Plasmon-Enhanced Hydrogen Evolution on Au-InVO4 Hybrid Microspheres†
Shao-Wen
Cao
a,
Jun
Fang
a,
Mohammad Mehdi
Shahjamali
a,
Freddy Y. C.
Boey
a,
James
Barber
ab,
Say Chye Joachim
Loo
*a and
Can
Xue
*a
aSolar Fuels Laboratory, School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798, Singapore. E-mail: cxue@ntu.edu.sg; joachimloo@ntu.edu.sg
bDivision of Molecular Biosciences, Imperial College London, South Kensington Campus London SW7 2AZ, U.K.
First published on 25th April 2012
Abstract
We demonstrate plasmon-enhanced hydrogen evolution from photocatalytic water reduction using Au-InVO4 hybrid microspheres. The surface plasmons of gold nanoparticles enhance the sub-band gap excitation of InVO4 and promote charge separation on its surface through plasmon-exciton coupling, thereby significantly improving their photocatalytic efficiency.
Solar-to-fuel conversion through photocatalytic water splitting has been recognized as highly important energy sources for the sustainable future.1 Researchers have made tremendous effort to develop various visible-light-active metal oxide photocatalysts, such as BiVO4,2,3 Fe2O3,4,5 and Cu2O,6,7 for water oxidation or reduction. Ye and co-workers have reported a new type of metal oxide InVO4 which is capable of producing H2 through visible-light driven photocatalytic water reduction.8,9 Recent studies revealed that the actual band-to-band energy gap of InVO4 could be more than 3 eV, while its visible-light activities were attributed to sub-band gap transitions from impurity states.10–12 Though it is certainly beneficial from extended absorption in the visible range, the impurity states will also promote the undesirable recombination of photogenerated electrons and holes, which may reduce the photocatalytic activities. Consistently, some researchers also found that their prepared InVO4 structures are not viable for visible-light-driven hydrogen production due to inefficient charge separation from the visible-light-induced sub-band gap transition.12–14
In order to achieve high photocatalytic activities, efficient charge separation is crucial for all photocatalysts. To date, researchers have developed many different methods to enhance the separation of photogenerated electrons and holes. One promising approach is depositing plasmonic particles of noble metals (e.g. Au and Ag) onto the photocatalysts.15–17 As is well known, gold and silver nanoparticles exhibit strong surface plasmon resonance (SPR) in the visible range.18,19 It has been demonstrated that the SPR of metal nanoparticles would greatly promote the photocatalytic efficiencies of metal oxide photocatalysts because the SPR-enhanced localized electric field can improve the charge separation near the metal-semiconductor interfaces.20,21
Herein, we report a new strategy to utilize gold SPR to enhance visible-light-driven hydrogen evolution through photocatalytic water reduction using Au-InVO4 hybrid microspheres prepared by growing gold nanoparticles on pre-synthesized InVO4 hollow microspheres. We demonstrate that the gold nanoparticle SPR can significantly enhance the sub-band gap transition of InVO4 microspheres and thereby their photocatalytic efficiencies. The detailed plasmonic effect on charge separation is also discussed in-depth.
The prepared InVO4 sample is identified as orthorhombic phase through XRD analyses (Fig. S1, ESI†). The absorption spectrum (Fig. 1a) shows that the InVO4 sample exhibits obvious absorption in the visible light range with a band gap estimated as 2.16 eV. The analyses by SEM and TEM (Fig. 1b–d) reveal that the sample consists of hollow microspheres that are built by the assembly of many InVO4 nanocrystals. The high-resolution TEM image (Fig. 1e) of the nanocrystal indicates a fringe spacing of 0.477 nm in accordance with the lattice spacing of the (110) plane of orthorhombic InVO4.
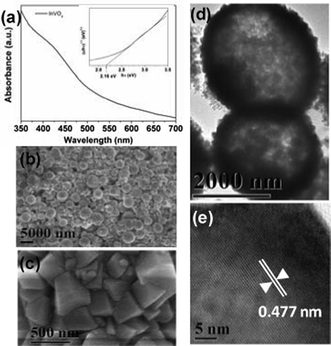 | ||
| Fig. 1 (a) UV-vis spectrum of InVO4 microspheres, inset is the plot of (αhν)1/2vs. photon energy (hν) to calculate the band gap; (b, c) SEM images and (d, e) TEM images of InVO4 microspheres. | ||
Three Au-InVO4 hybrid samples with different Au content were prepared by reducing HAuCl4 with L-ascorbic acid in the presence of InVO4 microspheres and L-cysteine that can bind to the InVO4 surface through its carboxylate group and captured gold onto InVO4 surfaces via the Au–S bonding.22,23 SEM and TEM images (Fig. 2a, and Fig. S2, S3, S4, ESI†) indicate that gold nanoparticles were successfully grown onto the InVO4 microspheres and the original microsphere morphology did not change during the gold nanoparticle growth process. The gold contents in these three samples were estimated as ∼ 2 wt%, 4 wt%, and 7.5 wt%, respectively, according to the analyses by energy-dispersive X-ray (EDX) spectroscopy. The high-magnification TEM images (Fig. S2–S4†) reveal that the sizes of the gold nanoparticles in these samples are 3.7 (± 0.9) nm, 6.7 (± 1.5) nm, and 13.0 (± 1.9) nm, respectively. The SPR feature of the gold nanoparticles can be clearly observed at ∼ 540 nm for the 4 wt% and 7.5% wt% Au-InVO4 samples (Fig. 2b). The absence of SPR feature for the 2 wt% Au-InVO4 sample may be due to the low concentration of gold nanoparticles. In addition, the SPR of small gold particles (< 5 nm) is usually very weak,24 and may be overwhelmed by InVO4 absorption.
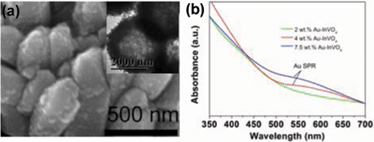 | ||
| Fig. 2 (a) SEM and TEM image (inset) of 4 wt% Au-InVO4 microspheres; (b) UV-vis spectra of 2, 4, 7.5 wt% Au-InVO4 samples. | ||
The prepared Au-InVO4 hybrid microspheres were then tested for photocatalytic H2 evolution under visible-light (λ > 420 nm) irradiation. L-ascorbic acid (0.1 M, pH = 4.0) was used as the electron donor. Fig. 3a shows the H2 evolution plots by different samples as a function of irradiation time. In comparison to pure InVO4 microspheres that showed no H2 evolution, all Au-InVO4 hybrid microspheres exhibit obvious visible-light-driven photocatalytic activity for H2 evolution. Among them, the 4 wt% Au-InVO4 sample showed the highest H2 evolution rate of 116.7 μmol h−1 g−1 which then dropped to 15.9 μmol h−1 g−1 after 90 min and became quite stable thereafter (Fig. S5). The stable H2 evolution rate (after 90 min) of the 2 wt% and 7.5 wt% Au-InVO4 samples were 12.5 μmol h−1 g−1 and 8.6 μmol h−1 g−1, respectively.
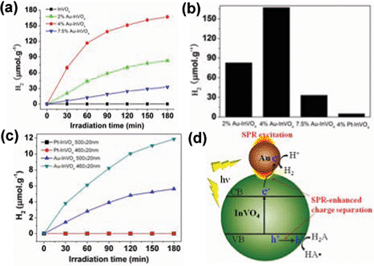 | ||
| Fig. 3 (a) Plots of photocatalytic H2 evolution amount versus irradiation (λ > 420 nm) time for different samples; (b) Comparison of H2 evolution amount of for Au-InVO4 and Pt-InVO4 samples after 3 h irradiation; (c) Photocatalytic H2 evolution for 4 wt% Au-InVO4 and 4 wt% Pt-InVO4 under irradiation at 460 ± 20 nm and 500 ± 20 nm; (d) Schematic illustration of the plasmon-enhanced photocatalytic process for H2 evolution on the Au-InVO4 hybrid structure, in which H2A and HA• refer to ascorbic acid and the ascorbate radical, respectively. | ||
As is well known, semiconductor photocatalysts can have improved efficiencies after loading with nanoparticles of noble metals such as Pt, Pd and Au.25,26 The metal nanoparticles can act as electron sinks to hold the excited electrons from the semiconductor band-gap excitation, which retard the charge recombination process and thereby increase its photocatalytic efficiency.27 In order to clarify whether the electron-sink effect dominates in the Au-InVO4 samples, we prepared Pt-InVO4 hybrid microspheres with 4 wt% Pt by similar methods (Fig. S6, ESI†). The XPS analyses (Fig. S7†) reveal the binding energy of Au 4f (84.1 and 87.8 eV) and Pt 4f (71.2 and 74.6 eV), indicating the zero-valent nature of the deposited gold and platinum. However, after visible-light irradiation for 3 h, the photocatalytic H2 evolution amount of this Pt-InVO4 sample (5.0 umol g−1) is much lower than that of the 4 wt% Au-InVO4 sample (167.1 umol g−1) (Fig. 3b). The Pt-InVO4 samples with less Pt loading (1 wt% and 2 wt%) showed even smaller H2 evolution amount (Fig. S8†). This comparison result suggests that the pure electron-sink effect plays only a minor role for the metal-InVO4 hybrids as it is well known that Pt is usually more active than Au for H2 evolution due to the lower H2 evolution overpotential on Pt surfaces.28
To strengthen the above conclusion, we carried out further comparison tests by controlling the irradiation wavelengths at 460 ± 20 nm and 500 ± 20 nm, respectively. Fig. 3c indicates that in both wavelength ranges, the 4 wt% Pt-InVO4 sample does not show observable H2 evolution, while the 4 wt% Au-InVO4 sample is still quite active. This means that even though InVO4 microspheres absorb photons in these two ranges through sub-band gap transition, the majority of photogenerated electrons would recombine rapidly with the holes rather than transfer to the contacted Pt nanoparticles for water reduction. Note that small Pt nanoparticles do not exhibit SPR absorption in the visible light range. So the SPR-absorption from the Au nanoparticles grown on the InVO4 microspheres should play important roles in the enhanced efficiency of Au-InVO4 samples.
It is known that the SPR-excitation of metal nanostructures can generate strongly enhanced localized electric fields with magnitudes up to 105 times of the incident energy.29 This way, the metal nanostructures act like optical antenna to concentrate the light energy at the near surfaces. Therefore the InVO4 nanocrystals near the Au-InVO4 interfaces would encounter much more intense sub-band gap excitation by this SPR-enhanced electric field, which can generate more photo-excited electrons for water reduction. Moreover, the visible-light absorption of InVO4 induced by the sub-bandgap transition has been ascribed to impurities or defects in the crystal structure. But these impurities and defects can also act as recombination sites of photogenerated electrons and holes. However, the wide overlap between the Au-plasmon band and InVO4 absorption in the visible light range enables intensive plasmon-exciton coupling that significantly increases the exciton dissociation efficiency,20,21 and thereby promotes the photocatalytic efficiency. As a result, the photogenerated holes can oxidize L-ascorbic acid (H2A) on the InVO4 surface; meanwhile the separated electrons will have relatively longer time to reduce H+ to H2 on the gold nanoparticle surface. This SPR-enhanced photocatalytic process is illustrated in Fig. 3d.
It is also noted that for the Au-InVO4 sample, the 460 ± 20 nm irradiation results in higher H2 evolution efficiency than the 500 ± 20 nm irradiation (Fig. 3c) even though the gold nanoparticles have stronger SPR at the latter irradiation range. This means that the major contribution of the photocatalytic activity still comes from the InVO4 excitation. Certainly the excitation at 460 ± 20 nm creates more excitons than the excitation at 500 ± 20 nm since the InVO4 microspheres have stronger absorption at 460 ± 20 nm. Even if the gold plasmon at 460 nm is relatively weak, it still can provide sufficient enhancement to achieve desirable exciton dissociation yield on InVO4. While under excitation at 500 nm, the stronger excitation of gold SPR at 500 nm does not show further improvement due to weaker excitation of InVO4 with less available photogenerated electrons for water reduction. Nevertheless, in order to achieve good photocatalytic performance, the gold surface plasmon is still indispensable to enhance the exciton dissociation on InVO4 since the Pt-InVO4 sample shows no activity for H2 evolution under the irradiation at the same wavelength range.
However, when the gold loading amount is too high, the excessive gold nanoparticles would serve as centers to promote electron-hole recombination and thereby reduce the photocatalytic activity. That's why the 7.5 wt% Au-InVO4 showed less H2 evolution activity compared with the other two Au-InVO4 samples. In addition, the decreased activities of the Au-InVO4 samples after 90 min (Fig. 3a) are attributed to increased size of the gold nanoparticles on InVO4 during the photocatalytic reaction. As shown in Fig. S9, ESI,† the gold particle size changes to above 15 nm, which may be ascribed to the structure re-construction of small Au nanoparticles induced by ascorbic acid.30 While larger gold particles have higher resistance to the etching by ascorbic acid, thus the photocatalytic activity becomes stable thereafter. Since the EDX analysis indicates no change in the gold percentage (Fig. S9e†), the conversion of gold nanoparticles from small to large size causes a decrease of Au-InVO4 interfaces. Therefore, the effective area of SPR enhancement is also reduced, leading to lower photocatalytic efficiency of Au-InVO4 samples.
In summary, we have demonstrated the plasmon-enhanced H2 evolution from photocatalytic water splitting by using Au-InVO4 hybrid microspheres. The surface plasmons of gold nanoparticles that were grown on InVO4 microspheres substantially improve the separation of photogenerated electrons and holes on InVO4 through plasmon-exciton coupling. In comparison, the Pt-InVO4 sample showed very little activity for H2 evolution due to the lack of visible light SPR from Pt nanoparticles. We believe that our studies will have considerable impact on future development of more efficient plasmonic metal-semiconductor photocatalysts for visible-light-driven water splitting.
The authors acknowledge financial support from NTU Start-Up Grant (SUG), MOE AcRF-Tier1 RG 44/11, NTU seed funding for Solar Fuels Laboratory, and Singapore NRF-CRP (NRF-CRP5-2009-04) .
References
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911 CrossRef CAS.
- A. Iwase and A. Kudo, J. Mater. Chem., 2010, 20, 7536 RSC.
- D. Wang, H. Jiang, X. Zong, Q. Xu, Y. Ma, G. Li and C. Li, Chem.–Eur. J., 2011, 17, 1275 CrossRef CAS.
- A. Duret and M. Grätzel, J. Phys. Chem. B, 2005, 109, 17184 CrossRef CAS.
- H. Xie, Y. Z. Li, S. F. Jin, J. J. Han and X. J. Zhao, J. Phys. Chem. C, 2010, 114, 9706 CAS.
- C. H. Kuo, C. H. Chen and M. H. Huang, Adv. Funct. Mater., 2007, 17, 3773 CrossRef CAS.
- L. N. Kong, W. Chen, D. K. Ma, Y. Yang, S. S. Liu and S. M. Huang, J. Mater. Chem., 2012, 22, 719 RSC.
- J. H. Ye, Z. G. Zou, M. Oshikiri, A. Matsushita, M. Shimoda, M. Imai and T. Shishido, Chem. Phys. Lett., 2002, 356, 221 CrossRef CAS.
- M. Oshikiri, M. Boero, J. H. Ye, Z. G. Zou and G. Kido, J. Chem. Phys., 2002, 117, 7313 CrossRef CAS.
- G. L. Li and Z. Yin, Phys. Chem. Chem. Phys., 2011, 13, 2824 RSC.
- C. S. Enache, D. Lloyd, M. R. Damen, J. Schoonman and R. van de Krol, J. Phys. Chem. C, 2009, 113, 19351 CAS.
- K. Rakesh, S. Khaire, D. Bhange, P. Dhanasekaran, S. S. Deshpande, S. V. Awate and N. M. Gupta, J. Mater. Sci., 2011, 46, 5466 CrossRef CAS.
- H. Y. Lin, Y. F. Chen and Y. W. Chen, Int. J. Hydrogen Energy, 2007, 32, 86 CrossRef CAS.
- L. X. Xu, L. X. Sang, C. F. Ma, Y. W. Lu, F. Wang, Q. W. Li, H. X. Dai, H. He and J. H. Sun, Chin. J. Catal., 2006, 27, 100 CrossRef CAS.
- Z. Liu, W. Hou, P. Pavaskar, M. Aykol and S. B. Cronin, Nano Lett., 2011, 11, 1111 CrossRef CAS.
- J.-J. Chen, J. C. S. Wu, P. C. Wu and D. P. Tsai, J. Phys. Chem. C, 2011, 115, 210 CAS.
- Z. Zheng, B. Huang, X. Qin, X. Zhang, Y. Dai and M.-H. Whangbo, J. Mater. Chem., 2011, 21, 9079 RSC.
- M. M. Shahjamali, M. Bosman, S. W. Cao, X. Huang, S. Saadat, E. Martinsson, D. Aili, Y. Y. Tay, B. Liedberg, S. C. J. Loo, H. Zhang, F. Boey and C. Xue, Adv. Funct. Mater., 2012, 22, 849–854 CrossRef CAS.
- X. Huang, X. Y. Qi, Y. Z. Huang, S. Z. Li, C. Xue, C. L. Gan, F. Boey and H. Zhang, ACS Nano, 2010, 4, 6196–6202 CrossRef CAS.
- H. W. Gao, C. Liu, H. E. Jeong and P. Yang, ACS Nano, 2012, 6, 234 CrossRef CAS.
- E. Thimsen, F. Le Formal, M. Gratzel and S. C. Warren, Nano Lett., 2011, 11, 35 CrossRef CAS.
- Y. Wang, Y. H. Shen, A. J. Xie, S. K. Li, X. F. Wang and Y. Cai, J. Phys. Chem. C, 2010, 114, 4297 CAS.
- S. W. Cao, Z. Yin, J. Barber, F. Y. Boey, S. C. Loo and C. Xue, ACS Appl. Mater. Interfaces, 2012, 4, 418 CAS.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212 CrossRef CAS.
- Y. H. Wei, S. B. Han, D. A. Walker, S. C. Warren and B. A. Grzybowski, Chem. Sci., 2012, 3, 1090 RSC.
- P. Li, Z. Wei, T. Wu, Q. Peng and Y. D. Li, J. Am. Chem. Soc., 2011, 133, 5660 CrossRef CAS.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211 CrossRef CAS.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS.
- S. Y. Gao, K. Ueno and H. Misawa, Acc. Chem. Res., 2011, 44, 251 CrossRef CAS.
- C. Novo and P. Mulvaney, Nano Lett., 2007, 7, 520 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional characterization data. See DOI: 10.1039/c2ra20405c |
| This journal is © The Royal Society of Chemistry 2012 |