Mutational analysis of phenolic acid decarboxylase from Bacillus subtilis (BsPAD), which converts bio-derived phenolic acids to styrene derivatives†
Annika
Frank
,
William
Eborall
,
Ralph
Hyde
,
Sam
Hart
,
Johan P.
Turkenburg
and
Gideon
Grogan
*
Department of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: gideon.grogan@york.ac.uk
First published on 21st March 2012
Abstract
Phenolic acid decarboxylase from Bacillus subtilis (BsPAD) catalyses the decarboxylation of phenolic acids such as coumaric acid to give vinyl phenols, which are of interest as possible polymer precursors and flavour/fragrance compounds. The structure of the Tyr19Ala mutant of BsPAD has been solved in complex with coumaric acid. In the active site, the substrate carboxylate is bound by Tyr11 and Tyr13, and the phenolic hydroxyl by the NE atom of Arg41. A comparison of the mutant complex with the wild-type apoenzyme reveals that the β1–β2 loop, running from Tyr11 to Ala19, closes over the active site in the presence of substrate, shielding it from bulk solvent. The complex structure, in conjunction with an activity study of point mutants of BsPAD, provides support for a mechanism for PADs, proposed by Mancheño and co-workers for the homologue from Lactobacillus plantarum [Proteins, 2010, 78, 1662–1676]. In this mechanism, a quinone methide intermediate results from deprotonation of the phenolic hydroxyl of the substrate by Glu64, assisted by Arg41. Decarboxylation of the substrate is effected through binding of the carboxylate by Tyr11 and Tyr13, the latter being brought into contact with the substrate as a result of the movement of the β1–β2 loop on substrate binding.
1. Introduction
The enzymatic transformation of organic molecules derived from renewable bio-based feedstocks has been the focus of considerable recent interest, as these processes can give rise to sustainable routes to commodity chemicals1 and also to high-value products of importance to the flavour and fragrance industry.2 One such feedstock is lignocellulose, a complex macromolecular structure consisting of both a carbohydrate component and lignin, which is itself largely composed of subunits derived from para-phenolic acids such as coumaric (CA), ferulic (FA) and sinapic (SA) acids. FA, for example, constitutes up to 4% dry weight of corn hulls,3 and thus has been identified as a cheap and abundant aromatic nucleus from which to derive such high-value compounds as ‘natural-labelled’ vanillin.4 Another common enzymatic transformation of phenolic acids is decarboxylation, which results in vinyl phenol derivatives. Enzymatic decarboxylation is thought to be employed by bacteria as a mechanism to mitigate the accumulation of toxic phenolic acids,5 and is also responsible for the generation of phenolic acid-derived phenols as flavours in alcoholic beverages.6 Enzymes catalysing decarboxylations may also be suitable biocatalysts for the transformation of renewables-based phenolic acids to styrene derivatives for the polymer industry.7 Since the description of a FA decarboxylase from Pseudomonas fluorescens by Rosazza and co-workers in 1994,8 several bacterial species have been identified that catalyse the transformation of phenolic acids such as CA and FA to their vinyl phenol derivatives and many genes encoding the relevant decarboxylases have been cloned and the enzymes characterised (Fig. 1).9–12 One of the best-studied is the phenolic acid decarboxylase from Bacillus subtilis (BsPAD),9 originally characterised by Cavin and co-workers. The enzyme has a molecular weight of 19 kDa and exists in solution as a homodimer. The decarboxylation activity was determined to be independent of metals or other cofactors and KM values of 1.1 and 1.3 mM and Vmax values of 280 and 265 μmol min−1 mg−1 were reported for the enzyme with FA and CA respectively. In subsequent work on the PAD from Klebsiella oxytoca, Hashidoko and Tahara used deuterium-labelled FA substrates to demonstrate that decarboxylation by such PAD-like enzymes proceeded with E selectivity and, given that it was a necessary feature of substrates that they possess a para-hydroxy group, suggested that the mechanism of the enzyme would almost certainly be initiated by deprotonation of the phenolic hydroxyl, to yield a quinone methide intermediate, which would subsequently undergo decarboxylation.13 Structural evidence for this mechanistic hypothesis has awaited X-ray crystallographic studies of PAD-like enzymes obtained in complex with their substrate.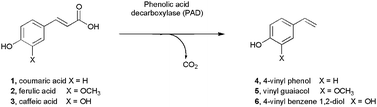 | ||
| Fig. 1 Reactions catalysed by phenolic acid decarboxylases. | ||
The substantial interest in PAD-like enzymes has prompted several structural and mechanistic studies of the enzymes (also known as FA decarboxylase, hydroxycinnamic acid decarboxylase) with a view to establishing the molecular determinants of substrate selectivity and mechanism. Structures of the BsPAD (2P8G) and also the enzyme from Lactobacillus plantarum (LpPAD, 2GC9) were deposited by the Joint Centre for Structural Genomics (JCSG) in 2006 and 2007 respectively. More recently, structures of the phenolic acid decarboxylase from Bacillus pumilus UI-670 (3NAD),14 and the FA decarboxylase from Enterobacter sp. Px6-4 (EFAD, 3NX1, 3NX2),15 have also been published. Each of these PAD-type enzymes shares a high level of amino acid sequence identity, and is structurally superimposable. The monomers each consists of a flattened β-barrel structure that envelops a hydrophobic core, in which are conserved a number of amino acid residues that have been implicated in the catalytic mechanism of the enzymes (Fig. 2). The most extensive structural and mechanistic studies have been carried out by Mancheño and co-workers on LpPAD (2W2A, 2W2B, 2WSJ and 2W2F)16 and Zhang and co-workers on EFAD.15 Mancheño and co-workers were unsuccessful in obtaining a structure of LpPAD in complex with a substrate, but by using a combination of activity assay and structural study of mutants coupled with in silico modelling, they proposed a mechanism for PAD-like enzymes (Fig. 3) in which: 1. The phenolic hydroxyl is deprotonated by Glu71 at the base of the active site to give a quinone methide, assisted by Arg48; 2. The carboxylate is secured by two tyrosine residues Tyr18 and Tyr20, near the entrance to the active site 3. a mobile loop, which closes over the active site in some mutants, must be active as a gating mechanism, in order to restrict entry of water to the cavity. In contrast, Zhang and- co-workers, using EFAD, obtained a complex of that enzyme in which the ligand FA has been trapped in a different conformation in the active site, which suggested that the roles in base-catalysed deprotonation of the phenol, and also in decarboxylation, are fulfilled by alternative residues. In that proposed mechanism, Glu134 (which is non-superimposable with Glu71 in LpPAD) effects phenol deprotonation, and tyrosine 27 (again, non-superimposable with either Tyr18 or Tyr20 in LpPAD) deprotonates the phenolic hydroxyl. In the case of both LpPAD and EFAD, assays of point mutants were used to support the evidence of in silico model or the FA complex structure respectively.
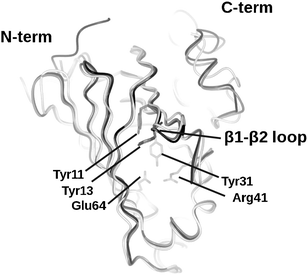 | ||
| Fig. 2 Superimposition of the structures of three PAD enzymes for which coordinates have been deposited within the PDB: BsPAD (2P8G, grey), LpPAD (2GC9, black), and EFAD (3NX2, white), illustrating high levels of structural homology and conservation of flattened barrel structure. Highlighted residues Tyr11, Tyr13, Tyr31, Arg41, Glu64, (numbered for 2P8G) are conserved amongst all the four enzymes within their active sites. | ||
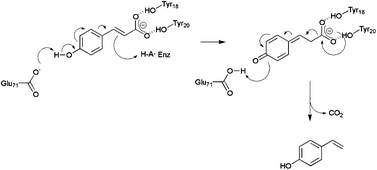 | ||
| Fig. 3 Mechanism of LpPAD-catalysed decarboxylation of CA proposed by Mancheño and co-workers.16 The phenolic carboxylate of the substrate is bound by Tyr18 and Tyr20 in that enzyme. Deprotonation of the phenolic hydroxyl by Glu71 leads to a quinone methide intermediate, which undergoes decarboxylation to form 4-vinyl phenol. | ||
Recent work by other groups has implicated a quinone methide intermediate in the enzymatic transformations of other para-hydroxylated aromatic substrates such as hydroxycinnamoyl CoA and 4-vinylphenol, by the enzymes hydroxycinnamoyl-CoA hydratase-lyase17 and vanillyl alcohol oxidase18 respectively. In a previous study, we showed that even in these two otherwise structurally unrelated enzymes, the molecular determinants of substrate recognition and mechanism were conserved in the active site,17 including a ‘pincer’ formed by two tyrosine residues, which secured the phenolic hydroxyl for deprotonation leading to the quinone methide and an arginine residue that stabilised the resulting phenolate anion. We were interested to see if this mode of catalysis were conserved in other enzymes capable of recognising para-hydroxy phenolics, and, observing the closely associated tyrosine residues in the BsPAD structure, identified this enzyme as a suitable target for investigation. In this study we have obtained a structure of the Tyr19Ala mutant of BsPAD with evidence of a complex with the substrate coumaric acid, which, in conjunction with an activity study of point mutants of the enzyme, appear to support the substrate orientation, role of residues and overall mechanism for PAD-like enzymes proposed by Mancheño for LpPAD.
2. Materials and methods
2.1 Chemicals
All chemicals were obtained from Sigma (Poole, Dorset, U.K.) unless otherwise specified. Purities exceeded 98% for all compounds used. Oligonucleotide primers for PCR were purchased from MWG-Biotech (Ebersburg, Germany). Cloning and expression strains of E. coli were purchased from Novagen/Merck (Nottingham U.K.).2.2 Gene cloning
The gene encoding wild-type (WT) BsPAD was amplified from the genomic DNA of Bacillus subtilis, obtained from Dr Mark Fogg of the York Structural Biology Laboratory, using oligonucleotide primers (Table S1, ESI†) designed for cloning the gene into the YSBLIC-3C vector, using a published procedure.19 PCR conditions were as follows: Initial denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 96 °C for 1 min, primer annealing at 55 °C for 30 s and extension at 72 °C for 90 s. A final extension was carried out for 5 min at 72 °C. The amplification product was purified using a Sigma GenElute™ gel extraction kit prior to cloning directly into the expression vector. The resultant recombinant plasmid contained the decarboxylase gene attached to an N-terminal hexa-histidine tag, allowing for Ni-affinity purification of the expressed protein and its mutants. Point mutants of the BsDCase gene were prepared from the WT gene as template using the Stratagene Quickchange™ kit and mutagenic primers detailed in Table S1 (ESI†). Eight mutant plasmids were prepared, encoding the active site mutants Tyr11Phe, Tyr13Phe, Tyr19Ala, Arg41Ala, Glu64Ala, Thr68Val, Thr98Ala and a Tyr11Phe/Tyr13Phe double mutant. The sequences of wild type and mutant genes were confirmed by nucleotide sequencing carried out in the Technology Facility at the University of York.2.3 Gene expression and protein purification
For gene expression and protein purification, cells of competent E.coli BL21 (DE3) were transformed using the plasmids carrying the WT or mutant decarboxylase genes. LB agar and LB broth used for solid and liquid phase growth routinely contained 30 μg mL−1 kanamycin as antibiotic. In the case of each variant, a single colony was picked and used to inoculate 5 mL of LB broth, which was grown overnight at 37 °C with shaking. The starter cultures were then used to inoculate 500 mL cultures of LB broth in which cells were grown until the optical density (OD600) of the culture had reached approximately 0.6, at which point the expression of BsPAD was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. These cultures were then incubated at 18 °C with in an orbital shaker overnight. After approximately 18 h growth, the cells in each case were harvested by centrifugation at 5000 r.p.m. for 15 min in a Sorvall RC5B Plus centrifuge and were then resuspended in 20 mL 50 mM Tris/HCl buffer pH 7.5 (henceforth referred to as ‘buffer’) containing 300 mM sodium chloride per litre of culture. Cells were disrupted by ultrasonication for 3 × 30 s bursts at 4 °C with 1 min intervals and the soluble and insoluble material fractions separated by centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. The supernatant, containing soluble enzyme(s), was loaded onto a 5ml His-Trap™ Chelating HP Nickel column. After washing with 10 column volumes of buffer containing 30 mM imidazole, enzyme was eluted using a gradient of 30–500 mM imidazole over 20 column volumes of eluant. Column fractions were analysed by SDS PAGE and those containing a pure 19 kDa protein, presumed to be the DCase or its variants, were pooled and concentrated using a 3kD cut-off Centricon® filter membrane. Concentrated proteins were loaded onto an S75 Superdex™ gel filtration column that had been equilibrated with buffer, and eluted 120 mL of the same buffer. Fractions containing pure protein were pooled and the enzyme concentrated to a final concentration of 10–20 mg ml−1 for crystallization experiments and stored at 4 °C. For kinetic assays of the enzymes, protein was flash-cooled in liquid nitrogen after purification, in concentrations ranging from 0.3 to 3 mg mL−1 and stored at −20 °C.
000 rpm for 30 min. The supernatant, containing soluble enzyme(s), was loaded onto a 5ml His-Trap™ Chelating HP Nickel column. After washing with 10 column volumes of buffer containing 30 mM imidazole, enzyme was eluted using a gradient of 30–500 mM imidazole over 20 column volumes of eluant. Column fractions were analysed by SDS PAGE and those containing a pure 19 kDa protein, presumed to be the DCase or its variants, were pooled and concentrated using a 3kD cut-off Centricon® filter membrane. Concentrated proteins were loaded onto an S75 Superdex™ gel filtration column that had been equilibrated with buffer, and eluted 120 mL of the same buffer. Fractions containing pure protein were pooled and the enzyme concentrated to a final concentration of 10–20 mg ml−1 for crystallization experiments and stored at 4 °C. For kinetic assays of the enzymes, protein was flash-cooled in liquid nitrogen after purification, in concentrations ranging from 0.3 to 3 mg mL−1 and stored at −20 °C.
2.4 Assays of WT BsPAD and mutants by HPLC
The activity of WT BsPAD and four of its variants (Tyr11Phe, Tyr13Phe, Tyr19Ala and Thr98Ala) was assessed by HPLC analysis as performed by Mancheño and co-workers.16 Samples were taken from reactions in which the enzymes were incubated with increasing concentrations of ferulic acid as substrate. Enzyme-free buffer solutions with ferulic acid at the same concentrations were used as a negative control and for preparation of a [substrate] calibration curve. Enzyme reactions were prepared by incubating 6 μg (0.35 nmol) of purified protein in 1 mL of buffer containing 0.25–2.5 mM ferulic acid. After incubation for 5 h at 30 °C, 500 μL aliquots were removed from each of the samples and extracted with 500 μL of ethyl acetate. The organic phases were retrieved and evaporated overnight to dryness. Samples were re-dissolved in 200 μL 50% 1:1 methanol:H2O. HPLC analysis was carried out on a Waters Alliance® high performance liquid chromatograph, 50 μL of each sample was injected onto a Techsphere™ C18 column (length 250 mm; internal diameter 4.6 mm; 5 μm particle size) at a temperature of 25 °C and using a flow rate of 1 mL min−1. Running conditions were optimized using substrate and product standards to achieve optimal separation of peaks. Calibration and reaction samples were eluted using an isocratic system of 1:1 water: methanol + 0.1% acetic acid. Each experiment was performed in duplicate. Peaks were detected in a wavelength range from 210–400 nm and evaluated using the Empower™ 3 chromatography data software. Peak heights of ferulic acid and vinyl guaiacol were determined at 280 and 260 nm respectively.
2.5 Protein crystallisation
Purified BsPAD in the absence of ligand was subjected to crystallization trials using a range of commercially available screens in 96-well plates using 300 nL drops at a range of protein concentrations. The best WT protein crystals were obtained using the Clear Strategy Screen (CSS)20 from Molecular Dimensions Ltd., and at a protein concentration of 20 mg mL−1. This screen was subsequently used for crystallisation studies on the mutants. Of the mutants, Tyr19Ala was clearly the most amenable to crystallisation, yielding crystals that, on scale-up, routinely diffracted to 2 Å on in-house detectors. For scale-up of crystallisation, the hanging-drop vapour diffusion method was used in 24-well plate Linbro dishes and using crystallisation drops of 2 μL, and with a protein concentration of 20 mg mL−1. In an attempt to obtain a ligand complex, Tyr19Ala crystals were grown first in the presence of 5 mM CA or 5 mM FA (added from stock solutions dissolved in DMSO). The best results were routinely obtained with crystals grown in the presence of CA, using crystal drops containing 0.2 M KSCN, 17% (w/v) PEG 1000, 17% (w/v) PEG 8000, in 0.1M Tris/HCl buffer at pH 9. Whilst the resulting crystals diffracted well, minimal ligand density was observed in electron density maps after data collection. Increasing the CA concentration to 15 mM resulted in crystals which were of poorer diffraction quality, but gave datasets which clearly showed CA in the active site of the enzyme. Crystals were flash-cooled in liquid nitrogen in a cryogenic solution containing the mother liquor plus 10% glycerol prior to data collection.2.6 Data collection and processing, structure solution and refinement
Crystals of the Tyr19Ala mutant co-crystallised with CA were tested for diffraction using an in-house Rigaku Micromax-007HF fitted with Osmic multilayer optics and a MARRESEARCH MAR345 imaging plate detector. Complete datasets were collected on beamline io-4 at the Diamond Light Source Synchrotron in Oxford on 23rd October 2010. Data were processed on the beamline using the expert data processing system Xia221 and the data collection statistics are given in Table S2, ESI.† The structure of WT BsPAD (pdb accession code 2P8G) deposited by the JCSG was used as a molecular replacement model for structure solution of the Tyr19Ala mutant using MOLREP22 in the CCP4 suite of programmes.23 The initial model had three molecules in the asymmetric unit, which corresponded to one and a half dimers. The model was improved using iterative rounds of building in Coot24 and refinement using REFMAC.25 In refinement, non-crystallographic symmetry (NCS) restraints were applied for chains B and C against chain A. After building the protein and water molecules, clear electron density was observed to persist throughout refinement in the active site of the enzyme, as observed in the Fo–Fc map at a level of 4σ. A coordinate file for CA for use in REFMAC5 was created using the PRODRG server26 and the ligand was modelled into the density in each subunit and refined. The final ligand-bound structure exhibited Rcryst and Rfree values of 20.1 and 26.3% respectively and was finally validated using PROCHECK.27 Refinement statistics are presented in Table S2. The Ramachandran plot showed 93.4% of residues to be situated in the most favoured regions, 5.1% in additional allowed and 1.5% in generously allowed regions. The structure factors and coordinates have been deposited in the Protein Databank with the accession code 4alb.3. Results
3.1 Cloning and expression of genes encoding BsPAD and site-directed mutants and assay of activity
An examination of the active site of BsPAD (2P8G), led to the identification of acid–base residues that might be mutated in order to study their contribution to the mechanism of decarboxylation in PAD-like enzymes. The mutations chosen were Tyr11Phe and Tyr13Phe (and also a variant in which both Tyr11 and Tyr13 were replaced by Phe), homologous to those tyrosines in LpPAD thought to bind the carboxylate of the phenolic acid substrate; Glu64Ala and Arg41Ala, thought to be responsible for securing the phenolic hydroxyl of FA/CA; Tyr19Ala, highlighted by Zhang et al.15 as the residue that binds the carboxylate of sodium ferulate; Thr68Val and Thr98Ala, which, in LpPAD, had been observed to bind a water molecule near to the proposed phenol binding site. Each of those residues was conserved amongst known PAD-like enzymes for which structures have been determined. Whilst attempts were made to determine kinetic constants for the mutants using a UV-spectrophotometric assay reported previously28 those studies were for the most part frustrated both by mutant activity that was too slow to be measurable in a reasonable time-frame or, for reasons of signal saturation, was not measurable owing to the high concentrations of substrate required to reach a maximal velocity (WT BsPAD had been reported to display a KM of 1.1. mM previously).9 In order to estimate the levels of activity of each mutant therefore, HPLC analysis was performed on reactions in which 500 μM of ferulic acid was incubated with 0.35 nmol of each variant for 6 h at 30 °C. An overlay of chromatograms for the reactions is shown in Fig. 4. After 6 h, the control reaction, performed in buffer with no enzyme, displayed no accumulation of the product, 4-vinyl guiacol (VG), whereas in the reaction catalysed by the WT, all the FA had been consumed. Of the other variants, the tyrosine mutants Tyr11Phe (3% VG of that produced by WT) and Tyr13Phe (2%) retained a small fraction of activity and the double tyrosine mutant Tyr11Phe/Tyr13Phe was inactive, consistent with a proposed mechanism in which these residues are essential for activity. Tyr19Ala also displayed low levels of activity, at 4% VG compared to WT. Both Thr68Val (52%) and Thr98Ala (76%) retained substantial, if reduced, activity compared to the WT. Glu64Ala and Arg41Ala mutants were inactive, again supporting a mechanism such as that proposed by Mancheño in which those residues are essential to catalysis. Using this HPLC assay as a basis, attempts were made to determine kinetic constants for the WT and the Tyr11Phe, Tyr13Phe, Tyr19Ala and Thr98Ala mutants (Fig. S1, ESI†), although again, the solubility of ferulic acid at concentrations higher than 2.5 mM prevented data points being recorded at higher concentrations and thus the determination of KM or kcat for any of the variants, as saturation kinetics were not reached in each case. However, the plots suggest that the Thr98Ala mutant possesses similar kinetic characteristics to the wild-type, and that the activity of the Tyr11Phe and Tyr13Phe mutants is severely compromised. The plot for the Tyr19Ala mutant suggests a maximum rate perhaps less than half that of the WT, although neither that quantity, nor the KM, could be absolutely determined for the reasons described above.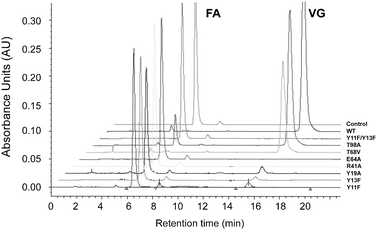 | ||
| Fig. 4 An overlay of chromatograms obtained from HPLC analysis of enzymatic transformations of ferulic acid (FA) to vinyl guaicol (VG) by WT BsPAD and point mutants. | ||
3.2 Structural analysis of the Tyr19Ala mutant of BsPAD
Attempts were made to crystallize the wild-type and mutants of BsPAD both in the presence and absence of either FA or CA as ligand. The poor catalytic activity of some mutants suggested that they might be good candidates for ligand complex studies directed towards trapping a substrate or intermediate in the active site. The best and most reproducible results were obtained using the Tyr19Ala mutant co-crystallised with CA. Crystals of the Y19A mutant grown in the presence of 5 mM CA diffracted routinely to approximately 2.0 Å, but the omit maps obtained from the relevant data did not reveal electron density in the active site of the enzyme. An increase of ligand concentration to 15 mM CA was thus used, and, whilst this led to crystals of inferior diffraction quality (2.7–3.0 Å routinely) gave superior results in respect of ligand occupancy. Thus, the structure of the Tyr19Ala mutant was solved using molecular replacement with the monomer of the 1.38 Å structure 2P8G as a model and refined with Rcryst and Rfree values of 20.1% and 26.3% respectively.The most notable differences between the 2P8G structure and the Tyr19Ala were as follows: First, the P3221 space group was associated with an unusual crystallographic arrangement for PAD-like enzymes studied thus far, and featured one and a half dimers in the asymmetric unit. The dimer structure was arranged as for the biological assembly proposed for the holoenzyme structure, in which the dimer interface was formed by one beta-sheet face of the barrel of each monomer , with an interfacial area of 1172 Å2 and in which the participating monomers interact directly through eight hydrogen bonds and three salt bridges as identified by structure analysis using PISA.29 Second, within the active site cavity, the Fo–Fc map at a level of 4σ clearly revealed the presence of additional electron density not attributable to the protein (Fig. 5). Coumaric acid was modelled into the density and refined. Residual density in the omit map adjacent to the phenolic hydroxyl, and within H-bonding distance of Thr68 and Glu64 was coincident with the water molecule observed in apoenzyme structures of LpPAD reported by Mancheño, albeit at lower resolution than in that study. The resulting substrate complex possessed many of the same structural features as that proposed by the in silico model of CA in the LpPAD active site reported previously.16 In the following description, the residues of the LpPAD are given in brackets to aid comparison. The phenolic hydroxyl of the substrate is H-bonded with the NE atom of Arg41(48) and with the water molecule that is bonded in a cavity formed by the side chains of Thr66 (73), Thr68 (Thr75) and Glu64 (Glu71). Glu64 itself is held in a specific orientation through H-bonds to the phenolic hydroxyl of Tyr31(38), the terminal amino group of Arg41 and the indole nitrogen of Trp62 (69). Hydrophobic residues, Ile33 (40), Val38 (45), Val70 (77), Ile85 (92) and Trp62 (69) surround the aromatic ring of the substrate, and would also include Tyr19 (26), which in this case has been mutated to alanine. The aromatic ring of Tyr31 (36) stacks against that of the substrate. The substrate carboxylate is secured by the phenolic hydroxyls of Tyr11 (18) and Tyr13 (20).
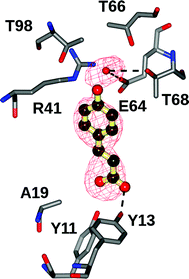 | ||
| Fig. 5 Active site of the Tyr19Ala mutant of BsPAD containing CA. Ligand electron density corresponds to the omit map (Fo–Fc map) contoured at a level of 4σ obtained after the final rounds of refinement in the absence of the ligand. The atom positions of the refined ligand have been added for clarity. | ||
The third major difference between the 2P8G structure and the complex is the movement of the β1–β2 loop running from Tyr11 to Ala19 in the complex structure, which serves both to cover the ligand in the active site, but also to bring Tyr13 into close association with the carboxylate of the substrate (Fig. 6). The other major effect of the loop movement is to bring Asn 15 (22) and Trp17 (24) to occlude the entrance to the active site (Fig. 6). Similar loop movements were observed for structures of the Arg41 (48) and Glu64 (71) mutants of LpPAD16 although residual density in those structures was not of sufficient quality to model the ligand within the active site.
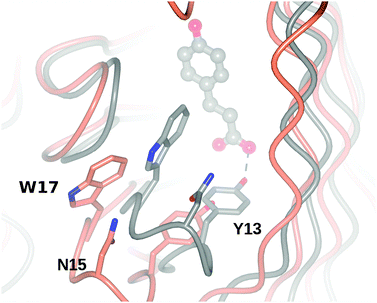 | ||
| Fig. 6 Superimposition of BsPAD apoenzyme (2P8G, coral) with ligand complex of Tyr19Ala mutant (grey), illustrating the movement of the β1–β2 loop that brings Asn15 and Trp17 over the entrance to the active site, and the phenolic hydroxyl of Tyr13 to contact the carboxylate of the substrate. | ||
4. Discussion
The structure and mechanism of PAD-like enzymes are interesting from the perspectives of fundamental microbial metabolism, flavour generation in the food industry and also industrial biotransformation reactions. Further information on structure and mechanism may thus help to explain kinetic phenomena in PADs, such as substrate inhibition, and also to inform protein engineering studies that might improve activity or alter substrate specificity.Mutation studies on PAD-like enzymes previously have yielded only limited quantitative kinetic data, as there are problems with continuous UV assays, and the mutation of key acid–base residues usually results in the massive reduction, or apparent elimination of enzyme activity. However, previous studies on LpPAD16 and BsPAD in this study have clearly demonstrated that the residues Tyr11 and Tyr13, Arg41 and Glu64 in BsPAD are essential to activity in that their hydrophobic point mutants are substantially or completely inactive. The ligand complex of BsPAD with CA in this study reinforces the conclusions drawn from both the results of mutation and the in silico model constructed previously and reveals that the orientation of the substrate, coumaric acid, is congruent with proposed roles for amino acids in the putative mechanism of PAD-like enzymes shown in Fig. 3.
In the mechanism, the phenolic hydroxyl must be deprotonated as a first step in the generation of the quinone methide intermediate. The phenol itself is H-bonded to the NE atom of Arg41, and is hence at this stage presumably still protonated, acting as an H-bond donor to the N atom of the arginine side-chain, which is presumably the first point of recognition for the phenol, and significant enough for mutation of this residue to result in removal of enzyme activity. The phenolic proton would thus be presented to the side-chain of Glu64, which must itself be negatively charged, in order to effect deprotonation. Mancheño has already observed that a negative charge on the carboxylate side-chain of the homologous residue in LpPAD would be stabilised by interactions with the side chains of Arg41 (48) and Trp62 (69), but also Tyr31 (38). The Glu64 carboxylate also contacts a water molecule that is held in a pocket formed by the hydroxyls of Thr66 and Thr68, although the mutation of Thr68 resulted in a mutant of only moderately reduced activity, consistent with a less significant role in the catalytic mechanism. Mutation of Thr98, which is more distant from this water molecule, had a less pronounced effect on activity. It may also be that the positive charge on the Arg41 side chain is able to fulfil a role in stabilization of the phenolate anion, as has been suggested for a structurally conserved arginine in both HCHL17 and VAO18 in the transformation of hydroxycinnamoyl-CoA and vanillyl alcohol respectively. It is interesting to note that in both HCHL and VAO, a pair of tyrosines in the active site has been recruited to carry out deprotonation of the phenolic hydroxyl. It may therefore have been anticipated that the proximal tyrosine side-chains in BsPAD might fulfil equivalent roles, and make contact with the phenol. However, the substrate complex shows that the CA is rotated 180° from that orientation, with Tyr11 and Tyr13 actually interacting with the carboxylate of the substrate. The quinone methide mechanism next requires the conjugated double bond to be protonated. Whilst there are no acid–base residues in the vicinity of the bond in the apo-structure, it has been suggested that the role of proton donor may be fulfilled by a water molecule.16 However, it is notable that the movement of the β1–β2 loop on substrate binding would bring the side chain of Tyr19 (in the wild-type) in close proximity to the double bond. This, together with the pronounced decrease in activity of the Tyr19Ala mutant (4% of wild-type) suggests that Tyr19 might be a candidate residue for investigation as the proton donor at this stage of the mechanism.
The carboxylate of the substrate is secured by Tyr11 and Tyr13. Mutation of one of these tyrosines to phenylalanine results in poor residual activity which must be achieved using only one tyrosine. Replacement of both these residues with Phe results in an inactive mutant, indicative of their fundamental contribution to catalysis. The exact mechanism of decarboxylation is not yet clear, although some indications might be drawn from work on other cofactor-independent decarboxylases. The arylmalonate decarboxylase from Bordetella bronchiseptica, for example, is thought to initiate decarboxylation of its substrates through the presentation of the departing, neutral CO2 to a hydrophobic pocket in the active site.30 The environment adjacent to the carboxyl in BsPAD, and excluding the two tyrosine residues, is certainly hydrophobic, formed by the side-chains of Ile21, the benzene ring of Trp62, Val60 and Leu72 and Phe 74, perhaps providing a hydrophobic pocket for recognition of the CO2 molecule once released.
Conclusions
The structure of BsPAD in complex with coumaric acid provides the first crystallographic evidence for the interactions between the substrate and active site residues in BsPAD in what appears to be a catalytically relevant context. Mutational analysis, coupled with the complex structure, reveals catalytic roles for Tyr11 and Tyr13 in carboxylate recognition and for Arg41 in binding the phenolic hydroxyl of the substrate. The complex structure provides support for the subtsrate orientation, mechanism and roles for active site residues in PAD-like enzymes proposed by Mancheño and co-workers. The complex structure may now be used to identify amino acid residues for further mutation, in order to tackle the problem of substrate inhibition by FA, or indeed to enable the structure to accommodate alternative substrates for extended applications of these enzymes in biocatalytic processes.This project was funded by the European Community under the Marie Curie International Training Network programme ‘BIOTRAINS’ (Project no 238531).
Notes and references
- B. G. Hermann, K. Blok and M. K. Patel, Environ. Sci. Technol., 2007, 41, 7915 CrossRef CAS.
- S. Serra, C. Fuganti and E. Brenna, Trends Biotechnol., 2005, 23, 193 CrossRef CAS.
- M. Hosny and J. P. N. Rosazza, J. Nat. Prod., 1997, 60, 219 CrossRef CAS.
- N. J. Walton, M. J. Mayer and A. Narbad, Phytochemistry, 2003, 63, 505 CrossRef CAS.
- N. P. Tran, J. Gury, V. Dartois, T. K. C. Nguyen, H. Seraut, L. Barthelmebs, P. Gervais and J.-F. Cavin, J. Bacteriol., 2008, 190, 3213 CrossRef CAS.
- N. Vanbeneden, F. Gils, F. Delvaux and F. R. Delvaux, Food Chemistry, 2008, 107, 221 CrossRef CAS.
- I.-Y. Lee, T. G. Volm and J. P. N. Rosazza, Enzym. Microb. Technol., 1998, 23, 261 CrossRef CAS.
- Z. Huang, L. Dostal and J. P. Rosazza, J. Bacteriol., 1994, 176, 5912 CAS.
- J.-F. Cavin, V. Dartois and C. Divies, Appl. Environ. Microbiol., 1998, 64, 1466 CAS.
- G. Degrassi, P. Polverino De Laureto and C. V. Bruschi, Appl. Environ. Microbiol., 1995, 61, 326 CAS.
- J.-F. Cavin, L. Barthelmebs, J. Guzzo, J. Van Beeumen, B. Samyn, J.-F. Travers and C. Diviès, FEMS Microbiol. Lett., 1997, 147, 291 CrossRef CAS.
- X. Li, J. Yang, X. Li, W. Gu, J. Huang and K.-Q. Zhang, Process Biochem., 2008, 43, 1132 CrossRef CAS.
- Y. Hashidoko and S. Tahara, Arch. Biochem. Biophys., 1998, 359, 225 CrossRef CAS.
- A. Matte, S. Grosse, H. Bergeron, K. Abokitse and P. C. K. Lau, Acta Crystallogr. Sect. F, 2010, 66, 1407 CrossRef.
- W. Gu, J. Yang, Z. Lou, L. Liang, Y. Sun, J. Huang, X. Li, Y. Cao, Z. Meng and K.-Q. Zhang, PLoS ONE, 2011, 6, e16262 CAS.
- H. Rodríguez, I. Angulo, B. de las Rivas, N. Campillo, J. A. Páez, R. Muñoz and J. M. Mancheño, Proteins, 2010, 78, 1662 Search PubMed.
- J. P. Bennett, L. Bertin, B. Moulton, I. J. S. Fairlamb, A. M. Brzozowski, N. J. Walton and G. Grogan, Biochem. J., 2008, 414, 281 CrossRef CAS.
- A. Mattevi, M. W. Fraaije, A. Mozzarelli, L. Olivi, A. Coda and W. J. H. van Berkel, 1997, 5, 907.
- K. E. Atkin, R. Reiss, N. J. Turner, A. M. Brzozowski and G. Grogan, Acta Crystallogr. Sect. F., 2008, 64, 182 CrossRef.
- A. M. Brzozowski and J. Walton, J. Appl. Crystallogr., 2001, 34, 97 CrossRef CAS.
- G. Winter, J. Appl. Crystallogr., 2010, 43, 186 CAS.
- A. Vagin and A. Teplyakov, J. Appl. Cryst., 1997, 30, 1022 CrossRef CAS.
- Collaborative Computational Project, Number 4. Acta Crystallogr., Sect D: Biol.Crystallogr., 1994, 50, 760.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect D: Biol. Crystallogr., 2004, 60, 2126 CrossRef.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect D: Biol. Crystallogr., 1997, 53, 240 CrossRef CAS.
- A. W. Schuettelkopf and D. M. F. van Aalten, Acta Crystallogr. Sect. D. Biol. Crystallogr., 2004, 60, 1355 CrossRef.
- R. A. Laskowski, M. W. Macarthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 26, 283 CrossRef CAS.
- A. Ben-bassat, S. L. Haynie, D. J. Lowe and L. L. Huang, U.S. Patent Application 20080182307.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774 CrossRef CAS.
- K. Okrasa, C. Levy, B. Hauer, N. Baudendistel, D. Leys and J. Micklefield, Chem. Eur. J., 2008, 14, 6609 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table containing sequences of primers for PCR and PCR-based site-directed mutagenesis; Table of Data Collection and Refinement statistics for Y19A mutant of BsPAD in complex with CA; Figure showing graph of HPLC conversions of ferulate versus [ferulate] for each variant. See DOI: 10.1039/c2cy20015e |
| This journal is © The Royal Society of Chemistry 2012 |