Natural deep eutectic salt promoted regioselective reduction of epoxides and carbonyl compounds
Najmadin
Azizi
*a,
Elham
Batebi
a,
Said
Bagherpour
a and
Hossein
Ghafuri
b
aChemistry and Chemical Engineering Research Centre of Iran, P.O. Box 14335-186, Tehran, Iran. E-mail: azizi@ccerci.ac.ir; Fax: +98 21 44580762
bDepartment of Chemistry, Payam Noor University-Damavand Branch, Tehran, Iran
First published on 18th January 2012
Abstract
A simple, efficient and green protocol for the regioselective and chemoselective reduction of epoxides and carbonyl compounds, with sodium borohydride in natural deep eutectic in good to excellent yields, is described.
Recent years have witnessed a major drive to increase the efficiency of organic transformations while lowering the amount of waste materials. Solvents play a central role in these efforts: solvents are often the largest sources of wastes in chemical syntheses and processes. Eliminating the use of solvents can dramatically reduce the amount of waste and volatile organic compound emissions that are produced in a process. Recent endeavors have focused on limiting the use of organic solvents and replacing them with new, environmentally benign media. Room temperature ionic liquids have attracted considerable attention as novel reaction media over the last decade. By virtue of their unique properties, such as non-flammability, chemical and thermal stability, and outstanding solvation ability and negligible vapour pressure, ionic liquids have been proposed as alternative solvents receiving serious consideration with the promise of both environmental and technological benefits. Unfortunately, cost, toxicity for some aquatic species and high purity are the main disadvantage of these green solvents which has limited chemists and industry. On the other hand, deep eutectic solvents (DESs)—eutectic mixtures of an ammonium salt and a hydrogen-bond donor compound such as urea, an acid, or an amine—which were developed by Abbott and co-workers, are alternatives to ionic liquids. These eutectic mixtures are attractive alternatives to room temperature ionic liquids (RTILs), as DESs can be less expensive, more synthetically accessible, nontoxic, and biodegradable. Furthermore, choline ([Ch][Cl]) and urea are both naturally occurring biocompatible compounds that are not hazardous if they are released back into nature. Besides, they are cheap and the processes that use these deep eutectic solvents are economically viable and green.1
The reduction of carbonyl compounds and reductive cleavage of epoxides to the corresponding alcohols are important transformations in organic synthesis2 and the total synthesis of biologically active compounds.3 A variety of reducing systems such as borohydride derivatives, organosilanes, organotin hydrides and a Hantzsch dihydropyridine system have been recently reported.4 Sodium borohydride and their derivatives are inexpensive, safe to handle, and environmentally friendly reducing agents in organic synthesis, which rapidly reduce aldehydes, ketones, and acid chlorides to alcohols.5 However, the reductions have disadvantages for practical utility, such as requiring long reaction times, limitation to carbonyl groups, and common employment of flammable solvents, like methanol and 2-propanol.
Throughout of our investigations to develop green organic chemistry by using water as reaction medium or by performing organic transformations under solvent-free conditions,6 herein we report the first example of an efficient and green procedure for chemoselective reduction of functionalized carbonyl compounds and epoxides with commercially available sodium borohydride in deep eutectic solvents as a novel and green catalyst and reaction media.
The experimental procedure is very simple and easy. In a typical experiment, benzaldehyde 1 (1 mmol) was treated with NaBH4 (2 mmol) in urea/choline chloride eutectic salt (1 mL) in the absence of any catalyst at room temperature. After 5 min, the benzaldehyde was consumed and the corresponding benzyl alcohol 2 was formed as the only detectable product and isolated in 99% yield (Scheme 1). To test the feasibility of a large-scale reaction, 1 (20 mmol) was treated with NaBH4 (30 mmol) urea/choline chloride eutectic salts (5 mL) at room temperature and the product was isolated in 95% yield after 20 min.
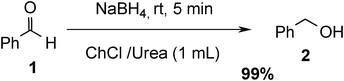 | ||
| Scheme 1 Optimization of reaction conditions. | ||
Under optimized reaction conditions, the scope and limitations of this simple process were explored by using a wide range of carbonyl compounds. A variety of structurally diverse aldehydes and ketones, including saturated, unsaturated, aromatic and heteroaryl aldehydes underwent green reduction smoothly without using any catalyst to afford the corresponding alcohol derivatives in excellent to quantitative yields. The results were summarized in Fig. 1. Aryl aldehydes substituted with various electron withdrawing and donating groups, as well as hetero-aryl aldehydes did not seem to influence the reduction time and yields, as revealed by the similarity of the results, and all carbonyl groups readily converted into their corresponding alcohols in the presence of a variety of functional groups, including carbon–carbon double bonds, halides, chlorides, epoxides and esters. Aliphatic aldehydes and simple ketones were also reduced to their corresponding alcohols efficiently. Furthermore, the presence of ortho-substituents did not hinder the reaction and yields of the reduction as manifested from the reaction conditions. Aryl ketones such as benzophenone and imine were not reduced under similar conditions despite a great deal of variation in experimental conditions.
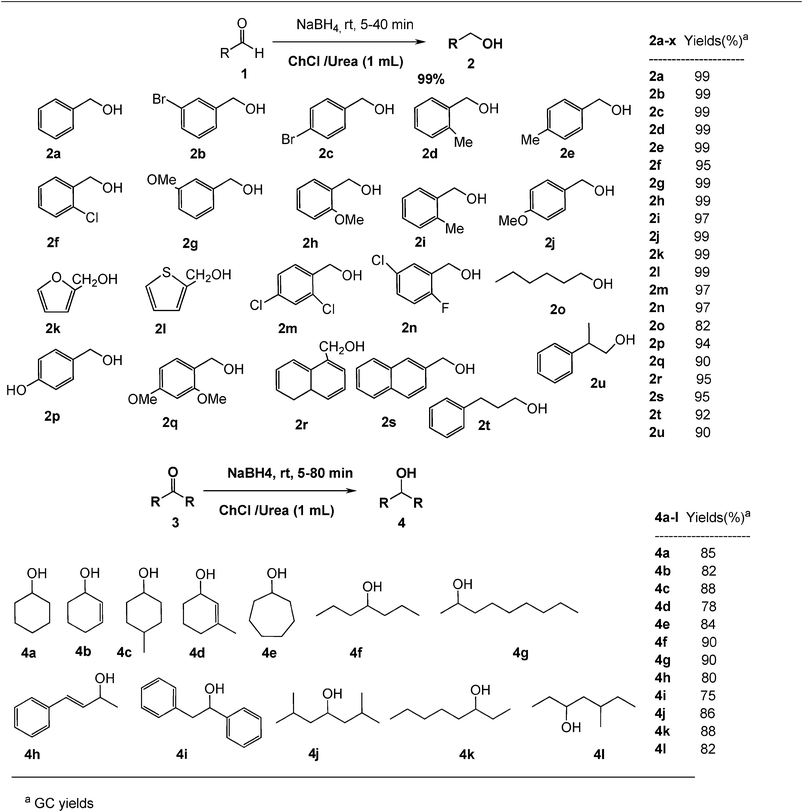 | ||
| Fig. 1 Green reduction of carbonyl compounds in deep eutectic solvent. | ||
We further explored the potential of this procedure for the synthesis of alcohol derivatives from epoxides. Catalytic regio-and stereo-selective reductive cleavage of epoxides to the corresponding alcohols is one of the most useful reactions in the organic synthesis7 and considerable effort has focused on the development of mild methods for regioselective cleavage of epoxide with conventional reducing agents in the literature.8 The reductive cleavage of epoxides requires the electrophilic assistance of a reagent, which can either be a Lewis acid or good reducing agent such as A1H3. Reduction of epoxides by borohydrides is very slow unless reactions were carried out in the presence of strong Lewis acid or promoter. The regioselectivity of the opening of dissymmetrical epoxides depends essentially on the strength of the Lewis acid–base interaction between the partners. If this interaction is rather weak, then the reduction takes place at the epoxide's least substituted carbon, and the mechanism of the reaction is SN2 assisted by the Lewis acid; with a stronger Lewis acid, the regioselectivity is reversed, and the reduction takes place at the most substituted epoxide carbon. The hydride preferentially attacks the carbon that is better able to stabilize a carbocation. As a literature survey shows, there are no reports concerning catalyst-free reduction of epoxides by borohydrides in green reaction media. Thus, the development of a novel and simple catalytic method for a mild direct reductive cleavage in deep eutectic solvent is an important research goal in today's research and development.
Treatment of glycidyl phenylether 5 (1 mmol) with NaBH4 (2 mmol) in urea/choline chloride eutectic salt (1 mL) resulted in reductive cleavage of epoxides to the corresponding alcohols in excellent yields. The reaction was carried out with a very simple procedure in deep eutectic solvent at 60 °C under mild reaction conditions and excellent yields (Scheme 2).
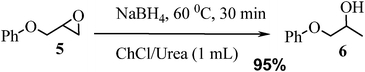
The high yield, simple reaction protocol and environmentally friendliness of this green process prompted us to explore the reaction for sterically, electronically and functionally diverse epoxides under the same reaction conditions, and the results of this investigation are shown in Table 1. The reactions proceeded smoothly with the almost all commercially available epoxides such as glycidyl phenyl ether, allyl glycidyl ether, isopropyl glycidyl ether, propylene oxide, butane oxide, cyclohexene oxide and styrene oxide. The results indicate the usefulness of this method. Unsymmetrical oxiranes such as glycidyl phenyl ether, propylene oxide, butane oxide and glycidyl 2-metheylphenyl ether underwent cleavage by NaBH4 with preferential attack at the less substituted carbon of the epoxide, affording a single product in high to quantitative yields. This process was also chemoselective for epichlorohydrin and epibromohydrin with two reaction positions, which resulted in the formation of the corresponding alcohols by the nucleophilic attack at the terminal carbon of the epoxides, Table 1. The only exception is styrene oxide, in which two regioisomers were formed in the ratio of 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12. The regioselectivity was determined by 1H NMR and by comparison with the known alcohols.9
:12. The regioselectivity was determined by 1H NMR and by comparison with the known alcohols.9
















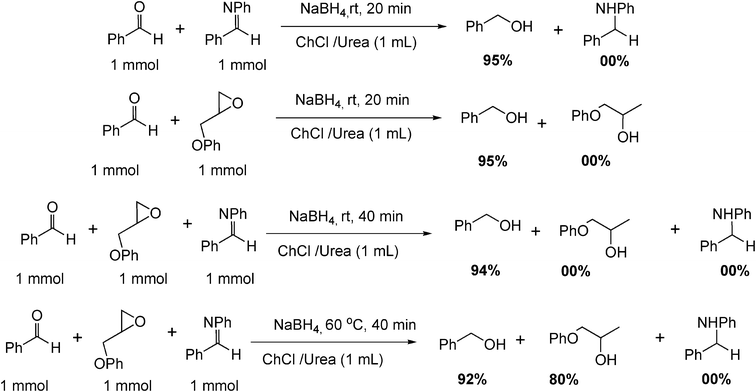
Differentiation in the reduction between aldehyde, imine and epoxide in competitive reactions is an important task in organic synthesis and in fact, only aldehyde reduction product was obtained in this reaction media, which demonstrated that the reaction in deep eutectic solvent had a good chemoselectivity (Scheme 3).
Conclusions
We have developed an efficient and mild method for the green reduction of epoxides and carbonyl compounds in deep eutectic solvent in good to excellent yields. The reductions provided not only high chemoselectivity for functionalized aldehydes and ketones including other reducible functional groups, but also high regioselectivity for α,β-unsaturated carbonyl compounds and imines to give only the corresponding allylic alcohols. Additional applications of deep eutectic solvent in organic synthesis are currently under investigation.10References
- (a) A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70 RSC; (b) I. Mamajanov, A. E. Engelhart, H. D. Bean and V. Hud, Angew. Chem., Int. Ed., 2010, 49, 6310 CrossRef CAS; (c) Z. Chen, B. Zhou, H. Cai, W. Zhu and X. Zou, Green Chem., 2009, 11, 275 RSC; (d) Z. Chen, W. Zhu, Z. Zheng and X. Zou, J. Fluorine Chem., 2010, 131, 340 CrossRef CAS; (e) K. Haerens, E. Matthijs, K. Binnemans and B. Van der Bruggen, Green Chem., 2009, 11, 1357 RSC; (f) J. T. Gorke, F. Srienc and R. J. Kazlauskas, Chem. Commun., 2008, 1235 RSC; (g) C. A. Angell, Y. Ansari and Z. F. Zhao, Faraday Discuss., 2012, 154, 9 RSC; (h) Z. Maugeri and P. Domínguez de María, RSC Adv., 2012, 2, 421 RSC; (i) A. P. Abbott, G. Frisch, H. Garrett and J. Hartley, Chem. Commun., 2011, 47, 11876 RSC.
- (a) N. Greeves, In Comprehensive Organic Synthesis; B. M. Trost, I. Fleming, ed. Pergamon Press: Oxford, 1991; vol. 9, pp. 1 Search PubMed; (b) B. C. Ranu, A. Sarkar, S. K. Guchhait and K. Ghosh, J. Ind. Chem. Soc., 1998, 690 CAS.
- (a) M. Kidwai, P. Mothsra, R. Mohan and S. Biswas, Bioorg. Med. Chem. Lett., 2005, 15, 915 CrossRef CAS; (b) M. Kidwai, S. Saxena, R. Mohan and R. Venkataramanan, J. Chem. Soc., Perkin Trans. 1, 2002, 1845 RSC.
- (a) B. C. Ranu and A. R. Das, J. Org. Chem., 1991, 56, 4796 CrossRef CAS; (b) A. P. Brogan, T. J. Dickerson and K. D. Janda, Chem. Commun., 2007, 4952 RSC; (c) M. Kidwai, V. Bansal, A. Saxena, R. Shankar and S. Mozumdar, Tetrahedron Lett., 2006, 47, 4161 CrossRef CAS; (d) K. Kamiura and M. Wada, Tetrahedron Lett., 1999, 40, 9059 CrossRef CAS; (e) B. T. Cho, S. K. Kang, M. S. Kim, S. R. Ryu and D. Keun An, Tetrahedron, 2006, 62, 8164 CrossRef CAS; (f) B. M. Choudary, M. L. Kantam, A. Rahman and C. V. Reddy, J. Mol. Catal. A: Chem., 2003, 206, 145 CrossRef CAS; (g) K. Yasutaka, M. Shuichi and T. Kazunobu, Synlett., 2008, 2523 Search PubMed; (h) B. C. Ranu and S. Samanta, J. Org. Chem., 2003, 68, 7130 CrossRef CAS; (i) B. C. Ranu, J. Dutta and S. K. Guchhait, Org. Lett., 2001, 3, 2603 CrossRef CAS; (j) B. C. Ranu and R. Chakraborty, Tetrahedron Lett., 1990, 31, 7663 CrossRef CAS; (k) D. C. Sarkar, A. R. Das and B. C. Ranu, J. Org. Chem., 1990, 55, 5799 CrossRef CAS.
- (a) L. Paquette, Reagents for Organic Synthesis; Ed.; Wiley: New York, NY, 1995; vol. 7, pp. 4522–4528 Search PubMed; (b) H. C. Brown and S. Krishnamurthy, Tetrahedron, 1979, 35, 567 CrossRef CAS.
- (a) N. Azizi and M. R. Saidi, Organometallics, 2004, 23, 1457 CrossRef CAS; (b) N. Azizi and M. R. Saidi, Org. Lett., 2005, 7, 3649 CrossRef CAS; (c) N. Azizi, F. Aryanasab, L. Torkiyan, A. Ziyaei and M. R. Saidi, J. Org. Chem., 2006, 71, 3634 CrossRef CAS; (d) N. Azizi, L. Torkiyan and M. R. Saidi, Org. Lett., 2006, 8, 2079 CrossRef CAS; (e) N. Azizi, F. Aryanasab and M. R. Saidi, Org. Biomol. Chem., 2006, 4, 4275 RSC.
- R. C. Larock, Comprehensive Organic Transformations, VCH, New York, 1989, pp. 505 Search PubMed.
- (a) V. N. Telvekar and R. A. Rane, Synth. Commun., 2010, 40, 2108 CrossRef CAS; (b) N. M. Yoon, H. C. Brown and W. E. Lamke, J. Org. Chem., 1967, 32, 537 CrossRef; (c) H. C. Brown and N. M. Yoon, Chem. Commun., 1968, 1549 RSC; (d) Y. Yamamoto, H. Toi, A. Sonoda and S. I. Murahashi, J. Chem. Soc., Chem. Commun., 1976, 672 RSC; (e) Y. D. Vankar, P. S. Arya and C. T. Rao, Synth. Commun., 1983, 13, 869 CrossRef CAS; (f) R. O. Hutchins, I. M. Taffer and W. Burgoyne, J. Org. Chem., 1981, 46, 5214 CrossRef CAS; (g) Y. R. Santosh and D. S. Iyengar, Synth. Commun., 1997, 27, 1731 CrossRef; (h) E. Thiery, J. L. Bras and J. Muzart, Green Chem., 2007, 9, 326 RSC; (i) J. Ekstrçm, J. Wettergren and H. Adolfsson, Adv. Synth. Catal., 2007, 349, 1609 CrossRef; (j) B. C. Ranu, Synlett, 1993, 885 CrossRef CAS; (k) B. C. Ranu and A. R. Das, J. Chem. Soc., Chem. Commun., 1990, 1334 RSC.
-
General procedure
: A mixture of carbonyl compounds or epoxides (1 mmol), in urea choline chloride (2:1) (1 mL) ionic liquid, NaBH4 (2 mmol) was added into a test tube with a magnetic stirring bar. The test tube was stirred at room temperature for carbonyl compounds at 5–80 min and heated in an oil bath at 60 °C for 30–180 min for epoxide and then was cooled to room temperature slowly and, the reaction was quenched with water and extracted with diethyl ether. The organic phase was washed with water and saturated aqueous sodium chloride. It was then dried over magnesium sulfate, and was concentrated under vacuum (rotary evaporator). The crude product was analyzed by NMR and GC and in the most cases pure products were obtained. In some cases the crude product was purified via chromatography to give the corresponding compounds. All compounds were characterized on the basis of their spectroscopic data (IR, NMR, and MS) and by comparison with those reported in the literature.
- The general route for the synthesis of the ionic liquids was as follows: Choline chloride (100 mmol) was mixed with urea (200 mmol) andheatedto ca. 100 °C in air with stirring until a clear colourless liquid was obtained.1.
| This journal is © The Royal Society of Chemistry 2012 |