Recent progress in the electrochemical conversion and utilization of CO2
Received
10th August 2011
, Accepted 19th October 2011
First published on 3rd November 2011
Abstract
Over the past several years, there has been a growing interest in the capture of carbon dioxide emissions and either their permanent immobilization or chemical conversion to industrially relevant products. Several processes have been developed and studied; however, many of these methods are quite expensive since they require either ultra high purity CO2 or are energy intensive. Also, many purely chemical methods show low product selectivity. To address these limitations, several researchers have initiated activities using electrochemical processes to increase reaction pathway selectivity and reduce cost since it allows for direct control of the surface free energy through the electrode potential, which has shown promise. This review article focuses on the advantages and disadvantages of current electrochemical, photoelectrochemical and bioelectrochemical processes for CO2 conversion, and future directions for research in this area are discussed.
1. Introduction
1.1 Environmental impact of CO2
Over the past century, our planet has experienced an unprecedented growth in energy consumption, which has been fueled by both population increase and technology advances. To meet this ever-growing demand, the global community has generally turned to the combustion of fossil resources including petroleum, natural gas, coal and wood. Though these resources have a proven track record of producing high quality energy at low cost, their use is unavoidably coupled with the release of many compounds toxic to our environment including nitrous oxides, sulfur oxides, radioactive compounds, heavy metals, volatile organic compounds and CO2.
On the surface, CO2 appears harmless. It is odorless and colorless. It is non-toxic to humans. It occurs naturally and serves as the primary source of carbon for photosynthesis of plants and crops. However, CO2 is a greenhouse gas that absorbs infrared heat that is reflected off the Earth from the Sun. This trapped excess heat has been shown to correlate very well with the average global temperature, which is shown in Fig. 1.1 The rise in atmospheric CO2 concentration has also been correlated with the onset of the industrial revolution at the end of the 19th century.2,3 This increase in atmospheric CO2 concentration has recently led many to be concerned about global warming trends, which could lead to severe global climate change.4–6
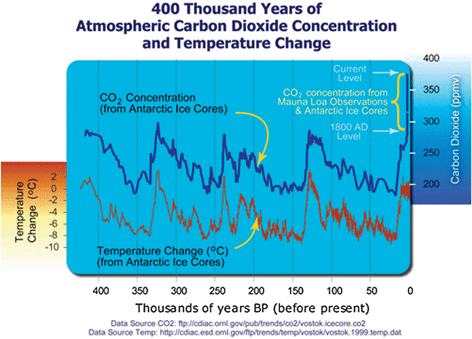 |
| | Fig. 1 Graph correlating atmospheric CO2 concentration and temperature change for the past 400 thousand years.1 | |
Therefore, recent years have seen a significant growing international interest in both limiting the emission of CO2 and reducing the atmospheric CO2 concentration to avoid a global catastrophe.7–10 There are several options that have emerged to control CO2 emissions: (i) development of, and conversion to, environmentally benign energy sources; (ii) enhancing the energy efficiency and utilization of existing processes; (iii) CO2 capture/sequestration; and (iv) conversion of CO2 to useful products. This review article will focus on the latter.
1.2 Current efforts to chemically utilize CO2
Using CO2 as a feedstock to produce a myriad of industrially relevant reaction precursors and directly usable organic materials represents a potentially sustainable, long-term resource when renewable sources of energy (i.e. solar, geothermal, wind, etc.) are used as the energy input for processing. Several researchers have recently begun to investigate carbon dioxide as a source of carbon for making industrially relevant organic chemicals including urea, salicylic acid, dimethyl carbonate, synthesis gas (syngas) and acetic acid, just to name a few, via heterogeneous catalysis.11–14 Reactions for the synthesis of each of these industrially relevant chemicals are presented in eqn (1)–(5), respectively.| | | Urea: CO2 + 2NH3 → NH2CONH2 + H2O | (1) |
| | | Salicylic Acid: C6H5OH + CO2 → C6H4OHCOOH | (2) |
| | | DMC: CO2 + 2CH3OH → CH3COCOOCH3 + H2O | (3) |
| | | CO (for syngas): CO2 + H2 → CO + H2O | (4) |
| | | Acetic Acid: CH4 + CO2 → CH3COOH | (5) |
Also, it has been suggested that the catalytic dissociation of carbon dioxide can produce active oxygen species, which can be used as a direct oxidant. For example, CO2 has been found to be beneficial for selective dehydrogenation of ethylbenzene to form styrene as well as the dehydrogenation of ethane, propane and butane to form ethylene, propylene and butene, respectively, on activated carbon-supported iron catalysts.15 CO2 utilization has also been reported via photocatalytic reduction of CO2 with H2O on highly dispersed TiO2 at 275 K to yield CH4, CH3OH and CO.16 Also, Sayama and Arakawa have reported the photocatalytic reduction of CO2 to CO over ZrO2 without any loaded metals under UV.17 However, there are several challenges to the chemical conversion of CO2 including: (i) costs of CO2 capture, separation, purification and transportation to the user site; and (ii) energy requirements of CO2 chemical conversion (eqn (1)–(5) all have ΔG ≫ 0).
1.3 Using redox processes to facilitate desired reactions
Electrochemical reactors provide three interesting advantages over pure heterogeneous chemical reactors: (i) electrochemical devices are not limited by traditional thermochemical cycles, meaning that their achievable efficiency is most often significantly higher than their chemical/combustion counterpart; (ii) direct control of the surface free energy of the catalyst through the electrode potential, allowing the reaction rate and pathway selectivity to be dialed in precisely; and (iii) non-direct reaction between precursors through complementary redox processes on two separate catalysts, which permits researchers to tailor the properties needed for each redox process independently. This can facilitate different reaction pathways depending on catalyst selection with identical precursors at the same temperature, etc. while minimizing competition between alternate pathways. This enables unique chemistries to occur that would not be possible in conventional systems.
Over the past several years, novel electrochemical synthesis methods have received considerable attention for the formation of high value products for the pharmaceutical and food industries. Two relevant, representative cases are detailed below that provide examples of electrochemical systems that have been designed to facilitate reactions that were either prohibitively energy intensive, had poor selectivity or were impossible from known, purely chemical routes.
An et al. reported the hydrogenation of oils in an electrochemical reactor with a proton exchange membrane separating the cathode and anode compartments.18 In this device, water electrolysis to O2 and H+ was employed at the anode. The protons were transported to the cathode where they were reduced to H2. The resulting hydrogen gas reacted in the cathode with flowing soybean, canola and cottonseed oils and other fatty acid solutions. This allowed three innovations. First, water was used as a hydrogen source for the hydrogenation, which is not possible using traditional routes. Second, the electrochemical reactor was run at lower temperatures than traditional hydrogenation reactors. Third, higher cis-isomer selectivity was demonstrated than achieved previously, which has important implications for food products and human health.
Moghaddam and coworkers reported an electrochemical route for the formation of new caffeic acid derivatives.19 Caffeic acid derivates are agriculturally and pharmacologically important as they have been shown to play a key role in the infection defense mechanism of several plant species and have favorable antioxidant qualities. Here, the authors performed an electrochemical partial oxidation of caffeic acid in an acetate-buffered acetonitrile/water binary electrolyte. The activated species further reacted with solvated sulfinic acids and further electrochemically oxidized to yield sulfone-functionalized caffeic acid derivatives. This electrochemical–chemical–electrochemical mechanism provided key advantages over existing systems by reducing the energy and time required for synthesis, eliminating the need for exotic and potentially environmentally harmful reagents (i.e. isooctane) and facilitated the high-selectivity formation of the target product.
1.4 Using electrochemical processes for the conversion and utilization of CO2
Recently, researchers have begun to take advantage of electrochemical approaches to the utilization and conversion of CO2.20 Several electrochemical, photoelectrochemical and bioelectrochemical approaches have been tried and have shown promise for the formation of various industrially relevant products from syngas all the way to short chain alcohols and hydrocarbon products. The purpose of this article is to summarize this body of work and to identify the most promising pathways for future research.
2. Electrochemical CO2 conversion
2.1 Background
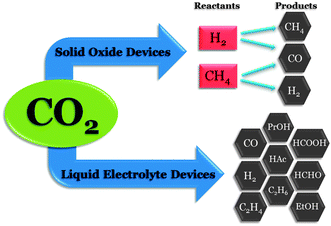
Multiple pathways have been investigated for the electrochemical conversion of CO2, including gaseous, aqueous, and non-aqueous phase techniques at both high and low temperatures. Fig. 2 shows a summary of these pathways, including a variety of possible products formed. High temperature CO2 conversions are typically carried out using variations of the solid oxide fuel cell (SOFC), whereas low temperature systems largely utilize transition metal electrodes in both aqueous and non-aqueous electrolytes, such as methanol, acetonitrile, propylene carbonate, or dimethyl sulfoxide. The scope of products formed at low temperatures is broader than at high temperatures, but the selectivity and performance of SOFC devices often exceed those observed in low temperature systems. Applied potentials of several volts are also necessary in low temperature aqueous and non-aqueous phase electroreductions, resulting in large power requirements. In this section, these systems and pathways for electrochemical CO2 reduction will be analyzed in terms of the products formed and the electrochemical performance and stability.
2.2 Syngas production
Conversion of CO2 electrochemically to syngas (CO and H2) is a highly promising pathway for CO2 utilization and mitigation. Syngas is an industrially-important precursor used in the synthesis of methanol and other hydrocarbons.21 Among the methods reported for syngas production, SOFCs are a popular choice due to high current densities and the potential for power-generating, rather than power-intensive, devices. Heat produced electrochemically through anodic oxidation is sufficient to sustain the SOFC operating temperature, and, as a result, the heat required to dissociate gaseous CO2 to CO and surface oxygen species is readily available. This heat utilization enables SOFCs to generate power, compared to low temperature, aqueous systems which require an applied current to electrochemically reduce CO2.22
As an alternative to traditional steam reforming of methane (CH4), which is a highly endothermic and energy-intensive process, dry reforming, or CO2 reforming of CH4, has been reported to produce syngas with more favorable H2/CO ratios (eqn (6)):21,23
Dry
reforming is a particularly attractive method since it not only addresses improved syngas formation, but also the elimination of greenhouse gases and utilization of cheap, abundant, industrially-relevant,
carbon-containing materials.
23 As illustrated in
Fig. 3, Kim
et al. showed that performance in an SOFC setup using CH
4 and CO
2 as reactants is also comparable to using H
2 as
fuel. These high temperature
cells typically utilize a yttria-stabilized
zirconia (YSZ) electrolyte or tube with various metal and/or mixed-metal
catalysts at the anode and cathode. Belyaev, Galvita, and Sobyanin varied inlet CO
2/CH
4 fuel ratios over porous
platinum electrodes and obtained syngas with a favorable composition for Fischer–Tropsch processes, which is reported to contain an H
2/CO ratio between 1 and 2.
21,24 Other researchers have produced syngas in SOFCs using
nickel-based
catalysts, such as ceria-doped nickel-YSZ or nickel oxide-calcium oxide mixed metal
electrodes, through CH
4 reforming with CO
2 and/or O
2.23,24 When CO
2 is used as the
fuel without CH
4, CO alone can be synthesized at the cathode (
eqn (7)) for use in syngas or for different processes, along with
oxygen ions, which are transformed to pure
oxygen gas at the anode (
eqn (8)):
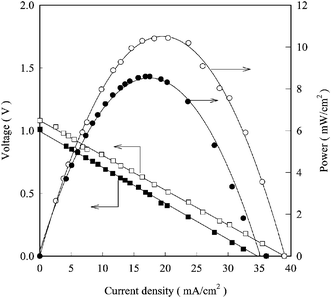
22,25Bidrawn
et al. demonstrated this CO
2 electrolysis with current densities over 1 A cm
−2 (
Fig. 4), and suggested this technology could rival efficiencies for similar
H2O electrolysis systems and make a significant impact on greenhouse gas mitigation.
25 H
2 fuel was applied by Furukawa and coworkers using a lower temperature (500 °C) SOFC
cell with
nickel/
zeolite and
silver electrodes to convert CO
2 into CH
4 and
H2O (
eqn (9)) with yields up to 80%:
26| | | CO2 + 4H2 → CH4 + 2H2O | (9) |
In all SOFC cases, gaseous CO
2 is readily reduced to usable products as a result of the heat available at temperatures as high as 800–900 °C.
22
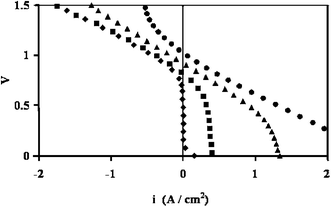 |
| | Fig. 4 Polarization curves for a solid oxide fuel cell with an LSCM-CZY-Pd in YSZ fuel side electrode operated at 800 °C with various CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO fuel stream ratios. (◆): 100:0; (■): 90:10; (▲): 50:50; (●): 10:90.25 :CO fuel stream ratios. (◆): 100:0; (■): 90:10; (▲): 50:50; (●): 10:90.25 | |
One of the biggest challenges facing prolonged SOFC usage is finding solutions to the electrode deactivation that occurs from coke formation and other mechanisms. Coke formation and deposition can occur through several different reactions, including CH4 decomposition (eqn (10)) and CO disproportion (eqn (11)):22–23,25
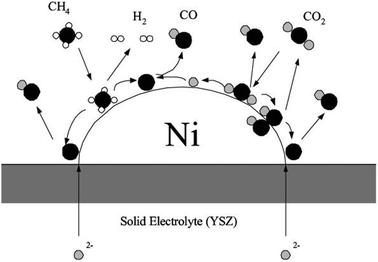
In
Fig. 5, Kim
et al. proposed molecular mechanisms for dry
reforming over a Ni-YSZ-CeO
2 SOFC
electrode, presenting potential pathways for
eqn (6), (10), and (11).
23 They also illustrate in
Fig. 6 that, fortunately, the rate of coke formation is lower than both the rates of reaction for CH
4 and CO
2 and the rate of formation of CO.
23 Over long periods of operation, however, coking is still a significant problem, and efforts have been made in attempts to minimize its impact. Choudhary
et al. reformed CH
4 with both CO
2 and O
2 to partially mitigate coking that stems from the use of CO
2 as a
reforming fuel.
24 Electrode modification
via alloying small fractions of additional metal or metal
oxide phases has also been shown to be highly beneficial to SOFC operation, such as the inclusion of 4% CeO
2 to a Ni-YSZ
electrode which improved the
cell performance and stability, and resulted in less coking.
23 Others have alloyed numerous materials including gadolinia-doped ceria (GDC),
copper,
palladium, and Ce
0.48Zr
0.48Y
0.04 (CZY) into various SOFC
electrodes in attempts to improve SOFC performance and stability.
22,25 However, high temperature operation still poses other problems for
electrode degradation, particularly for
nickel-based
electrodes. Formation of volatile
nickel-carbonyls deactivates the
electrode over time and imposes significant limitations on continuous, long-term operation of SOFC reactors.
25 Hence, to fully realize the potential for nonstop CO
2 utilization with SOFCs, permanent solutions to the problems of coking and
electrode degradation must be found.
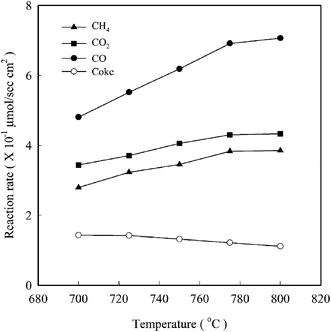 |
| | Fig. 6 Reaction rates for (▲) CH4 and (■) CO2, and rates of formation of (●) CO and (○) coke as a function of temperature for a solid oxide fuel cell with a Ni-YSZ-CeO2 electrode.23 | |
A few reports exist concerning syngas production under ambient conditions in aqueous solutions. Due to the lack of accessible heat at atmospheric temperatures and pressures, gaseous CO2 cannot be as easily electroreduced as with SOFCs. Dissociation of CO2 in aqueous solutions requires applied current, and therefore generally large applied (negative) potentials.22 Yano et al. converted CO2 to CO and H2 at applied potentials as high as −2.4 V over a silver mesh electrode, but conversions were much lower than those reported for SOFCs.27 Delacourt and coworkers achieved current densities up to 100 mA cm−2 over Ag and Pt–Ir gas-diffusion electrodes in fuel cell and modified aqueous-fuel cell experimental setups; however, they primarily reported H2 evolution with low CO formation, and insertion of an aqueous buffer layer increased cell resistance and lacks commercial feasibility.28 Low CO2 solubility in aqueous solutions under ambient conditions severely limits the output for these devices, and imposes the need for extremely large applied potentials to obtain a reasonable amount of product. Promising current densities as high as 50 mA cm−2 have been reported, however, for applied potentials up to −2.5 V over various metal-phthalocyanine (M-Pc) gas-diffusion electrodes.29 The primary product from these electrocatalysts was CO, though H2 evolution occurred in nearly all cases as well. Another potential pathway for electrochemical CO2 conversion is through the formation of carbonate anions (CO32−), which has been demonstrated over a Ca2Ru2O7−y pyrochlore cathode electrocatalyst in an anion exchange membrane fuel cell setup.30 CO32− could then be used as a high efficiency oxygen donator to convert incoming feedstocks to syngas and other useful products at the anode, completing the cell circuit. This could provide an alternative to traditional anion exchange membrane fuel cells operating on the hydroxide cycle since CO32− has exhibited improved exchange current densities compared to hydroxide, in addition to a substantially reduced membrane degradation rates.31,32 It also represents a unique system where the target product is obtained at the anode (oxidation) electrode, not the cathode (reduction).
The most commonly-used materials for electrochemical CO2 conversion under ambient temperatures and pressures are transition metals and metal oxides, specifically copper-based electrodes. Gattrell, Gupta and Co summarized aqueous electrochemical CO2 reduction methods using different copper electrodes, and found a variety of products formed depending on the electrode structure and reaction conditions.33 Disadvantages of CO2 reduction under ambient conditions are applied potentials as high as −4 V; however a wide range of hydrocarbon products can be synthesized in various aqueous electrolytes.34–40 While many low temperature systems also produce byproducts of CO and/or H2, compositions and purities are not ideal for syngas-related operations. In most cases, H2 forms at the cathode not directly from the CO2 reduction reaction, but rather from the concurrent hydrogen evolution reaction, in either acidic (eqn (12)) or alkaline (eqn (13)) media, due to extremely large overpotentials:41| | | 2H+ + 2e− → H2; E0 = 0.000 V vs. NHE | (12) |
| | | 2H2O + 2e− → H2 + 2OH−; E0 = −0.828 V vs. NHE | (13) |
With the high applied potentials necessary for aqueous-phase CO2 electroreduction, in virtually all cases hydrogen evolution is an unavoidable side-reaction and occurs spontaneously. As a result, in nearly all reports of hydrocarbon production, H2 current efficiencies are minimized as much as possible without compromising product selectivity.
High surface area and surface-modified copper electrodes have a high selectivity at low temperatures towards simple hydrocarbons like ethylene (C2H4) and CH4, but numerous other products have also been reported, such as CO, H2, formic acid (HCOOH), ethane, ethanol, propanol, acetic acid, and lactic acid.35–38 Others report larger current efficiencies for different species, such as CO or HCOOH, than for C2H4 or CH4, along with acetone, methanol, and aldehydes as additional minor species.39,40 Poisoning of these copper electrodes is a common issue that plagues long-term operation and limits their viability for commercialization. Use of acidic solutions can result in activity losses due to copper dissolution, evidenced by increased blue coloring of the solution from aqueous copper species over time.37,38 On the other hand, highly alkaline solutions cause copper oxides to form on the electrode surface, which are generally considered undesirable, though Yano et al. noted favorable C2H4 synthesis using copper oxide electrodes compared to pure a copper electrode.38 Other harmful species reported to form on copper electrodes are graphitic carbon and organic intermediates, such as formate ions, that can react either chemically (eqn (14)) or electrochemically (eqn (15)) to form unwanted surface species:37,40
| | | Cu + HCOO− → CuO + C + OH− | (14) |
| | | HCOO− + H2O + 2e− → C + 3OH− | (15) |
Among these mechanisms of
electrode poisoning, however, Smith and coworkers disputed coke deposition by closely examining repeated CO
2 reduction over a
copper electrode and finding only copper
oxides, copper
hydroxides, and surface tarnishing patina with no evidence of
carbon deposition.
42 Irrespective of the cause and identity, efforts have been made to mitigate surface poisons, including
copper halide-modification and use of a three-phase reaction
cell.
36,37,40
Researchers have also investigated the use of various other transition metals, alloys, and complexes as electrocatalysts for CO2 reduction in aqueous-phase cells. Shibata and Furuya studied a bevy of transition metals in M-Pc complexes as gas-diffusion electrodes and produced mostly CO and/or H2 products for the majority of metals.29Fig. 7 shows current efficiencies and densities for product formations from the reduction of aqueous phase CO2 as functions of potential gathered by Shibata and Furuya over cobalt-Pc catalysts.29 The main product was CO, with H2 formation suppressed as low as possible for potentials up to −2.5 V, and no HCOOH formation was observed for overall current densities up to around 50 mA cm−2. Some of these electrocatalysts also produced different products outside the scope of those seen with copper-based electrodes. Cobalt-, nickel-, and palladium-Pc electrodes showed the highest activity among all M-Pc electrodes, particularly for ammonia (NH3) formation from nitrite ions (NO2−) and urea synthesis from CO2 and NO2−.29,43Fig. 8 shows product current efficiencies over cobalt- and nickel-Pc electrodes for CO2 reduction with the addition of NO2−, and once again no HCOOH formation was detected for potentials up to −2.5 V.43 Formation of H2 likely occurred in all experiments as well, similar to those performed without NO2− (Fig. 7); however, that data was omitted in this case. Other electrode materials researchers have used include Monel metal (an alloy of nickel, cobalt, copper, and iron), stainless steel (chromium, nickel, and iron), numerous pure transition metals and powders, and boron-doped diamond (BDD).36,39,44,45 Various products were formed, including sodium formate and HCOOH reported by Narayanan and coworkers over indium and lead powders at the cathodes of Na+ and OH− exchange cells, and peroxycarbonate examined by Saha et al. using a BDD electrode in a CO2-saturated sodium hydroxide electrolyte at low temperature.44,45
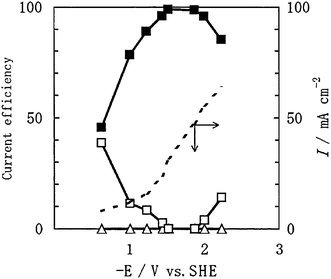 |
| | Fig. 7 Current efficiencies for (■) CO, (□) H2, and (△) HCOOH, and (- - -) overall current density (plotted on secondary y-axis) as functions of (negative) electrode potential for aqueous phase CO2 reductions over a cobalt-Pc gas-diffusion electrode.29 | |
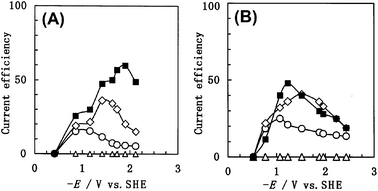 |
| | Fig. 8 Current efficiencies as a function of (negative) electrode potential for (■) CO, (○) NH3, (◇) urea, and (△) HCOOH products from aqueous phase CO2 reductions with NO2− ions over (A) cobalt-Pc and (B) nickel-Pc gas-diffusion electrodes. Edited from ref. 43. | |
In addition to varying the electrode material, some researchers have tried different reaction media to either improve the CO2 reduction reaction or tailor specific product selectivity. Ikeda et al. reduced CO2 to oxalic acid with faradaic efficiencies around 70–85% over lead and indium electrodes in propylene carbonate, acetonitrile, and dimethyl sulfoxide electrolytes at ambient conditions.39 Trifluoroacetic acid and trifluoromethane have also been produced from simultaneous bubbling of CO2 and CClF3 gases in a 0.1 M tetra(n-butyl ammonium) bromide in acetonitrile electrolyte over a silver wire electrode in a stainless steel autoclave.46 A review by Sanchez-Sanchez et al. of electrochemical CO2 conversions in various aqueous and non-aqueous media, as well as gas-diffusion electrodes, detailed the potential for electrocarboxylation of CO2 to useful pharmaceutical products, in addition to other low temperature methods, over transition metal catalysts.47 In general, higher current densities were observed in non-aqueous media and at higher pressures, but in all cases high overpotentials were required, there was significant competition with the H2 evolution reaction, and experiments showed poor product selectivity.47 Aside from liquid electrolytes, Yoshida, Yosue, and Nogami demonstrated a direct-current discharge plasma gas chamber with CO2 and H2 as fuels to synthesize CH4 and other hydrocarbon gases over copper and iron electrodes.48 However, due to the extreme applied potentials needed to create the excited plasma state, conventional efficiencies for this cell are very low, and these electrodes also suffer from poisoning caused by graphitic carbon deposition over prolonged usage.48
An especially popular choice of medium among non-aqueous electrolytes for use in electrochemical CO2 conversion devices is methanol. At temperatures at or below 15 °C, methanol is a much more effective solvent for CO2 dissolution than water. Kaneco and coworkers have studied CO2 reduction in methanol with various alkali metal salts over copper, lead, zinc and indium foil electrodes at temperatures ranging from −30 to 15 °C, and numerous hydrocarbon products were formed, such as CH4, HCOOH, C2H4, CO, and H2 (eqn (16)–(19)).49–52
| | | CO2 + 8H+ + 8e− → CH4 + 2H2O | (16) |
| | | CO2 + H+ + 2e− → HCOO− | (17) |
| | | 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | (18) |
| | | CO2 + 2H+ + 2e− → CO + H2O | (19) |
Similar applied potentials to aqueous-phase systems are still necessary (around −2 to −3 V), but the higher CO
2 solubility in
methanol compared to
water increases reactant availability at the
electrode surface which may improve overall product yields. It is unclear, however, how much of this product formation can be attributed directly to CO
2 electroreduction
versus the activity of the
methanol solvent itself, which could hypothetically act as an independent
carbon source regardless of CO
2 present in solution. H
2 formation is also generally unwanted in these
hydrocarbon synthesis systems, and current efficiencies were suppressed down to around 10%.
49–52 As an alternative to metal foil
electrodes, Aydin and Köleli performed electrocatalytic
reductions of CO
2 in
methanol at a pressure of 20 bar over
polypyrrole and
polyaniline film
electrodes.
53,54 Acetic acid was the main product formed, with
formaldehyde and HCOOH produced as minor products as well, but current densities were much lower when compared to similar systems using metal
electrodes. However, the scope of minor byproducts detected was much smaller using these
polymer film
electrodes compared to metal
electrodes, which could potentially
lead to better product selectivity and purity.
3. Photoelectrochemical CO2 Conversion
3.1 Background
Photoelectrochemical processes for CO2 conversion generally require more complex systems than their purely electrochemical counterparts, and are also much more energy intensive due to power requirements both for applied potentials and electrolyte illumination. Much like low temperature electrochemical methods, a diversity of hydrocarbon products can be synthesized depending on the choice of electrode and electrolyte. While the effects and/or participation of the electrolyte are typically nil in electrochemical systems, the electrolyte in photoelectrochemical devices typically contains active species, such as sacrificial reducing agents or a restorable electron relay. In this section, the various types of electrocatalysts and aqueous and non-aqueous electrolytes used are discussed.
3.2 Electrode/electrolyte selection
A variety of materials and solutions have been studied as photoelectrocatalysts and electrolytes for CO2 reduction. Aqueous and non-aqueous solutions typically consist of a sacrificial reducing agent/electron donor along with a photosensitizer as the photoelectrocatalyst, and CO2 is supplied as a gas to the irradiated solution under an applied potential. Although a few reports do show moderate current densities, overall performance of most photoelectrochemical systems is very low in terms of both current density and faradaic efficiency. Many commonly-used catalysts are bimetallic, n- or p-type electrodes, some of which are additionally doped. Bockris and Wass produced CO with methanol and H2 byproducts using phosphorus-doped p-type CdTe, boron-doped p-type Si, and deposited crown ether photoelectrocatalysts in both aqueous and non-aqueous media, but yields in aqueous solutions were very limited.55 On the other hand, current densities were the highest in dimethylformamide with tetraalkyl salts, although they only reached a maximum magnitude of around 3 mA cm−2.55 Other studies revealed methanol or HCOOH as the main products using aqueous media, and CO, HCOOH, and H2 using cold methanol electrolytes, over various bimetallic photoelectrocatalysts including n-type and p-type GaAs, p-type InP, and Zn-doped p-type GaP and p-type GaAs.56–58 Low current densities in methanol were observed between 4-6 mA cm−2, but they did not reach even 1 mA cm−2 for aqueous electrolytes and maximum faradaic efficiencies were only around 1%, causing the feasibility and viability of these setups to be questioned.
Other popular types of materials are complexes containing 2,2′-bipyridine (bipy) with transition metals. These photosensitizers work in conjunction with other species in solution to help carry out the reduction of CO2 under irradiation. One such complex that has been studied in both aqueous and non-aqueous electrolytes is Ru(bipy)32+.59–61 In acidic aqueous media, triethanolamine was added as a sacrificial electron donor and methylviologen was used as a restorable electron relay to synthesize HCOOH.59 With these aqueous solutions, water acts as a plentiful hydrogen source. In non-aqueous solutions, since there is not necessarily an available source of hydrogen, small concentrations of water can be added to facilitate the synthesis of products other than just CO. Using an acetonitrile electrolyte over Ru(bipy)32+ with CoCl2 as the reducing intermediate, Lehn and Ziessel varied the amount of H2O added to the electrolyte to create syngas with controllable CO/H2 ratios.60 In another report, using a similar setup with the addition of 1,10-phenanthroline–ruthenium(II) to the Ru(bipy)32+ photosensitizer, water content was altered to produce formate ions.61 Once again, overall yields suffer from the intense energy input required to apply high potentials while simultaneously illuminating the reaction cell.
Several other systems have shown the potential for photoelectrochemical CO2 reduction. Tazuke and Kitamura also used water as a hydrogen source in polar media with aromatic hydrocarbons and either 1,4-dicyanobenzene or 9,10-dicyanoanthracene to produce HCOOH and hydrogen peroxide at low concentrations.62 The reaction mechanism was not identical to many previously-described procedures, but did follow a similar multistep reaction pathway. Kaneco and coworkers used isopropanol as a non-aqueous electrolyte and positive hole scavenger in a TiO2 powder-catalyzed CO2 photoelectrochemical reduction.63 Low temperatures (20 °C) and high pressures (2–30 atm) under irradiation produced CH4 at energy conversion efficiencies almost double those obtained in aqueous media; however, the maximum for this isopropanol electrolyte was still low at 0.0065%.63 Finally, a combination of Fe(bipy)32+, Fe2+ aqueous solutions, pure Fe metal, and a Zn(Hg) amalgam was used to synthesize formaldehyde.64 Continuous irradiation with the addition of Cr2+ ions to the solution resulted in further transformation from formaldehyde to methanol, followed by a final conversion from methanol to CH4. However, initial production of formaldehyde was extremely limited, and each subsequent conversion resulted in even lower yields. While it seems non-aqueous electrolytes offer the potential for improved CO2 photoelectrochemical reductions, overall energy efficiencies and product yields are severely lacking compared to standard electrochemical methods, which may limit their commercial viability.
4. Bioelectrochemical CO2 conversion
4.1 Background
Though some success has been reported using metal electrodes for the reduction of CO2 to fuels, one of its main drawbacks remains the low selectivity of the process where multiple products are generally obtained. In response to low selectivity, several researchers have investigated electrolysis in CO2-saturated aqueous electrolytes using enzyme catalysts. The high selectivity of naturally occurring enzymes could lead to the formation of a specific fuel, while minimizing or completely eliminating secondary reactions. Most of these studies have focused on formate dehydrogenase (FDH), aldehyde dehydrogenase (AldDH) and alcohol dehydrogenase (ADH). CO2-saturated electrolytes contain bicarbonate anions that undergo a series of reactions to produce methanol. FDH, AldDH and ADH catalyze the formation of formate, formaldehyde and methanol, shown in eqn (20)–(22) respectively.| | | HCO3− + 2H+ + 2e− → HCO2− + H2O | (20) |
| | | HCO2− + 3H− + 2e− → CH2O + H2O | (21) |
| | | CH2O + 2H+ + 2e− → CH3OH | (22) |
4.2 Enzyme selectivity
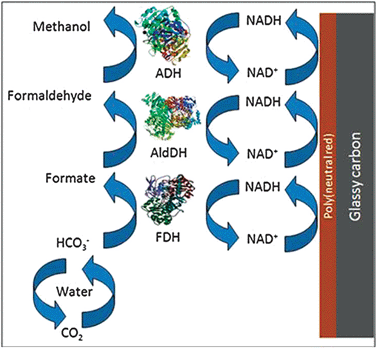
CO2 reduction products are highly dependent on the enzyme or enzyme series used, as shown in Fig. 9. Under normal conditions, enzymes are highly selective oxidation catalysts and several operating in series are normally needed to accomplish the complete conversion of a substrate, such as methanol, to CO2. In a similar manner, when the equilibrium is shifted by exposing the enzymes to concentrated CO2 environments and reduction occurs, the product will depend on the enzyme series used. In general, enzymes operating in series are required in order to convert CO2 to a high energy molecule, such as methanol (Fig. 9).
Most of the work regarding carbon dioxide reduction with enzymes is related to the schematic shown in Fig. 9, where formate, HCOOH or, ultimately, methanol are produced. FDH with a tungsten active site has been used to produce formate. The enzyme shows high reversibility for carbon dioxide—formate interconversion, and the reaction is carried out at mild conditions (pH ∼ 6). At low overpotentials (40 mV), the only reduction product detected has been formate, with small quantities of secondary products obtained at high overpotential (>400 mV).65 Current efficiencies for the production of formate as high as 93% have been observed with this enzyme using electrons photogenerated from a p-type indium phosphide cathode.66 Therefore, it is possible to reduce CO2 to higher energy substrates with high current efficiencies and at mild conditions.
The use of equal amounts of FDH and ADH in a CO2-saturated solution led to the formation of methanol with high current efficiencies of 90%.67 It was also observed that formate is first reduced to formaldehyde, which is subsequently reduced to methanol, and no methanol production is observed until formaldehyde accumulates.67,68 However, a threshold concentration of ADH is necessary for methanol production, since methanol formation is not observed for low concentrations of ADH.68 Also, this enzyme has been shown to lose its activity when exposed to visible light, forcing electroreduction to be carried out in the dark.68 Furthermore, conversion rates to methanol remain low, generally on the order of μmol day−1. Therefore, it appears that enzyme engineering is required to develop enzymes with high electrochemical activities, which will yield higher conversion rates.
Advances have been made in accelerating the production rate of methanol through the enzyme series method. Recently, it was shown that the addition of carbonic anhydrase accelerates the reduction of CO2 to methanol, causing an 82% increase in the reduction current.69 The first enzyme in the cascade, formate dehydrogenase, uses bicarbonate as its main substrate. Under normal conditions, bicarbonate in the aqueous electrolyte is produced by thermodynamical equilibrium between CO2, bicarbonate, carbonate, and carbonic acid. Addo et al. suggest that carbonic anhydrase catalyzes the hydration of CO2 to bicarbonate, therefore enhancing the carbon capture and rate of methanol production.69 Other factors unrelated to enzyme activity have been shown to provide production rates. The use of a sol–gel matrix enhances the production of methanol by over 2-fold, compared to the reaction in solution (Fig. 10).70 Obert and Dave suggested that the increase in methanol production was due to increases in local concentration of substrates, since these will be confined within the pores of the sol–gel, leading to yields as high as 91%. Therefore, this demonstrates that production rate improvements can be accomplished by other factors rather than enzyme engineering, such as precursor concentration and tailoring of the electrode microstructure.
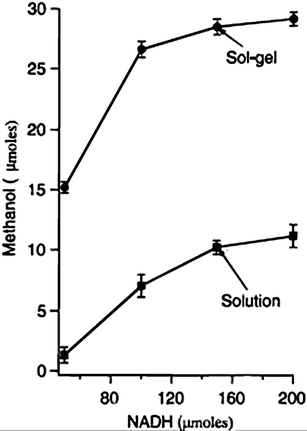 |
| | Fig. 10 Plot of methanol produced as a function of terminal electron donor (NADH) present in solution and sol–gel matrix.70 | |
A theoretical study on the thermodynamics of methanol synthesis from CO2 investigated the preferable conditions for the chain of reactions using enzymes.71 In general, it was found that low pH and high temperatures will improve the kinetics and lead to higher yields of reaction. pH was found to be the most sensitive factor, with a mild change of pH from 7 to 6 decreasing the Gibbs energy of formation to a negative value and increasing the equilibrium methanol concentration by 5 orders of magnitude. However, great care must be taken since most enzymes are intolerant of these conditions and will be deactivated at either low pH or high temperature. This finding stresses the need to engineer enzymes with high chemical and thermal stability which could yield significantly higher conversion rates.
Although most of the work has been focused on the production of methanol or its precursor substrates, other products can be obtained from enzymatic CO2 electroreduction. The use of acetyl-coenzyme A in a CO2-saturated phosphate buffer solution leads to the formation of pyruvic acid.72 However, the reaction is strongly inhibited after a certain amount of pyruvic acid is produced, attributed to the accumulation of products which shifts the reaction equilibrium. Isocitric acid can be obtained by the use of the enzyme isocitrate dehydrogenase as an electrocatalyst. The reaction had high current efficiencies (>80%) and low overpotentials were needed to accomplish CO2 fixation.73 Carbon monoxide production from CO2 can be observed using Carbon Monoxide Dehydrogenase.74,75 This enzyme has been shown to reduce CO2 to CO with low overpotentials (<100 mV) and high current efficiencies (∼100%) at a pH of 6.3. The high conversion rate is attributed to the enzyme structure, which may enhance carbon dioxide binding and electron transfer to the substrate.74 The reaction appears to be reversible and the catalyst has good activity, attributed to enzymes non-uniformly adsorbed in the surface of the graphite electrode.74 Therefore, the use of designated enzymes can lead to the formation of numerous high purity products which can be used in multiple applications. However, long times are normally required to produce small quantities of product due to the low activity of enzymes found in natural biological systems.
4.3 Electron mediators
In general, enzymes have a low electrical conductivity and, thus, limited interaction with electrodes used as current collectors. Therefore, the use of an electron mediator (EM), which facilitates electron transport from the enzyme to the electrode, is typically required to allow electrical interaction between the enzymes and the electrode. Methylviologen (MV) is the most generally used EM for CO2 reduction using enzymes. However, it has been observed that the use of pyrroloquinolinequinone (PQQ) affects the selectivity of the reaction.68 Kuwabata and coworkers found that when using MV with FDH and ADH, the products were a mixture of formate, formaldehyde and methanol in similar proportions. However, when PQQ is used as the EM, only methanol is obtained with current efficiencies greater than 90%. The increase in selectivity is attributed to PQQ adsorption on the surface of the electrode. Using thermodynamical arguments, it was demonstrated that adsorbed PQQ reduces formate to methanol, and not formaldehyde, since the formate to formaldehyde reduction potential lies outside the stability window of adsorbed PQQ.68 Therefore, this finding shows that the use of appropriate EMs can increase the selectivity and the synthesis rate of methanol. However, the work performed has been limited to the aforementioned EMs. Therefore, much work remains in finding appropriate EMs for the different reactions and at various reaction conditions. Mediators with high chemical and electrochemical stability windows and low electrical resistance should be found to increase the formation rate of high energy substrates while maintaining high selectivity.
5. Conclusions
Conversion of CO2 to industrially-relevant products is an area of research that has attracted global attention due to the potential threat of disaster as a result of climbing atmospheric CO2 concentrations. Electrochemical techniques are particularly attractive when compared to heterogeneous chemical methods in part due to greater potential efficiencies and more tailorable reaction pathways. Pure electrochemical conversions have been performed through a myriad of routes, including high temperature solid oxide fuel cell devices and low temperature aqueous and non-aqueous reactors. Among these methods, solid oxide fuel cell devices have exhibited the highest current densities and efficiencies, whereas many low temperature CO2 conversions resulted in comparatively diminished performance, low product selectivity, and in some cases high power requirements. Despite the added strain and electrode/electrolyte degradation that accompanies solid oxide devices due to operation at temperatures upwards of 500 °C, they show promise as a viable CO2 conversion technique. On the other hand, aqueous and non-aqueous methods, while appealing as a result of their facile operating conditions, will likely need drastic performance improvements and/or reductions in required power loads before they could become practical solutions to this problem. However, the diversity of products formed and relative ease of operation of low temperature electrochemical systems still make them attractive topics for further research.
Photoelectrochemical and bioelectrochemical methods typically result in high product selectivity while running under generally mild operating conditions. However, both types of systems encounter several obstacles that severely limit overall performance. The energy demand for photoelectrochemical systems is immense due to the need for simultaneous irradiation and large applied potentials, and this leads to efficiencies that are often orders of magnitude lower than purely electrochemical systems operating under similar temperatures and pressures. In addition, overall product yields are very low, which imposes long time frames if appreciable levels of product generation are desirable. Bioelectrochemical systems suffer from the same problem due to low activities and poor electrical conductivity of natural biological enzymes, and also experience stability constraints associated with thermal and chemical sensitivities to the reaction conditions. Without significant improvements, it is unlikely that photoelectrochemical or bioelectrochemical conversion techniques will approach a level of viability that can challenge that of pure electrochemical methods, specifically solid oxide technologies.
Acknowledgements
The authors would like to thank the National Science Foundation who supported the preparation of this manuscript under Grant Number CBET-1005303. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
References
- 〈ttp://www.whrc.org/resources/primer_fundamentals.html〉. Hole Woods Research Center, Climate Change Primer, Accessed July 25, 2011.
-
M. Etheridge, L. Steele, R. Langenfelds, R. Francey, J. Barnola, V. Morgan, Historical CO2 record from the Law Dome DE08, DE08-2, and DSS ice cores (atmospheric CO2 concentrations, Antarctic ice cores). in: Trends: A Compendium of Data on Global Change, online at the Carbon Dioxide Information Analysis Center, 〈http://cdiac.esd.ornl.gov/〉, June 1998.
-
C. Keeling, T. Whorf, Atmospheric CO2 records from sites in the SiO air sampling network, in: Trends: A Compendium of Data on Global Change. Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U. S. Department of Energy, Oak Ridge, TN, USA, 2005.
- S. Levin, Ecology, 1992, 73, 1943 CrossRef.
- J. Melillo, A. Mcguire, D. Kicklighter, B. Moore, C. Vorosmarty and A. Schloss, Nature, 1993, 363, 234 CrossRef CAS.
- D. Schimel, Global Change Biol., 1995, 1, 77 CrossRef.
-
Carbon Dioxide Chemistry: Environmental Issues, ed. J. Paul and C. Pradier, Royal Society of Chemistry, Cambridge, UK, 1994, p. 405 Search PubMed.
-
M. Halmann and M. Steinberg, Greenhouse Gas Carbon Dioxide Mitigation: Science and Technology, Lewis Publishers, Boca Raton, FL, 1999, p. 568 Search PubMed.
-
M. Maroto-Valer, C. Song and Y. Soong, Environmental Challenges and Greenhouse Gas Control for Fossil Fuel Utilization in the 21st Century, Kluwer Academic/Plenum Publishers, New York, 2002, p. 447 Search PubMed.
- DOE/NETL, Carbon Sequestration. Technology Roadmap and Program Plan 2005. US Department of Energy, National Energy Technology Laboratory, May 2005.
- M. Aresta and A. Dibenedetto, Catal. Today, 2004, 98, 455 CrossRef CAS.
-
T. Inui, M. Anpo, K. Izui, S. Yanagida and T. Yamaguchi, Editors, Stud. Surf. Sci. Catal., 1998, 114, 691.
- H. Arakawa, Stud. Surf. Sci. Catal., 1998, 114, 19 CrossRef CAS.
- S. Park, J. Yoo, J. Chang, K. Lee and M. Park, Preprint Paper Am. Chem. Soc. Div. Fuel Chem., 2001, 46, 115 CAS.
- M. Sugino, H. Shimada, T. Turuda, H. Miura, N. Ikenaga and T. Suzuki, Appl. Catal., A, 1995, 121, 125 CrossRef CAS.
- M. Anpo and K. Chiba, J. Mol. Catal., 1992, 74, 207 CrossRef CAS.
- K. Sayama and H. Arakawa, J. Phys. Chem., 1993, 97, 531 CrossRef CAS.
- W. An, J. Jong, P. Pintauro, K. Warner and W. Neff, J. Am. Oil Chem. Soc., 1998, 75, 917 CAS.
- A. Moghaddam, M. Ganjali, P. Norouzi and M. Niasari, J. Electroanal. Chem., 2007, 601, 205 CrossRef CAS.
- D. Whipple and P. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451 CrossRef CAS.
- V. Belyaev, V. Galvita and V. Sobyanin, React. Kinet. Catal. Lett., 1998, 63, 341 CrossRef CAS.
- T. Huang and C. Chou, Electrochem. Commun., 2009, 11, 1464 CrossRef CAS.
- T. Kim, S. Moon and S. Hong, Appl. Catal., A, 2002, 224, 111 CrossRef CAS.
- V. Choudhary, A. Rajput and B. Prabhakar, Catal. Lett., 1995, 32, 391 CrossRef CAS.
- F. Bidrawn, G. Kim, G. Corre, J. Irvine, J. Vohs and R. Gorte, Electrochem. Solid-State Lett., 2008, 11, B167 CrossRef CAS.
- S. Furukawa, M. Okada and Y. Suzuki, Energy Fuels, 1999, 13, 1074 CrossRef CAS.
- H. Yano, F. Shirai, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2002, 533, 113 CrossRef CAS.
- C. Delacourt, P. Ridgway, J. Kerr and J. Newman, J. Electrochem. Soc., 2008, 155, B42 CrossRef CAS.
- M. Shibata and N. Furuya, Electrochim. Acta, 2003, 48, 3953 CrossRef CAS.
- J. Vega, N. Spinner, M. Catanese and W. Mustain, J. Electrochem. Soc. Search PubMed , Accepted, in press.
- J. Vega, S. Smith and W. Mustain, J. Electrochem. Soc., 2011, 158, B349 CrossRef CAS.
- J. Vega, C. Chartier and W. Mustain, J. Power Sources, 2010, 195, 7176 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, Energy Convers. Manage., 2007, 48, 1255 CrossRef CAS.
- M. Gonçalves, A. Gomes, J. Condeço, R. Fernandes, T. Pardal, C. Sequeira and J. Branco, Energy Convers. Manage., 2010, 51, 30 CrossRef.
- K. Ogura, R. Oohara and Y. Kudo, J. Electrochem. Soc., 2005, 152, D213 CrossRef CAS.
- H. Yano, T. Tanaka, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2004, 565, 287 CrossRef CAS.
- J. Yano, T. Morita, K. Shimano, Y. Nagami and S. Yamasaki, J. Solid State Electrochem., 2007, 11, 554 CrossRef CAS.
- S. Ikeda, T. Takagi and K. Ito, Bull. Chem. Soc. Jpn., 1987, 60, 2517 CrossRef CAS.
- H. Yano, F. Shirai, M. Nakayama and K. Ogura, J. Electroanal. Chem., 2002, 519, 93 CrossRef CAS.
-
A. Bard and L. Faulkner, Electrochemical Methods, Fundamentals and Applications, John Wiley & Sons, New Jersey, Second edn, 2001 Search PubMed.
- B. Smith, D. Irish, P. Kedzierzawski and J. Augustynski, J. Electrochem. Soc., 1997, 144, 4288 CrossRef CAS.
- M. Shibata and N. Furuya, J. Electroanal. Chem., 2001, 507, 177 CrossRef CAS.
- S. Narayanan, B. Haines, J. Soler and T. Valdez, J. Electrochem. Soc., 2011, 158, A167 CrossRef CAS.
- M. Saha, T. Furuta and Y. Nishiki, Electrochem. Commun., 2004, 6, 201 CrossRef CAS.
- N. Sonoyama and T. Sakata, Chem. Lett., 2002, 31, 444 CrossRef.
- C. Sanchez-Sanchez, V. Montiel, D. Tryk, A. Aldaz and A. Fujishima, Pure Appl. Chem., 2001, 73, 1917 CrossRef CAS.
- Z. Yoshida, H. Yosue and G. Nogami, J. Electrochem. Soc., 2001, 148, D55 CrossRef CAS.
- S. Kaneco, K. Iiba, M. Yabuuchi, N. Nishio, H. Ohnishi, H. Katsumata, T. Suzuki and K. Ohta, Ind. Eng. Chem. Res., 2002, 41, 5165 CrossRef CAS.
- S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Energy Fuels, 2006, 20, 409 CrossRef CAS.
- S. Kaneco, Y. Ueno, H. Katsumata, T. Suzuki and K. Ohta, Chem. Eng. J., 2006, 119, 107 CrossRef CAS.
- S. Kaneco, R. Iwao, K. Iiba, S. Itoh, K. Ohta and T. Mizuno, Environ. Eng. Sci., 1999, 16, 131 CrossRef CAS.
- R. Aydin and F. Köleli, Synth. Met., 2004, 144, 75 CrossRef CAS.
- R. Aydin and F. Köleli, J. Electroanal. Chem., 2002, 535, 107 CrossRef CAS.
- J. Bockris and J. Wass, J. Electrochem. Soc., 1989, 136, 2521 CrossRef CAS.
- D. Canfield and K. Frese, J. Electrochem. Soc., 1983, 130, 1772 CrossRef CAS.
- H. Flaisher, R. Tenne and M. Halmann, J. Electroanal. Chem., 1996, 402, 97 CrossRef.
- S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Chem. Eng. J., 2006, 116, 227 CrossRef CAS.
- N. Kitamura and S. Tazuke, Chem. Lett., 1983, 12, 1109 CrossRef.
- J. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701 CrossRef CAS.
- J. Lehn and R. Ziessel, J. Organomet. Chem., 1990, 382, 157 CrossRef CAS.
- S. Tazuke and N. Kitamura, Nature, 1978, 275, 301 CrossRef CAS.
- S. Kaneco, Y. Shimizu, K. Ohta and T. Mizuno, J. Photochem. Photobiol., A, 1998, 115, 223 CrossRef CAS.
- B. Åkermark, U. Eklund-Westlin, P. Baeckström and R. Löf, Acta Chem. Scand., Ser. B, 1980, 34, 27 CrossRef.
- T. Reda, C. Plugge, N. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654 CrossRef CAS.
- B. Parkinson and B. Weaver, Nature, 1984, 309, 148 CrossRef CAS.
- S. Kuwabata, R. Tsuda, K. Nishida and H. Yoneyama, Chem. Lett., 1993, 22, 1631 CrossRef.
- S. Kuwabata, R. Tsuda and H. Yoneyama, J. Am. Chem. Soc., 1994, 116, 5437 CrossRef CAS.
- P. Addo, R. Arechederra, A. Waheed, J. Shoemaker, W. Sly and S. Minteer, Electrochem. Solid-State Lett., 2011, 14, E9 CrossRef CAS.
- R. Obert and B. Dave, J. Am. Chem. Soc., 1999, 121, 12192 CrossRef CAS.
- F. Baskaya, X. Zhao, M. Flickinger and P. Wang, Appl. Biochem. Biotechnol., 2010, 162, 391 CrossRef CAS.
- S. Kuwabata, N. Morishita and H. Yoneyama, Chem. Lett., 1990, 19, 1151 CrossRef.
- K. Sugimura, S. Kuwabata and H. Yoneyama, J. Am. Chem. Soc., 1989, 111, 2361 CrossRef CAS.
- W. Shin, S. Lee, J. Shin, S. Lee and Y. Kim, J. Am. Chem. Soc., 2003, 125, 14688 CrossRef CAS.
- A. Parkin, J. Seravalli, K. Vincent, S. Ragsdale and F. Armstrong, J. Am. Chem. Soc., 2007, 129, 10328 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.