Waste reduction in amide synthesis by a continuous method based on recycling of the reaction mixture†
Kishore
Thalluri
,
Krishna Chaitanya
Nadimpally
,
Ashim
Paul
and
Bhubaneswar
Mandal
*
Department of Chemistry, Indian Institute of technology Guwahati, Guwahati, Assam, India. E-mail: bmandal@iitg.ernet.in; Tel: 0361-2582319
First published on 21st May 2012
Abstract
In this article, a new one pot synthesis of amides is demonstrated. Potential of the recovery and reuse strategy for waste reduction is examined. Finally, a new strategy is proposed where the whole reaction mixture containing the catalyst, the solvent (auxiliary) and the unreacted starting material is recovered by precipitating the product using ethanol and reused for the next cycle in a continuous operation protocol.
Introduction
The first priority in green chemistry is to save the environment by reducing chemical waste generation. To achieve that in a systematic way, concepts such as, atom economy,1 step economy,2 pot economy,3 the twelve principles of green chemistry,4etc. have been developed and are in practice today. To evaluate how environmentally friendly a particular synthetic route is parameters such as E-factor5 and EQ are considered. The term, E-factor, implies the waste generated per kg of product in a chemical industry. Everything produced in a (multistep) process except the desired product is considered as waste including salts generated in the subsequent neutralization step. The challenge for the chemists today is to minimize the E-factor of a multistep chemical synthesis.To minimize the E-factor of a process, it is recommended in the Resource Conservation and Recovery Act (RCRA),6 which forms the legislative basis for the U.S. Environmental Protection Agency (EPA) regulations on the management of hazardous waste, that everything which adds to the waste should be recovered and reused. The issue of recovery and reuse is addressed separately for solvents and catalysts to date. To bypass the solvent issue, chemists are developing solvent free synthesis protocols,7 fluorous phase chemistry,8 reactions in water,9 ionic liquids10 and supercritical carbon dioxide.11 On the other hand, to reduce the E-factor of a chemical process, the usage of the catalytic amount of a reagent is recommended in stead of the usage of the stoichiometric amount of the same. As the recovery of homogeneous catalysts is cumbersome in general, scientists moved to the use of the heterogeneous catalysts. Moreover, biphasic homogeneous catalysis,12 supported liquid phase catalysis13 and thermo regulated biphasic catalysis14 have also been developed to improve the recyclability of the catalyst. Herein, we describe our effort to design a continuous operation protocol where a major portion of the product is separated out by precipitation using a specific green solvent and the homogeneous reaction mixture as a whole is reused in the same pool, thus, resulting in drastic waste reduction. The idea is well discussed. However, to the best of our knowledge, no such demonstration of recovery of the reaction mixture as a whole for amide synthesis has been described in the literature. It is also demonstrated that the recovery of the reagents and reuse in a batch process does not reduce the E-factor.
Results and discussion
The amide synthesis reaction is chosen as the target for the current discussion because the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable recently recognized amide synthesis as one of the most utilized but problematic syntheses in pharmaceutical industry and indicated it as a high priority research area.15Direct amide bond formation from free acid and amine is a high energy demanding method. Therefore, various strategies have been developed to reduce the energy barrier of the reaction by either making the acid function more reactive or by increasing the nucleophilicity at the amine end. Generally, the acid function is derivatized into more reactive functional group like anhydride or acid halide and that prohibits the method to be atom economic. All these methods suffer from many other drawbacks as well, e.g. azeotropic removal of water in the former case and the use of toxic reagents like thionyl chloride in the latter. Scientists employed various alternatives like microwave assisted synthesis directly from acids to solve these problems. Recently, Sheppard reported the preparation of amides using tris(2,2,2-trifluoroethyl) borate esters under microwave conditions.16 Although, the usage of molecular sieves,17 Fe3+–K–10 Montmorillonite Clay,18 K60 silica,19 silica gel,20 tetrabutylammonium salts,21para-toluene sulphonyl chloride,22 sulfated tungstate as a heterogeneous catalyst,23 phosphorus trichloride,24 from carboxylic acids and isocyanates,25 benzotriazole,26 benzene boronic acid and derivatives,27 boric acid,28 isobutyl chloroformates,29 diethyl chlorophosphate in pyridine,30 BDDI (1-tert-butoxy-2-tert-butoxycarbonyl-1,2-dihydroisoquinoline)31 made the synthesis simpler,32 still there is a demand for a methodology that is cost effective and environmentally friendly.
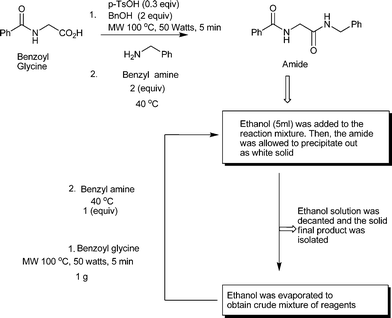
Our idea of reagent economized and waste minimized amide synthesis involves mild activation of the carboxylic acid group at the first step to a benzyl ester under microwave irradiation in the presence of catalytic amount of para-toluene sulphonic acid (pTsOH, 0.3 equiv.), which on treatment with the nucleophile, e.g. primary or secondary amines at 40 °C, results in an amide in a single pot (Scheme 1). At the beginning, preparation of ester was carried out using a microwave reactor; isolated yield of the ester was 77%. Next, the whole amount of the purified ester was incubated at 40 °C for 2.5 h in presence of 3 equiv. of benzyl amine, which resulted in the amide (isolated yield 70%). The procedure for conversion of the ester to amide was similar to that followed in our earlier report.7 Therefore, the final yield after two steps is about 53%. To improve the final yield we wanted to use the concept of pot economy. To our delight, we noticed that when benzoyl glycine was treated with pTsOH and benzyl alcohol in a microwave reactor for 1 min followed by addition of 3 equiv. of benzyl amine in the same pot, heating at 40 °C for 3 h resulted in a higher yield of the corresponding amide (isolated yield 75% after two steps, Scheme 1). After completion of the reaction, the reaction mixture was passed through a silica gel column. The percentage yields of the isolated amide, recovered unreacted amine and benzyl alcohol obtained in this way are reported in the respective tables.
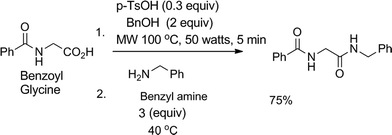 | ||
| Scheme 1 Model reaction with optimised conditions. | ||
After proper optimization of the reaction conditions, we started to explore the feasibility of the method for different N-protected amino acids with varying side chains, i.e. from small and simple glycine to quite bulky valine (Table 1). It was observed that N-protected glycine reacted faster than other N-protected amino acids. The reaction was not complete even after 36 h in the case of valine. However, unreacted benzyl ester of Cbz-Val-OH was recovered.
| Entry | Amino acid | Amine | Time | Isolated yield (%) | Amine rec/exp (%)b | Alcohol rec/exp (%)b |
|---|---|---|---|---|---|---|
| a Where, Bz = benzoyl, Cbz = N-benzyloxycarbonyl. b Amount recovered/expected (w/w, %, expected amine means the excess of the same used and expected benzyl alcohol means the whole amount of added benzyl alcohol, which in principle should get regenerated completely). | ||||||
| 1. | Bza–Gly–OH | n-Butyl | 20 min | 67 | 29 | 37 |
| 2. | Bza–Gly–OH | β-Hydroxyethyl | 20 min | 60 | 19 | 33 |
| 3. | Bza–Gly–OH | Benzyl | 3 h | 75 | 30 | 33 |
| 4. | Bza–Gly–OH | Cyclohexyl | 5 h | 64 | 27 | 30 |
| 5. | Cbza–Gly–OH | Benzyl | 2.5 h | 68 | 26 | 26 |
| 6. | Cbza–Gly–OH | Cyclohexyl | 5 h | 66 | 26 | 27 |
| 7. | Bza–Ala–OH | Benzyl | 10 h | 66 | 28 | 28 |
| 8. | Cbza–Val–OH | n-Butyl | 48 h | 30 | 27 | 26 |
| 9. | Cbza–Val–OH | Benzyl | 36 h | 36 | 24 | 27 |
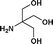
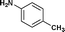
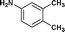
We further extended our work to aromatic and aliphatic amines, which generally exist in the solid state (Table 2). There are reports that describe aminolysis using aniline derivatives but all of them use harsh conditions. However, increasing the reaction temperature to 90 °C was necessary in the case of aniline derivatives to obtain good yield, whereas, 70 °C was sufficient in the case of TRIS in our method. Although the yield of the reaction was good, time required for completion was long for solid amines.
| Entry | Acid | Amine | Time (h) | Isolated yield (%) | Amine rec/exp (%)a | Alcohol rec/exp (%)a |
|---|---|---|---|---|---|---|
| a Amount recovered/expected (w/w). b Reaction was carried out at 70 °C in an oil bath. c Reaction was carried out at 90 °C in an oil bath. d Where, Bz = benzoyl, Cbz = N-benzyloxycarbonyl. | ||||||
| 10. | Bzd–Gly–OH |
|
16b | 68 | 36 | 38 |
| 11. | Bzd–Gly–OH |
|
24c | 65 | 42 | 31 |
| 12. | Bzd–Gly–OH |
|
24c | 67 | 44 | 33 |
| 13. | Bzd–Gly–OH |
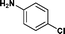
|
24c | 67 | 41 | 34 |
| 14. | Bzd–Ala–OH |
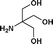
|
21b | 68 | 33 | 34 |
| 15. | PhCH2COOH |
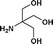
|
19b | 67 | 57 | 61 |
Next, we wanted to apply our methodology to long chain and simple carboxylic acids other than amino acids. All the tried reactions went smoothly (Table 3) resulting in very good yields, except the case of aniline (entry 19), where, 7 h heating at 100 °C in an oil bath was necessary. The isolated yield was also not very good for this reaction. However, the unreacted phenyl acetic acid (35 mg) and the corresponding ester (139 mg) intermediate were recovered. In the case of benzoic acid (entry 20) the reaction also did not progress very well (yield 31%) but, unreacted acid (57 mg) and ester (129 mg) were recovered. In both cases (entries 19 and 20) no side product was observed. In the case of long chain fatty acids (entry 21 and 22), good yields were obtained. However, heating for longer times at 40 °C was necessary. 200 mg of the starting carboxylic acid was used for each entry of Table 1, 2 and 3.
| Entry | Acid | Amine | Time (h) | Isolated yield (%)c | Amine rec/exp (%)a | Alcohol rec/exp (%)a | p-TsOH rec/exp (%) | Unreacted acid recovered (mg) | Ester recovered (mg) |
|---|---|---|---|---|---|---|---|---|---|
| a Amount recovered/expected (w/w, %). b Reaction was carried out at 100 °C. c Yields were calculated with respect to the initial carboxylic acid used (200 mg for each entry). | |||||||||
| 16. | PhCH2COOH | Benzyl | 5 | 84 | 48 | 51 | 52 | 36 | 0 |
| 17. | PhCH2COOH | Cyclohexyl | 5 | 48 | 58 | 54 | 60 | 37 | 125 |
| 18. | PhCH2COOH | n-Butyl | 3 | 93 | 49 | 69 | 60 | 36 | 0 |
| 19. | PhCH2COOH | Aniline | 7b | 26 | 53 | 51 | 58 | 35 | 139 |
| 20. | PhCOOH | Benzyl | 8 | 31 | 49 | 45 | 51 | 57 | 129 |
| 21. | C11H23COOH | n-Butyl | 12 | 87 | 47 | 58 | 38 | 0 | 0 |
| 22. | C11H23COOH | Benzyl | 24 | 89 | 49 | 62 | 26 | 0 | 0 |
All the amides were characterized using 1H NMR, 13C NMR, IR and HRMS. (Data provided in the experimental section. Original spectra provided in the ESI.†) Products obtained from entry 3 (Table 1) and 17 (Table 3) were also analyzed using X-ray crystallography. ORTEP representations with ellipsoids at 50% probability are presented in Fig. 1 and 2 and the crystallographic data in detail are furnished in the ESI.†
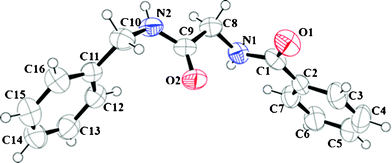 | ||
| Fig. 1 ORTEP molecular diagram with ellipsoid at 50% probability of N-2-benzyl amino-2-oxoethyl benzamide (3) CCDC # 837222. | ||
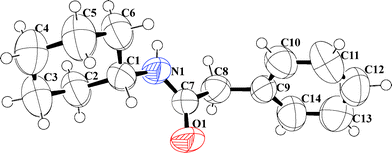 | ||
| Fig. 2 ORTEP molecular diagram with ellipsoid at 50% probability of N-cyclohexyl-2-phenyl-acetamide (17) CCDC # 837072. | ||
Our initial objective was to minimize waste generation by recovering the unreacted reagents and byproducts and reusing them in the next batch. For easy recovery of the product and the other reagents along with the unreacted starting material, the whole reaction mixture was loaded over a silica gel column and specific mixture of ethyl acetate (EtOAc) and hexane was passed through it. Benzyl alcohol was eluted with EtOAc–hexane mixture in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, whereas the product came out with EtOAc–hexane mixture of the ratio 4:6. Then 1% acetic acid (AcOH) was mixed with the eluant as additive and the unreacted benzoyl glycine was eluted with EtOAc–hexane mixture (1:1 with 1% AcOH), while the catalyst, p-TsOH came out of the same column with a mixture of EtOAc and 1% AcOH. During the process of purification the unreacted amine was sticking inside the column which was eluted with a 1:9 mixture of methanol and ethyl acetate. Finally, the quality of the recovered reagents, starting material and solvent was verified by NMR spectra (attached in the ESI†). It was noted that all the recovered materials were of comparable quality to the commercial material and therefore, used in the next batch.
:9, whereas the product came out with EtOAc–hexane mixture of the ratio 4:6. Then 1% acetic acid (AcOH) was mixed with the eluant as additive and the unreacted benzoyl glycine was eluted with EtOAc–hexane mixture (1:1 with 1% AcOH), while the catalyst, p-TsOH came out of the same column with a mixture of EtOAc and 1% AcOH. During the process of purification the unreacted amine was sticking inside the column which was eluted with a 1:9 mixture of methanol and ethyl acetate. Finally, the quality of the recovered reagents, starting material and solvent was verified by NMR spectra (attached in the ESI†). It was noted that all the recovered materials were of comparable quality to the commercial material and therefore, used in the next batch.
However, from Table 1, 2 and 3 it is clear that the percentage of recovery was not excellent. We reasoned that it is due to the small scale of operation. To establish that reasoning, we selected the reaction between benzoyl glycine and benzyl amine, and scaled it up to 1 g of the acid (entry 3, Table 4). In this case, we could recover 200 mg of the unreacted substrate, which could be reused, i.e. % of conversion was 80%. Considering only 800 mg was converted to the product, the final yield of the reaction becomes 97%. Percentage of recovery of the catalyst, p-TsOH increases to 78% which was 49% when the scale was smaller (entry 1, Table 4). Percentage of recovery of the solvent cum auxiliary, benzyl alcohol, improves to 74% (entry 3, Table 4) at a larger scale from 44% when the reaction scale is smaller (entry 1, Table 4). A similar incident was noticed for amine recovery as well. This result indicates that the observed loss in recovery depends on the reaction scale. Therefore, the percentage of recovery is supposed to improve at industrial scale. Most importantly, no side reactions were observed. In a few cases, intermediates also could be isolated (entries 17, 19 and 20).
| Entry | Amount of Bz–Gly–OH (mg) | Isolated yield (%)b | Amine rec/exp (%)a | Alcohol rec/exp (%)a | Acid rec (mg) | p-TsOH rec/exp (%)a |
|---|---|---|---|---|---|---|
| a Amount recovered/expected (w/w, %). b Yields were calculated considering the amount of the starting acid which could not be recovered. | ||||||
| 1. | 250 | 83 | 50 | 44 | 42 | 49 |
| 2. | 500 | 90 | 55 | 67 | 102 | 65 |
| 3. | 1000 | 97 | 70 | 74 | 200 | 78 |
Therefore, in principle, amide synthesis can be performed following the demonstrated homogenous reaction condition, where no waste will be generated, at least at the ideal situation, because all the so called undesired products, which usually add to the E-factor, will be recycled. However, part of the solvents which cannot be trapped and the silica gel in the flash column after several uses can be considered as waste in this protocol. Interestingly, we noticed that, in spite of the described optimization, when we recover and reuse the reagents in the next batch of the reaction, the E-factor of the process increases due to the solvents used for recovery of the reagents from 25.5 to 30.8 (see ESI† for a detailed method of E-factor calculation). It increases both the cost and the chemical waste.
Therefore, we considered whether a continuous version of the same reaction would be possible to design to make the process more efficient. More precisely, we were looking for a solvent that will allow the product to precipitate out in a way that the rest of the reaction mixture containing the catalyst along with the other reagents will stay in solution. Then, we can recover the reaction mixture by filtration followed by evaporation of the solvent and reuse them easily. Finally, we could find such a solvent which is ethanol, the green solvent. In the refined continuous procedure, the crude reaction mixture was washed with ethanol after completion of the reaction (monitored by TLC) which facilitates the amide to precipitate out and separate easily. The ethanol wash was later evaporated to obtain all the other reagents back as a mixture. After recovery of the reaction mixture, more benzoyl glycine (1 g) was added and it was subjected to microwave irradiation followed by the addition of an equivalent amount of benzyl amine and heated at 40 °C, as before, to obtain the amide product (Scheme 2). This process was continued up to three cycles without any problem. After three cycles the amide could not be precipitated and therefore the reaction mixture was diluted with ethyl acetate and washed with 5% HCl (one time) to remove benzyl amine. Ethyl acetate was evaporated and the crude reaction mixture was washed with ethanol to obtain the white solid amide product and continued as before up to six cycles. However, such cyclic operations can be continued many times depending on the necessity. The yield in each cyclic operation is demonstrated in Table 5. After considering that the recovered 500 mg of the starting carboxylic acid can be reused, the final yield after six cycles becomes 70%. However, as there is no side reaction noted, the yield can be increased with the utilization of expertise. Purity of the product in this new method was not compromised as it was noticed that the NMR spectrum of the precipitated products was as good as the NMR spectrum of the product purified by column chromatography. The E-factor of this continuous operation was found to be 3.87, which is much less than the E-factor of the batch process where the product precipitation protocol was not used (30.8, the E-factor of the new process was calculated considering up to 6 cycles, see ESI† for details). However, the E-factor of the batch process, considering the recovery of the reaction mixture by product precipitation protocol, comes down to 0.3.
 | ||
| Scheme 2 Continuous method for recovery and reuse of the reagents. | ||
| Run | Isolated yield (%) |
|---|---|
| a One time washing with 5% HCl solution was performed prior to precipitation. | |
| 1. | 80% |
| 2. | 72% |
| 3. | 70% |
| 4. | 60% |
| 5. | 54%a |
| 6. | 49%a |
Conclusions
In summary, we demonstrated a continuous operation protocol for the highly important amide synthesis reaction where chemical waste generation, evident from the E-factor calculation, is drastically reduced following a recover and reuse strategy. Here, the whole reaction mixture, containing the unreacted starting materials, catalyst and solvent was recovered very easily and reused for the next cycle. Yield is also not reduced. This is a reagent economized and waste minimized protocol for the preparation of amides from N-protected amino acids, aromatic acid and aliphatic acids with alkyl, aryl and some solid amines in one pot. In this strategy, although we do not lose any extra reagent and thus, do not generate much waste; a conversion from acid to amide is achieved following a homogeneous reaction condition. However, sterically hindered amino acid, e.g. valine and less nucleophilic amine, e.g. aniline, mark the limitation of the method. This new approach basically considers by products, which add to the E-factor of a chemical process not as “waste”, but rather possibilities to recycle, hence to render the whole procedure waste free. Column chromatography is avoided without compromising of the quality of the product, and environmentally unfriendly solvents such as hexane are not used. Such a reagent economized process development may complement the atom economy of a reaction. Devising such a reagent economized process after formulating a reaction protocol with the highest possible atom economy for each target is important for saving the environment and the global health.Experimental
All reagents were purchased from commercial sources and were used without any further treatment. Infrared (IR) spectra were recorded on a Perkin Elmer spectrometer. Melting points are determined with a Buchi-540 apparatus. Reactions were monitored using thin layer chromatography with silica gel G254 using the solvent system EtOAc–hexane. Solvents were removed under reduced pressure using a Buchi rotary evaporator. The products were purified by column chromatography (60–120) mesh silica gel. 1H-NMR (400 MHz) and 13C-NMR (100 MHz) were recorded using a DRX-400 Varian spectrometer using CDCl3 and MeOH-D4 as solvents. Chemical shifts are reported in parts per million (ppm) with an internal reference (0.05% to 1% tetramethyl silane for CDCl3). High resolution mass spectra (HRMS) were recorded on a Micromass Q-TOF ESI MS instrument (model HAB273) and Agilent 6500 Q-TOF LC/MS system. The deviation from the observed m/z value to the theoretical value is less than 5 ppm in each case. X-Ray data were collected on a Bruker SMART APEX equipped with a CCD area detector using Mo.General procedure for the preparation of amides using the batch protocol
Benzoyl glycine (1 g) was taken in sealed pressure tube (5 mL) and dissolved in benzyl alcohol (2 equiv.), p-toluene sulphonic acid (0.3 equiv.) was added and irradiated using a Biotage intiator microwave reactor at 50 W, 100 °C for 5 min. After getting the starting materials converted in to the respective esters, benzyl amine (2 equiv.) was added and stirring was continued at 40 °C. The reaction was monitored by TLC. The products were purified by column chromatography.General procedure for the preparation of amides using the continuous protocol
N-Protected amino acid or aromatic acid or aliphatic acid (1 g) was taken in a pressure glass tube (5 mL) and dissolved in benzyl alcohol (2 equiv.), p-toluene sulphonic acid (0.3 equiv.) was added and irradiated using a Biotage initiator microwave reactor at 50 W, 100 °C for 5 min. Ester formation was monitored by TLC. Then, the amine (2 equiv.) was added to the same vial and stirred at 40 °C. The reaction was monitored by TLC. Ethanol (5 ml) was added to the reaction mixture, which allowed the product (amide) to precipitate out as a white solid. The reaction mixture containing all the other reagents was recovered by filtration followed by subsequent evaporation of the ethanol wash and reused for the next cycle. When the precipitation of the product was difficult, the reaction mixture was dissolved in 10 ml of ethyl acetate and washed with 5 ml of the 5% HCl solution (one time). Then, the organic layer was evaporated and 5 ml of ethanol was used to precipitate the product. The reaction mixture was recovered by filtration and subsequent evaporation of the ethanol followed by reuse in the next cycle.:3). Yield 67%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.82–7.38 (m, 5H), 6.78 (br, 1H), 4.12–4.10 (d, 2H, J = 4.8 Hz), 3.27–3.22 (m, 2H), 1.51–1.43 (m, 2H), 1.36–1.28 (m, 2H), 0.89–0.85 (t, 3H, J = 7.4 Hz), 13C NMR (100 MHz, CDCl3) δ (ppm) 169.4, 168.2, 133.6, 132.1, 128.8, 127.4, 41.1, 39.7, 31.6, 20.2, 13.8; IR (KBr, ν/cm−1) 1539, 1635, 1652, 3298, 3320; LRMS m/z 235 [M + H]+ 257 [M + Na]+ HRMS (ESI) m/z [M + H]+ calculated for C13H19N2O2: 235.1447 found: 235.1458.
:9). Yield 60%; white solid; 1H NMR (400 MHz, CD3OD) δ (ppm) 7.79–7.35 (m, 5H), 3.94 (s, 2H), 3.52–3.46 (m, 2H), 3.25–3.16 (m, 2H), 13C NMR (100 MHz, CD3OD) δ (ppm) 170.8, 169.3, 133.8, 131.7, 128.3, 127.3, 60.3, 42.9, 41.7, 21.3; IR (KBr, ν/cm−1) 1638, 3437; LRMS m/z 245 [M + Na]+ HRMS (ESI) m/z [M + Na]+ calculated for C11H14N2NaO3: 245.0902 found: 245.0914.
:3). Yield 75%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.76–7.74 (d, 1H, J = 8.4 Hz, 1H), 7.50–7.24 (m, 10H), 6.98 (br, 1H), 4.45–4.43 (d, 2H, J = 6 Hz), 4.16–4.15 (d, 2H, J = 4.8 Hz). 13C NMR (100 MHz, CDCl3) δ (ppm) 169.2, 168.0, 138.0, 132.0, 128.8, 128.7, 127.9, 127.6, 127.3, 44.0, 43.8; IR (KBr, ν/cm−1) 1639, 1667, 3293; LRMS m/z 269 [M + H]+, 291 [M + Na]+ HRMS (ESI) m/z [M + H]+ calculated for C16H17N2O2: 269.1290 found: 269.1290.
:7). Yield 64%, white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.83–7.81 (d, 1H, J = 7.2 Hz, 1H), 7.53–7.37 (m, 5H), 6.77–6.75 (d, 1H, J = 7.6 Hz, 1H), 4.11–4.10 (d, 2H, J = 4.8 Hz), 3.74–3.72 (t, 1H, J = 4 Hz), 1.87–1.11 (m, 10H). 13C NMR (100 MHz, CDCl3) δ (ppm) 168.3, 168.0, 133.7, 131.9, 128.7, 127.3, 48.6, 44.1, 32.9, 25.6, 24.9; IR (KBr, ν/cm−1) 1542, 1644, 1665, 2934, 3303; LRMS m/z 261 [M + H]+, 283 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C15H21N2O2: 261.1603 found: 261.1611.
:3). Yield 78%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.33–7.23 (m, 10H), 5.09 (s, 2H), 4.44–4.42 (d, J = 5.6 Hz, 2H), 3.89–3.87 (d, J = 5.2 Hz, 2H), 13C NMR (100 MHz, CDCl3) δ (ppm) 169.2, 156.8, 137.9, 136.2, 128.7, 128.6, 128.3, 128.1, 127.7, 127.6, 67.2, 44.6, 43.5; IR (KBr, ν/cm−1) 1663, 1715, 3322; LRMS m/z 299 [M + H]+, 321 [M + Na]+, 337 [M + K]+ HRMS (ESI) m/z [M + H]+ calculated for C17H19N2O3 is 299.1396 found: 299.1406.
:9). Yield 66%, brown solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.31 (s, 5H), 6.1 (br, 1H), 5.6 (br, 1H), 5.0 (s, 2H), 3.79–3.78 (d, 2H, J = 4 Hz), 3.74–3.70 (m, 1H), 1.84–1.09 (m, 10H), 13C NMR (100 MHz, CDCl3) δ (ppm) 168.1, 156.8, 136.3, 128.7, 128.4, 128.2, 67.3, 48.5, 44.8, 33.0, 25.6, 24.9; IR (KBr, ν/cm−1) 1655, 1718, 2924, 3347; LRMS m/z 291 [M + H]+ 313 [M + Na]+ HRMS (ESI) m/z [M + H]+ calculated for C16H23N2O3: 291.1709 found: 291.1710.
:4). Yield 66%, white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.75–7.24 (10H), 7.13–7.14 (d, 1H, J = 4 Hz), 4.83–4.76 (m, 1H), 4.503–4.379 (dd, 2H, J = 6, 5.6 Hz), 1.49–1.47 (d, 3H, J = 6.8 Hz), 13C NMR (100 MHz, CDCl3) δ (ppm) 172.6, 167.4, 138.1, 138.8, 131.9, 128.8, 128.7, 127.8, 127.6, 127.3, 49.4, 43.7, 18.9; IR (KBr, ν/cm−1) 1632, 1657, 3305; LRMS m/z 283 [M + H]+ 305 [M + Na]+ HRMS (ESI) m/z [M + H]+ calculated for C17H19 N2O2: 283.1447 found: 283.1434.
:4). Yield 36%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.34 (s, 5H), 5.95 (br, 1H), 5.41–5.39 (d, 1H, J = 8 Hz, 1H), 5.1 (s, 2H), 3.92–3.82 (t, 1H, J = 8.4 Hz), 3.27–3.18 (m, 2H), 2.11–2.10 (m, 1H), 1.47–1.45(m, 2H), 1.35–1.29 (m, 2H), 0.96–0.83 (m, 9H), 13C NMR (100 MHz, CDCl3) δ (ppm) 171.4, 156.7, 136.4, 128.6, 128.2, 128.0, 67.0, 60.8, 39.3, 31.6, 31.2, 20.1, 19.3, 18.2, 13.8; IR (KBr, ν/cm−1) 1641, 1690, 329; LRMS m/z 307 [M + H]+, 329 [M + Na]+ HRMS (ESI) m/z [M + H]+ calculated for C17H27N2O3: 307.2022 found: 307.2032.
:4). Yield 30%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.31–7.24 (m, 10H), 6.29 (br, 1H), 5.37–5.35 (d, 1H, J = 7.6 Hz), 5.04 (s, 2H), 4.47–4.35 (dd, 2H, J = 5.2 Hz), 3.98–3.95 (t, 1H, J = 6.8 Hz), 2.14–2.12 (m, 2H), 0.95–0.93 (d, 3H, J = 6.4 Hz), 0.91–0.89 (d, 3H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ (ppm) 171.3, 156.7, 138.0, 136.3, 128.9, 128.7, 128.3, 128.1, 127.9, 127.7, 67.2, 60.8, 43.7, 31.2, 19.4, 18.1; IR (KBr, ν/cm−1) 1650, 1690, 3291; LRMS m/z 341[M + H]+, 363 [M + Na]+ HRMS (ESI) m/z [M + H]+ calculated for C20H25N2O3: 341.1865 found: 341.1872.
:4). Yield 84%; 1H NMR (400 MHz, CD3OD) δ (ppm) 7.87–7.44(m, 5H), 4.06(s, 2H), 3.74 (s, 6H), 13C NMR (100 MHz, CDCl3, CD3OD for solubility) δ (ppm) 172.4, 170.6, 133.1, 129.6, 128.5, 126.9, 63.5, 62.2, 44.6; IR (KBr, ν/cm−1) 1544, 1641, 2918, 3432; LRMS m/z 283 [M + H]+, 305 [M + Na]+ HRMS (ESI) m/z [M + H]+ calculated for C13H18N2O5 is 305.1113 found 305.1117.
:3). Yield 65%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 9.04 (br, 1H), 7.83–7.06 (m, 10H), 5.68 (br, 1H), 4.326–4.312 (d, 2H, J = 5.6, Hz), 2.27 (s, 3H), 13C NMR (100 MHz, CDCl3, CD3OD for solubility) δ (ppm) 167.8, 167.4, 135.4, 133.4, 131.5, 130.8, 129.1, 128.3, 127.2, 119.8, 44.2, 20.7; IR (KBr, ν/cm−1) 1638, 1676, 2925, 3269; LRMS 269 [M + H]+, 291 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C16H16N2O2 is 269.1290 found: 269.1285.
:3). Yield 67%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.76 (br, 1H), 7.87–7.03 (m, 10H), 4.356–4.343 (d, 2H, J = 5.2 Hz), 2.19 (s, 6H), 13C NMR (100 MHz, CDCl3) δ (ppm) 167.9, 167.4, 136.9, 135.7, 131.6, 129.7, 128.4, 127.2, 121.2, 117.4; 44.3, 19.8, 19.1 IR (KBr, ν/cm−1) 1638, 1693, 2921, 3283; LRMS 283 [M + H]+, 305 [M + Na]+, HRMS (ESI) m/z [M + Na]+ calculated for C17H18N2NaO2 is 305.1266 found: 305.1251.
:4). Yield 68%; 1H NMR (400 MHz, CD3OD) δ (ppm) 7.76–7.7.35(m, 5H), 4.51–4.44(m, 1H), 3.62 (s, 6H), 1.37–1.36 (d, 3H, J = 7.2) 13C NMR (100 MHz, CDCl3) δ (ppm) 176.0, 170.5, 135.1, 133.1, 129.7, 128.6, 63.5, 62.3, 51.8, 18.1; IR (KBr, ν/cm−1) 1640, 1671, 2922, 3436; LRMS m/z 297 [M + H]+, 319[M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C14H20N2O5 is 297.1450 found 297.1455.
:4). Yield 67%; 1H NMR (400 MHz, CD3OD) δ (ppm) 7.19–7.13 (m, 5H), 3.59 (s, 6H), 3.46 (s, 2H), 13C NMR (100 MHz, CDCl3) δ (ppm) 175.1, 136.7, 130.3, 129.7, 128.0, 63.6, 62.5, 44.3; IR (KBr, ν/cm−1) 1542, 1644, 2934, 3283; LRMS m/z 240 [M + H]+, 262 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C12H17NO4 is 240.1236 found 240.1233.
:4). Yield 68%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.36–7.16(m, 10H), 5.76 (br, 1H), 4.41–4.39 (d, 2H, J = 6 Hz), 3.62 (s, 2H), 13C NMR (100 MHz, CDCl3) δ (ppm) 171.1, 138.3, 135.0, 129.4, 129.0, 128.7, 127.5, 127.4, 127.3, 43.7, 43.6; IR (KBr, ν/cm−1) 1552, 1638, 2925, 3289; LRMS 226 [M + H]+, 248 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C16H15NO is 226.1232 found 226.1231.
:4). Yield 48%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.37–7.23 (m, 5H), 5.23 (br,1H), 3.76–3.74 (t, 1H, J = 5.2 Hz), 3.54 (s, 2H), 1.70 (s, 2H), 1.83–1.54 (m, 4H), 1.41–1.25 (m, 4H), 1.13–0.88 (m, 2H), 13C NMR (100 MHz, CDCl3) δ (ppm) 170.2, 135.3, 129.4, 129.0, 127.3, 48.3, 44.0, 32.9, 25.5, 24.8; IR (KBr, ν/cm−1); 1556, 1635, 2929, 3273; LRMS m/z 218 [M + H]+, 240 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C14H19NO is 218.1545 found 218.1540.
:4). Yield 93%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.37–7.26 (m, 5H), 5.40 (br, 1H), 3.56 (s, 2H), 3.22–317 (m, 2H), 1.41–1.35 (m, 2H), 1.27–1.21 (m, 2H), 0.88–0.85 (t, 3H, J = 7.2 Hz), 13C NMR (100 MHz, CDCl3) δ (ppm) 171.2, 135.4, 128.9, 128.3, 128.0, 126.6, 43.1, 39.1, 31.2, 19.8, 13.5; IR (KBr, ν/cm−1) 1559, 1655, 1630, 2957, 3253; LRMS m/z 192 [M + H]+, 214 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C12H17NO is 192.1388 found 192.1387.
:4). Yield 26%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.42–7.06 (m, 10H), 3.73 (s, 2H), 13C NMR (100 MHz, CDCl3) δ (ppm) 169.8, 137.9, 134.7, 129.5, 129.1, 128.9, 127.5, 124.5, 120.2, 44.6; IR (KBr, ν/cm−1) 1601, 1657, 3256, 3285; LRMS m/z 212 [M + H]+, 234 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C14H13NO is 212.1075 found 212.1071.
:4). Yield 31%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.79–7.77 (m, 2H), 7.43–7.25 (m, 8H) 4.648–4.634 (d, 2H, J = 5.6 Hz), 13C NMR (100 MHz, CDCl3) δ (ppm) 167.6, 138.4, 134.4, 131.6, 128.8, 128.6, 127.9, 127.5, 127.1, 44.1; IR (KBr, ν/cm−1) 1551, 1637, 2924, 3290; LRMS m/z 212 [M + H]+, 234 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C14H13NO is 212.1075 found 212.1073.
:4). Yield 87%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 5.52 (br, 1H), 3.23–3.18 (q, 2H, J = 6.8 Hz), 2.141–2.103 (t, 2H, J = 7.2), 1.60–1.56 (m, 2H), 1.48–1.41(m, 2H), 1.22(s, 18H), 0.90–0.82(m, 6H), 13C-NMR (100 MHz, CDCl3) δ (ppm) 173.5, 39.2, 36.7, 31.9, 31.7, 29.6, 29.5, 29.4, 29.4, 25.9, 22.7, 20.1, 14.1, 13.7; IR (KBr, ν/cm−1) 1552, 1638, 2919, 3296; LRMS m/z 256 [M + H]+, 278 [M + Na]+ HRMS (ESI) m/z [M + H]+ calculated for C16H33NO is 256.2640 found 256.2638.
:3). Yield 89%; white solid; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.30–7.19 (m, 5H), 5.69 (br, 1H), 4.378–4.364 (d, 2H, J = 5.6 Hz), 2.157–2.119 (t, 2H, J = 7.6 Hz), 1.59–1.54 (m, 2H), 1.18 (s,16H), 0.82–0.79(t, 3H, J = 6 Hz), 13C NMR (100 MHz, CDCl3), δ (ppm) 173.5, 138.6, 128.5, 128.3, 127.6, 43.3, 36.6, 31.9, 29.6, 29.5, 29.4, 29.3, 25.8, 22.7, 14.1; IR (KBr, ν/cm−1) 1555, 1632, 2916, 3292; LRMS 290 [M + H]+, 312 [M + Na]+, HRMS (ESI) m/z [M + H]+ calculated for C19H31NO is 290.2484 found 290.2482
Acknowledgements
We are thankful to DST for XRD facility; CIF, IITG for NMR and HR-MS facility; BRNS-DAE (Sanction No. 2008/20/37/6/BRNS) for research grant. TK, NKC and AP thank IITG for the fellowship. We are thankful to Prof. A. T. Khan, Dr Md Lokman H. Choudhury (IIT Patna) and his students for their generous help to perform the reactions in their Biotage, Initiator™ microwave reactor.References
- (a) B. M. Trost, Science, 1991, 254, 1471 CAS; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- (a) P. A. Wender, F. C. Bi, G. G. Gamber, F. Gosselin, R. D. Hubbard, M. J. C. Scanio, R. Sun, T. J. Williams and L. Zhang, Pure Appl. Chem., 2002, 74, 25 CrossRef CAS; (b) P. A. Wender, S. T. Handy, D. L. Wright and Chem. Ind, London, 1997, 765 Search PubMed; (c) P. A. Wender and B. L. Miller, Organic Synthesis: Theory and Applications, ed. T. Hudlicky, JAI, Greenwich 1993, vol. 2, pp. 27–66 Search PubMed; (d) P. A. Clarke, S. Santos and W. H. C. Martin, Green Chem., 2007, 9, 438 RSC.
- P. A. Clarke, S. Santos and W. H. C. Martin, Green Chem., 2007, 9, 438 RSC.
- Green Chemistry: Theory and Practice, ed. P. Anastas and J. C. Warner, Oxford University Press, Oxford, 1998 Search PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- NRC Committee on Hazardous Substances in the Laboratory, Prudent Practices for Disposals of Chemicals from Laboratory, National Academy Press, Washington, D.C., 1981,pp. 44–50 Search PubMed.
- K. C. Nadimpally, K. Thalluri, N. Palakurthy, A. Saha and B. Mandal, Tetrahedron Lett., 2011, 52, 2579 CrossRef CAS.
- W. Zhang and D. P. Curran, Tetrahedron, 2006, 62, 11837 CrossRef CAS.
- Organic reactions in water: Principles, Strategies and applications, U. M. Lindstrom, Wiley-Blackwell, April 2007 Search PubMed.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391 CrossRef CAS.
- Chemical Synthesis using Super critical Fluids, ed. P. G. Jessop and W. Leitner, Wiley-VCH, Weinheim, 1999 Search PubMed.
- P. J. Dyson, D. J. Ellis and T. Welton, Platinum Metals Rev., 1998, 42, 135 CAS.
- L. F. Liotta, A. M. Venezia, G. Deganello, A. Longo, A. Martorana, Z. Schay and L. Guczi, Catal. Today, 2001, 66, 271 CrossRef CAS.
- H. Azoui, K. Baczko, S. Cassel and C. Larpent, Green Chem., 2008, 10, 1197 RSC.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- P. Starkov and T. D. Sheppard, Org. Biomol. Chem., 2011, 9, 1320 CAS.
- (a) J. Cossy and C. Pale-Grosdemange, Tetrahedron Lett., 1989, 30, 2771 CrossRef CAS; (b) L. J. Gooben, D. M. Ohlmann and P. P. Lange, Synthesis, 2009, 1, 160 Search PubMed.
- K. V. N. S. Srinivas and B. Das, J. Org. Chem., 2003, 68, 1165 CrossRef CAS.
- J. W. Comerford, J. H. Clark, D. J. Macquarrie and S. W. Breeden, Chem. Commun., 2009, 2562 RSC.
- X. D. Yang, X. H. Zeng, Y. H. Zhao, X. Q. Wang, Z. Q. Pan, L. Li and H. B. Zhang, J. Comb. Chem., 2010, 12, 307 CrossRef CAS.
- Y. Watanabe and T. Mukaiyama, Chem. Lett., 1981, 285 CrossRef CAS.
- K. Wakasugi, A. Iida, T. Misaki, Y. Nishii and Y. Tanabe, Adv. Synth. Catal., 2003, 345, 1209 CrossRef CAS.
- P. S. Chaudari, S. D. Salim, R. V. Sawant and K. G. Akamanchi, Green Chem., 2010, 12, 1707 RSC.
- M. Colombo, S. Bossolo and A. Aramini, J. Comb. Chem., 2009, 11, 335 CrossRef CAS.
- K. Sasaki and D. Crich, Org. Lett., 2011, 13, 2256 CrossRef CAS.
- A. R. Katritzky, H. Y. He and K. Suzuki, J. Org. Chem., 2000, 65, 8210 CrossRef CAS.
- (a) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196 CrossRef CAS; (b) T. Maki, K. Ishihara and H. Yamamoto, Tetrahedron, 2007, 63, 8645 CrossRef CAS; (c) K. Arnold, B. Davies, D. Herault and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 2673 CrossRef CAS.
- P. Tang, Org. Synth., 2005, 81, 262 CAS.
- D. M. Shendage, R. Frohlich and G. Haufe, Org. Lett., 2004, 6, 3675 CrossRef CAS.
- J. McNulty, V. Krishnamoorthy and A. Robertson, Tetrahedron Lett., 2008, 49, 6344 CrossRef CAS.
- Y. Saito, H. Ouchi and H. Takahata, Tetrahedron, 2008, 64, 11129 CrossRef CAS.
- (a) for few current reviews, see: A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS; (b) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC; (c) M. M. Joullié and K. M. Lassen, ARKIVOC, 2010,(viii), 189 Search PubMed; (d) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC; (e) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- C. Sabot, Tetrahedron Lett., 2007, 48, 3863 CrossRef CAS.
- G. A. Molander, J. Raushel and N. M. Ellis, J. Org. Chem., 2010, 75, 4304 CrossRef CAS.
- G. Pelletier, W. S. Bechara and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 12817 CrossRef CAS.
- K. N. Kumar, K. Sreeramamurthy, S. Palle, K. Mukkanti and P. Das, Tetrahedron Lett., 2010, 51, 899 CrossRef CAS.
- B. Roberts, D. Liptrot, L. Alcaraz, T. Luker and M. J. Stocks, Org. Lett., 2010, 12, 4280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: detailed method of E-factor calculation, copies of 1H and 13C NMR and HRMS data of each final compound and crystallographic data of compound 3 and 17 respectively. CCDC reference numbers 837072, 837222. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20643a |
| This journal is © The Royal Society of Chemistry 2012 |