 16 | [PP P P HE]2[B12Cl12] | nm | nm | m | m | m | pm | pm | nm | nm |
It can be seen from Table 2 that the salts prepared, determined by the nature of the [closo-B12Cl12]2− dianion, were all strongly hydrophobic compounds and even insoluble in the polar organic solvent of ethanol. However, these salts were completely miscible with other polar organic solvents such as acetonitrile, dimethylsulphoxide and acetone. Unexpectedly, the solubility of the salts prepared in less polar organic solvents including chloroform, dichloromethane and ethyl acetate was affected by the length of the alkyl chain on the cation. With elongation of the alkyl chain on the cation, the solubility of the salts, including [C8mim]2[B12Cl12], [C10mim]2[B12Cl12] and [N1 1 1 16]2[B12Cl12], changed from non-miscible or partially miscible to fully miscible (Table 2, entries 5, 6 and 12). However, just as expected, the above salts were all insoluble in the non-polar organic solvent of diethyl ether. Interestingly, the introduction of a hydroxyl-substituent on the cation has little influence on the solubility of the prepared [closo-B12Cl12]2− dianion containing salts (Table 2, entries 2 vs. 9, 10 vs. 13 and 15 vs. 16).
These phenomena indicated that the polarities of the [closo-B12Cl12]2− dianion containing salts decrease with increasing the alkyl chain length on the cation, thus resulting in an increase of the solubility in less polar solvent.15 which follows the principle of ‘like dissolves like’.16
Infrared spectroscopy
Dominant infrared bands observed in the range of 50–4000 cm−1 for the newly prepared [closo-B12Cl12]2− dianion containing salts are listed in Table 3. Values offered distinctive information about the structures of the salts prepared originating from the stretching mode of various chemical bonds. Some weak bands and shoulders that can be variously assigned to overtones, solid state splitting or multiplicities arising from crystallographic disorder were not listed in the table. The actual spectra are available in the ESI‡ for the selected samples.| Entry | Materials | vC–Ha | vC(2)–Hb | vC(3)–Hb | vC(4)–H,vC(5)–Hb | vO–Hc | vB–Cld |
|---|---|---|---|---|---|---|---|
| a C–H stretching vibration of alkyl chain on cation. b C–H stretching vibration of aromatic rings, carbon atoms on aromatic rings were labeled in Scheme 1. c O–H stretching vibration of cation. d B-Cl stretching vibration of the [closo-B12Cl12]2− dianion. | |||||||
| 1 | [Hmim]2[B12Cl12] | 2959(m) | — | — | 3162(m) | — | 1031(s) |
| 2 | [C2mim]2[B12Cl12] | 2989(m) | 3116(m) | — | 3165(m) | — | 1029(s) |
| 3 | [C3mim]2[B12Cl12] | 2877(m), 2969(m) | 3114(m) | — | 3164(m) | — | 1031(s) |
| 4 | [C4mim]2[B12Cl12] | 2878(m), 2963(m) | 3117(m) | — | 3151(s), 3166(m) | — | 1030(s) |
| 5 | [C8mim]2[B12Cl12] | 2856(m), 2927(m) | 3117(m) | — | 3153(m) | — | 1031(s) |
| 6 | [C10mim]2[B12Cl12] | 2855(m), 2926(m) | 3116(m) | — | 3159(m) | — | 1032(s) |
| 7 | [Bnmim]2[B12Cl12] | — | 3120(m) | — | 3142(m) | — | 1030(s) |
| 8 | [C4C1mim]2[B12Cl12] | 2871(m), 2964(m) | — | — | 3174(m), 3138(m) | — | 1030(s) |
| 9 | [HEmim]2[B12Cl12] | 2934(m) | 3114(m) | — | 3166(m), 3149(m) | 3566(s) | 1031(s) |
| 10 | [N2 2 2 4]2[B12Cl12] | 2876(m), 2964(m) | — | — | — | — | 1031(s) |
| 11 | [N2 2 2 6]2[B12Cl12] | 2862(m), 2959(m) | — | — | — | — | 1030(s) |
| 12 | [N1 1 1 16]2[B12Cl12] | 2851(m), 2921(m) | — | — | — | — | 1032(s) |
| 13 | [N2 2 2 HE]2[B12Cl12] | 2951(m), 2989(m) | — | — | — | 3558(s) | 1030(s) |
| 14 | [PyC4]2[B12Cl12] | 2877(m), 2963(m) | 3065(m) | 3088(m) | 3130(m) | — | 1028(s) |
| 15 | [PP P P 2]2[B12Cl12] | 2913(m), 2945(m) | 3055(m) | — | — | — | 1028(s) |
| 16 | [PP P P HE]2[B12Cl12] | 2884(m), 2912(m) | 3067(m) | — | — | 3573(s) | 1030(s) |
As seen from the corresponding infrared spectra, the C–H stretching vibration of the alkyl chain on the cation mainly appeared in the range of 2851–2989 cm−1, whatever the cation type. Besides, the frequency bands within 1028–1032 cm−1 range can be assigned to the B–Cl stretches of the [closo-B12Cl12]2− dianion. Trivial variations of the B–Cl frequency bands, at most 4 cm−1, indicated that there only existed weak H-bond interactions between the cation and the chlorine atoms of the [closo-B12Cl12]2− dianion containing salts.
With respect to the frequency bands above 3000 cm−1, they can be assigned to the diagnostic of the two (or three) aromatic C–H bonds on cation. Taking the imidazolium based salts as an example, the highest frequency bands were firstly assigned to the C(4)–H and C(5)–H stretches,5,8 and the corresponding frequency bands lay within the 3174–3138 cm−1 range (Table 3, entries 1–9). The above mentioned small range of 36 cm−1 similarly illustrated the weak H-bond interactions between the C–H hydrogen atom on the cation with the chlorine atoms on the dianion, which varied only in a trivial manner. This was further proved by the data from C(2)–H hydrogen atom (only 4 cm−1 variations), which reinforced the notion of weak H-bonds interactions of [closo-B12Cl12]2− dianion containing salts. Interestingly, the frequency bands for C(4)–H and C(5)–H stretches showed a downward trend with the elongation of the alkyl chain on the cation (Table 3, entries 1–6). Additionally, the frequency band for C(2)–H as expected is absent with respect for 2-methylated imidazolium salts (entry 8).
For the salts with hydroxyl-substituent on the cation, the O–H stretching vibration appeared at 3600 cm−1 nearby, further indicating the weak (O–H⋯Cl) H-bond interactions. Moreover, data of aromatic C–H bonds for pyridinium and phosphonium based salts were assigned in the range of 3055–3130 cm−1 (Table 3, entries 14–16).
Trends in melting point
The phase transition temperatures for the prepared [closo-B12Cl12]2− dianion containing salts were measured by differential scanning calorimetry (DSC). Table 1 shows the determined melting point calculated from the second heating cycle in which a first order melting transition with broadened onset to melt was observed in all cases for the salts synthesized. Besides, most samples showed only one transition, namely from the solid state to the isotropic liquid, but some formed intermediate thermotropic mesophases. Super-cooling behavior is also typical for these salts. Just as shown in Fig. 4 (see later), the crystallization of [N1 1 1 16]2[B12Cl12] occurs at about 82 °C degrees below its melting point. These phenomena mentioned above have been investigated previously and were identified as an indicator of pre-melting alkyl chain mobility and the formation of disorder plastic phases as part of the melting transition.10The melting points of the newly synthesized imidazolium, ammonium, phosphonium and pyridinium based salts with [closo-B12Cl12]2− dianion were particularly cation dependent. This will be explained in the following paragraphs.
As for the 1,3-alkylmethylimidazolium based salts of the type [Cnmim]2[B12Cl12], the corresponding melting point values mainly showed a downward trend with increasing the alkyl chain length on cation (Fig. 2 and Table 1, entries 2–8). This variation trend is consistent with the common rules observed for other simple ILs in the field of ILs chemistry.17 Thereby, we can conclude that the alkylation pattern on cation is the main determinant of the melting point which affected the packing efficiency of the salts prepared. Additionally, it was reported that the imidazolium ILs have the lowest melting points when their side chain contains an odd number of carbon atoms and particularly one of the most interesting cation in this respect is 1,3-propylmethylimidazolium.18 Inspired by this, we have further investigated the thermal property of the 1,3-propylmethyl-imidazolium salt with the [closo-B12Cl12]2− dianion. However, the [C3mim]2[B12Cl12] melt at a substantial higher temperature than their neighbor salts of [C2mim]2[B12Cl12] and [C4mim]2[B12Cl12] (entries 2–4), which does not support the theory mentioned above. Besides, the substitution of C(2)–H proton on imidazolium rings by a methyl group usually results in a significant decrease of the melting point if the acidic protons engaged in strong H-bonds with the anion. As for the compound of [C4C1mim]2[B12Cl12] (Table 1, entry 10), the substitution of C(2)–H proton on imidazolium rings by methyl group, on the contrary, resulted in an increased melting point some 105 °C higher than [C4mim]2[B12Cl12] (Table 1, entry 4). This result may on the other hand better illustrate the fact of only weak H-bonds interactions between imidazolium C(2)–H and the [closo-B12Cl12]2− dianion. Similar phenomena was also observed for the carborane anion salts.5
![Melting point as a function of cation for [closo-B12Cl12]2− dianion containing salts. From left to right: [Hmim]2[B12Cl12], [Cnmim]2[B12Cl12] (n = 2, 3, 4, 8, 10, 16, 18), [Bnmim]2[B12Cl12] and [C4C1mim]2[B12Cl12].](/image/article/2012/RA/c2ra21700g/c2ra21700g-f2.gif) | ||
| Fig. 2 Melting point as a function of cation for [closo-B12Cl12]2− dianion containing salts. From left to right: [Hmim]2[B12Cl12], [Cnmim]2[B12Cl12] (n = 2, 3, 4, 8, 10, 16, 18), [Bnmim]2[B12Cl12] and [C4C1mim]2[B12Cl12]. | ||
For the pyridinium based salts, we only verified [PyC4]2[B12Cl12] which melts at a substantially higher temperature. It was reported the fact that little variations of the melting point would happen with the modification of alkyl chain length on cation for the pyridinium based salts.10 Thereby, it made little sense to investigate the other pyridinium based ILs with [closo-B12Cl12]2− dianion.
With regard to the ammonium based salts, the above mentioned rule, namely the dependence of melting point on alkyl chain, was also applied to their situation (Table 1, entries 12–14). This can be rationalized by the increased interionic separation and the consequent weakening of the electrostatic forces as well as the packing efficiently.19 Characteristic polarizing optical microscope (POM) texture of [N1 1 1 16]2[B12Cl12] obtained on cooling of the isotropic liquids between two glass microscope slides is shown in Fig. 3. It presents small but characteristic focal conic texture in the dark homeotropic regions. This phenomenon is common for the salts with at least one long unbranched alkyl chain that displays liquid crystalline behaviors. However, it should be noted here the samples used for the test should be heated on the microscope heating stage to above its melting point and then rapidly cooled down to avoid excessive mixing. The DSC heating and cooling curves for [N1 1 1 16]2[B12Cl12] are illustrated in Fig. 4, from which can be seen that the transition from the solid to the liquid crystalline phase was of much lower energy than the transition from the liquid crystal to the isotropic liquid.
![Textures observed of the mesophase for [N1 1 1 16]2[B12Cl12] using POM (100 × magnification) under crossed polarizers at (a) 38 °C and (b) 78 °C. Both photographs were taken after cooling of the samples from the isotropic phase.](/image/article/2012/RA/c2ra21700g/c2ra21700g-f3.gif) | ||
| Fig. 3 Textures observed of the mesophase for [N1 1 1 16]2[B12Cl12] using POM (100 × magnification) under crossed polarizers at (a) 38 °C and (b) 78 °C. Both photographs were taken after cooling of the samples from the isotropic phase. | ||
![DSC traces observed upon heating (lower) and cooling (upper) for the [N1 1 1 16]2[B12Cl12]. The peak labels indicate the type of phase transition occurring: S = solid ↔ smectic; I = smectic ↔ isotropic.](/image/article/2012/RA/c2ra21700g/c2ra21700g-f4.gif) | ||
| Fig. 4 DSC traces observed upon heating (lower) and cooling (upper) for the [N1 1 1 16]2[B12Cl12]. The peak labels indicate the type of phase transition occurring: S = solid ↔ smectic; I = smectic ↔ isotropic. | ||
The tetraalkylphosphonium salts of [closo-B12Cl12]2− dianion, owning to their similar structures, could be expected to follow similar melting rules as the tetraalkylammonium ones, namely the melting points decrease in general when increasing the cation size for a given anion. However, it is worth noting that the quaternary phosphonium cation [P6 6 6 14]+ based salts, known from the literature,8,20,21 are of the lowest possible melting points in combination with a series of anions like the sample of [P6 6 6 14]2[B10Cl10] with a melting point of 53 °C. Thus, the combination of the [P6 6 6 14]+ cation with the [closo-B12Cl12]2− dianion would have the potential to form phosphonium ILs of the type [P6 6 6 14]2[B12Cl12] with the lowest possible melting points. However up to date we have been unable to prepare entirely analytically pure samples of the precursor [P6 6 6 14]Br for the preparative metathesis reactions. Thereby, we focus our research on the easily available alkyltriphenylphosphonium salts. The purpose of this was to observe whether the incorporation of hydroxy-substituent onto phosphonium cation namely [PP P P HE]2[B12Cl12] would lead to a significantly decreased melting points.
Interestingly, the incorporation of hydroxy-substituent onto the ammonium and phosphonium cations led to a decrease in the phase transition temperatures evident for the corresponding [closo-B12Cl12]2− dianion containing salts (Fig. 5 and Table 1, entries 12 vs. 15, 17 vs. 18), while for the imidazolium based salts only a slight increase can be observed in this context (Fig. 5 and Table 1, entries 2 vs. 11). Comparison of the melting points for imidazolium, ammonium and phosphonium based salts with hydroxy-substituent and those without hydroxy-substituent on cation are shown graphically in Fig. 5.
![Comparison of melting points for imidazolium, ammonium and phosphonium based [closo-B12Cl12]2− dianion containing salts. Hashed bars represent cation with hydroxy-substituent and empty bars represent cation without hydroxy-substituent. From left to right: [C2mim]2[B12Cl12] vs. [HEmim]2[B12Cl12], [N2 2 2 4]2[B12Cl12] vs. [N2 2 2 HE]2[B12Cl12], [PP P P 2]2[B12Cl12] vs. [PP P P HE]2[B12Cl12].](/image/article/2012/RA/c2ra21700g/c2ra21700g-f5.gif) | ||
| Fig. 5 Comparison of melting points for imidazolium, ammonium and phosphonium based [closo-B12Cl12]2− dianion containing salts. Hashed bars represent cation with hydroxy-substituent and empty bars represent cation without hydroxy-substituent. From left to right: [C2mim]2[B12Cl12] vs. [HEmim]2[B12Cl12], [N2 2 2 4]2[B12Cl12] vs. [N2 2 2 HE]2[B12Cl12], [PP P P 2]2[B12Cl12] vs. [PP P P HE]2[B12Cl12]. | ||
In view of the above mentioned changes in melting point, we hypothesized that the formed O–H⋯Cl H-bonds of [closo-B12Cl12]2− dianion containing salts would have an influence on their melting points. A linear correlation between the melting points and interaction energies obtained by DFT calculation for imidazolium and ammonium based salts with the typical dianion was reported.22,23 For the ion-pairs, the interaction energies between an dianion (B) and two cations (A) were calculated by eqn (1).
| ΔE(A2B) = E(A2B) − 2E(A) − E(B) | (1) |
The interaction energies are 720.77 kJ mol−1 for [N2 2 2 HE]2[B12Cl12] ion-pair and 755.85 kJ mol−1 for [HEmim]2[B12Cl12] ion-pair, so a similar thermal stability can be predicted, which was discussed above. Furthermore in view of the structures, we found that there exists H-bonds between the cation and the dianion in the ion-pairs, and Fig. 6 exhibits the geometric distances of these H-bonds. In the [N2 2 2 HE]2[B12Cl12] ion-pair the only H-bond is Cl⋯H–O, while for the [HEmim]2[B12Cl12] ion-pair there exist Cl⋯C–H H-bonds in addition to the Cl⋯H–O H-bonds.
![The optimized structures of (a) [N2 2 2 HE]2[B12Cl12] and (b) [HEmim]2[B12Cl12] at the B3LYP/3-21+G* level where the dashed lines showed the H-bonds. With regard to [N2 2 2 HE]2[B12Cl12] ion-pair, the distances for the left cation: Cl⋯H–O = 2.78 Å, while for the right cation: Cl⋯H–O = 2.70 Å. With regard to [HEmim]2[B12Cl12] ion-pair, the distances for the left cation: Cl⋯H–C = 2.36 Å, Cl⋯H–O = 2.53 Å, while for the right cation: Cl⋯H–C = 2.45 Å, Cl⋯H–O = 2.40 Å.](/image/article/2012/RA/c2ra21700g/c2ra21700g-f6.gif) | ||
| Fig. 6 The optimized structures of (a) [N2 2 2 HE]2[B12Cl12] and (b) [HEmim]2[B12Cl12] at the B3LYP/3-21+G* level where the dashed lines showed the H-bonds. With regard to [N2 2 2 HE]2[B12Cl12] ion-pair, the distances for the left cation: Cl⋯H–O = 2.78 Å, while for the right cation: Cl⋯H–O = 2.70 Å. With regard to [HEmim]2[B12Cl12] ion-pair, the distances for the left cation: Cl⋯H–C = 2.36 Å, Cl⋯H–O = 2.53 Å, while for the right cation: Cl⋯H–C = 2.45 Å, Cl⋯H–O = 2.40 Å. | ||
The general nY→σXH* donor–acceptor H-bond interaction (XY⋯Y) can be evaluated by the following perturbation theory of NBO to calculate the delocalization energies E2:
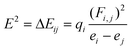 | (2) |
In addition to the factors mentioned above, the valence influence of the dianion was particularly noteworthy since it has a significant effect on the melting points for the [closo-B12Cl12]2− dianion containing salts. There is a nice example of ILs with [SO4]2− and [CH3SO4]− anions, when going from [SO4]2− to [CH3SO4]− resulted in the salts with melting points below 25 °C. Similarly, it also applies to the situation of boron cluster anion salts which can be divided into two distinguishable groups. Those with monovalent anion e.g. [CB11H6Cl6]− were found to melt at relatively lower temperatures, around or below 100 °C. In particular, [C8mim][CB11H6Cl6] liquefied at 67 °C, lower than the analogues with anions based on icosahedral structures.5 On the contrary, the salts with dinegative perchlorinated closo-borate dianions e.g. [B12Cl12]2−, melt at temperatures substantially higher (in general) than the ones with monovalent anions. This, of course, is in accordance with the Kapustinskii equation, namely higher Coulombic attraction for doubly charged ions and, in turn, higher melting enthalpy.35 Thereby, we can conclude that both the higher symmetry and the enhanced electrostatic forces in [closo-B12Cl12]2− dianion containing salts led to an increased lattice energy, and of course, the higher melting points. Inspired by this, we may predict that the monocharged analog of [closo-B12Cl12]2− dianion, namely the [CHB11Cl11]− anion,24 would have the potential to form the corresponding lowest melting point boron cluster anion salts due to its monovalent anion.
[closo-B12Cl12]2− dianion containing ‘quasi-eutectic ILs’
Recently, the so-called ‘deep eutectic ILs’,25–27 the mixture of a solid organic salt with a suitable complexing agent which can be liquified at temperatures below 100 °C, has fascinated scholars. The phase behavior of the mixture can be simply modeled by taking account of the mole fraction of the agent in the mixture. The mechanism put forth is that the complexing agent (typically a H-bond donor) interacts with the anion, thereby increasing the effective size of anion and shielding its interaction with the cation, which in turn induced a depression in the melting point of the mixture.28 An evident example is the mixture of choline chloride (Tm = 302 °C) and urea (Tm = 133 °C) in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, resulting in a room-temperature deep eutectic IL (Tf = 12 °C).
:2, resulting in a room-temperature deep eutectic IL (Tf = 12 °C).
Here we have shown that the above mentioned principle can be applied to the mixture of [closo-B12Cl12]2− dianion containing salts with a series of H-bonds donors (e.g. lower melting point ILs) to form ‘quasi-eutectic ILs’. Unlike the room-temperature ILs, these ‘quasi-eutectic’ mixtures are easy to prepare in a pure state. For example, by mixing the [N1 1 1 16]2[B12Cl12] with [C4mim]Cl at a temperature of 60 °C, the ‘quasi-eutectic ILs’ with [closo-B12Cl12]2− dianion can be successfully synthesized and their phase behaviors were characterized based on the analysis of two consecutive DSC cooling and heating cycles. The melting point of the mixture (m1 = [N1 1 1 16]2[B12Cl12] and m2 = [C4mim]Cl) was detected by changing their quantity ratio (m1:m2 = 1:0, 3:1, 1:1, 1:3, 0:1). Data in Fig. 7 suggests that the melting points of the ‘quasi-eutectic ILs’ decreased along with the increased [C4mim]Cl loading. To be emphasized, when it comes to the quantity ratio of m1:m2 = 1:3, the phase transition temperature of the mixture decreased to 31 °C and it kept viscous liquids at temperatures as low as 0 °C. The DSC scans gave no evidence for a freezing exotherm on the cooling segment, suggesting that the newly synthesized ‘quasi-eutectic ILs’ existed as stable supercooled liquids at sub-ambient temperature.
![Melting points of the mixture between [N1 1 1 16]2[B12Cl12] (1) and [C4mim]Cl (2) were detected by changing their quantity ratio (m1 : m2 = 1 : 0, 3 : 1, 1 : 1, 1 : 3, 0 : 1). Heating rate of 10 °C min−1 was employed.](/image/article/2012/RA/c2ra21700g/c2ra21700g-f7.gif) | ||
| Fig. 7 Melting points of the mixture between [N1 1 1 16]2[B12Cl12] (1) and [C4mim]Cl (2) were detected by changing their quantity ratio (m1:m2 = 1:0, 3:1, 1:1, 1:3, 0:1). Heating rate of 10 °C min−1 was employed. | ||
A major advantage of the above mentioned approach is that inexpensive and bulk commodity as well as sample ILs can frequently be employed. Besides, the liquid properties can be fine-tuned by combining various [closo-B12Cl12]2− dianion containing salts with H-bonds donors in different ratios, thus the generation of [closo-B12Cl12]2− dianion containing ILs with melting point below 100 °C is completely under control.
Conclusions
A new family of salts by combining [closo-B12Cl12]2− dianion with imidazolium, ammonium, phosphonium and pyridinium cations in straightforward metathetic reactions have been synthesized and structurally characterized. The melting point of each salt with the [closo-B12Cl12]2− dianion was measured to investigate if it can be classified to the field of ILs. Results showed that with the elongation of the alkyl chain on cation, the melting points of the imidazolium and ammonium based salts experienced a gradual decline. It suggested that the alkylation pattern on the cation is the main determinant of the melting point and that packing inefficiency is the intrinsic cause of a decreased melting point by increasing the alkyl chain. In addition, the ‘quasi-eutectic ILs’ with [closo-B12Cl12]2− dianion can be formed by introducing the ‘H-bond donor’ and in turn shielding the dianion's interactions with the cation. Just as for the compound [N1 1 1 16]2[B12Cl12], the resulted ‘quasi-eutectic ILs’ of [N1 1 1 16]2[B12Cl12] and [C4mim]Cl can melt at 31 °C with the quantity ratio of 1:3, and furthermore the presence of liquid crystal phase for [N1 1 1 16]2[B12Cl12] can be observed between the solid and liquid states. In terms of the influence of H-bonds on melting points for the salts synthesized, there existed weak H-bond interactions between the hydroxy protons with the chlorine atoms, which only have modest effects on their melting points.
Future work will be directed at optimizing the structure of the salts so as to further control their melting points and explore their catalytic applications.
Experimental section
General considerations
All metathetic reactions were carried out under standard laboratory conditions. Air and moisture sensitive solid reagents were manipulated using standard Schlenk techniques or in a glove box with an atmosphere of dry argon (H2O and O2 < 1 ppm). Elemental analyses was performed using a Vario EL cube microanalyser. Electron spray mass spectrometry was recorded on a Bruker 300 spectrophotometer in acetonitrile/water mixture (1:1 by volume). 1H NMR (600.17 MHz) spectra were recorded on a JEOL ECA-600 spectrometer and chemical shift values for 1H NMR spectra in DMSO-d6 solutions were referenced to SiMe4. For solid samples, FT-IR spectra were recorded from KBr pellets in a Nicolet FTIR-380 spectrophotometer. The melting points for the samples prepared were determined by differential scanning calorimetry (DSC), at 10 °C min−1 heating/cooling rates on 4–8 mg samples using a Mettler-Toledo DSC instrument, and further determined by heated-stage polarizing optical microscopy using an Olympus BX 51 microscope equipped with a Linkam THMS 600 hot stage. DSC apparatus was calibrated using indium as standards. Quoted values were taken from the first peak of the second heating cycle (i.e. all samples were first melted and re-solidified before taking data) in the DSC traces. Some samples showed more than one peak on the heating trace, indicative of polymorphic crystal phase formation which was confirmed by heated-stage polarizing optical microscopy. Thermogravimetric analyses (TGA) was performed for 5–10 mg samples in platinum pans, using a TGA Q5000 thermogravimetric analyzer under a dinitrogen atmosphere, at 10 °C min−1 heating rate. Density values were determined using an AccuPyc 1340 densimeter.
[Rmim]Cl, [Nn n n m]Br, [PP P P m]Br and [PyC4]Br were synthesized by previously reported methods29–33. Cs2[B12Cl12] was synthesized as described elsewhere10,34. Organic solvents were purchased as reagent grade and used as received. Isolated yields of the boron salts were greater than 90%. The synthesis of the Cs2[B12Cl12], elemental analyses (C,H,N,Cl), electron spray mass spectrometry data and 1H NMR spectroscopic data are presented in the ESI.‡ Melting points, decomposition temperatures and density values (where determined) are given in Table 1, while the solubility of the prepared salts in organic solvents and the relevant FT-IR spectra data are presented in Table 2 and Table 3, respectively.
General synthesis of [Rmim]2[B12Cl12]
[Rmim]Cl (0.4 mmol) was dissolved in acetonitrile (5 mL) and then added to a stirred solution of Cs2B12Cl12 (0.2 mmol) in acetonitrile (10 mL). The solution immediately became cloudy. After the mixture was stirred for 12 h, the white solid formed (mainly CsCl) was removed by filtration through celite and the filtrate was evaporated to dryness under vacuum. The resulting solid was washed with water (3 × 2.5 mL) and dried in vacuum for 12 h at 100 °C, yielding [Rmim]2[B12Cl12] (Yield: > 96%).General synthesis of [Nn n n m]2[B12Cl12]
[Nn n n m]Br (0.4 mmol) was dissolved in acetonitrile/water mixture (1:1 by volume, 10 mL) and then added to a stirred solution of Cs2[B12Cl12] (0.2 mmol) in acetonitrile (10 mL). After 12 h stirring, the filtrate was evaporated to dryness under vacuum. The remaining solid was redissolved in acetonitrile (15 mL) and filtered through celite. The filtrate was evaporated to dryness, then the remaining solid was washed with water (3 × 2.5 mL) and dried in vacuum for 12 h at 70 °C, yielding [Nn n n m]2[B12Cl12] (Yield: > 94%).
General synthesis of [Pp p p n]2[B12Cl12]
[PP P P n]Br (0.4 mmol) was dissolved in acetonitrile (5 mL) and then added to a stirred solution of Cs2B12Cl12 (0.2 mmol) in acetonitrile (10 mL). After the mixture was stirred for 12 h, the white solid formed (mainly CsCl/Br) was removed by filtration through celite and the filtrate was evaporated to dryness under vacuum. The resulting solid was washed with water (3 × 2.5 mL) and dried in vacuo for 12 h at 100 °C, yielding [PP P P n]2[B12Cl12] (Yield: > 92%).General synthesis of [PyCn]2[B12Cl12]
[PyCn]Br (0.4 mmol) was dissolved in acetonitrile (5 mL) and then added to a stirred solution of Cs2[B12Cl12] (0.2 mmol) in acetonitrile (10 mL). After the mixture was stirred for 12 h, the white solid formed (mainly CsCl/Br) was removed by filtration through celite and the filtrate was evaporated to dryness under vacuum. The resulting solid was washed with water (3 × 2.5 mL) and dried in vacuum for 12 h at 100 °C, yielding [PyCn]2[B12Cl12] (Yield: > 90%).Computational description
Ab initio calculations were performed with Gaussian0935 and the geometry optimizations were performed at B3LYP/3-21G* theoretical level. In the optimizing process, cations ion-pairs relaxed the symmetry constraint, while the [closo-B12Cl12]2− dianion was considered the structural symmetry. All of the optimized geometries were confirmed by frequency to ensure no imaginary frequencies. The initial configurations of the ion-pairs were mainly based on the charge distribution and electrostatic potential on the isolated cations and anion. Natural bond orbital (NBO) analysis of the conformers was carried out by version 3 in Gaussion09.36,37Acknowledgements
The work was supported by the National Basic Research Program of China (2009CB219901), the Key Program of National Natural Science of Foundation of China (grant No. 21036007) and National Natural Science of Foundation of China (grant No. 51004093). In addition, we thank Professor Suojiang Zhang (Institute of Process Engineering, Chinese Academy of Sciences) for helpful discussions.References
- (a) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS; (b) R. Giernoth, Angew. Chem., Int. Ed., 2010, 49, 2834–2839 CrossRef CAS; (c) T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS; (d) V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef; (e) T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS.
- J. Dupont, Acc. Chem. Res., 2011, 44, 1223–1231 CrossRef CAS.
- (a) C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS; (b) M. J. Nava and C. A. Reed, Inorg. Chem., 2010, 49, 4726–4728 CrossRef CAS; (c) S. Koerbe, P. J. Schreiber and J. Michl, Chem. Rev., 2006, 106, 5208–5249 CrossRef CAS; (d) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS; (e) C. A. Reed, K.-C. Kim, R. D. Bolskar and L. J. Mueller, Science, 2000, 289, 101–103 CrossRef CAS.
- (a) R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC; (b) P. Wasserscheid, R. V. Hal and A. Bösmann, Green Chem., 2002, 4, 400–404 RSC.
- A. S. Larsen, J. D. Holbrey, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 2000, 122, 7264–7272 CrossRef CAS.
- J. Dymon, R. Wibby, J. Kleingardner, J. M. Tanski, I. A. Guzei, J. D. Holbrey and A. S. Larsen, Dalton Trans., 2008, 2999–3006 RSC.
- E. Justus, K. Rischka, J. F. Wishart, K. Werner and D. Gabel, Chem.–Eur. J., 2008, 14, 1918–1923 CrossRef CAS.
- M. Nieuwenhuyzen, K. R. Seddon, F. Teixidor, A. V. Puga and C. Viñas, Inorg. Chem., 2009, 48, 889–901 CrossRef CAS.
- (a) C. Knapp and C. Schulz, Chem. Commun., 2009, 4991–4993 RSC; (b) M. Keßler, C. C. Knapp, V. Sagawe, H. Scherer and R. Uzun, Inorg. Chem., 2010, 49, 5223–5230 CrossRef; (c) J. Derendorf, M. Keßler, C. Knapp, M. Rühle and C. Schulz, Dalton Trans., 2010, 39, 8671–8678 RSC.
- V. Geis, K. Guttsche, C. Knapp, H. Scherer and R. Uzun, Dalton Trans., 2009, 2687–2694 RSC.
- A. Avelar, F. S. Tham and C. A. Reed, Angew. Chem., Int. Ed., 2009, 48, 3491–3493 CrossRef CAS.
- C. Bolli, J. Derendorf, M. Keßler, C. Knapp, C. Scherer, C. Schulz and J. Warneke, Angew. Chem., Int. Ed., 2010, 49, 3536–3538 CrossRef CAS.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- D. R. MacFarlane and K. R. Seddon, Aust. J. Chem., 2007, 60, 3–5 CrossRef CAS.
- J. Broeke, F. Winter, B.-J. Deelman and G. Koten, Org. Lett., 2002, 4, 3851–3854 CrossRef.
- G.-H. Tao, L. He, W.-S. Liu, L. Xu, W. Xiong, T. Wang and Y. Kou, Green Chem., 2006, 8, 639–646 RSC.
- J. D. Holbrey and K. R. Seddon, J. Chem. Soc., Dalton Trans., 1999, 2133 RSC.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2nd edn, 2007 Search PubMed.
- A. F. Kapustinskii, Q. Rev. Chem. Soc., 1956, 10, 283–294 RSC.
- A. Cieniecka-Roslonkiewicz, J. Pernak, J. Kubis-Feder, A. Ramani, A. J. Robertson and K. R. Seddon, Green Chem., 2005, 7, 855–862 RSC.
- R. E. Del Sesto, C. Corley, A. J. Robertson and J. S. Wilkes, J. Organomet. Chem., 2005, 690, 2536–2542 CrossRef CAS.
- K. Dong, S. Zhang, D. Wang and X. Yao, J. Phys. Chem. A, 2006, 110, 9775–9782 CrossRef CAS.
- K. Dong, Y. Song, X. Liu, W. Cheng, X. Yao and S. Zhang, J. Phys. Chem. B, 2012, 116, 1007–1017 CrossRef CAS.
- M. Juhasz, S. Hoffmann, E. Stoyanov, K.-C. Kim and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 5352–5355 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS.
- A. P. Abbott, G. Capper and S. Gray, ChemPhysChem, 2006, 7, 803–806 CrossRef CAS.
- H. Zhao, G. A. Baker and S. Holmes, Org. Biomol. Chem., 2011, 9, 1908–1916 CAS.
- (a) J. S. Wilkes, J. A. Levisky, R. A. Wilson and C. L. Hussey, Inorg. Chem., 1982, 21, 1263–1264 CrossRef CAS; (b) M. Deetlefs and K. R. Seddon, Green Chem., 2003, 5, 181–186 RSC; (c) P. J. Dyson, M. C. Grossel, N. Srinivasan, T. Vine, T. Welton, D. J. Williams, A. J. P. White and T. Zigras, J. Chem. Soc., Dalton Trans., 1997, 3465–3469 RSC.
- (a) A. G. Avent, P. A. Chaloner, M. P. Day, K. R. Seddon and T. Welton, J. Chem. Soc., Dalton Trans., 1994, 3405–3413 RSC; (b) Q. B. Liu, M. H. A. Janssen, F. V. Rantwijk and R. A. Sheldon, Green Chem., 2005, 7, 39–42 RSC.
- J. Z. Sun, D. R. MacFarlane and M. Forsyth, Ionics, 1997, 3, 356–362 CrossRef CAS.
- C. J. Bradaric, A. Downard, C. Kennedy, A. J. Robertson and Y. H. Zhou, Green Chem., 2003, 5, 143–152 RSC.
- M. C. Law, K. Y. Wong and T. H. Chan, Green Chem., 2002, 4, 328–330 RSC.
- W. X. Gu and O. V. Ozerov, Inorg. Chem., 2011, 50, 2726–2728 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.01), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
Footnotes |
| † Dedicated to Professor David Cole-Hamilton on the occasion of his retirement and for his outstanding contribution to transition metal catalysis. |
| ‡ Electronic Supplementary Information (ESI) available: Tables listing C, H, N, Cl microanalysis data, 1H NMR chemical shifts in DMSO-d6 for the newly synthesized salts, 1H NMR and FT-IR spectra of selected samples for this paper. See DOI: 10.1039/c2ra21700g |
| This journal is © The Royal Society of Chemistry 2012 |