 Open Access Article
Open Access ArticleContrasting cellular uptake pathways for chlorido and iodido iminopyridine ruthenium arene anticancer complexes†
Isolda
Romero-Canelón
,
Ana M.
Pizarro
,
Abraha
Habtemariam
and
Peter J.
Sadler
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: p.j.sadler@warwick.ac.uk; Fax: +44 (0)24 7652 3819; Tel: +44 (0)24 7652 3818
First published on 9th November 2012
Abstract
The pathways involved in cellular uptake and accumulation of iminopyridine complexes of general formula [Ru(η6-p-cymene)(N,N-dimethyl-N′-[(E)-pyridine-2-ylmethylidene]benzene-1,4-diamine)X]PF6 bearing two different halido ligands X = Cl or I, have been explored. The ratio of passive/active cellular accumulation of Ru in A2780 human ovarian cancer cells is compared and contrasted with cisplatin. Also, saturation of cellular uptake, time-dependence of cellular influx/efflux equilibria, together with endocytotic pathways such as caveolae and facilitated diffusion are investigated and discussed. Temperature dependence studies of Ru accumulation in the A2780 cells show that in contrast to cisplatin (CDDP) and chlorido complex 1, which are taken up largely through active transport, the iodido complex 2 enters cells via passive transport. The cellular efflux of Ru is slow (ca. 25% retained after 72 h) and is partially inhibited by verapamil, implicating the P-gp protein in the efflux mechanism. Ouabain inhibition experiments suggest that the cellular uptake of these ruthenium complexes relies at least in part on facilitated diffusion, and in particular is dependent on the membrane potential. In addition the finding that depletion of cellular ATP with antimycin A had little effect on cellular Ru accumulation from iodido complex 2 is consistent with passive diffusion. In contrast, ATP depletion caused a major increase in cellular accumulation of ruthenium from chlorido complex 1.
Introduction
Ruthenium arene anticancer complexes are being widely studied as potential alternatives to platinum chemotherapeutics,1–9 especially because acquired resistance to platinum-based drugs represents a major clinical drawback for compounds such as cisplatin (CDDP)10,11 and oxaliplatin (OXA).12,13 This type of resistance usually develops as a consequence of impaired cellular accumulation that can be caused by lower cellular uptake or increased cellular efflux.10,14,15 There is much current interest in understanding the mechanisms of cellular uptake for CDDP and platinum accumulation in cells, including the role of the copper transport protein CTR1.16–18 However, much less is known regarding the mechanisms involved in the cellular accumulation of ruthenium anti-cancer drugs. Deeper understanding of these pathways could contribute to rational design and further improvement of such complexes. In general, although not always, cell accumulation of a drug is directly related to potency. In the work reported here, the cellular uptake and metal accumulation mechanisms are compared for chlorido and iodido iminopyridine ruthenium(II) arene anticancer complexes, including time-, concentration- and temperature-dependence as well as extent of metal efflux. The effects on metal accumulation investigated include (a) the role of the Na+/K+ pump, as a facilitated diffusion endocytosis pathway, (b) membrane disruption by amphotericin B as a model for protein-mediated transport, (c) the role of the caveolae endocytosis pathway, as well as (d) the role of the copper uptake protein CTR1, and (e) ATP depletion. We have also investigated P-gp glycoprotein-mediated efflux. For these studies, two isostructural complexes 1 [Ru(p-cym)(ImpyNMe2)Cl]PF6 and 2 [Ru(p-cym)(ImpyNMe2)I]PF6 which only differ in their monodentate halido ligand were selected so that the specific effect on the cellular uptake processes of complexes bearing either a chloride or iodide as a monodentate ligand could be studied.Experimental
Materials
The dimer [Ru(η6-p-cymene)Cl2]Cl2,19,20 and complexes 1 and 2 (Fig. 1) were synthesised and characterised as reported.21 Verapamil hydrochloride (≥99.0%), ouabain octahydrate (≥95%), antimycin A from Streptomyces sp., amphotericin B from Streptomyces sp., methyl β-cyclodextrin and copper(II) chloride dihydrate (≥99.0%) were purchased from Sigma-Aldrich. | ||
| Fig. 1 Structures of complexes 1 and 2. | ||
Cell culture
The A2780 ovarian cancer cell line was obtained from the ECACC (European Collection of Animal Cell Culture, Salisbury, UK). The cells were maintained in RPMI 1640 medium which was supplemented with 10% fetal calf serum, 1% L-glutamine and 1% penicillin/streptomycin. All cells were grown at 310 K in an humidified atmosphere containing 5% CO2.In vitro growth inhibition assay
The antiproliferative activity of complexes 1, 2 and CDDP was determined in the A2780 ovarian cancer cell line. Briefly, 96-well plates were used to seed 5000 cells per well; they were left to pre-incubate in drug-free medium at 310 K for 48 h before adding various concentrations of the compounds to be tested. A drug exposure period of 24 h was allowed. After this, the supernatants were removed by suction and each well was washed with PBS. A further 72 h was allowed for the cells to recover in drug-free medium at 310 K. The SRB assay was used to determine cell viability. This assay, first developed by Skeham et al. in 1990,22 is based on the ability of the sulforhodamine B to bind electrostatically to basic amino acid residues of proteins from fixed cells. Absorbance measurements of solubilised dye allow the determination of the amount of viable treated cells against an untreated control. These measurements were carried on a BioRad iMark microplate reader using a 470 nm filter. IC50 values, as the concentration which cause 50% inhibition of cell growth, were determined as duplicates of triplicates in two independent sets of experiments, and their standard deviations calculated.Metal accumulation in cancer cells
Metal accumulation studies for complexes 1, 2 and CDDP were conducted on the A2780 ovarian carcinoma cell line. Briefly, 4 × 106 cells were seeded on a Petri dish; after 24 h of pre-incubation time in drug-free medium, at 310 K in a 5% CO2 humidified atmosphere, the test complexes were added to give final concentrations equal to IC50/3 and then allowed a further 24 h of drug exposure under similar conditions. After this time, cells were treated with trypsin, counted, and cell pellets were collected. Each pellet was digested overnight in concentrated nitric acid (73%) at 353 K; the resulting solutions were diluted to 5% v/v HNO3 using doubly deionised water, and the amount of ruthenium/platinum taken up by the cells was determined by ICP-MS, using an Agilent Technologies 7500 series instrument. The solvent used for all ICP-MS experiments was double deionised water (DDW) with 5% HNO3. Metal standards (Ru/Pt) were freshly prepared before each experiment. The concentrations used for the calibration curve were in all cases 0, 5, 10, 50, 200, 500, 1 × 103, 5 × 103, 10 × 103, 50 × 103, 200 × 103 ppt. The isotopes detected were 101Ru and 195Pt; readings were made in duplicate (He gas and no-gas mode). These experiments did not include any cell recovery time in drug-free medium. They were all carried out in triplicate in two set of independent experiments and the standard deviations were calculated. The statistical significance of all cellular accumulation values was determined as P < 0.05. Results are compared to the corresponding data for CDDP.Further metal accumulation experiments were carried out as described above including the following experimental variations. In all cases, 4 × 106 A2780 cells were seeded in Petri dishes, the pre-incubation time in drug-free medium at 310 K was 24 h, and the drug concentrations used were equipotent and equal to IC50/3 (CDDP = 0.4 μM, complex 1 = 5 μM and complex 2 = 1 μM) unless otherwise stated.
Results
Synthesis and characterization
Ruthenium arene complexes 1 [Ru(p-cym)(ImpyNMe2)Cl]PF6 and 2 [Ru(p-cym)(ImpyNMe2)I]PF6 were prepared via their respective I- or Cl-bridged dimers, as previously reported and were characterized by standard methods.21In vitro growth inhibition assay
The antiproliferative activity of complexes 1 and 2 towards A2780 human ovarian cancer cells was determined using the SRB assay. The iodido complex 2 shows similar potency to CDDP and is ca. 5× more active than the chlorido complex 1 (Table 1).| Compound | IC50a (μM) | Cellular accumulationb (ng Ru/Pt × 106 cells) |
|---|---|---|
| a IC50 is expressed as the concentration of each complex which causes 50% cancer cell growth inhibition. b Total accumulation of Ru/Pt in A2780 cells for complexes 1, 2 and CDDP after 24 h of drug exposure at 310 K with no recovery time. Equipotent concentrations used were 1 = 5 μM, 2 = 1 μM and CDDP = 0.4 μM. | ||
| 1 | 16.2 ± 0.9 | 7.5 ± 0.5 |
| 2 | 3.0 ± 0.2 | 11.9 ± 0.8 |
| CDDP | 1.2 ± 0.2 | 0.25 ± 0.03 |
Metal accumulation and distribution in cancer cells
Total cellular accumulation of ruthenium from complexes 1 and 2 was determined for A2780 ovarian cells using equipotent concentrations at IC50/3. Ruthenium accumulation after 24 h of drug exposure at 310 K in a 5% CO2 humidified atmosphere was lower for complex 1 (7.5 ± 0.5 ng Ru per million cells) than for complex 2 (11.9 ± 0.8 ng per million cells). In contrast the accumulation of Pt from CDDP at an equivalent dose (IC50/3) is ca. 90× less on a molar basis (Table 1).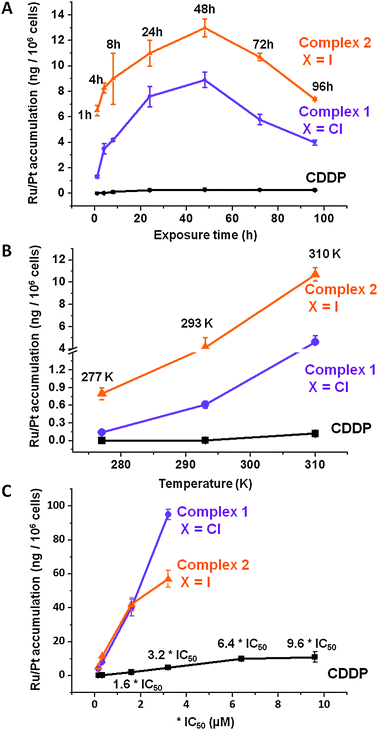 | ||
Fig. 2 (A) Time, (B) temperature, and (C) concentration dependence of Ru/Pt accumulation in A2780 cells for complexes 1 ( ), 2 ( ), 2 ( ) and CDDP (–■–) at 310 K. In all cases pre-incubation time before adding the drug was 24 h with no cell recovery time in drug-free medium, at 310 K, in a 5% CO2 humidified atmosphere. ) and CDDP (–■–) at 310 K. In all cases pre-incubation time before adding the drug was 24 h with no cell recovery time in drug-free medium, at 310 K, in a 5% CO2 humidified atmosphere. | ||
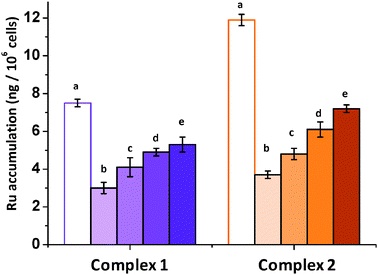 | ||
Fig. 3 Accumulation of Ru/Pt in A2780 cells after co-incubation with complexes 1 ( ), 2 ( ), 2 ( ) and various concentrations of verapamil at 310 K. Results are expressed as ng of metal per 106 cells. Equipotent concentrations used were 1 = 5 μM and 2 = 1 μM. For both complexes, (a) metal accumulation with no recovery time (full extent of efflux), (b) metal accumulation with 24 h recovery time and 0 μM verapamil, (c) 5 μM, (d) 10 μM and (e) 20 μM of verapamil. ) and various concentrations of verapamil at 310 K. Results are expressed as ng of metal per 106 cells. Equipotent concentrations used were 1 = 5 μM and 2 = 1 μM. For both complexes, (a) metal accumulation with no recovery time (full extent of efflux), (b) metal accumulation with 24 h recovery time and 0 μM verapamil, (c) 5 μM, (d) 10 μM and (e) 20 μM of verapamil. | ||
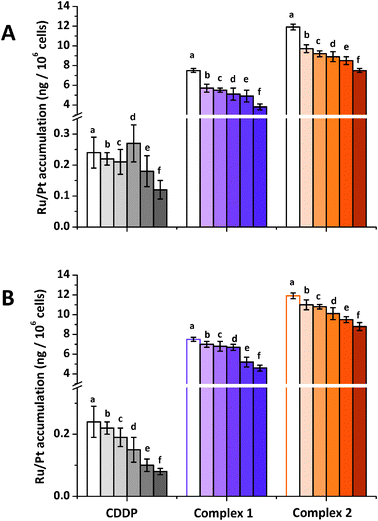 | ||
Fig. 4 Accumulation of Ru/Pt in A2780 cells after co-incubation with CDDP ( ), complexes 1 ( ), complexes 1 ( ) and 2 ( ) and 2 ( ) and various concentrations of (A) ouabain (a) 20 μM, (b) 5 μM, (c) 10 μM, (d) 20 μM, (e) 0.1 mM and (f) 0.2 mM (B) copper(II) (a) 0 μM, (b) 5 μM, (c) 10 μM, (d) 20 μM, (e) 0.1 mM and (f) 0.2 mM. In all cases equipotent concentrations used were CDDP = 0.4 μM, 1 = 5 μM and 2 = 1 μM. ) and various concentrations of (A) ouabain (a) 20 μM, (b) 5 μM, (c) 10 μM, (d) 20 μM, (e) 0.1 mM and (f) 0.2 mM (B) copper(II) (a) 0 μM, (b) 5 μM, (c) 10 μM, (d) 20 μM, (e) 0.1 mM and (f) 0.2 mM. In all cases equipotent concentrations used were CDDP = 0.4 μM, 1 = 5 μM and 2 = 1 μM. | ||
 | ||
Fig. 5 Accumulation of Ru/Pt in A2780 cells after co-incubation with CDDP ( ), complexes 1 ( ), complexes 1 ( ), 2 ( ), 2 ( ) and antimycin at 310 K. Results are expressed as ng of metal per 106 cells. Equipotent concentrations used were CDDP = 0.4 μM, 1 = 5 μM and 2 = 1 μM. For all complexes the antimycin concentrations were (a) 0 μM, (b) 5 μM. ) and antimycin at 310 K. Results are expressed as ng of metal per 106 cells. Equipotent concentrations used were CDDP = 0.4 μM, 1 = 5 μM and 2 = 1 μM. For all complexes the antimycin concentrations were (a) 0 μM, (b) 5 μM. | ||
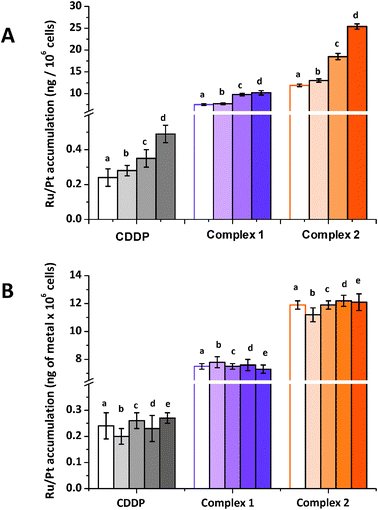 | ||
Fig. 6 Accumulation of Ru/Pt in A2780 cells after co-incubation with CDDP ( ), complexes 1 ( ), complexes 1 ( ), 2 ( ), 2 ( ) and various concentrations of (A) amphotericin B (a) 0 μM, (b) 1 μM, (c) 5 μM and (d) 10 μM, or (B) β-methyl cyclodextrin (a) 0 μM, (b) 10 μM (c) 20 μM, (d) 0.5 mM and (e) 1 mM. All the experiments were carried out at 310 K in a 5% CO2 humidified atmosphere and the equipotent concentrations used were CDDP = 0.4 μM, 1 = 5 μM and 2 = 1 μM. ) and various concentrations of (A) amphotericin B (a) 0 μM, (b) 1 μM, (c) 5 μM and (d) 10 μM, or (B) β-methyl cyclodextrin (a) 0 μM, (b) 10 μM (c) 20 μM, (d) 0.5 mM and (e) 1 mM. All the experiments were carried out at 310 K in a 5% CO2 humidified atmosphere and the equipotent concentrations used were CDDP = 0.4 μM, 1 = 5 μM and 2 = 1 μM. | ||
Discussion
First it is important to consider the aqueous solution behavior of the chlorido complex 1 [Ru(p-cym)(ImpyNMe2)Cl]+ and iodido complex 2 [Ru(p-cym)(ImpyNMe2)I]+. Our investigations by 1H-NMR (reported elsewhere), show that both complexes can undergo aquation; 2 mM solutions generate ca. 64% of the aqua adduct after 24 h at 310 K (Table S1, ESI†).21 However, surprisingly, we found that the iodido complex 2 does not undergo conversion to its chlorido analogue in the presence of physiologically relevant NaCl concentrations and is stable up to 48 h in cell culture medium (NaCl concentration of 108 mM).21 Hence the differences in biological behavior studied here can be reasonably attributed largely to differences in cell recognition and transport of the intact chlorido and iodido complexes. Indeed they exhibit marked differences in their antiproliferative activity. Complex 2 is 5× more potent than complex 1 in A2780 ovarian carcinoma; furthermore, its activity is of similar potency to cisplatin. If the iodido complex was converted into the chlorido complex, this would not be the case. We now discuss the differences in their cell uptake and accumulation pathways investigated in the present work which are likely to contribute greatly to the difference in their potency and mechanism of action. The main emphasis in the present work is on uptake pathways. Species formed by transformation of the complexes in the cytosol, by e.g. aquation and reaction with glutathione, may be involved in the efflux pathways but these are not considered in detail here.Pathways involved in cellular uptake and accumulation
The extent of the Ru accumulation in A2780 ovarian cancer cells was determined using equipotent concentrations (IC50/3) of complexes 1, 2 and CDDP. Results in Table 1 indicate that the accumulation Ru from complexes 1 and 2 is more than one order of magnitude higher than Pt accumulated from CDDP, indicating that Ru accumulation pathways are more efficient than those involved in Pt uptake.Mechanisms of Pt cellular uptake from cisplatin and cellular accumulation have been widely investigated,11,14 however little is known about the analogous pathways involved in the uptake of half-sandwich ruthenium anticancer complexes.24 One of the aims of this study was to gain a more in-depth understanding of this crucial step. It has been reported that CDDP uptake is linear during the first 60 min of drug exposure,25 however the present investigation involves a longer time frame. A2780 ovarian cells were exposed to complex 1, its iodido analogue complex 2 and CDDP over seven different periods of time ranging from 1 to 96 h. In all cases, maximum cellular metal accumulation occurs between 24–48 h of drug exposure, after this time, the amount of accumulated Pt/Ru decreases slightly. The accumulation of Pt from CDDP appears to reach a plateau after 96 h (Fig. 2A).
Active transport, as the movement of a substance across a cell membrane that involves energy consumption, is considered responsible for the uptake of numerous drugs.26,27 Therefore, the temperature dependence of the cellular uptake and accumulation of Ru was also investigated. Experiments were carried out at three different temperatures, 277 K, 295 K and 310 K. CDDP influx does not occur at the lowest temperature (277 K), which is consistent with previous reports that CDDP uptake is energy-dependent.28 As expected for an energy-dependent process, CDDP influx begins at 295 K and increases as the temperature is raised to 310 K (from 0.005 ± 0.002 ng of Pt to 0.12 ± 0.03 ng of Pt per 106 cells). The temperature dependence of Ru accumulation for complexes 1 and 2 is very different. Although an increased uptake accompanies an increase in temperature, only complex 2 exhibited energy-independent uptake at 277 K (0.8 ± 0.1 ng of Ru per 106 cells) (Fig. 2B). Another important factor investigated was the saturation of the cellular uptake with increasing drug concentration (Fig. 2C). Experiments were carried out at equipotent concentrations of complexes 1, 2 and CDDP. Platinum uptake slows down after reaching 6.4 × IC50 concentration values (7.6 μM), but does not reach a plateau. This is consistent with previous reports indicating that CDDP accumulation does not saturate up to 100 μM CDDP.17 Meanwhile, ruthenium complexes 1 and 2 exhibit a much sharper gradient; concentrations of up to 3.2 × IC50 values do not cause saturation of the accumulation pathways. It is also relevant that at concentrations above 6.4 × IC50, total cell death was observed for both Ru complexes 1 and 2. Therefore the concentration of Ru at which saturation occurs could not be established.
Cellular accumulation of metal (Ru/Pt) arises as the result of two important processes: cellular influx and efflux. The latter is especially important in antiproliferative activity determinations that involve a cell recovery period in drug-free medium. The extent of the efflux of Ru for chlorido complex 1 and its iodido analogue 2 was investigated over time (Fig. 3). A2780 ovarian cells were exposed to the ruthenium complexes for 24 h and then left to recover for various periods of time. Although there is a significant efflux during the first 24 h of recovery, the concentration of metal retained in the cells reaches a plateau after 48 h, with no marked difference between 24 and 48 h. Notably, at the longest time point used, at least 25% of the original ruthenium is retained.
One of the most important mechanisms of resistance to anticancer pharmaceuticals involves impaired cellular accumulation as a result of an increased extent of efflux.29 Therefore investigating the mechanism of efflux of a drug can provide insights into the mechanism of resistance. Verapamil, an L-type calcium channel blocker, effectively abrogates P-gp mediated active efflux of anticancer drugs in ovarian cancer cells by competitive inhibition of drug transport, and is capable of reversing multi-drug resistance.30–32 Although it is not fully understood how verapamil interacts with P-gp to decrease cellular efflux, it has been reported that 50 μM of verapamil is capable of restoring doxorubicin sensitivity in MDR cell lines30 by blocking active efflux.33 Accordingly, complexes 1 and 2 were used to investigate the extent of Ru efflux when cells are allowed to recover in drug-free medium that contains verapamil.
By increasing the concentration of verapamil it is possible to impair the efflux of ruthenium complexes 1 and 2 (Fig. 3). This is especially important for chlorido complex 1 which is retained by more than 70% in the presence of 20 μM of this calcium channel blocker. This result is consistent with P-gp involvement in the efflux of complex 1. However, it is remarkable that preliminary molecular docking calculations34 carried out for the N,N-iminopyridine chelating ligand seem to indicate that this is not a P-gp substrate. The use of verapamil to restore the sensitivity of cancer cells to ruthenium arene complexes and involvement of P-gp in resistance has been demonstrated previously.6 In particular, sensitivity to [Ru(η6-p-bip)(en)Cl]PF6 (RM175), is restored by verapamil in adriamycin-resistant A2780AD cells.30 In contrast, verapamil does not restore CDDP sensitivity, as the it is not recognized by P-gp.35–37
Polar molecules are not usually thought to diffuse freely through the cell membrane; instead, they need to rely on membrane proteins or membrane channels to reach the interior of cell compartments. One of these proteins in the plasma membrane is the sodium–potassium adenosine triphosphatase enzyme or Na+/K+ pump which is responsible for maintaining cellular volume and, most importantly, for maintaining the resting potential of the cell.14,17,38,39 The function of this pump is altered by ouabain which reduces the sodium gradient across the cell membrane causing the membrane potential to change.40 There are no previous reports that investigate the effect of co-administering ouabain with ruthenium drugs. To analyse cellular accumulation of Ru under these conditions, A2780 cells were co-incubated with complexes 1, 2 and 5–200 μM of ouabain. Corresponding data for CDDP were also obtained (Fig. 4A). In the case of complexes 1 and 2, co-administration with the cardiac glycoside ouabain impaired cellular Ru accumulation. Ruthenium accumulation from complex 1 is almost halved from 7.5 ± 0.2 ng of Ru to 3.8 ± 0.3 ng of Ru per 106 cells when co-administered with 200 μM of ouabain. Similarly, for complex 2, Ru accumulation decreases by ca. 40% from 11.9 ± 0.3 ng of Ru to 7.2 ± 0.2 ng of Ru per 106 cells when co-administered with the same concentration of ouabain. These results suggest that the cellular uptake of these ruthenium complexes relies at least in part on facilitated diffusion, and in particular is dependent on the membrane potential. As expected from previous reports,17,25 cisplatin accumulation is also reduced with increasing concentrations of ouabain, by about 50% from 0.24 ± 0.05 ng of Pt to 0.12 ± 0.05 ng of Pt per 106 cells when co-administered with 200 μM of the glycoside. Although the mechanism of inhibition is still not clear, it has been proposed that the sodium gradient in the membrane determines the facilitated transport of CDDP into the cells.14,41 This suggests that CDDP transport is dependent on the membrane potential, therefore any agent that affects the electrochemical gradient in the cell could potentially modify CDDP uptake.28 The decrease in CDDP cellular accumulation caused by the action of ouabain on the Na+/K+-ATPase pump,11 is caused by changes in the electrochemical gradient and not because the pump itself transports the drug into the cell.28
Cellular accumulation of CDDP has been linked to copper transport pathways in mammalian cells.42,43 Hence, A2780 cells were co-incubated with complexes 1, 2 and various concentrations of CuII with the aim of investigating whether CTR1 is also involved in the transport of these complexes across the cellular membrane. Corresponding data for CDDP were also obtained for comparison (Fig. 4B). Results suggest that the CTR1 pathway may also be involved in the uptake of ruthenium complexes 1 and 2. Fig. 4A shows that the accumulation of Ru from chlorido complex 1 decreases by 26% in the presence of the highest concentration of copper used (0.2 mM). The results for complex 2 are more striking as Ru accumulation is lowered by 33% when co-administered with 200 μM of CuII (from 11.9 ± 0.3 ng of Ru per 106 cells to 8.8 ± 0.4 ng of Ru per 106). A2780 ovarian cancer cells were also co-incubated with CDDP together with various concentrations of CuII that ranged between 10 μM, and 0.2 mM. Under similar conditions Pt accumulation from CDDP was reduced by ca. 67%, from 0.24 ± 0.05 ng of Pt to 0.08 ± 0.01 ng of Pt per 106 cells in the presence of 0.2 mM CuII, a 40% reduction in accumulation of Pt. These data are consistent with previous reports which indicate that CTR1 regulates CDDP toxicity by regulating CDDP uptake44 and that the expression of CTR1 alters sensitivity to CDDP and other platinum-containing anticancer drugs.45
Although the nature of the interaction of CDDP with CTR1, which facilitates Pt accumulation is poorly understood, recent NMR data show that CDDP binds to the methionine sulfur atoms on extracellular CTR1, to form monosulfur adducts (cis-[PtCl(Met)(NH3)2]+) that may facilitate the transport and activation of the drug.46 Such an interaction may also be involved in the activation and antiproliferative mechanism of other metal based-chemotherapeutic drugs such as ruthenium arene complexes since it is known that complexes such as [Ru(η6-p-bip)(en)Cl]PF6 (RM175) can form cysteine and methionine adducts.47
Some energy dependent pathways can be inhibited by lowering the levels of ATP. Such reduction of ATP concentrations can be achieved by co-administering antimycin A, a mitochondrial ATP synthesis inhibitor that interferes with oxidative phosphorylation by binding to the Qi site of cytochrome c reductase.48–50 Antimycin (1.5 μM) has been reported to achieve 90% depletion of ATP in LLC-PK cells (pig kidney cells) when exposed for 5 h.48 In order to investigate the role of ATP depletion on cellular accumulation of Ru, A2780 cells were co-incubated with complexes 1 or 2 and 5 μM antimycin A. Similar experiments were also carried out using CDDP (Fig. 5).
It is expected that if the cellular uptake of complexes 1 and 2 is ATP-dependent, its depletion should cause a decrease in Ru cellular accumulation. However, only a small, non-significant variation was observed in the cellular accumulation of Ru from complex 2 [Ru(η6-p-cym)(p-Impy-NMe2)I]PF6 (Fig. 5). These results suggest that Ru accumulation from 2 is largely ATP-independent. This is consistent with the significant cellular accumulation observed at low temperatures (277 K, vide supra). In contrast, Fig. 4 also shows that there is a major increase in cellular accumulation of ruthenium from complex 1 [Ru(η6-p-cym)(p-Impy-NMe2)Cl]PF6. This behaviour may be due to the involvement of an ATP-dependent efflux pump in the detoxification of this complex. This could be an ABC transporter, such as the MRP2 pump. The MRP2 pump is inhibited by antimicin-induced ATP depletion; therefore its inhibition could allow intracellular Ru concentrations to increase. The ATP-dependent pump has been reported to be involved in the efflux of CDDP conjugated to glutathione and to multidrug resistance mechanisms.51
Fig. 5 also shows that co-incubation of A2780 cells with CDDP and 5 μM of antimycin A does not reduce Pt accumulation significantly (from 0.24 ± 0.05 ng of Pt per 106 cells to 0.22 ± 0.02 ng of Pt per 106 cells). Even more striking is the report that carboplatin accumulation is inhibited by 90% in the BEL-7404 cell line treated with 50 μg mL−1 of antimycin A.23
Enhanced protein-mediated transport across cell membranes has been reported as a means of increasing cellular accumulation.52 Consequently, the role of protein-mediated transport in the cellular accumulation of Pt and Ru drugs was investigated. A2780 cells were co-incubated with complexes 1, 2 or CDDP and variable concentrations of amphotericin B, which forms pores in the cellular membrane.36,53–55
These pores, permeable to water and non-electrolytes, may give rise to increased drug influx and therefore higher cellular accumulation. Experimental results from co-incubation with amphotericin B (Fig. 6A) show that there is no significant variation in the cellular accumulation of Ru from complex 1, suggesting that facilitated diffusion is not involved in the uptake pathway of this complex. In contrast, cellular accumulation of Ru from complex 2 is doubled from 11.9 ± 0.3 ng of Ru per 106 cells to 25.4 ± 0.6 ng of Ru per 106 cells by the use of 10 μM of amphotericin B. This result is consistent with the results of the temperature-dependent uptake studies (vide supra) that passive diffusion of this complex through the cell membrane is involved.
Fig. 6A also shows that co-incubation with amphotericin B causes Pt accumulation from CDDP to double, from 0.24 ± 0.05 ng to 0.49 ± 0.05 ng of Pt per 106 cells. This is consistent with previous reports that amphotericin B increases CDDP cellular accumulation.14,36 This effect of amphotericin B on CDDP accumulation has been used to reverse Pt resistance in non-small cell lung cancer cells.17 However, when CDDP resistance develops, cells may also develop resistance to amphotericin B, 5-fluorouracil and aphidicolin.56
Finally, the role of the caveolae endocytosis pathway in cellular metal accumulation was explored (Fig. 6B). A2780 cells were co-incubated with complexes 1, 2 or CDDP and increasing concentrations of β-methyl cyclodextrin, a cholesterol-extracting agent which disrupts caveolae.57,58 Results in Fig. 6B, indicate that this endocytosis pathway is not involved either in the uptake of Pt from CDDP nor in the uptake of Ru for complexes 1, 2 as there are no significant changes in cellular metal concentrations with increasing concentrations of β-methyl cyclodextrin.
Conclusions
Possible pathways for the uptake of the organometallic ruthenium arene anticancer complexes 1 [Ru(η6-p-cym)(p-Impy-NMe2)Cl]PF6 and 2 [Ru(η6-p-cym)(p-Impy-NMe2)I]PF6 which differ only in the monodentate halido ligand, have been investigated for A2780 ovarian cancer cells and compared to the clinical platinum drug CDDP. It was demonstrated that maximum Ru accumulation from both Ru complexes occurs between 24 h and 48 h of exposure. Also, complex 2 exhibits partial energy-independent uptake. In contrast CDDP is not taken up at low temperatures (277 K), and its accumulation is greatly enhanced by amphotericin B, a facilitative diffusion agent. Cellular accumulation of ruthenium in A2780 cells was enhanced by inhibition of efflux pathways by verapamil, suggesting that a MDR protein, such as P-gp, could be involved in ruthenium efflux and detoxification. This is also supported by results of co-incubation with antimycin A, which show enhanced accumulation of chlorido complex 1 consistent with inhibition of the MRP2 pump which is ATP-dependent. Changes in the resting membrane potential induced by ouabain were shown to reduce Ru accumulation in A2780 ovarian cancer cells, which suggests that electrochemical gradients can modulate uptake. The CTR1 copper transporter, which is involved in the cellular uptake of CDDP, is likely to be involved as well in the uptake of iodido complex 2. Finally it was shown that the caveolae endocytosis pathway is not involved in the uptake of either of the ruthenium complexes 1 or 2.Although these Ru complexes exhibit very similar aqueous reactivity characteristics, achieving similar extents of hydrolysis after a 24 h period (63–66%), we have shown that both complexes are stable in cell culture medium and that they do not undergo aquation in the presence of high NaCl concentrations.21 Therefore, the differences observed in their uptake pathways are likely to be related to the properties of the halido structures themselves, and not to the common aqua adduct. DFT calculations show that the iodido face of the pseudo-octahedral half sandwich complex 2 is more positively-charged than for the chlorido complex 1.21 This change in polarization may play a major role in the remarkable difference in uptake.
Efflux of the complexes is likely to be affected by possible chemical transformations inside the cells. For example, reactions with glutathione may lead to thiolate as well as sulfenate and sulfinate derivatives,59,60 which may be recognized by efflux transporters, as in the case of products from reaction of CDDP with GSH.61,62 Hence aqua, GSH and complexes with other intracellular biomolecules may play a role in efflux, and will be interesting to investigate in future work.
Acknowledgements
We thank Dr Michael Khan for assistance with cell culture. Ms Ruth McQuitty for assistance with HPLC and members of COST Actions D39 and CM1105 for stimulating discussions. This research was supported by the ERC (grant no. 24363), AWM/ERDF, University of Los Andes, Venezuela (studentship for IR) and IAS University of Warwick, UK.References
- A. Casini, C. G. Hartinger, A. A. Nazarov and P. J. Dyson, Top. Organomet. Chem., 2010, 32, 57–80 CrossRef CAS.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- A. Peacock and P. J. Sadler, Chem.–Asian J., 2008, 3, 1890–1899 CrossRef CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS.
- L. Ronconi and P. J. Sadler, Coord. Chem. Rev., 2007, 251, 1633–1648 CrossRef CAS.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- J. Reedijk, Macromol. Symp., 2008, 270, 193–201 CrossRef CAS.
- P. C. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS.
- C. G. Hartinger, A. D. Phillips and A. Nazarov, Curr. Top. Med. Chem., 2011, 11, 2688–2702 CrossRef CAS.
- H. Timmer-Bosscha, N. H. Mulder and E. G. de Vries, Br. J. Cancer, 1992, 66, 227–238 CrossRef CAS.
- M. Kartalou and J. M. Essigmann, Mutat. Res., 2001, 478, 23–43 CrossRef CAS.
- M. Jones, J. Siracky, L. R. Kelland and K. R. Harrap, Br. J. Cancer, 1993, 67, 24–29 CrossRef CAS.
- D. Trachootham, W. Zhang and P. Huang, Oxidative stress and drug resistance in cancer, Springer US, New York, NY, 2009 Search PubMed.
- M. D. Hall, M. Okabe, D. W. Shen, X. Liang and M. M. Gottesman, Annu. Rev. Pharmacol. Toxicol., 2008, 48, 495–535 CrossRef CAS.
- A. M. Florea and D. Büsselberg, Cancer, 2011, 3, 1351–1371 CrossRef CAS.
- S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298–14302 CrossRef CAS.
- D. P. Gately and S. B. Howell, Br. J. Cancer, 1993, 67, 1171–1176 CrossRef CAS.
- D. Sinani, D. J. Adle, H. Kim and J. Lee, J. Biol. Chem., 2007, 282, 26775–26785 CrossRef CAS.
- M. Bennett and A. K. Smith, Dalton Trans., 1974, 233–241 RSC.
- E. S. Sk, R. A. Zelonka and M. C. Baird, J. Organomet. Chem., 1972, 35, 43–46 CrossRef.
- I. Romero-Canelón, L. Salassa and P. J. Sadler, Submitted.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS.
- D. W. Shen, S. Goldenberg and I. R. A. Pastan, J. Cell. Physiol., 2000, 116, 108–116 CrossRef.
- M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 3, 591–599 RSC.
- V. Cepeda, M. Fuertes, J. Castilla, C. Alonso, C. Quevedo and J. M. Pérez, Anti-Cancer Agents Med. Chem., 2007, 7, 3–18 CrossRef CAS.
- J. Thomas, L. Wang, R. E. Clark and M. Pirmohamed, Blood, 2004, 104, 3739–3745 CrossRef CAS.
- P. D. Dobson and D. B. Kell, Nat. Rev. Drug Discovery, 2008, 7, 205–220 CrossRef CAS.
- P. A. Andrews, S. Velury, S. C. Mann and B. I. Stephen, Cancer Res., 1988, 48, 68–73 CAS.
- M. M. Gottesman, S. V. Ambudkar and D. Xia, Nat. Biotechnol., 2009, 27, 546–547 CrossRef CAS.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS.
- S. G. Aller, J. Yu, A. Ward, Y. Weng, S. Chittaboina, P. M. Harrell, Y. T. Trinh, Q. Zhang, I. L. Urbatsch and G. Chang, Science, 2009, 323, 1718–1722 CrossRef CAS.
- J. Ford and W. Hait, Pharmacol. Rev., 1990, 42, 155–199 CAS.
- J. Cummings, J. S. Macpherson, I. Meikle and J. F. Smyth, Biochem. Pharmacol., 1996, 52, 979–990 CrossRef CAS.
- Z. Bikadi, I. Hazai, D. Malik, K. Jemnitz, Z. Veres, P. Hari, Z. Ni, T. W. Loo, D. M. Clarke, E. Hazai and Q. Mao, PLoS One, 2011, 6, e25815 CAS.
- J. D. Allen, R. F. Brinkhuis, L. V. Deemter, J. Wijnholds and A. H. Schinkel, Cancer Res., 2000, 60, 5761–5766 CAS.
- S. Y. Sharp, P. Mistry, M. R. Valenti, P. Bryant and L. R. Kelland, Cancer Chemother Pharmacol., 1994, 35, 137–143 CrossRef CAS.
- L. Gibalová, M. Sereš, A. Rusnák, P. Ditte, M. Labudová, B. Uhrík, J. Pastorek, J. Sedlák, A. Breier and Z. Sulová, Toxicol. in Vitro, 2012, 26, 435–444 CrossRef.
- J. Q. Chen, R. G. Contreras, R. Wang, S. V. Fernandez, L. Shoshani, I. H. Russo, M. Cereijido and J. Russo, Breast Cancer Res. Treat., 2006, 96, 1–15 CrossRef CAS.
- W. Zhang, H. Takeuchi, M. Kurono and M. Emaduddin, Gen. Pharmacol., 1997, 29, 625–632 CrossRef CAS.
- N. Ramu and R. Gorodetsky, Biochem. Pharmacol., 1991, 42, 1699–1704 CrossRef.
- J. Uozumi and C. L. Litterst, Cancer Chemother Pharmacol., 1995, 15, 93–96 Search PubMed.
- J. Lee, M. J. Petris and D. J. Thiele, J. Biol. Chem., 2002, 277, 40253–40259 CrossRef CAS.
- A. K. Holzer, K. Katano, L. W. J. Klomp and S. B. Howell, Clin. Cancer Res., 2004, 10, 6744–6749 CrossRef CAS.
- A. K. Holzer, G. H. Manorek and S. B. Howell, Mol. Pharmacol., 2006, 70, 1390–1394 CrossRef CAS.
- U. N. Kommuguri, S. Bodiga, S. Sankuru and V. L. Bodiga, J. Trace Elem. Med Biol., 2012, 26, 13–19 CAS.
- X. Wang, X. Du, H. Li, D. S. B. Chan and H. Sun, Angew. Chem., Int. Ed., 2011, 50, 2706–2711 CrossRef CAS.
- F. Wang, H. Chen, J. Parkinson, P. D. S. Murdoch and P. J. Sadler, Inorg. Chem., 2002, 41, 4509–4523 CrossRef CAS.
- M. Venkatachalam, Y. Patel, J. Kreisberg and J. Weingberg, J. Clin. Invest., 1988, 81, 745–758 CrossRef CAS.
- J. Wilson, M. Winter and D. M. Shasby, Blood, 1990, 76, 2578–2582 CAS.
- S. J. Atkinson, M. Hosford and B. Molitoris, J. Biol. Chem., 2004, 279, 5194–5199 CrossRef CAS.
- K. Taniguchi, M. Wada, K. Kohno and M. Kawakami, Cancer Res., 1996, 56, 4124–4129 CAS.
- H. Resat and M. Baginski, Eur. Biophys. J., 2002, 31, 294–305 CrossRef CAS.
- B. Venegas, J. González-Damián, H. Celis and I. Ortega-Blake, Biophys. J., 2003, 85, 2323–2332 CrossRef CAS.
- G. Fujii, J. E. Chang, T. Coley and B. Steere, Biochemistry, 1997, 36, 4959–4968 CrossRef CAS.
- E. Romero, E. Valdivieso and B. E. Cohen, J. Membr. Biol., 2009, 230, 69–81 CrossRef CAS.
- T. Tanaka, H. Kurokawa, K. Matsuno, S. Matsumoto and Y. Hayashida, Anticancer Res., 2008, 28, 2663–2668 CAS.
- D. Garmann, A. Warnecke, G. V. Kalayda, F. Kratz and U. Jaehde, J. Controlled Release, 2008, 131, 100–106 CrossRef CAS.
- R. Yumoto, H. Nishikawa, M. Okamoto, H. Katayama, J. Nagai and M. Takano, Am. J. Physiol-Lung C., 2006, 290, L946–L955 CrossRef CAS.
- F. Wang, J. Xu, A. Habtemariam, J. Bella and P. J. Sadler, J. Am. Chem. Soc., 2005, 127, 17734–17743 CrossRef CAS.
- F. Wang, S. Weidt, J. Xu, C. L. Mackay, P. R. R. Langridge-Smith and P. J. Sadler, J. Am. Soc. Mass Spectrom., 2008, 19, 544–549 CrossRef CAS.
- X. Y. Hao, J. Bergh, O. Brodin, U. Hellman and B. Mannervik, Carcinogenesis, 1994, 15, 1167–1173 CrossRef CAS.
- H. H. W. Chen and M. T. Kuo, Met.-Based Drugs, 2010, 2010 CrossRef , pii:430939.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2mt20189e |
| This journal is © The Royal Society of Chemistry 2012 |