Research highlights
Šeila
Selimović
ab,
Gulden
Camci-Unal
ab,
Mehmet R.
Dokmeci
abc and
Ali
Khademhosseini
*abcd
aCenter for Biomedical Engineering, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, Massachusetts 02139, USA. E-mail: alik@rics.bwh.harvard.edu
bHarvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
cWyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts 02115, USA
dWorld Premier International – Advanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai, 980-8577, Japan
First published on 28th June 2012
Cellular deformability
There is an increasing demand for the identification of healthy or diseased states of cells,1,2 which can be determined by analysing biophysical and biochemical markers or by measuring cellular deformations in response to an external force.3 As such, mechanical tests are traditionally performed on cells by means of bulk or single cell measurements.1 Bulk measurements may yield inaccurate data due to cell size heterogeneity,4 whereas individual cell measurements are more sensitive, yet time-consuming. One way to address these issues is to use cell deformation as an intrinsic marker for determining the state of single cells in a high-throughput manner. In this context, cellular deformability could potentially be used to determine the extent of stem cell differentiation, cellular state of activation, integrity of cytoskeleton, phenotypic changes and diagnosis/prediction of the state of diseases.5 To enable the analysis of single cell mechanics, Di Carlo and colleagues4 have recently developed a robust microfluidic system that can automatically analyze single cell deformations using a label-free approach. The resulting data could be used to determine the disease state of cells, differentiation state of stem cells and rearrangement of organelles.Di Carlo’s cell deformation set-up, termed deformability cytometer, consisted of a poly(dimethylsiloxane) (PDMS) microfluidic device in which cells that were flowing inside a stream were exposed to high strain conditions that induced cellular deformation (Fig. 1). The resulting deformations could be visualized in real time by a high speed video camera that was connected to a computer for sample analysis and characterization. The major advantage of this device is that it performs mechanical measurements two to five orders of magnitude faster than currently available set-ups, significantly decreasing the duration of experiments. In addition, the device operates based on a label-free strategy eliminating the need for expensive, laborious and time consuming antibody labelling steps.
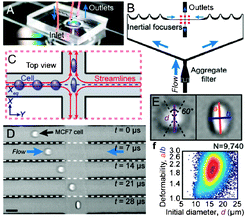 | ||
| Fig. 1 Operating principle of the deformability cytometer. a) Photograph of the device, b) schematic of inertial focusing, c) schematic and d) photographs of focused, deformed cells, e) calculation of shape parameters, and f) cell deformability plot. Figure adapted and reprinted with permission from Gossett et al.4 | ||
The approach used by Gossett et al.4 was based on a simplified version of cytometry principles. The cell suspensions were homogeneously delivered by inertial focusing to the extensional stretching flow area. The high-speed camera was then used to record images of the deformed cells. Initial cell dimensions and deformations were quantified by the image analysis algorithm, followed by automated calculations of the shape parameters. This approach resulted in more reliable and accurate measurements than those obtained using traditional techniques.
The deformability cytometer was able to detect deformation differences in objects with different viscoelasticities. This device was successfully used to identify the differentiation state of embryonic stem cells, diagnose the metastatic nature of leukocytes, observe the activation of peripheral blood mononuclear cells, visualize changes in cytoskeletal arrangement of organelles, and, lastly, identify disease states using blood or pleural fluids cells. Because this cytometer was used to measure an intrinsic property of the cells, the results were not dependent on the sensitivity or power of a detector. Moreover, this device decreased user bias and provided reliable and repeatable results with high statistical confidence.
In summary, the authors developed a deformability cytometer to analyse different cellular states. The results from this work could be used in a clinical setting for identification of diseases, malignancy states, and differentiation of progenitor cells. This method offers a number of advantages over current approaches, including high sensitivity, cost-efficiency, as well as rapid, label-free and high-throughput cell detection. In the future, this could be used for detection of cell deformation upon exposure to hypoxia, identification of blood diseases such as sickle-cell anaemia, or regulation of leukocyte adhesion, capture of endothelial progenitor cells from circulating blood.
An integrated sensor for detection of drug-resistant Tubercle bacilli
Among infectious diseases, tuberculosis (TB) is noteworthy due to the recent emergence of several bacterial strains that are resistant to standard medical drugs. Hence, Tubercullus bacillus has experienced a resurgence both in developed and developing countries, and unfortunately less than 1% of all estimated TB infections are correctly diagnosed and treated.6–8 One of the reasons for such low clinical efficiency is the long time associated with culturing the bacteria from infected cell samples and testing their response to drugs.Soper and colleagues have utilized a combination of microfluidics and nucleic acid amplification tests (NAAT) to develop a genosensing platform, which enables fast and reliable identification of drug-resistant bacterial strains. While such approaches have been utilized previously to detect MDR-TB (multi-drug resistance tuberculosis), in particular in conjunction with glass/silicon microfluidic devices, Wang et al.9 have instead chosen a thermoplastic material and a modular testing platform.
The microfluidic chip was fabricated by heat-embossing polycarbonate (PC). This manufacturing method and the material were cost-effective and yielded microfluidic chips affordable enough to justify one-time use. In addition, their versatile technology enabled the fabrication of chips with different designs, when necessary. Furthermore, PC is compatible with elevated temperatures (65 °C–95 °C), like those required for polymerase chain reactions (PCR). The type of PC used here was also sufficiently flexible to be used for molding microfluidic valves, and strong enough to withstand pressures of up to 105 psi. One drawback of using PC as a material is the relatively strong background during fluorescence imaging, limiting the sensitivity of the detector. For this reason, the testing platform containing the DNA extraction area, solenoid valves, the thermal control area, and other elements was made by heat-embossing poly(methyl methacrylate) (PMMA). PMMA is optically clear and does not promote nonspecific adsorption of molecules, making it useful for fluorescence microscopy. However, specific adsorption via covalent attachment to probe molecules can be induced by plasma-treating PMMA, as done in this study. The assay probes were printed on the platform using a robotic stage for DNA spotting, and the samples – untreated sputum – were loaded manually into the microfluidic chip. The assembled platform was 1 ft3 in size and was completely automated, with the exception of the sample loading stage.
The testing platform was designed to be compatible with both laser-induced fluorescence and colorimetric analysis. In the first case, the presence of drug-resistant bacteria strains was determined by the observation of a fluorescence signal. In heterogeneous populations consisting of both drug-susceptible and -resistant strains, the signal strength was proportional to the percentage of resistant bacteria in the culture. For example, sample populations containing only 1% of resistant bacteria emitted a weaker fluorescence signal than fully resistant populations, yet even this signal was 10-times stronger than the non-specific signal from a fully drug-susceptible bacterial population. In the case of colorimetric assays, nanogold-labeled primers were utilized. When put in contact with the probes, they facilitated the deposition of silver during the assay, generating a visible change in color. This colorimetric version of the platform was sensitive enough to detect resistant bacteria populations as small as 50 cells, similar to its fluorescence counterpart. This level of sensitivity exceeded the precision and accuracy of clinical smear tests by several orders of magnitude, which commonly require at least 5000 bacteria in a 1 ml sample.
The high sensitivity of the presented platform, the simple fabrication process, and the ability to fully automate and complete the assay in 30 min are the most important advantages of this particular microfluidic approach to cell detection and identification. To date, the bacterial resistance to two major TB drugs has been analyzed using this platform. However, it seems straightforward to conduct a variety of assays simultaneously that will help detect and identify bacterial samples resistant to a wider range of drugs, so-called extremely drug-resistant TB (XDR-TB). This application would not require a redesign of the testing platform, only an extended, modular PCR platform and a slightly modified microfluidic chip. Furthermore, this high-throughput multiplexed and automated approach could be useful for detection of a variety of pathogens, such as environmental agents, cancer cells, and even viruses.
High-throughput, high-resolution screening
Screening of drug candidates in the pharmaceutical industry usually begins with a coarse primary screen, in which a set of promising samples are screened; followed by a finer dose-response screen, which is designed to identify the range of drug concentrations suitable for the treatment of diseases. However, the high costs involved in developing these compounds and time constraints limit the number of conditions that can be screened.10–12Droplet-based microfluidic approaches are an advantageous alternative to address issues associated with high costs and long times associated with drug screening.13,14 Namely, each test reaction can be contained in a single pl-sized droplet, which reduces the amount of costly reagents needed for each condition. Also, diffusion-based processes can be completed faster in such small volume reactors than in μl or larger reactors. A team led by Link and Griffiths has recently employed their established droplet-generation technique to screen a library of over 700 compounds against a diabetes and cancer marker, the protein tyrosine phosphatase 1B.
The droplet-generation method used by Miller et al.15 utilized a flow-focusing fluidic structure in combination with a nozzle, such that aqueous droplets could be formed and separated by plugs of fluorinated oil. The droplets contained a mixture of enzymes, a fluorogenic substrate solution, and the compound of interest, such that the concentrations of the first two components were kept constant, but the relative amount of the compounds were varied. The compound was injected into the microfluidic device through a capillary, using a high-performance liquid chromatography autosampler (Fig. 2a). Namely, the autosampler transferred samples from a well-plate into the capillary in a pulsed fashion, such that different samples were separated in the capillary by the buffer stream. As the samples flowed inside the capillary, their interaction with the buffer was subject to Taylor-Aris dispersion (Fig. 2b). In this way, shear flow was utilized to increase the effective diffusion constant of the compounds and aided in their mixing with the buffer. As a consequence, different dilutions of the sample in the buffer were formed and propelled towards the droplet-generating region of the device (Fig. 2c). Here, each of these spread-out pulses of the sample were sheared off into thousands of roughly 140 pl-sized droplets. In this manner, a series of different compound concentrations could be generated in a short amount of time, while utilizing a relatively short fluidic path. In contrast, most other concentration gradient generators that are based on molecular diffusion require dedicated mixing channels and can only supply a limited number of distinct concentrations. Finally, the droplets passed through several analysis points where they were probed with a laser based optical setup. The analysis points are placed such that they were reached after an appropriate reaction time. The different compound concentrations are fluorescently encoded, so that the strength of the signal can be correlated to the sample concentration. Similarly, as the compound reacted with the enzyme, a fluorescent product could be detected, however, at a different wavelength. The collected data was plotted to display enzyme inhibition as a function of sample concentration, yielding at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 data points.
000 data points.
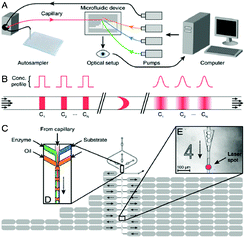 | ||
| Fig. 2 Drug screening system. a) Schematic of the experimental setup, b) concentration profiles of the pulsed compounds in the microfluidic channel, and c) device structure. Figure reprinted with permission from Miller et al.15 | ||
A validation study was conducted by determining the dose–response curve for β-galactosidase and one known inhibitor. The IC50 value, which indicates the compound concentration required to inhibit a biological process by 50%, matched well with the corresponding microplate result. Then, a chemical library containing over 700 different compounds was screened to determine inhibitors of tyrosine phosphatase. Seven inhibitors were identified with IC50 values of less than 20 μM.
Currently, this screening system can analyze 1 compound in 2.5 min, while consuming less than 20 μl. A standard screen consumes 18 times more reagents, yet has a much lower data resolution. In the future, this high-throughput, high-resolution screening system could be adapted to cell-based assays, with only one cell encapsulated in each reaction droplet.
References
- D. H. Kim, P. K. Wong, J. Park, A. Levchenko and Y. Sun, Annu. Rev. Biomed. Eng., 2009, 11, 203–233 CrossRef CAS.
- S. E. Cross, Y. S. Jin, J. Rao and J. K. Gimzewski, Nat. Nanotechnol., 2007, 2, 780–783 CrossRef CAS.
- B. Lincoln, H. M. Erickson, S. Schinkinger, F. Wottawah, D. Mitchell, S. Ulvick, C. Bilby and J. Guck, Cytometry, Part B, 2004, 59A, 203–209 CrossRef.
- D. R. Gossett, H. T. K. Tse, S. A. Lee, Y. Ying, A. G. Lindgren, O. O. Yang, J. Rao, A. T. Clark and D. D. Carlo, Proc. Natl. Acad. Sci. U. S. A., 2012 DOI:10.1073/pnas.1200107109.
- J. Guck, R. Ananthakrishnan, H. Mahmood, T. J. Moon, C. C. Cunningham and J. Kas, Biophys. J., 2001, 81, 767–784 CrossRef CAS.
- D. S. Hadzibegovic, S. A. Maloney, S. T. Cookson and A. Oladele, The International Journal of Tuberculosis and Lung Disease, 2005, 9, 409–414 CAS.
- N. S. Shah, A. Wright, G.-H. Bai, L. Barrera, F. Boulahbal, N. Martín-Casabona, F. Drobniewski, C. Gilpin, M. Havelková, R. Lepe, R. Lumb, B. Metchock, F. Portaels, M. F. Rodrigues, S. Rüsch-Gerdes, A. V. Deun, V. Vincent, K. Laserson, C. Wells and J. P. Cegielski, Emerging Infect. Dis., 2007, 13, 380–387 CrossRef CAS.
- M. Moore, I. M. Onorato, E. McCray and K. G. Castro, J. Am. Med. Assoc., 1997, 278, 833–837 CrossRef CAS.
- H. Wang, H.-W. Chen, M. L. Hupert, P.-C. Chen, P. Datta, T. L. Pittman, J. Goettert, M. C. Murphy, D. Williams, F. Barany and S. A. Soper, Angew. Chem., Int. Ed., 2012, 51, 4349–4353 CrossRef CAS.
- J. Drews, Science, 2000, 287, 1960–1964 CrossRef CAS.
- R. Macarron, Drug Discovery Today, 2006, 11, 277–279 CrossRef.
- A. Dove, Nat. Biotechnol., 1999, 17, 859–863 CrossRef CAS.
- P. S. Dittrich and A. Manz, Nat. Rev. Drug Discovery, 2006, 5, 210–218 CrossRef CAS.
- Y. Wen and S.-T. Yang, Expert Opin. Drug Discovery, 2008, 3, 1237–1253 CrossRef CAS.
- O. J. Miller, A. E. Harrak, T. Mangeat, J.-C. Baret, L. Frenz, B. E. Debs, E. Mayot, M. L. Samuels, E. K. Rooney, P. Dieu, M. Galvan, D. R. Link and A. D. Griffiths, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 378–383 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |