Verification and implications of the dissolution–electrodeposition process during the electro-reduction of solid silica in molten CaCl2
Wei
Xiao
a,
Xin
Wang
a,
Huayi
Yin
a,
Hua
Zhu
a,
Xuhui
Mao
a and
Dihua
Wang
*ab
aSchool of Resource and Environmental Sciences, Wuhan University, Wuhan, 430072, P. R. China. E-mail: wangdh@whu.edu.cn; Fax: +86 27 68775799; Tel: +86 27 68775799
bState Key Laboratory for Corrosion and Protection, Institute of Metal Research, Chinese Academy of Sciences, Shenyang, 110016, P. R. China.
First published on 13th June 2012
Abstract
With the verification of the existence of the dissolution–electrodeposition mechanism during the electro-reduction of solid silica in molten CaCl2, the present study not only provides direct scientific support for the controllable electrolytic extraction of nanostructured silicon in molten salts but it also opens an avenue to a continuous silicon extraction process via the electro-deposition of dissolved silicates in molten CaCl2. In addition, the present study increases the general understanding of the versatile material extraction route via the electro-deoxidization process of solid oxides in molten salts, which also provokes reconsiderations on the electrochemistry of insulating compounds.
Introduction
Silicon is the building block for the solar, semiconductor, optic-fiber and metallurgical industries. In light of the drawbacks of the old-fashioned carbothermic Si production, in terms of the low energy efficiency and large carbon footprint, innovations in green and affordable silicon production is desired. Recently, electrolytic silicon extraction via direct electro-reduction of solid silica in molten chlorides emerged.1 A solid-to-solid electro-reduction mechanism was proposed to address the electro-reduction of solid silica in molten CaCl2, expressed as reaction 1,1–3 in which solid SiO2 is directly electro-reduced into solid silicon.3–6| SiO2(s) + 4e = Si(s) + O2− | (1) |
In fact, the electrochemistry of insulating solid compounds possesses crucial roles in sustaining many electrochemical processes with important applications ranging from energy storage (i.e., secondary batteries) to experimental technologies (e.g. Ag/AgCl reference electrodes).7 The basic electrode process of solid PbSO4 (solubility product, Ksp is 1.6 × 10−8), which mediates the reversible charge–discharge process of lead-acid batteries,8 involves a dissolution–electrodeposition mechanism as depicted in the consecutive reactions of reaction (2 and 3). While a solid-state ion transport mechanism (reaction 4) is believed to regulate the redox reaction of the solid Ni(OH)2 (Ksp = 2.0 × 10−15) electrode which is used as the positive electrode material in nickel–cadmium or nickel–metal hydride batteries.8
| PbSO4(s) = Pb2+ + SO42− | (2) |
| Pb2+ + 2e = Pb(s) | (3) |
| Ni(OH)2(s) + OH− = NiOOH(s) + e + H2O(l) | (4) |
Although the solubility of SiO2 in molten CaCl2 was too low to trigger a dissolution–electrodeposition mechanism, the situation might be changed with CaSiO3 as an intermediate product during the reduction process. Practically, the CaCl2 melt always contains some CaO due to the hydrolysis reaction of CaCl2·xH2O during the conventional CaCl2 preparation process, and inevitably, the in situ generated CaO during the reduction of SiO2 on the cathode. Therefore, reactions (5 and 6) might take place and generate soluble silicates in the cathodic region and/or inside the porous cathode since the Gibbs free energy change of reaction 5 at 850 °C is −139.76 kJ mol−1. This is understandable if one considers the basic nature of CaO and the acidic characteristic of silica. A recent reference reported that reaction 5 becomes significant in molten CaCl2 with the addition of 4.8 mol% CaO.9 The increased solubility of silica in molten CaCl2 with adscititious CaO also facilitates the electrorefining of silicon in CaCl2–CaO–SiO2 melts via the dissolution–electrodeposition mechanism.10
Furthermore, it was estimated in our previous work that the concentration of in situ generated oxygen ions at the electrochemical interfaces during the electro-reduction of solid silica in molten CaCl2 could be higher than 1 mol L−1.5 Such a high concentration is believed to be adequate to trigger the formation of silicates in the silica cathode. Therefore, the presence of CaSiO3 at the cathode is inevitable during the electro-reduction of solid silica in molten CaCl2, regardless of the addition of CaO in the melt. The involvement of CaSiO3 at the cathode during the reduction process was proposed and reported before.6,11 If the solubility of CaSiO3 in the melt is high enough, Si could also be produced through the electrodeposition of the dissolved silicate as described in reactions (5–8).
| SiO2(s) + CaO(l) = CaSiO3(s) | (5) |
| CaSiO3(s) = Ca2+ + SiO32− | (6) |
| SiO2(s) + O2− = SiO32− | (7) |
| SiO32− + 4e = Si(s) + 3O2− | (8) |
However, the above mechanism has remained far from being well-addressed in the past literature concerning the electrochemical reduction of solid silica in molten CaCl2,1–6,12 although the liquid process was recently speculated to be present during the course of the reaction.11,13,14 Herein, we provide verification of the dissolution–electrodeposition mechanism during the electro-reduction of solid silica in molten chlorides through systematical investigations on the solubility and electrochemical properties of silicates, which not only form the scientific base for a controllable electrolytic extraction of nanostructured silicon in molten salts, but also opens an avenue to a continuous silicon extraction process via electro-deposition of dissolved silicates in molten CaCl2. In addition, the present study increases the general understanding of the versatile material extraction route via the electro-deoxidization process of solid oxides in molten salts, which also provokes reconsiderations on the electrochemistry of insulating compounds.
Experimental section
All chemicals are of analytical grade and used as received. The electrochemical equipment and treatment of CaCl2-based molten salts for this work were similar to those described previously.5,6,15 A graphite crucible containing anhydrous molten CaCl2 electrolyte was sealed in a steel tube under an argon flow. Before undertaking electrochemical measurements, pre-electrolysis of the molten salt was carried out at 2.5 V between a graphite anode and a nickel sheet cathode for about 12 h to eliminate moisture and other impurities in the molten salt. The reference electrode was a silver wire dipped in a 45 mol% NaCl + 45 mol% KCl + 10 mol% AgCl mixture in a mullite cylinder (11 and 7 mm in outer and inner diameter, respectively). The equilibrium potential of the Ca/Ca2+ couple was employed as the internal reference. In all electrochemical tests, a graphite rod with a diameter of 20 mm was used as the counter electrode and a CHI 1140 electrochemical workstation equipped with software and a PC was used as the test instrument.Static dissolution experiments of silica in CaCl2–CaO melts
A quartz rod (about 10 g) with a diameter of 5 mm was immersed in 450 g of molten CaCl2 with the consecutive addition of differing amounts of CaO (0, 2, 3 and 5 mol%) at 850 °C for 5 h. The experiments were conducted in one bath and a new quartz rod was employed for each CaO concentration. The weight loss and change in size of the quartz rods were recorded. Note that all added CaO can be dissolved well in the melt since the saturation solubility of CaO in molten CaCl2 at 850 °C is 20 mol%.5Solubility of CaSiO3 in molten CaCl2 at 850 °C
Excessive CaSiO3 pellets (50 g) were added into 500 g of molten CaCl2. The mixture was kept still at 850 °C for 9 h to ensure thorough dissolution. Considering the higher density of CaSiO3 (2.9 g cm−3) compared to molten CaCl2 (2.0 g cm−3), insoluble silicates should accumulate in the bottom part of the melts, leaving an even solution in the upper. A fraction of the upper solution was then sampled from the melts and cooled to room temperature. The mass of the resulting solid mixture was quickly measured and then thoroughly rinsed in diluted HCl. CaSiO3 does not dissolve in the rinse solution. The remaining solid was then collected, dried and weighed and the solubility of CaSiO3 was then calculated based on the mass of CaSiO3 and the melt.Cyclic voltammetry (CV) measurements
CV measurements were performed to investigate the electrochemical properties of molten CaCl2 (434 g) at 850 °C with the addition of different amounts of SiO2 (0.1 mol), CaO (0.1 mol) or CaSiO3 (0.1, 0.2 and 0.5 mol). Before the CV tests, the mixture was kept still for 1 h. A molybdenum wire (1 mm in diameter) was immersed in the melts with a depth of 2.3 mm and was employed as the working electrode.Molten-salt electrolysis
Potentiostatic (three-electrode configuration) and constant-voltage (two-electrode configuration) electrolysis of solid silica (spheres with diameters ranging from 2 to 5 μm) were performed at 850 °C by using a nickel foam-sandwiched porous silica pellet (16 mm diameter, 2.3–3.3 mm thickness, 0.7–1.0 g mass, about 35% porosity, prepared by die-pressing silica powder and annealing at 900 °C for 2 h) assembled cathode. The graphite rod was used as the counter electrode.Electrodeposition experiments were performed in molten CaCl2 at 900 °C with 10 wt% CaSiO3 (CaSiO3 was supersaturated). A nickel foil was used as the deposition substrate.
The electrolytic samples were thoroughly rinsed in water and then collected after centrifugation and vacuum dried at 60 °C.
Electrolytic product characterization
The electrolytic powder was characterized by using powder X-ray diffraction (XRD, Shimadzu XRD-7000, Cu-Kα, λ = 1.5406 Å) and scanning electron microscopy (SEM, FEI sirion field-emission).Results and discussion
In situ dissolution–electrodeposition of SiO2 in molten CaCl2
The static dissolution of silica in molten CaCl2 was executed by measuring the weight loss of a quartz rod in the melt. The results are shown in Fig. 1a. After immersion in molten CaCl2, the quartz rod shows minor weight loss (0.17 g), indicating negligible solubility of silica in molten CaCl2 (1 #). A steeply increased weight loss (1.90 g) was observed when the quartz was immersed in the melt containing 2 mol% CaO (2 #), resulting in the decreased size of the quartz rod after static dissolution (Fig. 1a). Therefore, the mass of dissolved species is 2.07 g with the presence of 2 mol% CaO. Weight loss for new quartz rods immersed in the melts with further consecutive addition of 3 (3 #) and 5 (4 #) mol% CaO was measured as 1.1 and 1.41 g, meaning 3.17 and 4.58 g of silica dissolved in the melts containing 5 and 10 mol% CaO, respectively. The solubility of quartz increases with CaO concentration, indicating that CaO is relevant to the dissolution of silica. It demonstrates that CaO can function as a promoter to facilitate the dissolution of silica via reaction 5, allowing the formation of silicates. It was also found recently that the solubility of Fe2O3 in molten CaCl2 can be improved by adding CaO.15 The generated silicates then dissolve into the electrolyte (reaction 6); otherwise, CaSiO3 would cover the surface of the quartz rods preventing further dissolution.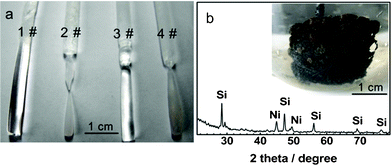 | ||
| Fig. 1 (a) Images of quartz rods after being immersed in molten CaCl2 (1 #) and CaCl2–CaO (2–4 #); (b) the XRD pattern for the brown powder collected from the outer surface of the nickel foam. The inset shows an image of the Ni-sandwiched porous silica pellet cathode after electrolysis at 0.4 V (vs. Ca/Ca2+) at 850 °C in CaCl2 with 2 mol% CaO. | ||
The dissolution of SiO2 with the assistance of CaO was also confirmed by the cyclic voltammetry (CV) measurements. As shown in Fig. 2a, the cyclic voltammogram of the Mo electrode in molten CaCl2 (dotted curve) only exhibited cathodic peak C1 and its anodic counterpart A1 near the negative scan limit, ascribed to the formation and re-dissolution of Ca. With the addition of 0.1 mol silica, the cyclic voltammogram (dashed curve) remains unchanged at the potential range positive than that of Ca formation (onset of C1), indicating a low SiO2 concentration in the melt. With the consecutive addition of 0.1 mol CaO, another two pairs of redox peaks (C2/A2 and C3/A3) in the corresponding cyclic voltammogram (solid curve) were observed, which are ascribed to the redox reactions of the dissolved silicates.
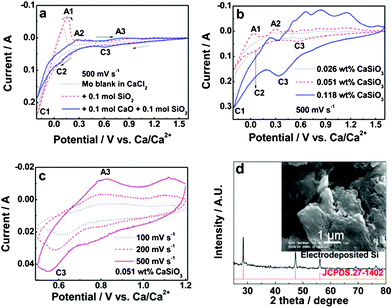 | ||
| Fig. 2 (a) Cyclic voltammograms of the Mo electrode in molten CaCl2 (434 g) (dotted), CaCl2 + 0.1 mol SiO2 (dashed) and CaCl2 + 0.1 mol SiO2 + 0.1 mol CaO (solid) at 850 °C; (b) Cyclic voltammograms of the Mo electrode in molten CaCl2 at 850 °C with the addition of different amounts of CaSiO3; (c) Cyclic voltammograms at different scan rates of the Mo electrode in molten CaCl2 at 850 °C with the presence of 0.051 wt% CaSiO3; (d) XRD pattern and SEM image of the powder electrodeposited on the Ni substrate via potentiostatic electrolysis at 0.6 V for 10 h in the CaSiO3-saturated molten CaCl2. | ||
The dissolution–electrodeposition of SiO2 was further verified by a potentiostatic electrolysis experiment. Using a silica pellet (wrapped in nickel foam) cathode, potentiostatic electrolysis was carried out in molten CaCl2 containing ∼2 mol% CaO. The inset of Fig. 1b shows the picture of the cathode after 12 h of electrolysis. The observed brown powder on the outer surface of the nickel foam which had no direct contact with the silica feedstock tells a different story from the solid-to-solid electro-reduction mechanism. This phenomenon was also found to exist in the same experiment performed in pure CaCl2 melt (without the addition of CaO). As per the solid-state electro-reduction mechanism, the cathode would retain its original integrity within the nickel foam. The XRD pattern shown in Fig. 1b suggests that the obtained brown powder contains crystalline silicon. Such anomalous deposited silicon provides evidence for the existence of the dissolution–electrodeposition mechanism. Upon cathodic polarizations, the dissolved silicates tend to be electro-reduced to solid silicon on the current collector, incurring the formation of silicon on the outer surface of the nickel foams, as depicted in the inset of Fig. 1b (reaction 8).
Implications on the electrochemistry of insulating solid compounds
The solubility of CaSiO3 in molten CaCl2 was measured as 1.56 wt% at 850 °C in the present work. Since the process of direct electrochemical reduction of solid oxides to their relative metals was reported and patented in 1998 (the so-called FFC-Cambridge process),16 the solid-to-solid mechanism has been widely accepted. The present study has clearly shown that the dissolution–electrodeposition mechanism is playing an important role. Due to the favourable reaction between the acidic silica and basic CaO, and the relatively high solubility of CaSiO3 in molten CaCl2 (1.56 wt%, ∼750 times that of PbSO4 in an H2SO4 aqueous solution), the electrodeposition of Si from the liquid phase should be significant. It is well recognized that the reduction of PbSO4 in H2SO4 aqueous solution follows the dissolution–electrodeposition mechanism, although the obvious deposition phenomenon shown in Fig. 1b was never reported or observed before for the negative electrode of a lead-acid battery. Naturally it then follows that the dissolution–electrodeposition mechanism should contribute a lot during the electro-reduction of solid silica in molten CaCl2, providing the co-existence of the solid-state reaction mechanism (as illustrated in Scheme 1). The new mechanism can provide a good explanation for Si nanowire formation by the electrolysis of solid silica in molten salt and stimulate a novel process for silicon extraction through electrodeposition, which are discussed in the following sections.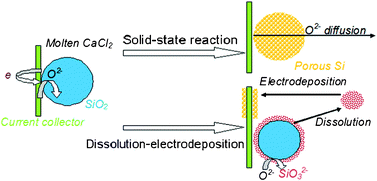 | ||
| Scheme 1 A schematic representation of the electro-reduction process of solid silica in molten CaCl2 governed by both the solid-state reaction and dissolution–electrodeposition mechanisms. | ||
Implications on controllable electrolytic extraction of nanostructured silicon
It is well-known that nanoarchitectures offer unprecedented opportunities to arm nanostructured silicon with unusual and enhanced functionalities compared to its bulk counterparts. A case in point lies in the reported enhanced lithium storage capability of silicon nanowires which are capable of accommodating large local strains, and providing short lithium diffusion paths and good electrical connections.17 However, its molten-salt synthesis may be challenging since the presented high temperature facilitates the overgrowth and violent aggregation of nuclei or building blocks. Such a situation was verified by the formation of over-aggregated even sintered or dense solid metallic products such as sponge-like Ti,16 Fe18 and consolidated Zr alloys19 generated from molten-salt electrolysis of corresponding solid oxides. Metal-based materials contain large amounts of delocalized electrons, which can drive thermal-induced strain. The thermal-induced strain governed by the mobility of the delocalized electrons tends to facilitate the agglomeration of the generated metallic species. For those with relatively low melting points such as Ti (m.p. 1668 °C), Zr (m.p. 1855 °C) and Fe (m.p. 1538 °C), the presented highly mobile delocalized electrons facilitate the coalescence of metal species, allowing the formation of interconnected even sintered or dense solid products. For refractory metals such as Ta (m.p. 3017 °C) and Nb (m.p. 2477 °C), the mobility of contained delocalized electrons is highly restrained due to their high melting points, resulting in the significantly retarded agglomeration of electro-generated metal species and the formation of less-interconnected nanopowders.20When it comes to electrolytic silicon, Si is merely a semi-metal, if not a non-metal. Without sufficient delocalized electrons to prompt thermal-induced strain, the aggregation of generated Si seeds is not accelerated, facilitating the formation of ultrafine products. Such an ultra-refining mechanism is verified in the present study. As exhibited in Fig. 3, the precursory amorphous micro-spherical silica with diameters ranging from 2 to 5 μm was transformed into pure and crystalline silicon nanowires or silicon nanoparticles, upon constant-voltage electrolysis at 850 °C in molten CaCl2 for 10 h. The sample prepared via electrolysis at 2.2 V consists of high-aspect-ratio Si nanowires with diverse diameters ranging from 50 nm to 100 nm. While the sample formed at 2.4 V is composed of interconnected Si nanoparticles with diameters less than 80 nm. The above argument on retarded thermal-induced strain in less-metallic silicon is rational to address the formation of Si nanoparticles, but yet not adequate to interpret the generation of high-aspect-ratio Si nanowires. The formation of silicon nanowires through molten-salt electrolysis of solid silica was recently reported and the utilization of nano-sized silica precursors was claimed to be a prerequisite on the generation of silicon nanowires.13 Our present study demonstrates that molten-salt electrolysis is a controllable electrolytic extraction method of nanostructured silicon, which is powerful enough to ultrarefine micro-sized silica.
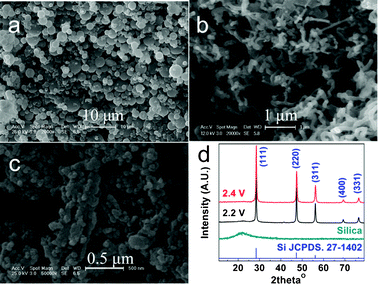 | ||
| Fig. 3 SEM images (a–c) and XRD patterns (d) of micro-sized silica spheres before (a) and after constant-voltage electrolysis at 850 °C in molten CaCl2 at 2.2 V (b) and 2.4 V (c) for 10 h. | ||
Based on the solid-to-solid electro-reduction mechanism, the electro-deoxidation process leaves voids in the electrolytic samples due to the decrease in the molar volume from SiO2 (22.6 cm3 mol−1) to Si (12.1 cm3 mol−1). The observed ultrarefining phenomenon (Fig. 3) is two orders of magnitude higher than that of the inherent volume shrinkage from silica to silicon. Such an anomalous shrinkage therefore cannot be addressed by the solid-to-solid electroreduction mechanism, which however can be well-explained by the dissolution–electrodeposition mechanism.
During molten-salt electrolysis of solid silica, the sluggish kinetics of O2− diffusion and its high concentration at electrochemical interfaces allow a fast combination reaction between solid silica and in situ ionized O2−, causing the formation of silicates at the cathode (reaction 5). Both dissolved silicates and solid precursors (including solid silicates and solid silica) can be electro-reduced to silicon upon cathodic electrolysis. It should be noted that the proportion of the influence from the two coincident mechanisms should depend on the concentration of O2− ions in the vicinity of the reaction zone (reaction 7). The influence of dissolution–electrodeposition will be enhanced with the increased concentration of O2−. Many parameters can influence the O2− concentration. For example, the electrolysis potential can influence the concentration of O2− at the electrochemical interface. Upon electrolysis at higher overpotentials, the concentration of O2− at the electrochemical interface is higher.5 Therefore, a more significant influence of the dissolution–deposition mechanism occurs during the electrolysis at more negative potentials. The reported correlation between the electrode potential and the activity of O2− in the melts is a good reference for assessing contributions from the two mechanisms.11 The electrodeposition of dissolved silicates results in the formation of solid silicon, which can function as nucleation centers for the growth of silicon generated from solid feedstock and might lead to the growth of nano-silicon.13,21
Grain growth follows the nucleation process, during which competition between nucleation and growth also exists. The homogeneous nucleation process is relevant to overpotentials, indicating the possibilities of engineering the morphologies of products by tuning the overpotential. With a relatively low cathodic overpotential (for example, constant-voltage electrolysis at 2.2 V), the generation rate of nuclei is reasonably low, allowing the anisotropic growth and formation of Si nanowires (Fig. 3b). Upon electrolysis at higher cathodic overpotentials (more negative potentials), i.e. 2.4 V, the generation rate of Si nuclei is accelerated, facilitating the formation of Si nanoparticles (Fig. 3c). In addition, the growth process is speculated to be governed by both Ostwald-ripening and oriented-attachment mechanisms.22 The presence of high-aspect-ratio nanowires (Fig. 3b) indicates the existence of the classical Ostwald-ripening process, in which bigger crystals grow at the expense of smaller ones. The existence of the coincident oriented attachment process is evidenced from the generation of interconnected nanoparticles (Fig. 3c), in which smaller particles with matched orientations directly coalesce into larger ones. Despite its high-temperature nature, the molten-salt featuring strong polarity and high surface tension is capable of retarding the agglomeration of the tiny particles.23 The above justifications suggest that molten-salt electrolysis is a generic and self-templated preparation of nanostructured materials.
Continuous silicon extraction via the electrodeposition of dissolved silicates in molten salts
The measured solubility of CaSiO3 in molten CaCl2 (1.56 wt% at 850 °C) is reasonably high enough to sustain the continuous electrodeposition production of silicon if the formation of silicon is not interfered with by the generation of Ca–Si intermetallics.The cyclic voltammograms of the Mo electrode in molten CaCl2 at 850 °C with different amounts of dissolved CaSiO3 are shown in Fig. 2b. Peak C3 indicating the formation of electrodeposited Si appears in the cyclic voltammograms. The onset potential of C3 is 0.75 V, in agreement with Fig. 2a. Peaks C2 and A2 related to Si–Ca are shown in the cyclic voltammograms with small silicate concentrations (dashed and dotted curves) and overlap other peaks with a high silicate concentration (solid curve). Peak C3 intensifies in the cyclic voltammograms recorded at faster scan rates, indicating the electrodeposition mechanism. The cyclic voltammograms collected at different scan rates (Fig. 2c) exhibit increased currents with scan rates, which is a typical characteristic of electrochemistry for dissolved species.7 While the cyclic voltammograms of the direct electro-reduction of solid silica display decreased cathodic currents with increased scan rate, which is an intrinsic part of the nature of the solid-state electro-reduction mechanism (second-type electrode process).6
Potentiostatic electro-deposition on the Ni substrate at 0.6 or 0.4 V (vs. Ca/Ca2+) for 10 h was then performed in the CaSiO3-saturated molten CaCl2 at 850 °C. The corresponding deposits show the morphology of the agglomerated granules (inset of Fig. 2d). The XRD pattern of the sample can be well indexed to cubic silicon (JSPDS. 27-1402), indicating the formation of high-purity crystalline silicon. It was found that deposits with thicknesses of ca. 3 mm can be produced upon electrolysis at 0.4 V for 10 h (Fig. 4, right), which was coherently connected with the Ni substrate. Upon electrolysis at 0.6 V for 10 h, only a thin-layer deposit can be generated (Fig. 4, left), which promises the fabrication of silicon film through the proposed method. This preliminary study promises the electro-deposition of dissolved silicates as a continuous silicon production method, however, the detailed information on the energy efficiency, productivity and property of the deposits deserves further investigation.
 | ||
| Fig. 4 Typical photos of the deposits on the Ni substrate prepared via potentiostatic electrolysis at 0.4 V (vs. Ca/Ca2+) (left) or 0.6 V (right) for 10 h in the CaSiO3-saturated molten CaCl2 at 850 °C. The dashed line in the right-hand image indicates the liquid level of the melt. | ||
Conclusions
The existence of the dissolution–electrodeposition mechanism during the electro-reduction of solid silica in molten CaCl2 was verified. It was found that the in situ ionized oxygen can combine with solid silica, bringing about the formation of calcium silicates. Solubility tests showed that CaSiO3 possesses a solubility of 1.56 wt% in molten CaCl2.On the one hand, the dissolved silicates can be electro-reduced into silicon which is capable of functioning as a nucleation centre for the growth of nanostructured silicon. Fewer nuclei are generated upon the electrolysis of porous silica at low overpotentials, facilitating the formation of high-aspect-ratio silicon nanowires. While silicon nanoparticles are produced upon the electrolysis of porous silica at high overpotentials, due to the presence of more nuclei.
On the other hand, electrochemical tests showed that the electrodeposition of silicon from dissolved silicates started at a potential of 0.75 V (vs. Ca/Ca2+), which is far more positive than the formation potential of Si–Ca intermetallics. This result provokes a continuous silicon production process via the electrodeposition of CaSiO3 in molten CaCl2, which was verified in the present study.
Acknowledgements
Financial support from NSFC (20873093, 50934001), MOE (NCET-08-0416), MOST (2009DFA62190) and Wuhan University (121075) is acknowledged.References
- (a) T. Nohira, K. Yasuda and Y. Ito, Nat. Mater., 2003, 2, 397–401 CrossRef CAS; (b) X. B. Jin, P. Gao, D. H. Wang, X. H. Hu and G. Z. Chen, Angew. Chem., Int. Ed., 2004, 43, 733–736 CrossRef CAS; (c) K. Yasuda, T. Nohira, K. Amezawa, Y. H. Ogata and Y. Ito, J. Electrochem. Soc., 2005, 152, D69–D74 CrossRef CAS; (d) K. Yasuda, T. Nohira, Y. H. Ogata and Y. Ito, J. Electrochem. Soc., 2005, 152, D208–D212 CrossRef.
- (a) P. C. Pistorins and D. J. Fray, J. S. Afr. Inst. Min. Metall., 2006, 106, 31–41 Search PubMed; (b) E. Ergul, I. Karakaya and M. Erdogan, J. Alloys Compd., 2011, 509, 899–903 CrossRef; (c) E. Juzeliunas, A. Cox and D. J. Fray, Electrochem. Commun., 2010, 12, 1270–1274 CrossRef CAS.
- W. Xiao, X. B. Jin, Y. Deng, D. H. Wang and G. Z. Chen, Chem.–Eur. J., 2007, 13, 604–612 CrossRef CAS.
- (a) Y. Deng, D. H. Wang, W. Xiao, X. B. Jin, X. H. Hu and G. Z. Chen, J. Phys. Chem. B, 2005, 109, 14043–14051 CrossRef CAS; (b) D. H. Wang, X. B. Jin and G. Z. Chen, Annu. Rep. Prog. Chem., Sect. C, 2008, 104, 189–234 RSC.
- W. Xiao, X. B. Jin, Y. Deng, D. H. Wang, X. H. Hu and G. Z. Chen, ChemPhysChem, 2006, 7, 1750–1758 CrossRef CAS.
- W. Xiao, X. B. Jin, Y. Deng, D. H. Wang and G. Z. Chen, J. Electroanal. Chem., 2010, 639, 130–140 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, 2001 Search PubMed.
- D. Linden and T. B. Reddy, Handbook of Batteries, McGraw-Hill, 3rd edn, 2002 Search PubMed.
- O. E. Kongstein, C. Wollan, S. Sultana and G. M. Haarberg, ECS Trans., 2007, 3, 357–361 CrossRef CAS.
- J. Cai, X. T. Luo, G. M. Haarberg, O. E. Kongstein and S. L. Wang, J. Electrochem. Soc., 2012, 159, D155–D158 CrossRef CAS.
- K. Yasuda, T. Nohira, R. Hagiwara and Y. H. Ogata, J. Electrochem. Soc., 2007, 154, E95–E101 CrossRef CAS.
- S. K. Cho, F. R. F. Fan and A. J. Bard, Electrochim. Acta, 2012, 65, 57–63 CrossRef CAS.
- J. Y. Yang, S. G. Lu, S. R. Kan, X. J. Zhang and J. Du, Chem. Commun., 2009, 3273–3275 RSC.
- (a) J. Y. Yang, S. G. Lu, H. Y. Ding, X. J. Zhang and S. R. Kan, Chin. J. Inorg. Chem., 2010, 26, 1837–1843 CAS; (b) J. Y. Yang, S. G. Lu, S. R. Kan, X. J. Zhang and H. Y. Ding, Chin. J. Inorg. Chem., 2009, 25, 756–760 CAS.
- H. Y. Yin, L. L. Gao, H. Zhu, X. H. Mao, F. X. Gan and D. H. Wang, Electrochim. Acta, 2011, 56, 3296–3302 CrossRef CAS.
- (a) G. Z. Chen, D. J. Fray and T. W. Farthing, Nature, 2000, 407, 361–364 CrossRef CAS; (b) D. J. Fray, T. W. Farthing and G. Z. Chen, PCT patent, WO9964638, priority data, 5th June 1998.
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS.
- H. Y. Yin, D. Y. Tang, H. Zhu, Y. Zhang and D. H. Wang, Electrochem. Commun., 2011, 13, 1521–1524 CrossRef CAS.
- J. J. Peng, K. Jiang, W. Xiao, D. H. Wang, X. B. Jin and G. Z. Chen, Chem. Mater., 2008, 20, 7274–7280 CrossRef CAS.
- (a) T. Wu, X. B. Jin, W. Xiao, X. H. Hu, D. H. Wang and G. Z. Chen, Chem. Mater., 2007, 19, 153–160 CrossRef CAS; (b) T. Wu, W. Xiao, X. B. Jin, C. Liu, D. H. Wang and G. Z. Chen, Phys. Chem. Chem. Phys., 2008, 10, 1809–1818 RSC.
- Y. Nishimura, T. Nohira, K. Kobayashi and R. Hagiwara, J. Electrochem. Soc., 2011, 158, E55–E59 CrossRef CAS.
- H. C. Zeng, J. Mater. Chem., 2006, 16, 649–662 RSC.
- Y. Tian, D. R. Chen, X. L. Jiao and Y. Z. Duan, Chem. Commun., 2007, 2072–2074 RSC.
| This journal is © The Royal Society of Chemistry 2012 |