Ionic conductance of synthetic channels: analysis, lessons, and recommendations
Jonathan K. W.
Chui
and
Thomas M.
Fyles
*
Department of Chemistry, University of Victoria, Victoria B.C., Canada. E-mail: tmf@uvic.ca; Fax: +1 250 721 7147; Tel: +1 250 721 7192
First published on 21st June 2011
Abstract
Synthetic ion channels have been known for nearly three decades, but it is only in the past decade that analysis of the currents these ionic conductors carry has become a standard technique. A broad range of structural types have been explored and these reports have produced a very diverse collection of ion channel conductance behaviours. In this critical review we describe a notational method to extract salient information from reported ion channel experiments. We use an activity grid to represent quantitative information on conductance and opening duration with a five-level colour code to represent qualitative information on the nature of the conductance–time profile. Analysis of the cumulative dataset suggests that the reported conductance data can reflect the structural features of the compounds prepared, but does also reflect the energetic landscape of the bilayer membrane in which synthetic ion channels function (143 references).
1. Introduction
Natural ion channels are essential to cellular life. They are efficient and remarkably selective gate keepers that mediate the traffic of ions across the insulating cell membrane. These features are coupled with controlling elements sensitive to external stimuli such as mechanical, potential, or chemical signals, and this makes them regulating elements in diverse physiological phenomena.1 Ion channels are the elementary components of our nervous system; they are transistors to the computer between our ears. Understanding their mechanism potentially allows us to tailor their function, with implications in both biomedical and technological fields.Natural ion channels are high-molecular weight proteins whose functions depend upon being embedded in bilayer membranes. They are challenging to express in quantities needed for mechanistic studies by chemical methods, and difficult to reconstitute in functional forms when available. Early electrophysiology studies were done on selected tissues (e.g. giant axons from squid),2 and a general molecular understanding would come only with the advent of the voltage clamp technique with which single channel activities of a variety of ion channels can be observed.3,4 In recent years the natural ion channel field has accumulated a solid mechanistic understanding of both small peptidic channels (e.g. gramicidin)5 and large protein channels6 based upon a combination of solid-state structures and computational modelling.
Parallel to the investigation of natural ion channels are efforts to create ion channels using synthetic chemistry. The premise of this reductionist approach is that the ion channel activity can be mimicked with substantially simpler compounds than those evident in nature. The goals of such studies vary from explicitly biomimetic and mechanistic, to directed at biomedical or technological applications. Whatever the goal, the main emphasis is on function—the ability of the compound to mediate ion transfer through a bilayer membrane.
It is almost three decades since Tabushi et al. published the first report of a synthetic ion channel.7 Since that time, synthetic chemists have shown tremendous imagination in the preparation of a diverse array of compounds that actively exhibit ion channel functions. Synthetic ion channels are often characterized by vesicle-based techniques, in which a probe molecule reports on the creation or collapse of ionic gradients across a vesicle membrane.8 Such techniques are ideal for survey purposes in which structure–activity relationships are deduced within a single set of related molecules. However, the specialized synthetic efforts required to prepare each class of compounds inevitably segregates the understanding of these activities from which a general mechanistic comparison is difficult.
In principle, the voltage-clamp technique offers a means to report ion channel activity in a way that is generally comparable across a range of structures. The technique is based on direct observation of the current carried by an ion channel embedded in a planar bilayer membrane.4,9 Some details of the experiment are discussed in the next section. The high conductance of ion channels and the sensitivity of current measurement instruments allow detection of events arising at the “single molecule” level.1 The nature of those events—the current carried and their duration—can provide information on the nature of the ion channel.
However, the observations are of transient phenomena occurring randomly, so the analyses of the records are statistically based and frequently restricted to relatively few observations. More problematic is the general observation that even for compounds that have single-molecule voltage-clamp data available, the activities vary qualitatively. It is often unclear how one should report traces with very disparate activities represented and the literature has numerous references to “erratic” phenomena. In addition, conductance and opening durations vary over orders of magnitude, and are dependent on experimental conditions such as the lipid used to construct the bilayer and the bathing electrolyte. The voltage-clamp data for synthetic ion channels reported to date are therefore largely qualitative and no systematic comparison has been attempted. This review is our attempt to make sense of this large body of work. To make this possible, we propose a visualization method that enables side-by-side comparisons in a framework that gives a coarse but unifying view.
Reviews on membrane transport by synthetic ion channels are widely available: a series of reviews spanning the entire period is believed to be comprehensive,10–13 and additional reviews cover tutorial aspects,8,14 together with different structural or substrate perspectives.15–20 We limit ourselves to synthetic ion channels for which voltage-clamp data were published as part of the primary report, either as a figure in the text or as part of the supporting information; compounds whose activity was solely characterized by vesicle techniques are outside the scope of this review.
No clear line divides synthetic and modified natural ion channels, and we have chosen to include only compounds that use synthetic elements in their (assumed) ion translocation unit. This review will thus include THF-modified gramicidin channels,21 but not hybrid channels that are formed by tethering gramicidin or alamethicin units.22–24 We also do not discuss ion channels that are solely peptides, as these are usually related to natural products or sequences within ion channel proteins.
This review begins with a discussion of the voltage-clamp experiment and the ways in which the data can be interpreted written for a non-specialist reader. Section 3 describes a notational system that permits the essential features of the data to be represented using an “activity grid”. The actual review of the literature, believed to be comprehensive to March 2011, appears in Section 4; a non-specialist reader could skip this section and proceed directly to the summary sections, 5 and 6, that follow.
2. Measurement and interpretation of voltage clamp data
The voltage clamp experiment is a specialized technique, and in this section, we will briefly discuss the types of information that can be gleaned from such an experiment, and their implications for the mechanism of ion transport. More details can be found in other specialized monographs.4,252.1 Experimental methods
A schematic of the voltage clamp experiment is shown in Fig. 1. Two electrolyte solutions are contained in separate chambers, each of which is connected to a Ag/AgCl electrode through a salt-bridge. Between the two chambers is a small aperture of 25–250 μm in diameter. At the beginning of the experiment, a solution of lipid in a solvent (typically decane or hexadecane) is painted across this aperture. The lipid molecules self-assemble into a bilayer, whereupon the two electrolyte solutions are electrically insulated from one another. This thin film of hydrophobic material presents a high kinetic barrier for ion flow, and the conductance between the electrodes is reduced to near zero.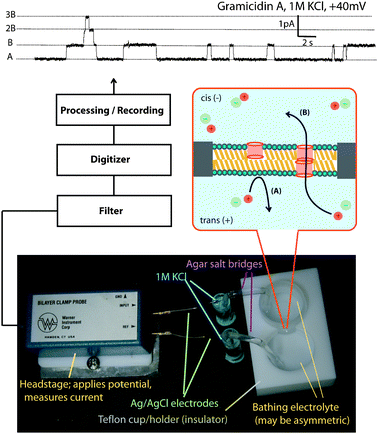 | ||
| Fig. 1 Voltage clamp experiment. The bottom image shows the cell with the information flow leading to a typical trace for gramicidin at the top. The insert schematic shows the formation of a conducting dimer. | ||
A decrease in conductance is necessary but not sufficient evidence of the formation of a bilayer. The formation of a bilayer at the molecular level is observed by measuring the membrane capacitance, which correlates the surface area with thickness of the insulator. A value of approximately 0.6 pF cm−2 translates into a membrane thickness of 4 nm indicating a well-formed bilayer. A bilayer capacitance test function is available on some commercial instruments.
A putative ion channel compound is then introduced to the bilayer. This can be achieved in a variety of ways: (i) fusion of compound-containing vesicles to the bilayer; (ii) direct injection into the electrolyte solution with gentle stirring whereby the compound diffuses to the bilayer and inserts; (iii) physical transfer by brushing a solution containing the compound across the bilayer; (iv) pre-mixing the compound into the lipid solution before formation of the bilayer. It can be difficult to establish the integrity of the bilayer when the latter two methods are required, and these are typically used only when the compound is very insoluble.
The putative ion channel is now partitioned between the aqueous solution and the lipid bilayer. Depending on the properties of the compound, a complex set of equilibria may be present, including species of different stoichiometries, conformations, and orientations with respect to the bilayer. One or more of the states in this complex mixture may provide an energetically feasible pathway for ion translocation through the bilayer, referred to as “conducting”, “active”, or “productive” states. Fig. 1 shows two possible states for a gramicidin molecule—a productive dimer channel whose conductance can be detected, and an isolated “half-channel” that is invisible to the technique.
The experimentalist observes the conducting states by holding a constant potential across the bilayer (hence the name “voltage clamp”) and directly observes the fluctuations in current as the active state alternately conducts ions and then closes. Typically, each conducting ion channel passes 106 to 107 ions across the membrane each second. The instrument is typically able to measure pA fluctuations in current, and experimental conditions are often chosen such that activity from only one or two ion channels is seen at one time. The current is monitored over time, is digitized, and low-pass filtered to give “traces” or “voltage-clamp records” from which molecular properties of the active state can be inferred. Fig. 1 shows a typical trace of gramicidin; many synthetic ion channels “look” exactly like this trace. The trace of Fig. 1 suggests that there are three channels active during the duration of this experiment; level A corresponds to no active states, level B to one active state, etc. Parameters of interest are the conductance of the active state (B–A; 2B–B; etc.) and the duration of the active state (∼0.3–3 seconds in Fig. 1).
These traces represent fundamentally different information from that obtained in conventional chemical analyses. Conventional analytical techniques report the average behaviour of the ensemble present in the system, where the contribution of each state is weighed according to its proportion within the mixture. This is the case because most techniques operate with femtomoles or more molecules available, and within this large population (>108 molecules), even very rare states are represented in the populations. When the analysis scales down to one involving only a few molecules, the statistics of large ensembles do not apply. In this situation a molecule occupies only one state at any time, and shifting between states is a probabilistic event. Individual features in a voltage-clamp trace therefore arise from stochastic phenomena. These random processes are usually modelled as Markov processes, where the states are assumed to be separated by static barriers and transitions between them are history-independent.25
In addition to single-channel behaviours, a number of other parameters are important. Foremost is the conductance of the channel, assessed from the slope of the current as a function of applied potential (i–ε plot). This can be built from an analysis of single-channel records, or directly by application of a voltage ramp to a membrane containing a number of active channels. Frequently channels simply follow Ohm's law; non-linearity in the i–ε curve can be related to voltage-gating of the system. It is common to use the same electrolytes on either side of the membrane, but the i–ε plot under electrolyte gradients can be used to determine ionic selectivity. The potential of zero current (reversal potential εrev) is related to the permeabilities of the cations (PM) and anions (PX) and the activities of the cations and anions in the contacting solutions (aM and aX respectively) on the two sides of the cell (cis- and trans-) via the Goldman–Hodgkin–Katz (GHK) equation:
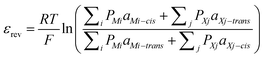 | (1) |
2.2 Significance of the time dimension
While the typical instrument can acquire currents on the tens of microseconds timescale, data are usually only reliable at greater than milli-second resolution. There are several reasons for this much slower time scale. First, low-pass filters with corner frequencies of 20–1000 Hz are needed to remove the high frequency electrical noise, but this filtering also rejects short duration signals. Secondly, large digital storage space is required to store high-resolution data, and the experimenter may decimate the data to make storage practical. Lastly, no automated procedures are available to analyze the variegated behaviour represented in typical voltage-clamp traces, and it is difficult to manually process hours of data recorded at microsecond resolution. Thus reliable data are usually available for events of millisecond or longer durations. At the other extreme, some events last for tens of minutes.The length of the event, also referred to as the “open duration” or the “open dwell time”, correlates to the kinetic stability of an open state. That these are ever observed for longer than milliseconds is remarkable, considering that the membrane is not a static environment but rather a dynamic liquid crystal that undergoes a range of molecular motions at different time and length scales. Individual lipids change conformations, diffuse laterally, and flip to the other leaflet; leaflets swell and thin, and the entire membrane undulates.26 Apart from lipid flip-flop, all these processes are in the pico- to tens of nano-second time range. The observation of a molecular conducting event of millisecond duration implies that the conducting state has resisted decomposition during uncounted collisions, and has been able to diffuse laterally within the bilayer.
Over the course of a long acquisition, an average open dwell time may be observed. While the average lifetime hints to the overall kinetic stability of the active state, a far more interesting parameter is in the distribution of lifetimes. In unique events described by a single closing process, lifetimes can be described by a mono-exponential; events that cannot be described by a single exponential can often be fitted to a combination of Markov processes.27 After fitting the data to an adequate model, parameters pertaining to the molecular mechanism can be extracted. However, it should be noted that some open durations are better described by a power law,28 suggesting that alternative classes of mechanisms may be underlying the particular observed behaviour.
2.3 Significance of the conductance dimension
The function of an ion channel is to pass ions, hence the conductance is an important attribute of any active state. Conductance is simply defined by Ohm's law: g = i/ε where g is the conductance (units of Siemens), i is the measured current (Amperes), and ε is the applied potential (Volts). The unit conductances of ion channels are usually of the order of picoSiemens (pS). Depending on the noise characteristics of the instrument, a range of conductances from pS up to several nS can be measured.In one sense, the ion channel conductance is derived from a continuum phenomenon, where the channel is simply treated as an aqueous, cylindrical electrical shunt within an otherwise impermeable membrane. The resistance of the conducting structure then predicts the maximal possible rate of flux. This relationship is often referred to as the Hille equation:1
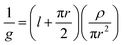 | (2) |
An alternative way to conceptualize conductance is as a rate parameter reflecting the ability of the active structure to catalyse ion translocation across the membrane. This can be discussed in the framework of an Eyring model,1,4 in which the ion on its trajectory encounters a series of stabilizing energy wells separated by energy barriers. High throughput (high conductance) requires that the passing ions face neither significant barriers nor deep wells, as the latter would trap the ion and impede translocation. Many of the motifs used in synthetic ion channels are inspired by moderately strong ion complexing structures such as crown-ethers. However, here their function is not to bind and hold the ion but simply to lower the kinetic barrier by solvating the ion within the bilayer. Within an Eyring model, different ions could be solvated differently, and the model provides a direct account of ion selectivity.
Ion selectivity in transport can occur by a balance of two distinct mechanisms. The first is by a size-exclusion mechanism, where larger (partially solvated) ions are physically excluded from entry (higher barrier for larger ions). The other stems from the need for ions to be partially desolvated during the bilayer crossing. An ion with a lower solvation energy is thus transported more frequently, and with all else equal, will have higher permeability than an ion with a higher solvation energy. This results in a “natural selectivity” of Cs+ > Rb+ > K+ > Na+ ≫ Li+ for cations, also known as the Eisenman sequence I.30 The higher Eisenman sequences (II–XI) represent increasing contribution by the size-exclusion mechanism. The Hofmeister series relates to an analogous solvation sequence for anions.31
2.4 Significance of the shape of the trace
The visually most distinct shape observed in voltage-clamp traces is the classical “square-top” behaviour, where the channel switches abruptly from an “off”-state to an “on”-state, during which it maintains a constant current (Fig. 1). At some later time it abruptly shifts from “on” to “off”. This corresponds to a single active state being formed at a time-scale faster than the observing resolution, the active state maintaining a defined conducting structure for the duration of the event, followed by a subsequent fast collapse.As illustrated in Fig. 1, gramicidin is a well-studied ion channel that exhibits this behaviour. Gramicidin is a short peptide with 15 alternating D- and L-amino acids; when partitioned into a membrane, it forms β6,3 helices that approximately span a single leaflet.5 The transition to the “on” state occurs when two helixes in opposite leaflets assemble at the leaflet interface, thereby forming a transmembrane tunnel. The antiparallel dimers are held together by 6 hydrogen bonds, and lifetimes of up to several seconds can be observed. Ion passage ceases when the dimers drift apart. Multiple copies of the dimer can be present at the same time, and as these gate independently of one another, they appear as binomially distributed stacks of square-tops.5 This observed signal shape is affected by filtering, and the lack of a “square-top” behaviour cannot be taken as evidence for the lack of a discrete conducting structure. This is particularly applicable when the short event intersects the rise-time of the low-pass filter.
If all ion channel conductance traces were as simple as those in Fig. 1, there would be little need of this review. The reality is that many, perhaps a majority of, ion channel recordings contain other shapes in the traces, corresponding to more complex molecular behaviours of the productive states. A selection of commonly observed shapes is given in Fig. 2. The top panel (Fig. 2A) shows step-like behaviour but the transitions between the steps are very rapid (“flicker”) and the steps are unequal in conductance. This must be due to pairs of different open states that rapidly interconvert. A similar situation of multiple open states is illustrated in Fig. 2B. Here the “event” starts and stops clearly, with a large number of discrete states accessed during the event. It is possible to discern (in this example) a recurring level, but there are a large number of other states encompassed within this event. Fig. 2C shows some clear steps, and then an odd slowly evolving structure which shows no discrete levels. It is clear from the time span that these are events associated with reorganization of the compound and the lipids of the bilayer as these fall in the time range expected for lipid flip-flop and other slow processes. Fig. 2C also shows “spike” behaviour—transient events that are typically unresolved by the digitizer but not removed by the filter. These are compound related, and in some cases can be the sole observed activity. Finally, there are truly “erratic” events such as those illustrated in Fig. 2D. The molecular basis for this type of behaviour is unclear, but it is certainly compound related in many cases, and a few examples have been shown to exhibit apparent power-law scaling in the durations of the current excursions above a threshold value.28
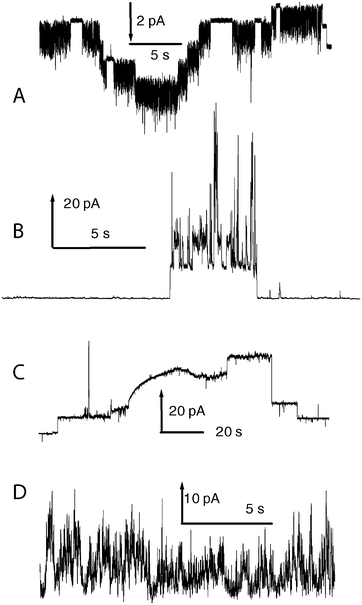 | ||
| Fig. 2 Representative traces showing the range of observed signals from voltage-clamp experiments. | ||
3. A notational scheme for voltage-clamp data
As discussed above, the conductance activity of ion channels varies substantially. This variation is both qualitative—in the shape of the conductance over time—and quantitative—in the magnitude of the current and the duration of the event. Literature data report ion channel conductances spanning three orders of magnitude (pS to nS) and events spanning at least five orders of magnitude in the time dimension (milliseconds to hours). In order to compare behaviours we require a system to represent both quantitative and qualitative information reported.The quantitative aspects are readily represented by location on an activity grid of conductance/duration (Fig. 3, left). The grid is necessarily logarithmic to accommodate the thousand-fold differences in both current and duration. The scale of the axis was chosen to cover the extent of all reported data. Divisions are made for every order of magnitude along the duration, and half order of magnitude in the conductance dimension. This logarithmic classification reveals the big picture between disparate compounds. It also eliminates the continuum distinction between different electrolytes; there is only a twofold difference in solution conductivity between sodium and caesium salts at the same activity. The logarithmic scale masks relatively small but significant differences in activity; these are noted in the text and a finer grid may be appropriate as our understanding improves.
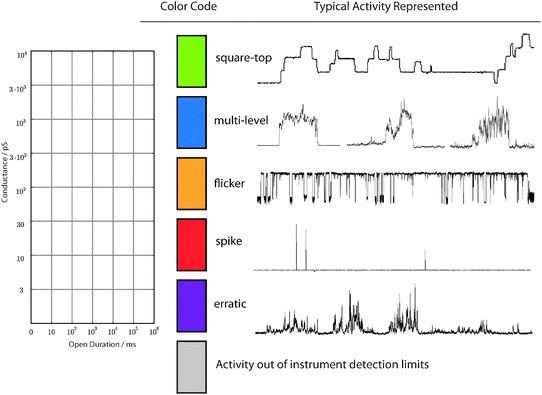 | ||
| Fig. 3 (Left) Activity grid showing the log–log scaling of the conductance and duration dimensions; (centre) colour codes for the classes of activity; (right) typical conductance–time traces representative of the classes of activity. | ||
Within this activity grid, the qualitative information is displayed as colour coding according to the type of activity observed. The colour code emerged from surveying experimental data from the literature and our own work. It appears that a large majority of reported (and unreported) data may be reconstructed by five prominent classes of behaviour. The colour codes, together with representative traces, are shown in Fig. 3 (right panel). Green behaviours describe the stepwise transitions between an “off” and an “on” state (“square-top”). Note that the baseline noise level is comparable to the noise level of the “open” state for this type of behaviour. Blue behaviours are stepwise transitions that are irregular or show multiple opening levels in the open state, i.e. the “noise band” of the open state is large with respect to the baseline noise. Yellow behaviours are stepwise transitions where the open state “flickers” or transits rapidly between the two open states, or between an open and a closed state. Red behaviours involve a “spike” or burst of current with no defined step. They also include slowly evolving events without defined steps such as those illustrated in Fig. 2C and reported as “shark-fins”.32Purple behaviours are erratic clusters and bursts that to the eye show no apparent structure. The colour grey denotes a region of a grid that is unknowable due to experimental constraints such as heavy filtering that precludes observation of short lifetimes, or a conductance that would lie below the baseline noise level.
Classifying qualitative data is a subjective act: regular steps (“square-tops”) blend into irregular steps in a continuum, and the binning into one or other depends on observer judgment. The classification scheme is therefore coarse grained to avoid being overwhelmed by the fine divisions and distinctions between different behaviours. A single trace can contain many different behaviours, and these can be represented as different colours on a single activity grid. Where overlaps of duration or conductance occur, separate grids can be generated for clarity.
The survey of the literature which follows is based upon the use of activity grids to represent the reported activity. Shapes (and thus colour) were assessed by visual inspection of traces presented in the original publication. If reported, lifetimes and conductances were used directly; when these are not reported, they were measured from the supplied traces, assuming that the traces are representative of the compound's behaviour. When a compound was reported to behave differently over a range of conditions (e.g. a pH dependence), each condition generates an individual activity grid. In isolated cases, multiple papers attribute different sets of behaviour to the same compound. For these, all reported activities are given on the same grid, with any exceptions noted in the text.
4. Voltage clamp data from the literature
The previous section proposes a framework to describe voltage clamp data. This is necessary but not sufficient for a subsequent discussion of structure–activity relationships and we require in addition meaningful structural descriptors. As the tabulation of the activity grids required a database of compounds in order to facilitate production and sorting of the activities, we added a number of structure-related descriptors with each record. Reported channel-forming molecules differ in multiple structural dimensions. Few (if any) metrics allow clean demarcations between classes of compounds and overlapping inevitably occurs. Whatever classes are chosen, the divisions need to each contain enough compounds (i.e. the classes cannot be too small), and there should be sufficient categories to allow comparisons (i.e. the classes cannot be too big). To optimize the divisions, we tagged each compound in the database by both low-level features (e.g. functional groups present) as well as high-level features (e.g. whether the compound is expected to span a single or both leaflets of the bilayer; rotational degrees of freedom as a surrogate for rigidity, etc.).Fundamentally, the divisions reflect chemical intuition and the review is loosely based on our sense that overall topology matters. This is certainly reflected in the ways in which structures are reported, as there is an underlying sense that a molecule needs to achieve some tubular or column-like structure within the membrane and this in turn requires a particular topology at the outset. In discussing topology, we made a distinction between compounds of the following types: (i) Circular compounds contain a single macrocyclic core which may also bear radiating units; (ii) Beaded string describes compounds which contain at least one macrocyclic unit connected to one or more linkers. The majority of these linkers are connected to another macrocyclic unit (bead); (iii) Tree describes compounds which contain appendages connected along a linear scaffold unit. Sometimes the “tree” is explicit; occasionally the structure might alternatively be described as a “brush”; (iv) Linear compounds can be traced from one end to the other; and (v) Complex topologies are those which do not neatly rest in one of the other categories.
4.1 Circular topology/macrocycle-based
A common mental image of synthetic ion channels consists of an aqueous tube through the membrane, and a face-to-face stack of macrocycles features prominently as a possible structural implementation. This design strategy requires some type of interaction between the macrocycles, such as hydrogen bonding. Another image is of a channel “mouth” based on a macrocycle with appended “throat” components that make up the transmembrane structure. In this section, we describe compounds which are constructed around a macrocycle, further classified by the type of macrocycle used.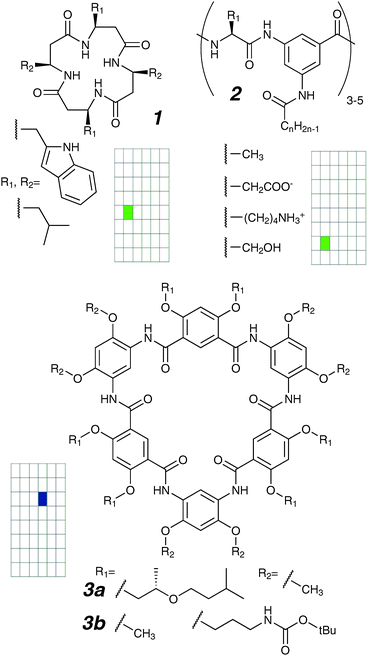 | ||
| Fig. 4 Peptidic and amidic macrocycles. | ||
The conductances switch off stochastically with typical lifetimes in the millisecond range, and the authors proposed disassembly of a monomer from the stack followed by ring collapse as a likely mechanism for this all-or-none transition.33 It is noteworthy that these lifetimes are orders of magnitude longer than even the slowest of membrane processes, suggesting conducting structures that persist in the chaotic membrane environment. Not all peptides of type 1 are active; the tetra-Leu derivative was reported as inactive.
Using the same strategy, Ishida and co-workers reported ion channels formed by compounds of type 2, described as “cyclic peptides containing an non-natural amino acid” (3-amino-5-N-acylbenzoic acid).36,37 Each of the individual elements were modified systematically: ring size (hexa- to deca-peptide), length of alkyl chain (10, 16, 18 carbons total), and amino acid side-chain on the macrocycle. Regular openings were observed for all of the compounds, and these openings were amenable to blocking by Ca2+ ions. Surprisingly, the lifetimes and durations exhibited by all these compounds are identical despite the large structural variations. This is difficult to rationalize under the proposed model where the macrocycle lies at the membrane–aqueous interface juncture with the alkyl chains anchored perpendicularly into the membrane.
Gong and co-workers reported a class of macrocycles constructed by one-step condensation of diamines and diacyl chlorides.38 These molecules form non-collapsible rings of 0.8 nm inside diameter, with the interior lined with polar carbonyls. They are known to aggregate into fibres, likely through extensive contact of the aromatic surfaces. Certain members of the family (3a and 3b) have suitable solubility for voltage clamp, and like compound 1, they showed stepwise activities presumably through a similar stack assembly process. These channels share similarly high conductances of >700 pS (1 M KCl), consistent with a large diameter pore, and lifetimes are in excess of 10 seconds, reflecting a high kinetic stability of the conducting state.
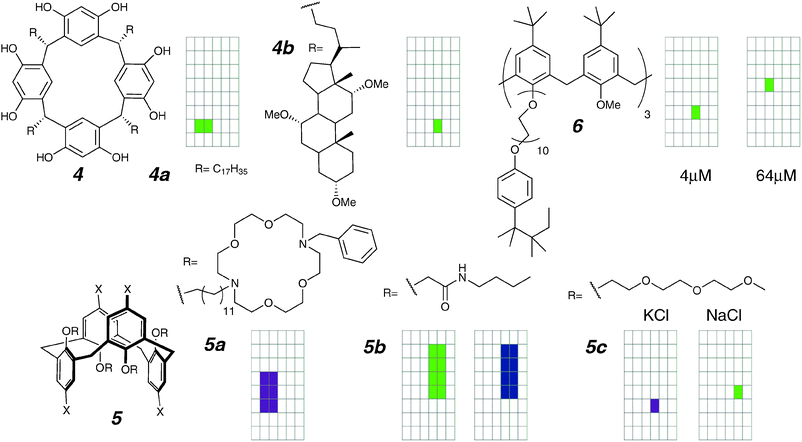 | ||
| Fig. 5 Resorcinarene and calixarene channels and activity grids. | ||
A detailed study of 4a showed a K+/Cl− selectivity of ∼20 and a K+/Na+ selectivity of ∼3. Surprisingly, the K+ current is entirely blocked by Rb+, suggesting a single conducting portal. The authors proposed that a desolvated cation would squeeze through the resorcinarene ring, even though this process is not observed in cavitands.40 They also illustrated a desolvated K+ ion within the confines of the alkyl tails. It is not clear how ions would be stabilized in that configuration, and it is unclear what specific interaction allows molecules in each leaflet to dimerize for times in excess of one second. Recent reports of pyrogallarene capsules as channels (Section 4.5) offer an alternative structural model that may apply in this system as well.
In subsequent exploration, Kobuke's group would replace the flexible alkyl chain with rigid steroidal anchors to give compound 4b.41 These cholate derivatives contain polar methoxy groups, and the expectation is that they can both better sustain a stable structure (longer lifetimes) and better stabilize an ion-in-transit (higher conductance). The results partially conform with this expectation: the open state is more defined, the lifetimes are tenfold longer, and the conductance is also increased (9.4 pS, up from 6.1 pS).39,41 The K+/Cl− selectivity also decreased two-fold relative to 4a. Clearly selectivity cannot be a function of the macrocycle, but a property of the entire structure.
Calixarenes and resorcinarenes are structurally related macrocycles, with the significant difference that several conformations in calixarenes are both accessible and can be locked in. How does the large difference in the geometry affect the activities of calixarene-derived channels? Gokel and co-workers attempted to address this with a series of calix[4]arenes substituted with pendant crown ethers (e.g.5a; 1,3-alt conformation illustrated).42 For these compounds, it was noted that both the 1,3-alt (shown) and 1,2-alt conformers (not shown) gave erratic burst-type activity, whereas the cone conformer is completely inactive. This is attributed to the inability of the cone conformer to span both leaflets on its own, whereas in the alt-conformers, the alternating arms would allow the calixarenes to be buried in the centre of the bilayer. Compound 5a can be compared with the hydraphile class of compounds (Section 4.2.1), in which the calixarene is replaced by a crown ether.
Potentially self-associating 1,3-alt-calix[4]arene derivatives were reported by Davis and co-workers, and the most promising compound 5b was examined for its activity in planar bilayers.43 Compound 5b forms chloride-bridged dimers in the solid state, and chloride-driven dimerization in the solution state was demonstrated by NMR-monitored titrations. This 1,3-alt-calix[4]arene shows a rich collection of conductance behaviours. Both discrete opening and closing events are observed; however, there is a spread in the conductance of these events from 100 to 2000 pS. Most of the open states are not of uniform conductance and fluctuations are seen over periods of seconds before abrupt collapse to the closed state.
More recently, Cragg and co-workers reported a related 1,3-alt-calix[4]arene compound 5c which replaces the primarily alkyl pendants of previous examples with highly hydrophilic polyethers.44 It was reported that two types of activity can be seen, and that the type of activity seen is governed by the choice of electrolyte. In Na+ solutions, discrete openings of 30–100 pS were observed, whereas in K+ solutions, only bursts of <10 pS were seen. While not a formal proof of selectivity (e.g. based on reversal potential), compound 5c nonetheless remains unique in showing higher conductance of Na+ over K+, somehow reversing the dehydration governed Eisenman sequence I usually observed for synthetic ion channels.
The same group had previously reported the calix[6]arene derivative 6 with polyether chains related to the surfactant Triton.45 The smaller homolog calix[4]arene was reported to be completely inactive, whereas the larger analogue shows different types of regular openings that are concentration dependent. These detergent-derived structures are reminiscent of the relatively inactive “bouquet” channels (in vesicles only; no voltage-clamp data reported).46,47 Comparing the two classes of compounds, the reported activity of the quite hydrophilic 6 with the low activity of the more hydrophobic bouquets is puzzling and indicates that a more refined understanding of partitioning is likely required.
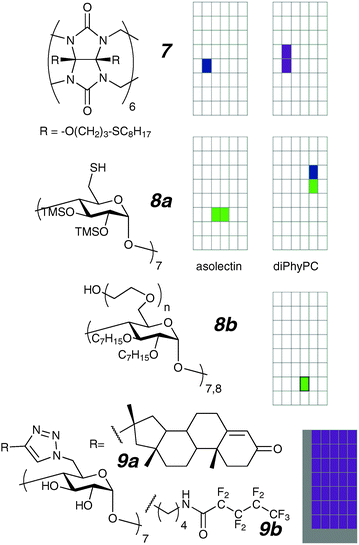 | ||
| Fig. 6 Cucurbituril and cyclodextrin ion channels. The grey colour on the 9a,b grid indicates that low conductance states are not observable in the presence of much higher conductance openings, and that instrument filtering precludes observations of short duration events. | ||
While the first synthetic ion channel was based on β-cyclodextrin,50 and a number of other vesicle-active cyclodextrin channel compounds have been prepared,51,52 few members of this class of compounds have been examined in planar bilayers using the voltage-clamp technique. The first report is of the activity of a protected β-cyclodextrin (8a).53 Due to its simple structure the compound activity was described as “unexpected” and moreover exhibited completely different behaviour in different lipids. The openings in diPhyPC were an order of magnitude larger and longer-lived than those seen in asolectin.‡ Subsequent experiments suggest this to be a function of membrane thickness, where the compound is more active in thinner membranes than thicker ones. It is postulated that the conducting structure may be small micellar structures, possibly made easier to generate by partially oxidized, disulfide-linked oligomers. In low concentrations, these oligomers would be difficult to identify with conventional techniques. Subsequent work from this group reported the starburst oligomers 8b prepared by cyclodextrin initiated polymerization of ethylene oxide. Despite the very wide range of lengths of poly-ethyleneoxide chains reported (n = 0 to 50) and the use of both β- and γ-cyclodextrin cores as initiators, the conductance traces are essentially identical in character.54 The authors discuss an alternative approach to estimating channel radius, and propose that an end-to-end dimer lined with polyethylene oxide is the active species. Given that the n = 0 species is active, it seems more likely that the common denominator heptyl units reside in the membrane with the polyethylene oxide towards the aqueous phase.
We have recently reported on two cyclodextrin-based channels (9a–b) that show long-lived and “stable” erratic behaviour.28 The activity is “self-similar” with highly erratic behaviours over widely different observation scales in both conductance and time. This behaviour is intractable at the level of a single “event”, but collectively can be shown to follow a power law in the time domain. This methodology may be applicable to analysis of erratic behaviours shown by other compounds.
4.2 Beaded strings topology
In this section we continue our description of compounds incorporating macrocycles. The macrocycles here, however, act as a subsidiary components connected by various types of linkers.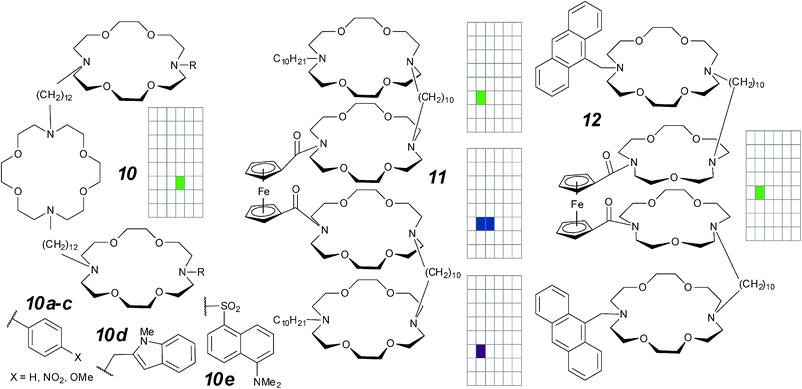 | ||
| Fig. 7 Hydraphile-type compounds and activity grids. | ||
In vesicle studies, the macroscopic ion flux is measured. Under those conditions, compounds 10a–c show transport activity that correlates with the electron-donating effect of the aromatic substituents; this is attributed to stabilization of an ion-in-transit by the terminal crowns.60 In single-molecule voltage-clamp studies, the same trio of compounds show only square-top transitions with unit conductances around 12–15 pS (0.4 M NaCl); no electronic effect is evident. Note that the activity observed in vesicles may be due to preferential partitioning into, or orientation within the bilayer. Multiple conducting states of about the same magnitude in the same electrolyte are clearly seen in compounds 10d–e.58 It is possible that these are different orientations within a unimolecular structure, but the conductance is too small for a full-length channel of the dimensions of an 18-crown-6.
In the quest for a redox-active artificial ion channel, Hall and co-workers linked the aza-crown ethers of the hydraphiles with a ferrocene unit to give compounds 1161 and 12.62 These compounds were examined under patch clamp conditions with lipids from hamster brain. Compound 11 displays two different semi-regular transitions of 50 pS and 30 pS (0.125 M KCl); irregular spikes of similar magnitude were observed when the polarity of the current was switched. Compound 12 substitutes an anthracene unit for the dodecyl group of compound 11 together with a 15-crown-5 for 18-crown-6 substitution of the ferrocenyl crowns. The dual substitution gives rise to short-lived (10–100 millisecond) regular transitions with an unusual Na+/K+ selectivity.62 Equally unusually, the activity is blocked by addition of Ce4+ ion. The opening probability of the channel was found to be potential-dependent but the channels themselves are not rectified. A compound in which all crown ethers are 18-crown-6 is described as having “similar” behaviour, but this compound is unselective between Na+ and K+.62
Gramicidin is one of the on-going inspirations to the design of synthetic ion channels. As noted in Section 2.4, gramicidin coils into a β6,3-helix with a hydrophilic interior of 0.49 nm diameter. Each monomer of gramicidin spans the length of a single leaflet of the bilayer; dimerization across both leaflets then generates a tunnel through which ion transit is energetically favourable. The conductance and selectivity of gramicidin in planar bilayers are well established by the biophysical community.5
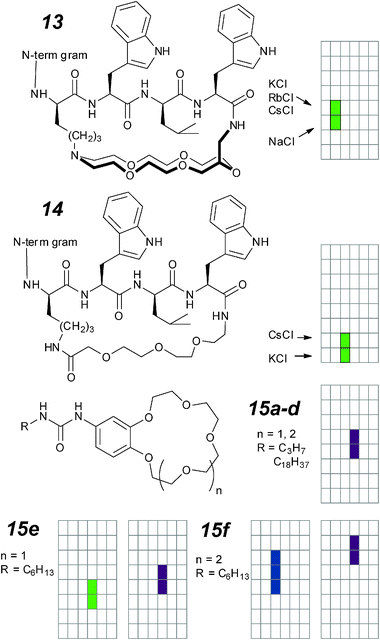
Based on this very well-studied structure, Koert and co-workers systematically modified the topologically linear gramicidin to explore the effects of structural changes. One of these attempts consists of spanning a macrocycle across the mouth of the ion portal, with the expectation that this can act as an additional filter to perturb the base selectivity (Fig. 8; 13, 14).63 Gramicidin itself shows selectivity for Cs+ > K+ > Na+ > Li+; this Eisenman I selectivity sequence corresponds to the energetic penalty for partially removing the ion hydration shell. Compound 14 shows severely attenuated conductances for Cs+ and K+ but unchanged selectivity ratios; this is the usual observation for modified gramicidins.63 On the other hand, the crown-ether guarded compound 13 preserves the conductance of native gramicidin with reversed Cs+∶K+ selectivity. This is one of the few synthetic ion channels that can select for K+ over Cs+.
 | ||
| Fig. 8 Macrocyclic gramicidins and ureido-crown ethers. | ||
This comes from both an increase in conductance of K+ as well as a decrease in conductance of Cs+. The former is attributed to dehydration assistance by the 18-crown-6, well-known to bind K+ preferentially; the latter is due to restricted passage of the larger ion in the vicinity of the crown ether at the mouth of the channel. The dwell times of both ions are substantially lower than in native gramicidin, suggesting that the active dimers are destabilized by modifications at the other end of the helical segment.
In an attempt to prepare minimalist, self-assembled structures that can act as ion channels in bilayer membranes, the Barboiu group prepared a number of urea-linked crown ethers of which 15a–f are active as ion channels.64 The compounds differ by the size of crown ether as well as in the ureido alkyl tail. The “string” in this instance is the solid-state and solution evidence of urea self-assembly into stacks supported by a linear array of urea–urea hydrogen bonds. All of the active compounds are sparingly soluble, and typically show erratic behaviour up to 300 pS in conductance. Hexyl substituted 15-crown-5 (15e) and 18-crown-6 (15f) derivatives appear to form more regular openings with defined on–off events when applied directly to the bilayer in low concentrations.
 | ||
| Fig. 9 Bis-macrocyclic bolaamphiphiles and activity grids. | ||
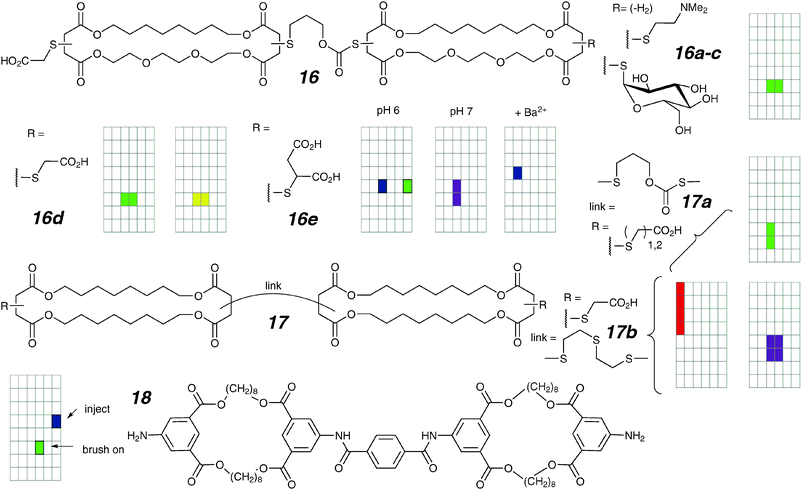
The initial group of compounds features side-discriminated macrocycles comprised of poly-methylene on one edge and polyether on the other (16a–e).65 This was expected to allow for both lipid anchoring (through the lipophilic alkanes) and stabilization of the ion-in-transit (by the donor ether oxygens). Despite their different head-groups, compounds 16a–d formed similar step-transitions in planar bilayers; in addition, compound 16d shows flickering behaviour. It seems clear that the channel structure does not depend very strongly on the symmetrical head-group type.
The dissymmetric compound 16e is markedly different and gives rise to voltage dependent activity (“voltage-gating”).66 This compound under single-channel conditions displays irregular burst clusters instead of regular openings. This is attributed to repulsion of the highly charged thiosuccinate head-groups, as semi-regular behaviour is seen upon attenuation of the charge by addition of Ba2+ or by protonation. The asymmetric charge distribution on either end of the molecule induces a preferential orientation for insertion into the bilayer. This is manifested in an initial rectifying current when samples are introduced to a single side of the bilayer; the asymmetry dissipates over several cycles of current ramp as the molecules equilibrate their orientation in the bilayer.
How important are the “conducting elements” to the function of an ion channel? This question was explored by the preparation of analogues 17a,b where methylenes replace the oxygens within the macrocycle.32 These compounds behaved qualitatively differently from the facially dissymmetric compounds of type 16; they show bursts of varying conductances from 100 to 8000 pS, as well as gradually rising conductances followed by rapid collapse (“shark-fins”). While the magnitude and duration of each individual observation is random, the overall behaviour is reproducible and stable over time. A comparison of isosteric 16 and 17 suggests that polar elements within the membrane contribute to the aggregation into stable structures. However, the fact that these transmembrane structures can be partially modulated by changes in headgroup repulsion indicates that the conducting structure is inherently unstable. The same conclusion can be drawn from vesicle studies which focus on the magnitude of the Hill cooperativity coefficient.8
In an evolution towards structurally similar but more readily accessible channels, compound 18 was prepared.67 This compound differs from the previous succinate macrocycles by replacing both the head-groups and the linker between macrocyclic rings with more rigid aromatic derivatives, and the connecting functional group with amide bonds. An unforeseen side-effect is the substantially decreased solubility; 18 is sparingly soluble even in hot DMSO and can only be introduced to the bilayer by contact injection or brushing the compound directly onto the aperture. Surprisingly, each of these two methods resulted in different observed behaviours; this may be a result of different compound concentrations (and thus degrees of aggregation) in the membrane. Large (300 pS in 1 M KCl), hour-long irregular openings were observed with contact injection, whereas small (14 pS in 1 M KCl), seconds-long regular openings were found when the compound was brushed on. The observation of regular channels despite the presence of a fully alkyl macrocycle suggests the importance of the rigid linker capable of self-aggregation. This is a theme well-explored in the class of linear compounds (Section 4.4).
4.3 Tree topology
In this section we describe compounds comprised of similar branches radiating from a linear trunk. For example, a dendritic folate rosette (Fig. 10; 19) has been reported to act as an ion channel in planar bilayers.68 Multiple units were expected to self-assemble through the N-protected folate, exposing the lipophilic tails to the non-polar interior of the membrane. Stepwise openings of multiple conductances are seen, suggesting several defined assembly sizes are accessible; the most frequent state exhibits conductance of 21 pS (2 M KCl). The permeability ratios follow Eisenman sequence I, and a K+/Cl− selectivity ratio of 4.9 was observed.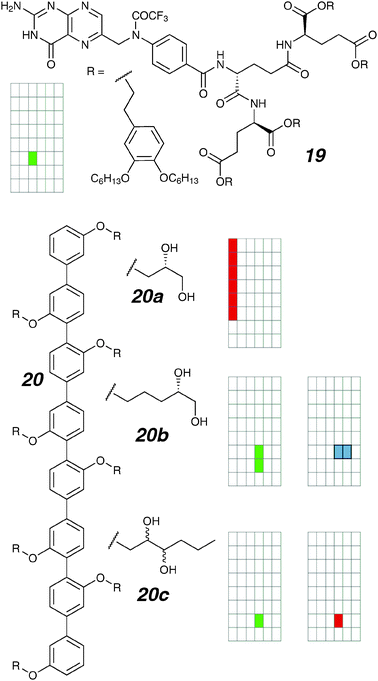 | ||
| Fig. 10 Tree topology channels and activity grids. | ||
The first generation of these rigid rods 20a–c incorporated polyols in their side-chains.70 While the shorter hexiphenyl homolog is completely inactive, the otherwise identical octiphenyl rod 20a shows conductance spikes of variable magnitude. The frequency of spike appearance correlates exponentially with potential, and the total current increases e-fold with every 27 millivolt increase in applied potential across the membrane. Extending the side-chain by two carbons alters the activity considerably; the location of the polyol also matters. In all cases the activity is predominantly irregular with occasional small square-tops. This is expected as the rotationally-flexible polyols are not expected to self-assemble with defined directionality, and thus many aggregation stoichiometries are equally favourable in energy.
A later generation of octiphenyl rigid rods (Fig. 11; 21a–d) incorporated peptides in the side-chain.71 The difference in activity between these and the polyols 20a–c is remarkable; the openings are now more than an order of magnitude larger. The authors attribute the active structure to tetramers whose side-chains interdigitate and hydrogen bond in an anti-parallel β-sheet fashion to form an artificial “β-barrel” for which there is considerable structural evidence.69 While compound 21a with symmetric methoxy caps to the octiphenyl rods shows stable “infinitely” long openings with conductance of 880 pS (1 M KCl), the “push–pull” rod 21b asymmetrically capped with methoxy and methylsulfonyl groups shows voltage-dependent bursts similar to compound 20a (14.4 mV for an e-fold increase). The qualitative differences in opening as well as voltage-dependent behaviour can be attributed to the overall dipole present in 21b but absent in 21a. Channels formed by 21b are anion selective with a X−/K+ selectivity of 2.7 to 3.7 depending upon the anion; an overall selectivity sequence of Cl− > acetate− ≈ F− > K+ was deduced.
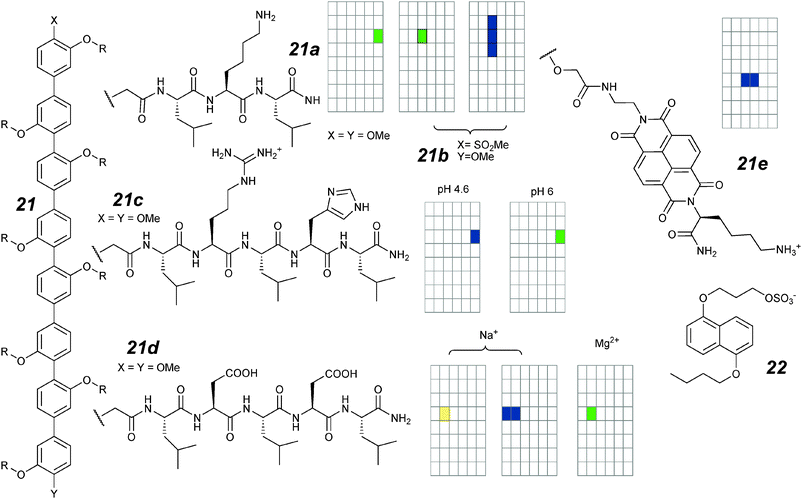 | ||
| Fig. 11 Octiphenyl ion channels and activity grids. | ||
The β-barrel structure places lipophilic side chains to the exterior of the barrel, and hydrophilic side chains within the barrel. This leads to the possibility of control of ion currents through manipulation of the charge state within the channel as explored with compounds 21c–d.72 The arginine–histidine compound 21c was shown to be pH-sensitive; at pH 4.6, the histidines are protonated, and this results in shorter mean single channel dwell-times than at pH 6.0. This is accompanied by a shift from moderate cation selectivity at pH 6 (K+/Cl− = 2) to more marked anion selectivity at pH 4.6 (Cl−/K+ = 3.7). At pH 6, the aspartate derivative 21d compound shows only short, unresolved (<0.5 millisecond) spikes. Addition of Mg2+ attenuates the repulsion between the side-chains. As a result, the openings are more stable (lifetime of 12 milliseconds) and also show improved permeability for anions, as would be expected if the ion transit path is lined with partial positive charges.
With these octiphenyl rods, more sophisticated ligand-gating is also possible. It is known that naphthalenediimide (NDI) acceptors and dialkoxynaphthalene (DAN) donors π-stack into charge-transfer complexes; by incorporating an NDI into the side-chain of an octiphenyl (21e), Matile's group was able to make use of the motif to generate an ion channel switched on by the DAN derivative 22.73 The native octiphenyl rigid-rods were expected to self-assemble into the bilayer as a “π-helix” which has no free space for ion passage. Upon addition of 22 and its subsequent partitioning into the bilayer, the embedded assembly is untwisted, opening up a passage for ion-flow. Experimentally, the assembly is active in planar bilayers only when both 21e and 22 were added together. The dominant single-channel currents have life-times of the order of seconds, and conductance of 94 pS (2 M KCl). Data from modelling and structural studies corroborated the account. Given that an active structure may contain up to 20 molecules, the single molecule traces are remarkably homogenous.
For “ease of synthesis”, the octiphenyl rods described up to this point all contained identically substituted side-chains. After an heroic synthetic effort, a compound with mixed side-chains was prepared.74 Compound 23 (Fig. 12) contains an arginine–histidine (RH) motif in its four inner chains and asparagine–asparagine on the four terminal chains. The single-channel conductance records show discrete events with a number of conducting states. The RH motif was known to bind anionic guests such as ADP; the localization in the centre of the membrane was expected to give localized (“in-depth”) pore blockage. Under voltage-clamp conditions, a large potential gradient exists across the membrane. Together with the specifically localized binding site, this was expected to give voltage-dependent molecular recognition. Experimentally this can be examined by determining the binding constant of a guest to the channel over a range of potentials; experimentally this is seen as the voltage dependence of a multichannel current in the presence of a channel blocker. An equation due to Woodhull75 (eqn (3)) describes the binding constant at a potential (KD) as a function of the binding constant at zero applied potential and an exponential term that contains terms for the depth of binding within the pore (lW), the pore length (l), the charge of the blocking guest (z), the applied potential (ε), and fundamental constants:
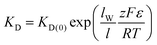 | (3) |
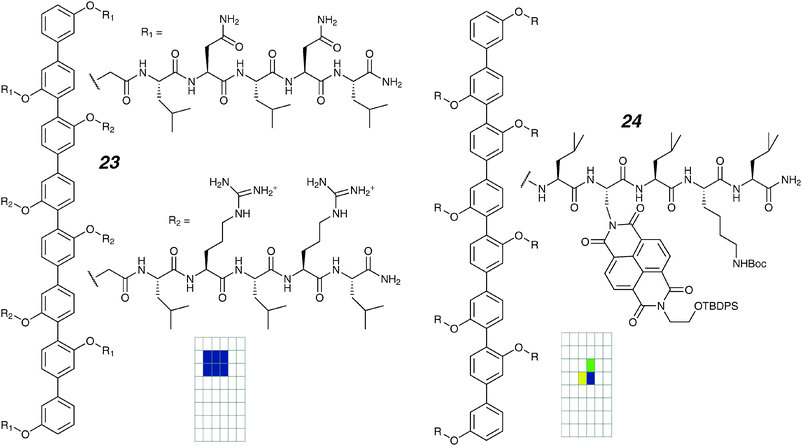 | ||
| Fig. 12 Octiphenyl ion channels and activity grids. | ||
Although charge recognition is interesting, the functionality of these types of pores is greatly extended by means of a π-complexing character within the pore, leading to discrimination among nucleotides as one application. Compound 24 contains NDI units within the pore;76 molecular modelling suggests a large diameter of the parent assembly. This is confirmed by the observation of large conductance (∼500 pS in 2 M KCl) openings associated with long lifetimes. At the same time, two other conducting states (lower conductance levels; frequent high conductance spikes with short lifetimes) were also seen. The mixed conducting states would complicate a direct analysis of different analyte binding events by voltage-clamp methods and the authors chose to work with vesicle assays to reveal the multi-analyte capabilities of these pores. Nonetheless, this type of system should be amenable to direct stochastic analyte sensing.
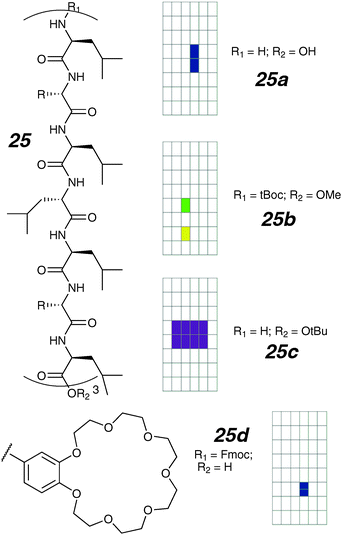 | ||
| Fig. 13 Peptide scaffold ion channels and activity grids. | ||
Using the regular channel 25b as a starting point, systematic variation of the side-chain crown-ether size was undertaken.78 The activities were determined with 23Na-NMR techniques in vesicles, and rates of transport can be compared between compounds. The finding here is that when smaller crown-ethers are present, the relative rates of transport decreased over 300-fold. Presumably these compounds would either be of low single-channel conductance, or open infrequently as to make single-channel measurements difficult or impossible.
Non-helical peptides were used in a series of anion transporters as headgroups.79 Compound 26 (Fig. 14) features twin alkyl chains attached to a Gly3-Pro-Gly3 unit, and this structurally simple molecule shows several types of behaviour (0.45 M KCl). Type 1 are “square-top” openings of 12 pS conductance up to 2 seconds in duration; Type 2 are 30 pS conductance and flicker over a 100 millisecond to 10 second range. These are accompanied by sub-second clusters of ∼40 pS current bursts. If the different types of activity are the result of alternative aggregating states in the membrane, covalently tethering the components together may restrict the energetics of aggregation and lead to both reduced variety of conductance behaviours, and longer living channels. This is a strategy well-tested from work on modifying alamethicin, a depsipeptide ion channel whose active states consist of various sized clusters.22,80 For a Gly3-Pro-Gly3 dimer compound 27, the strategy appears to improve the stability of some active states as the single-channel currents of the pseudo-dimer are more regular than in the parent compound. Burst clusters seem to be completely absent for this molecule.
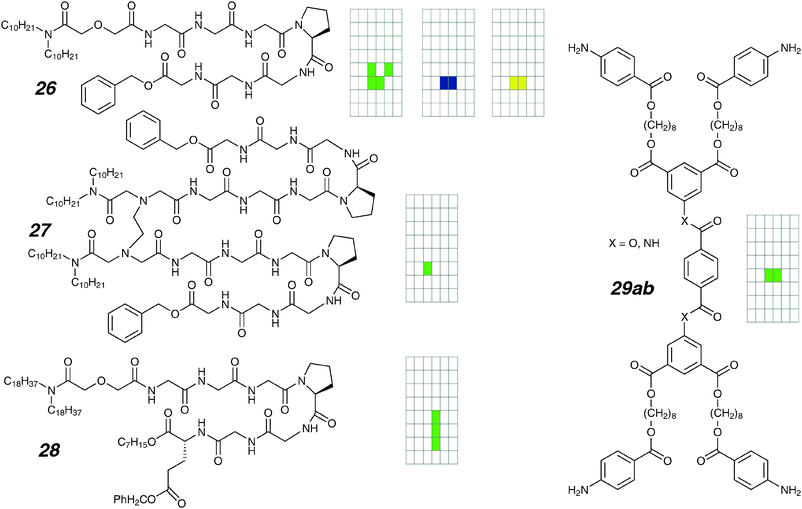 | ||
| Fig. 14 Branched channels and activity grids. | ||
A large suite of related compounds were later prepared, all of which contained longer alkyl chains to facilitate membrane-anchoring, and replacement of the terminal benzyl-ester group in 26.81 A voltage-clamp study was undertaken for compound 28. Like the pseudo-dimer 27, several types of regular openings were again found. The dominant state has a conductance of 28 pS (0.45 M KCl), and appears to be longer lived than those of 27. Other conductance states of 6 pS and ∼100 pS were also observed.
Related to 18 are topologically distinct, ring-opened analogues 29a,b.82 The compounds are centrosymmetric and relatively easy to prepare. The pair differs in the central linkers: amides were used in 29a whereas esters were used as linkers in 29b. From the practical point of view, the ester has better solubility and can be handled more conveniently; as ion channels, both compounds are essentially identical, with regular openings of 40 pS conductance (1 M CsCl) and half-lives about 300 milliseconds. For this pair of compounds, the stability of active channels does not seem to be a function of the potential amide–amide hydrogen bond.
4.4 Linear topologies
In this section, synthetic ion channels with a linear topology are considered. “Linear” here refers to the ability to trace from one end of the structure to another. We ignore the multiple paths through five- and six-membered rings, and in more complex cases through rigid structures such as steroidal fragments. These compounds comprise over half of the synthetic channels whose activity has been examined using the voltage clamp method.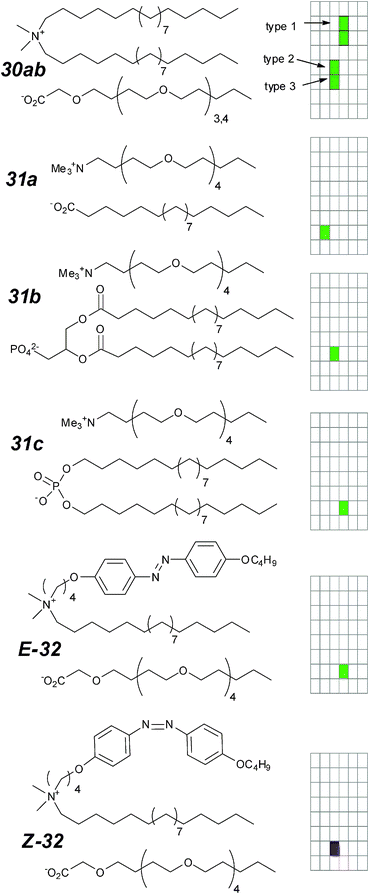 | ||
| Fig. 15 Ion-pair ion channels and activity grids. | ||
The active states of these compounds were postulated to involve an aggregate where the lipophilic component contacts the lipid, shielding the hydrophilic oligoether. The clustering of the oligoethers would then provide a passage for ion flow. This hypothesis suggests that the relative volume of the ion-pair would play a critical role in the activity, and this was initially consistent with the complete inactivity of some bulky analogues (structures not shown);83 subsequent reports (31a–c) are not so obviously consistent with this hypothesis.84 The postulated active state structure further predicts that swapping the identities of the head-groups should not affect the activity, as the function of the head-group is merely to anchor the structure at the surface of the membrane. This is not entirely true; compare the range of conductance and lifetimes represented for compound 30a,b and 31a–c. Even within this set of simple compounds there are substantial differences in K+/Cl− selectivity (17 for 30avs. 5 for 31c).
Even though the correlation of structure with function may not always be predictable for these simple compounds, the geometry of the compound in the anisotropic lipid environment is critical to its function. This line of thought leads to the development of a photo-switchable ion-pair 32 in which an azobenzene replaces one of the alkyl units.85 As with previously reported compounds, the individual components of the ion pair are inactive. Photo-isomerization is reversible, and the isomers behave differently when examined under the voltage-clamp experiment. While the Z-isomer shows erratic bursts, the E-isomer shows a range of square-top conducting states which persist for up to 10 seconds. The selectivity of the E-32 open states follows the Eisenman I sequence.
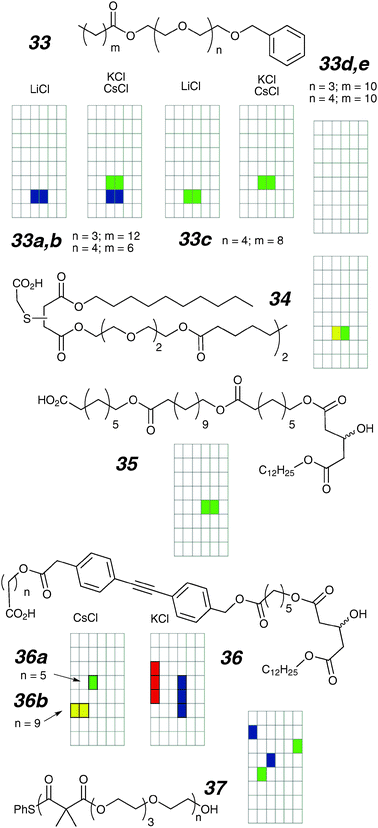 | ||
| Fig. 16 Oligoester ion channels and activity grids. | ||
“De-macrocyclization” of an active channel was noted previously (18 gives 29 by this strategy) and the same strategy produced 34 starting from the bis-macrocyclic ester 17.87 In this case, despite the completely different topology, the activity of the linear compound is essentially indistinguishable from that of the bis-macrocycle; if anything the acyclic 34 shows somewhat fewer low conductance events than the parent.
Further work on this family of compounds involved inverting the polarity of the ester bonds such that a class of compounds would be amenable to solid-phase synthesis, which in turn allowed a large library to be prepared.88–90 Screening using vesicle methods uncovered active compounds that were then examined by voltage-clamp. This strategy is fruitful to the extent that activity in curved membranes is commensurate with activity in planar membranes. While this is generally true, exceptions are common.91 Using this methodology, compound 35 was identified to be highly active in both vesicles and planar bilayers,92 making it a useful lead compound.
Addition of a rigidifying diphenylacetylene unit within this lead structure produced compounds 36a,b and related sequence isomers in which the hydroxy-acid subunits appear in different sequences within a linear structure.93 In vesicles, compound 36a and its sequence isomers are about five-fold more active than 35 while 36b is completely inactive. In planar bilayers, 36a forms well behaved square-top channels (140 pS; 700 millisecond lifetime; 1 M CsCl) with the sequence isomers showing similar behaviours. In contrast, the “inactive” compound 36b forms short duration bursts of flickers between a closed and an open state of conductance 26 pS. These compounds are quite insoluble in water so activity was most reliably observed when membranes were formed which contained the compounds pre-mixed with the lipids. In other electrolytes (KCl shown) all compounds show a mix of short-duration spikes and longer lived events that have multiple conductance states. The diphenylacetylene provided an intrinsic environment-sensitive probe, and the poor activity of 36b in vesicles was related to kinetically slow partitioning.
A polymer having similar composition to the other compounds in this section has been recently reported.94 A “beaded string” polymer with pendant crown ethers was expected, but the polymerization occurred with ring opening to produce 37. While starting monomers are reported to be inactive, polymer 37 (M.Wt. 6000; PDI 1.5) is active when incorporated during the membrane formation stage. Heterogeneous activities were observed, ranging from short, large openings to long-lived (but rarer) openings. The presence of well-defined conducting structures is surprising for the limited degree of structural integrity associated with this flexible polymer. Further linear polymeric examples are discussed in Section 4.4.5.
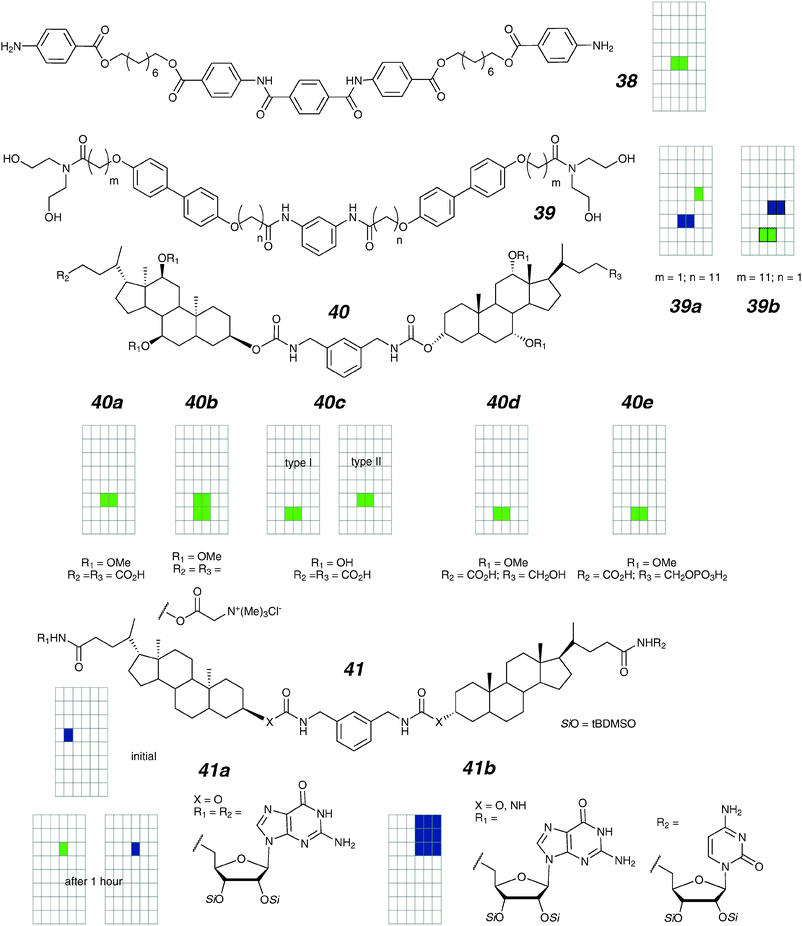 | ||
| Fig. 17 meta-Phenyl-linked channels and activity grids. | ||
Gokel and co-workers designed the “aplosspan” family of compounds (39) which bear structural resemblance to the compounds described above.95,96 Two of the compounds (39a,b; differing in the location of the biphenyl within the arms) have suitable solubility for voltage-clamp studies. A long-living, low conductance open-state was initially reported for both compounds,96 with a K+/Cl− selectivity of 2.3. It was later reported that even though the openings of 39a remain more-or-less regular, the conductance gradually increases by 10-fold over a two-hour period.95 If both the initial and the later structures are amenable to description by the Hille equation (2), this would suggest an expansion in the water tunnel from 0.3 to 0.9 nm. The gradual (non-discrete) enlargement precludes an alamethicin-like assembly, which predicts distinct conducting states for each discrete stoichiometry within an aggregate.
While alkyl chains are convenient linkers between different polar elements, they are rotationally flexible. Macrocycles can act as covalent constraints to limit the degrees of freedom (see Section 4.2), but a synthetically simpler approach is through the use of steroidal subunits. Regen's research group pioneered the use of cholates as facially-amphiphilic membrane-spanning units, and functional transport was found with vesicle techniques.97–101 Covalently linking multiple units also gives rise to improved activity by increasing the effective concentration for oligomerization within the lipid. Voltage-clamp studies of linked cholates were reported by Kobuke and co-workers (40a–e).102–104 The first generation of these compounds (40a,b) were fully centrosymmetric. These form regular step open–close behaviours in bilayers; small variations of conductances were seen (9 to 17 pS in 500 mM KCl). The cation selectivity was much higher for the negatively-charged compound 40a than the positively-charged 40b (K+/Cl−: 17 vs. 8). The published trace shows frequent openings but no multiple openings; given that appearance of multiple copies in the bilayer is independent of one another, it is suggestive that the opening probability of each single conducting state is high.
If we assume that the methoxy groups at C-12 and C-7 serve as ion-portals in 40a,b, it would be useful to find out the effect of deprotection at these sites; 40c is the direct analogue of 40a.103 Under identical conditions, 40c shows two types of stepwise open–close behaviours. Type I has a unit conductance of 10 pS and a selectivity of 8; type II has slightly larger conductance (25 pS and a similar selectivity ratio K+/Cl− of 7). The overall effect of the substitution seems to be an increase in chloride permeability, with reduction of accessible conducting states. The latter may relate to the directionality inherent to hydrogen bonds favouring specific geometries over others.
Given that facially-amphiphilic compounds are easily accessible through the use of steroidal scaffolds, it may be possible to de-symmetrize the molecule by functionalizing the head-groups differently. In particular, placing different charges on each end may effect a rectifying behaviour as it does for the 16d,e pair. This is borne out by experiment: the centro-dissymmetric compounds 40d,e both show rectifying behaviour when introduced to the bilayer on a single side.104
In later studies, the Davis group explored different aspects of the membrane-spanning steroidal framework coupled to the self-assembly of “G-quartets”.105,106 Locating guanosines in place of simple charged head-groups effectively replaces a non-directional, repulsive element with a directional, self-assembling one. The comparisons are not direct with earlier compounds; this suite of compounds has a deletion of polar elements from the steroidal framework, which may have a significant effect on the behaviour. Nonetheless, the importance of the guanosine head-group can be readily seen by a comparison of the step-like activity of 41a with a compound having N-methyl in place of the guanosines which is completely inactive. The activity is initially ill-defined, but evolves over one hour to larger, more regular openings. The openings are substantially stabilized and expanded by improved intermolecular hydrogen bonding, as seen by a comparison of the urea-linked 41b to 41a, although the higher conductance activity shows multiple conductance states within an opening event.
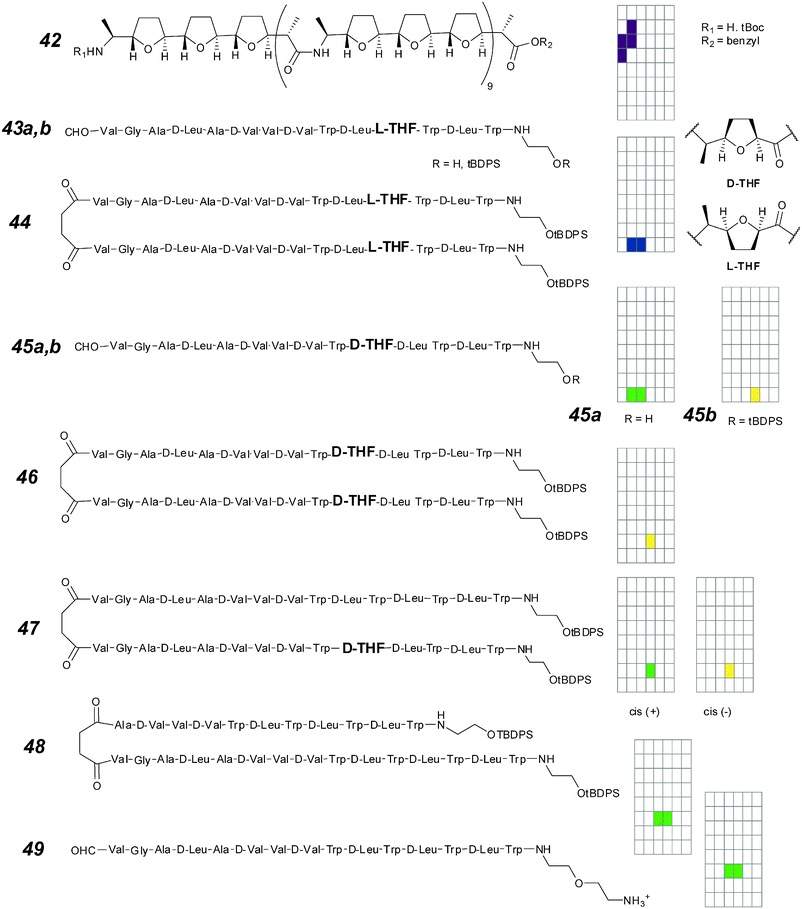 | ||
| Fig. 18 Modified gramicidin ion channels and activity grids. | ||
More subtle perturbations of gramicidin involve the use of a tetrahydrofuranyl unit in place of two of the three residues at positions 10 to 12 of the peptide (D-Leu-Trp-D-Leu). These are at the “dehydration” region of the native gramicidin channel; two of the possible stereoisomers (D-THF and L-THF 43–45) preserve the overall amino acid structure but replace an amide oxygen with an ether oxygen in this critical region.63 A substantial variety of behaviours can be observed. The insertion of L-THF at positions 11 and 12 in monomer (43a,b) and the head-to-head dimer (44) all show significantly attenuated conductance (3 pS) relative to parent gramicidin. The profile of the conductances is also altered; the square-top behaviour is replaced by irregular openings. The monomers (43a,b) also need to be added in high concentration for activity to be observed, and this may be explained by a decreased propensity to insert into the bilayer and/or a decreased propensity to form a productive conformation after membrane insertion. Covalently linking at the C-terminus (44) allows activities to be observed both at lower concentrations and by single-sided addition. The conductance remains low, but lies closer to the expected square-top behaviour.
The insertion of D-THF at positions 10 and 11 results in behaviour that is more gramicidin-like.63 The resulting channels are of low conductance but defined structure; the deprotected compound 45a shows small square-top behaviours, while the protected compound 45b shows rapid flickering between open and closed states in the range of 10–20 milliseconds. This is unlikely the result of multiple channels as independent copies would show a binomial distribution in opening levels. The covalent dimer 46 shows similar flickering behaviour, indicating that this is not a result of a destabilized dimer, but rather of conformational changes that occur in the millisecond timescale. Compounds 45a,b and 46 also show a perturbed selectivity sequence equivalent to Eisenman III (Rb+ > K+ > Cs+ > Na+ > Li+), suggesting that a different binding site is presented by the ether oxygen to incoming cations.
Does the conformational gating (flickering behaviour) only affect incoming ions, or does it affect both the incoming ions and ions being ejected? The hybrid compound 47 introduced to one side of the bilayer inserts asymmetrically, resulting in different behaviours related to the sense of the applied potential. The more “gramicidin-like” behaviour of the “cis- +” condition indicates that the conformational gating affects only the incoming ion. One implication is that it may be possible to construct rectifying behaviour by selectively sabotaging one of the interaction sites at the entrance of an otherwise symmetrical ion channel.
While a gramicidin hybrid may be a suitable test for the above hypothesis, the compound is unknown to date. However, a different type of “inactive” gramicidin is known.107 So-called “mini-gramicidins” are active only in thinner bilayers (e.g. DMPC) but inactive in normal bilayers. A dimer mini-gramicidin–gramicidin 48 may then be expected to show asymmetric conductances.108 This was not the case; the asymmetric dimer is fully Ohmic over a range of applied potentials (±200 mV). This may be attributed to the attenuation of conductance in mini-gramicidins as a structural phenomenon that is only partially compensated by being joined to the longer regular gramicidin.
A different strategy for de-symmetrizing gramicidin consists of potential-dependent blockage. This “ball-and-chain” type of rectifier was pioneered by Woolley in a series of modified alamethicins.109 In the same fashion, an ammonium ion was covalently linked to the N-terminus of gramicidin with a flexible linker to give compound 49.110 The positive charge is expected to be repelled by a positive potential and thread into the mouth of the channel; this may preclude other cations from flowing through the same portal. The “plugging” occurs insofar as a positive potential is held, and the current flow is thus expected to be non-linear. The expectations were borne out by experiment; the current flow follows an Eisenman I sequence, but is non-Ohmic and attenuated up to 50% when positive potential is applied on the side cis to compound addition.
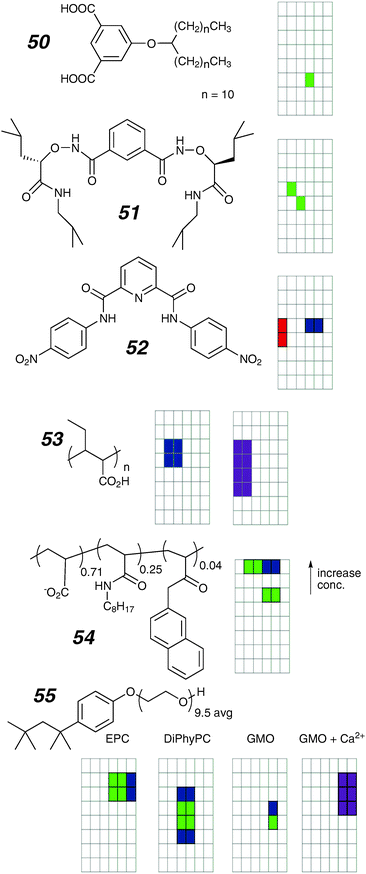 | ||
| Fig. 19 Linear ion channels and activity grids. | ||
Another active small molecule 51 prepared by Yang and co-workers112,113 forms host–guest complexes with chloride in CDCl3 and also shows discrete activity in planar bilayers. The compound shows two distinct conducting states with step-wise transitions; the conductances of the states are 54 and 108 pS respectively (0.2 M KCl). The conductance and reversal potentials do not depend on the identity of the cation; NaCl, KCl, N-methylglucamine hydrochloride are all reported to give identical values. The stepwise conductance behaviour rules out the possibility of a ferrying mechanism. As such, the cation-invariant behaviour is highly puzzling, as it implies perfect selectivity for chloride over all three cations. The mechanism of transport is unclear, but the compound shows clear antibacterial function related to collapse of membrane potential.113
Compound 52, reported by Gokel and co-workers114 also binds chloride and also produces conductance events in planar bilayers. The most commonly observed state had a conductance of 117 pS (0.45 M KCl) although other states were obvious. Vesicle studies suggest that a carrier mechanism is also possible. A model proposed involves stacking of the aromatic rings to form the wall of a channel; fluorescence evidence suggests that stacking occurs in the bilayer environment, and stacks are observed in the solid-state structure.
At the other end of the molecular weight spectrum are polymeric materials for potential application as gene-delivery and anti-bacterial agents related to their pore-forming ability.115 Two of these have been explored using the voltage-clamp technique. Tirrell and co-workers reported that polydisperse, stereo-irregular poly(2-ethylacrylic acid) of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 average molecular weight (53) shows heterogeneous behaviour in planar bilayers.116 Some activity is sub-second discrete steps that are irregular in the open-state; others are erratic bursts. The conductance of each of these classes of activity varies over two orders of magnitude from ∼10 to 1000 pS. There is evidence that the activity can be switched on and off by altering the acidity of the bathing electrolyte, as the activity is observed only in acidic solutions. Another polydisperse and random acrylate copolymer 54, reported by Tribet and coworkers,117 shows the formation of well-defined channels. The magnitude of the conductance depends on the polymer concentration. The authors propose that the lower concentration events are due to a threaded disruption of the bilayer that opens to a large (>5 nm) pore as the amount of polymer available is sufficient to line the walls of this large structure.
000 average molecular weight (53) shows heterogeneous behaviour in planar bilayers.116 Some activity is sub-second discrete steps that are irregular in the open-state; others are erratic bursts. The conductance of each of these classes of activity varies over two orders of magnitude from ∼10 to 1000 pS. There is evidence that the activity can be switched on and off by altering the acidity of the bathing electrolyte, as the activity is observed only in acidic solutions. Another polydisperse and random acrylate copolymer 54, reported by Tribet and coworkers,117 shows the formation of well-defined channels. The magnitude of the conductance depends on the polymer concentration. The authors propose that the lower concentration events are due to a threaded disruption of the bilayer that opens to a large (>5 nm) pore as the amount of polymer available is sufficient to line the walls of this large structure.
Finally for this class of linear topologies, we would remiss if we did not attempt to summarize the channels that are formed by the detergent Triton X-100 (55). The first report by Schlieper and De Robertis from 1977 predates all others within the scope of this review and was intended to sound a cautionary note for those using detergent solubilisation of membrane proteins as a means to isolate and study natural ion channels.118 Nonetheless it does contain a surprisingly modern account of the essential features of a minimalist ion channel. Long-lived and relatively well-structured channels were observed in EPC with longer-lived openings showing more evidence of multiple states. Later work was centred on the effects of pH and divalent ions on these channels as a means to establish a general behaviour for detergent-like channels formed from peptide toxins.119 In DiPhyPC and GMO membranes the channels form over a range of conductances and are somewhat longer lived. Divalent cations destabilize the channels leading to erratic behaviours over periods up to minutes; the GMO/calcium case illustrated is typical.
4.5 Complex topologies
This section contains structures known to have more complex topologies at the outset, or which are shown experimentally to involve more complex structures within the bilayer membrane. Many of these structures are the result of self-assembly processes, either pre- or post-membrane insertion. In order to generate a structure of sufficient stability to compete at the concentrations of a typical voltage-clamp experiment a pairwise association constant of the components in excess of 104 M−1 in a predominantly aqueous environment is required. Ion channels that make use of the G-quartet and DAN–NDI donor–acceptor pair have been described above (see Fig. 17; 41 and Fig. 11; 21e). An appealing alternative is the use of (reversible) ligand–metal bonding, a well-developed theme in the self-assembly literature.120–122 The first attempt to utilize this approach explored the possibility of preparing channels by self-assembly using the well-established Fujita square motif,120 where a square planar palladium with two exchangeable ligands forms dative bonds with bipyridine (Fig. 20; 56 plus 57).123 The long alkyl tail on 56 was expected to facilitate insertion into the bilayer. The activity is time dependent. Bursts of activity of ∼100 pS were initially seen when an equimolar mixture of 56 and 57 was added to the electrolyte. These erratic bursts evolve over time to very long-lived channels that are apparently a “square-top” based on a few observations of opening and closing. Similar activity can occasionally be seen to evolve in the absence of 57. Clearly a self-assembled square is not the only mechanism for conductance.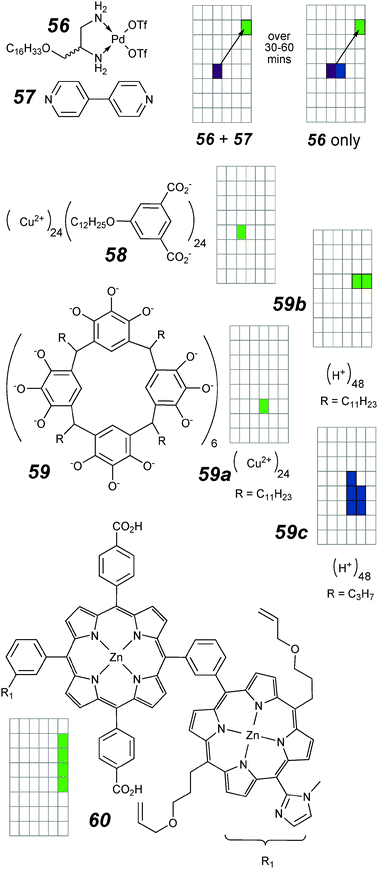 | ||
| Fig. 20 Ion channels with complex topologies and activity grids. | ||
A more clearly self-assembled structure as an ion channel is the rhombicuboctahedral MOP-18 structure (58)124 whose membrane activity was reported by Kim and co-workers.125 The structure is based on bis-Cu-tetracarboxylate “paddle wheels” that define the core structure; the pendant dodecyl groups are on the outside, leaving a large cavity accessible via two different portals. The overall dimension exceeds 5 nm in the solid-state, but the core is only roughly half this diameter.124 The membrane activity is remarkably uniform and shows a very unusual Eisenman XI selectivity (Li+ ≫ Na+ > K+ > Rb+ > Cs+) in both planar bilayer as well as vesicle transport experiments. This selectivity sequence is possible only if all cations are selected by size of the completely dehydrated ion. It may then be expected that a high activation barrier exists (due to the dehydration) but the unit conductance of each open channel is in line with other channels (36 pS in 2 M KCl). It is not clear how the structures can give rise to the discrete transitions observed.
Another example of this type is the copper-complexed pyrogallarene 59a. The complex is a hexamer with copper bound in 24 square-planar sites along the edges of the pyrogallarene units.126 Gokel and co-workers initially reported that the protonated pyrogallarene (59b) was inactive, but that the copper-bound form (59a) gave low conductance channels (12 pS in 450 mM KCl) with regular transitions.127 This system is also selective for smaller cations over larger cations (Na+ > K+ ≫ Cs+). Furthermore, it was reported that with Cs+, transport shut down shortly after the experiment began; the authors attributed this to pore blockage by the larger ion. The channels are selective for K+ over Cl− by a 2.4∶1 ratio. In a subsequent report, the activity of 59b was clarified.128 Compounds of this type crystallize in two forms: a bilayer-like extended structure, and a capsular structure consisting of hydrogen-bonded hexameric capsules. Sonicated suspensions of samples of the bilayer structure in DMSO applied to asolectin bilayers can give rise to channel activity while samples from the capsular structure are inactive. The compound from dodecanal (59b) forms long-lived channels that are apparently square-tops of 190 pS conductance (0.45 M KCl). The channels are unselective between cations and anions and the authors speculate that a monomer is required for membrane insertion, followed by oligomerization (“to at least a hexamer”) to form the channels. The compound from butanal (59c) similarly crystallizes as a bilayer structure, and similarly forms channels, although these are of the multiple opening type.
Kobuke's group has reported a zinc tris-porphyrin-based ion channel (60).129 The structure assembles in solution to a cyclic trimer in which the pendant imidazoles coordinate to the terminal Zn-porphyrin units. This is followed by olefin metathesis to freeze the structure via covalent linkages between the monomer units. Although the channel appears to be perpetually open, it was possible to block the ion flow with a 4th-generation dendrimer. No blockage is observed when smaller dendrimers were used as the blocker, suggesting a large but defined diameter. As multiple copies of the channel are permanently open, determination of unit conductance is challenging; the minimal conductance observed is several tens of pS with a wide range of conductance at higher levels. The conducting structure shows only moderate cation selectivity; all of these observations corroborated a stable, large-diameter conducting structure.
5. Overview of literature data
The activity grids represent an abstraction of the original data that in principle can be used to probe the original literature for trends. We begin with an overview as shown in the 3D-bar chart plot of Fig. 21 (top). In this plot the activity within the different types (colours) has been summed over particular values of conductance and duration of the activity grid. Although useful as an overview, any chosen perspective on this plot obscures smaller features. An alternative representation of the same dataset is given as a series of 2D density plots for each of the types of activities (Fig. 21, bottom). In these plots the shading density reflects the proportion of the total of this type of observation which occurs at a particular grid location of conductance and duration. The scale is coarse as there are relatively few observations in any grid location.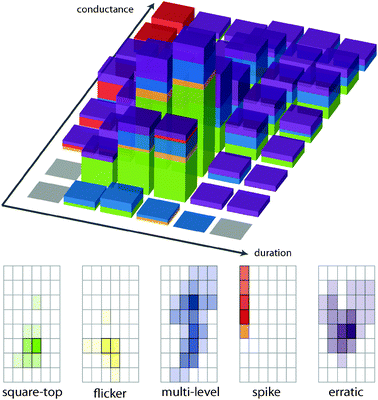 | ||
| Fig. 21 (Top) Bar chart representation of the cumulative activity grids of compounds surveyed. (Bottom) Activity grids in which colour intensity reflects the proportion of activities that occur with a given conductance/duration, separated by type of activity. Each type is normalized to assign the maximum intensity to the most frequent occurrence within that class; see text. | ||
The immediately striking point from the 3D-bar chart view is that: (i) well-defined square-tops form the majority of reported activity for synthetic ion channels, and (ii) that these frequently observed behaviours are strongly clustered in a few cells. In numerical terms, over 50% of the active compounds display some type of square-top activity, followed by 25% which show multiple levels during an opening, 15% are reported to show erratic behaviours, while 5% each show spikes or flickering behaviours. The clustering of the square-top openings is clearly evident in the density plot; the dominant reported square-top activity has a conductance of 10–30 pS with an opening duration of the order of 1–10 seconds. Other types of activity are clustered as well. Multiple-level opening events and erratic activities occur to somewhat longer opening durations and somewhat higher conductances than square-tops while the rarely reported spike activities are—as expected—of short duration but centred at even higher conductance. The flicker behaviour is rare relative to the dominant behaviours but appears to be clustered as the square-top openings. The “shark-fin” or slow evolving behaviour illustrated in Fig. 2C is exceedingly rare.
In addition to the activity grids, the dataset also included a number of structural descriptors: overall topology, rigidity, functional group, size. We were therefore surprised and disappointed that no matter which sub-set of structural descriptors is chosen, there is no discernable difference between the selected subset and the population as a whole. This is partially a statistical issue as the population consists of only 165 independent observations of the activity type exhibited by approximately 125 unique compound/conditions. Compounds are prepared in groups, and even though some marked effects of different activities between homologs were noted above, in general, homologs tend to behave similarly. There are fewer than 50 truly independent types of compounds. This is too few on which to attempt to derive a general structure–activity picture of any statistical force.
Despite the lack of a statistically sound analysis, some issues explored by many groups do admit of some general conclusions. For example, the thrust in many research groups has been to prepare compounds that are as simple as possible; the decreased effort in synthesis allows more compounds to be made and examined. While this is a sound approach, there appears to be a trade-off between easy syntheses and tractable analyses. There are two factors that complicate the analysis of smaller molecules: (i) as the size of the individual components decrease, more units are necessary to assemble the conducting state. Other aggregation stoichiometries and conformations may also be possible (“supramolecular polymorphism”),8 and the result is a mixture of structures at the molecular level and mixture of activities in the voltage-clamp traces. (ii) Lower molecular weights mean that each structural perturbation is relatively large. Examples abound in Section 4 where the activity is sensitive to subtle changes in large structures (e.g. compounds 21a,b). By making molecules even smaller, each incremental change in the structure is more likely to simultaneously change several properties of the resulting ion channels. In these cases no changes are innocent (e.g.33a–e).
In the cases of “simpler” compounds, the types of behaviours are frequently indistinguishable from larger or more complex compounds, but the ability of the experimenter to optimize for a particular activity through synthesis is therefore limited.
5.1 Structure–activity optimization
While the synthetic ion channel field has long focused on binary square-top behaviour, it should be clear from this survey that many other types of activity are possible and prevalent. Many of these individually erratic activities are collectively structured and the collective behaviour sometimes can be controlled. Some examples include the exponential potential dependence of compound 20a, the rectifying behaviour of compound 16e, and apparently fractal conductances observed with compounds 9a,b. At the present state of development very little is known about how to select or optimize these types of behaviours, and this is certainly an open area for investigation.We know more about optimizing for “regular” behaviour, but many competing factors are in play. First and foremost, the compound should be partitioned into the bilayer; with few exceptions (e.g.5c) compounds that are too hydrophilic show no activity. In some cases, deprotecting some polar elements is enough to switch off activities completely (e.g.42) or significantly perturb activity (e.g.25a–e). Contrary to expectations, a cylindrical shape (of the monomer) is not critical to regular activity; regular activity was seen from every topology and many different shape parameters.111 Shape parameter here refers to the volume ratio of the membrane–surface element to the membrane-embedded element, which is presupposed to constrain the packing modes available within the lipid. Moreover, a cylindrical shape (of the monomer) does not predict regular activity (e.g. compound 21a). The length of a transmembrane segment needs to approximate the bilayer thickness but the match is not stringent. The clearest indications come from the mini-gramicidins and their hybrids (48); it is apparent that the bilayer can dimple to some extent in order to accommodate shorter compounds, or bulge to accommodate longer ones. While a polar element within the membrane embedded portion is not essential for ion flow (e.g. compound 50), it appears that all regular channels with extended stretches of hydrocarbon share similar conductance behaviours that are difficult to shift with structural changes.
If the active structure is an aggregate, then regular activity of substantial duration can only be observed when the charge on the head groups does not exceed two charges per monomer; with higher charge the aggregate will be sufficiently destabilized that regular activity can only be observed when the charges are screened. Alignment of non-ion-conducting parts is important only if the alignment affects the ion path; regular activity is seen with the rotationally flexible tails in compound 19, the MOP compounds 58 or 59a, and the collection of tails present in macrocycle 2. Within aggregate channels, improving the stability of the aggregate, hence prolonging the event duration, can be done at the head-group or in the membrane-embedded layer. Substitution of functional groups that can self-associate (e.g. octiphenyl β-barrels of Fig. 10 and 11, facially amphiphilic steroids and urea–carbamate exchanges of Fig. 17) may improve the stability, but this may depend on the geometry of the molecule when inserted into the bilayer (e.g. the effect is negligible for the 29a,b pair).
Several types of voltage-response have been demonstrated in the literature. These include voltage-dependent binding of analyte, voltage-dependence of conductance, as well as rectifying behaviour. The latter have been sufficiently well studied that several strategies to desymmetrize a channel within a bilayer can be recognized: (i) the overall dipole of the molecule affects its alignment to an external applied field, and is sufficiently sensitive to be used as a voltage-gating element (21a,b).71 (ii) Plugging is possible with a covalently tethered ion (49).110 (iii) Preferential insertion can create asymmetry in the bilayer by having a strongly charged headgroup that does not readily penetrate the bilayer (16e66 or 40d,e).104
5.2 Why are activities clustered?
We return to Fig. 21 from a different perspective. If the activity grids genuinely do reflect the characteristics of the reported data, why should the literature data be so strongly clustered on one type of activity (square top) and so small a range of conductance–duration (10–30 pS; 1–10 seconds)? A wide range of compounds has been explored; if these explorations were sampling the potential diversity randomly, should not the observed activities be more randomly distributed? Stated another way—why should so many markedly different structures behave so similarly? We note as well that is the normal range for gramicidin. How can all these compounds behave like gramicidin?There are a number of possible responses to these questions. At the outset we should acknowledge that perhaps the activity grids have some inherent biases; we don't think they do but that is a possibility. We do record the many channels that lie outside the predominant behaviour, so we do not believe that the grids themselves are the cause of the clustering.
Perhaps there is a selective reporting bias. We recognize from our own work that it is much more difficult to summarize and report erratic behaviours than it is to report regular ones. The tools to analyse and summarize regular square-top behaviours are accessible and well-developed while the corresponding tools to analyse and report more complex behaviours are less developed. This could result in a “file drawer effect”130 in which data that are seen as “not of good quality” lie unreported in investigators' files. If true, this is a serious issue as it inevitably leads to Bacon's “contract of error”.§ Speaking only on our own behalf, we recognize that this is possible, perhaps even plausible; there are certainly many unreported files in our laboratory. But we regard reporting bias, even if present, as a secondary issue. It might explain why square-top behaviours dominate the reports, but it cannot explain why the data are clustered. It is just as simple to detect and report a 250 pS 500 millisecond square top as a 25 pS 5 second square top.
We regard the clustering evident in all types of activity as evidence of an underlying energetic landscape that governs the behaviour of small molecule additives in bilayer membranes. As noted above, conductance values in the range of 10–30 pS lie below the range where a continuum model such as the Hille equation (eqn (2)) is reliable. This is the conductance range of the gramicidin channel which is known to require ion dehydration for passage down the axis of the 0.5 nm diameter of the β-helix.5 For other, simpler compounds to produce similar conductance values, implies that the channel environment is similar in character i.e. an array of donors that directly solvate a predominantly dehydrated ion in transit through close intermolecular contacts in a confined tube. Many of the compounds discussed above lack explicit donors, so water must play a role as well, and many authors invoke a mixture of lipid, compound, water, and the ion as a means to describe the channel.
Although we have insisted several times that the bilayer is a good insulator and ionic permeability is low, it has been recognized for many years that some lipids near their phase transition temperatures allow significantly enhanced water and ionic permeabilities.131,132 Although much of this area is known from studies on vesicles, single-channel observations have also been reported. The earliest reported example used “oxidized cholesterol” membranes133 but this was eventually followed by reports of pure synthetic lipids, such as DSPC.134 The voltage-clamp traces do closely resemble those discussed in this review, but it is their apparent similarity to traces from naturally occurring peptides and proteins that are the main concern of workers in this area. A recent review argues that the close similarities require a reassessment of the role of proteins as the sole mediators of ionic conduction in biological membranes.132 This may be correct, but we also need to take a closer look at the supposed similarities as well. Fig. 22 summarizes a few examples of the activities of lipids reported. With the exception of the DiPhyPC grid, all these were observed at or near the phase transition temperature of the bilayer. We are sceptical of the DiPhyPC report135 (cited in ref. 132) as this lipid is widely used for voltage clamp studies. One reason for this is precisely because it does not have a detectible phase transition136 and is known to be free of spurious “channel-like” conductance events.87,137 Nonetheless, Fig. 22 does show that a range of conductance behaviours are accessible from lipids alone. Another feature of the channels from DPPC138 is that they show Eisenman X or XI selectivity (Li+ > Na+ > K+ > Cs+).138 This is significantly different from the usual selectivity of synthetic ion channels (except 58).125
 | ||
| Fig. 22 Activity grids of “lipid ion channels” reported: DSPC;134 DPPC;138 DOPC;139 DC15PC;140 DiPhyPC.135 | ||
What Fig. 22 does not support is the idea that lipid channels alone are the origin of the clustering evident in the data summarized in Fig. 21. The reason is simple—the activity grids from lipids do not appear to fall in the same grouping as the compounds reviewed here. We acknowledge that this argument has no statistical weight whatsoever, but we point out that the activity grids force more precision in a discussion of “similarity”, and as a consequence the published data on lipid channels do not show much similarity between themselves.
Where the analysis of lipid channels has greater relevance is in the physical understanding of these channels. There is considerable evidence that underlying near-equilibrium fluctuations of thermodynamic parameters govern the probability that an unfavourable state, like an aqueous defect through the bilayer, will be accessible.132 In a lipid-only system, these fluctuations are most pronounced at the phase transition temperature and at phase boundaries between co-existing domains and it is precisely these conditions that produce channel-like activity.
In the presence of an additive—protein, peptide, or synthetic ion channel—there will certainly be interfacial contacts between the lipid and the additive where fluctuations in the thermodynamic sense will be more pronounced than in the lipid domains alone. We therefore view the clustering evident in Fig. 21, as the signature of an additive in a bilayer membrane. Simply placing compounds in bilayers gives rise to conditions where formation of channels via defects adjacent to the inserted compound becomes more probable. This is the explicit strategy of minimalist channels, but is inevitably associated with all compounds.
5.3 Implications and recommendations
A voltage-clamp trace is frequently presented as an unequivocal proof that a certain compound acts as an ion channel, and to the extent that channels form in the presence of the compound, the trace has significant weight. However, if there is a general landscape of channel-like behaviour, it will be progressively more difficult to argue a particular structural model the closer the observed behaviour lies to the typical. This places much higher weight on unusual or rare events—such as flickering behaviours—as the basis on which to move from function to structure. It also places more weight on unusual channel selectivity, gating characteristics, or lifetimes which might be related to the structure.We are not arguing that the observation of a behaviour that is associated with one of the clusters constitutes evidence that the additive is “only” acting as a perturbing species in the bilayer and that a particular designed structure is not present. Gramicidin channels certainly involve explicit ion transfer down the axis of the helix and they fall directly in the square top cluster. But the gramicidin mechanism is buttressed by large numbers of complementary studies and many lines of evidence. In the absence of such buttressing information, we simply point out that the observations might simply reflect the underlying thermodynamics of lipids and additives; mechanistic and structural proposals should be approached cautiously.
In the course of preparing this review, we have noted recurring gaps in the reported information; life-times, lipid, and applied potential are often omitted. From this perspective, we suggest the following features that will make voltage-clamp data more informative than it is currently:
Longer acquisition times. Earlier work was limited by data storage considerations and is often only 60 seconds in duration. Digital storage is no longer a critical consideration and many useful analyses (dwell times, power-law) have power proportional to the sample size. Acquisition times of tens of minutes is achievable and desirable.
Standard reporting format for experimental conditions. Just as NMR benefited from a standardized reporting format, the same may be expected for voltage-clamp data. An example may be “lipid, cis-electrolyte, [trans-electrolyte if different], method of compound addition, [amount of compound added if known], range of potentials.” Reporting in figures should use a conductance–time format (as opposed to current–time) whenever possible.
Complete sampling. In our experience, erratic behaviour is a very common activity and is probably more prevalent than square-top behaviours. A complete, representative sampling of compound activity is important to getting the picture right.
Standard representation of activities. While not applicable in all cases, we think that the activity grids presented here can be useful in facilitating comparisons within and between families of compounds.
6. Conclusions and prospects
The collection of reports surveyed represents a tremendous investment of effort in the creation of novel architectures and in the observation of “single molecule” events. That there is lots of diversity at the bottom should come as no surprise. In contrast, the observation of regular channel opening and closing events is truly remarkable, whatever be the structural origin. But it is the still tenuous relationship between function and structure that is the central issue. We echo a recent comprehensive review of the past four years of this field10 in encouraging innovation to bring new types of transport systems to fruition. We have the sense that we still have far too few examples on which to base a general discussion of the type we have undertaken here. We require genuinely new types of systems as opposed to additional examples that lie close to the known territory.As noted above, there is significance in the regions of the activity grid that lie remote from the clustering associated with additives to bilayers, and we have the (statistically unsound) sense that these regions have in the past been accessed with structures containing well-defined cavities or enforced openings such as the β-barrel structures. Such structures have been prepared by arduous synthetic routes and an on-going challenge is to create structures of this type more efficiently, for example via self-assembly routes. A few such examples have been reported, but the potential in this area is very high.
Another area of significant challenge and opportunity is in the analysis and eventual control of the erratic behaviours that are prevalent. As noted, some of these have been shown to exhibit underlying regularities. Even without more detailed insight, we note that these events carry significant currents and in many cases these are the most efficient “catalysts of translocation” known. We also note that explorations of the biological activity of synthetic ion channels have started from systems that exhibit regular behaviours;112,113,141 if gradient collapse is a significant component of the biological activity, then erratic channels make equal sense as a starting point. More generally, understanding the nature of erratic events, likely on a statistical and thermodynamic level, will require significant ingenuity to translate back to new targets for synthesis and new experimental approaches to control activity.
Finally we note that the notation is substrate-agnostic, and can be readily applied to lipid ion channels as shown above, as well to the many natural transporters whose activities also vary qualitatively and quantitatively. More generally, there may be other areas of contemporary supramolecular chemistry where the type of merging of quantitative and qualitative information represented by our activity grids may have some value. For example, many fields use imaging on scales of nm to mm to reveal a variety of morphologies that are described qualitatively (rods, wires, brushes, spheres, etc.). Such observations are usually presented in the text, or as a table that combines qualitative and quantitative information (for example).142 Like a voltage-clamp trace, an image is a single snapshot of limited statistical power. Whenever qualitative morphological information produced by underlying chemistry is related to specific quantitative functional performance, then the type of metadata analysis we present here may have a role in understanding how the structure influences the function.
Abbreviations
| DAN | dialkoxynaphthalene |
| DC15PC | dipentadecanoyl phosphatidylcholine (1,2-diacyl-sn-glycero-3-phosphocholine) |
| DiPhyPC | diphytanoyl phosphatidylcholine (1,2-diacyl-sn-glycero-3-phosphocholine) |
| DMPC | dimyristanoyl phosphatidylcholine (1,2-diacyl-sn-glycero-3-phosphocholine) |
| DOPC | dioleoyl phosphatidylcholine (1,2-diacyl-sn-glycero-3-phosphocholine) |
| DPPC | dipalmitoyl phosphatidylcholine (1,2-diacyl-sn-glycero-3-phosphocholine) |
| DSPC | disteroyl phosphtidylcholine(1,2-diacyl-sn-glycero-3-phosphocholine) |
| EPC | phosphatidylcholine from egg |
| MOP/F | metal–organic polyhedron/framework |
| NDI | naphthalenedimide |
| RH | arginine–histidine motif |
| a M , ax | activity of cation, anion |
| cis, trans | sides of cell; typically trans is electrical ground |
| ε, εrev | potential, reversal potential (eqn (2)) |
| g | conductance |
| i | current |
| l, lw | pore length, distance to blocking site (eqn (3)) |
| K D, KD0 | binding constant within a pore, at zero potential |
| P M , PX | permeability of action, anion |
| r | channel radius |
| RT/zF | gas constant, temperature, charge, Faraday |
| ρ | solution resistivity |
Acknowledgements
We thank authors of unintentionally overlooked publications for accepting our sincere apologies, and the University of Victoria and the Natural Sciences and Engineering Research Council of Canada for on-going financial support.Notes and references
- B. Hille, Ionic Channels of Excitable Membranes, Sinauer Associates, Incorporated, Sunderland, 3rd edn, 2001 Search PubMed.
- A. L. Hodgkin and A. F. Huxley, J. Physiol., 1952, 117, 500–544 CAS.
- E. Neher and B. Sakmann, Nature, 1976, 260, 799–802 CAS.
- Single-Channel Recording, ed. B. Sakmann and E. Neher, Springer, New York, 2nd edn, 1994 Search PubMed.
- O. S. Andersen, R. E. Koeppe and B. Roux, in Biological Membrane Ion Channels, ed. S.-H. Chung, O. S. Andersen and V. Krishnamurty, Springer, New York, 2007, p. 658 Search PubMed.
- E. Gouaux and R. MacKinnon, Science, 2005, 310, 1461–1465 CrossRef CAS.
- I. Tabushi, Y. Kuroda and K. Yokota, Tetrahedron Lett., 1982, 23, 4601–4604 CrossRef CAS.
- S. Matile and N. Sakai, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley -VCH, Weinheim, 2007, pp. 381–418 Search PubMed.
- M. Montal and P. Mueller, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 3561–3566 CAS.
- S. Matile, A. Vargas, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- A. L. Sisson, M. R. Shah, S. Bhosale and S. Matile, Chem. Soc. Rev., 2006, 35, 1269–1286 RSC.
- S. Matile, A. Som and N. Sorde, Tetrahedron, 2004, 60, 6405–6435 CrossRef CAS.
- T. M. Fyles and W. F. van Straaten-Nijenhuis, in Comprehensive Supramolecular Chemistry, ed. D. N. Reinhoudt, Elsevier Science, Amsterdam/New York, 1996, vol. 10, pp. 53–77 Search PubMed.
- T. M. Fyles, Chem. Soc. Rev., 2007, 36, 335–347 RSC.
- G. W. Gokel and M. M. Daschbach, Coord. Chem. Rev., 2008, 252, 886–902 CrossRef CAS.
- G. W. Gokel and I. A. Carasel, Chem. Soc. Rev., 2007, 36, 378–389 RSC.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- L. Mutihac, Anal. Lett., 2010, 43, 1355–1366 CrossRef CAS.
- F. Sansone, L. Baldini, A. Casnati and R. Ungaro, New J. Chem., 2010, 34, 2715–2728 RSC.
- G. W. Gokel and N. Barkley, New J. Chem., 2009, 33, 947–963 RSC.
- H. Wagner, K. Harms, U. Koert, S. Meder and G. Boheim, Angew. Chem., Int. Ed. Engl., 1996, 35, 2643–2646 CrossRef CAS.
- S. Futaki, M. Fukuda, M. Omote, K. Yamauchi, T. Yagami, M. Niwa and Y. Sugiura, J. Am. Chem. Soc., 2001, 123, 12127–12134 Search PubMed.
- M. X. Macrae, S. Blake, M. Mayer and J. Yang, J. Am. Chem. Soc., 2010, 132, 1766–1767 Search PubMed.
- D. Noshiro, K. Asami and S. Futaki, Biophys. J., 2010, 98, 1801–1808 Search PubMed.
- R. H. Ashley, Ion Channels: A Practical Approach, Oxford University Press, Oxford, 1995 Search PubMed.
- The structure of biological membranes, ed. P. Yeagle, CRC Press, Boca Raton, 2005 Search PubMed.
- F. Qin and L. Li, Biophys. J., 2004, 87, 1657–1671 Search PubMed.
- J. K. W. Chui and T. M. Fyles, Chem. Commun., 2010, 46, 4169–4171 RSC.
- O. S. Smart, J. Breed, G. R. Smith and M. S. P. Sansom, Biophys. J., 1997, 72, 1109–1126 CrossRef CAS.
- G. Eisenman, Biophys. J., 1962, 2, 259–323 CAS.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS.
- T. M. Fyles, D. Loock and X. Zhou, Can. J. Chem., 1998, 76, 1015–1026 CrossRef CAS.
- T. D. Clark, L. K. Buehler and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 651–656 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS.
- H. S. Kim, J. D. Hartgerink and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 4417–4424 CrossRef CAS.
- H. Ishida, Z. Qi, M. Sokabe, K. Donowaki and Y. Inoue, J. Org. Chem., 2001, 66, 2978–2989 CrossRef CAS.
- H. Ishida, K. Donowaki, Y. Inoue, Z. Qi and M. Sokabe, Chem. Lett., 1997, 953–954 CrossRef CAS.
- B. Gong, A. J. Helsel, A. L. Brown, K. Yamato, W. Feng, L. Yuan, A. J. Clements, S. V. Harding and Z. Shao, J. Am. Chem. Soc., 2008, 130, 15784–15785 CrossRef CAS.
- Y. Tanaka, Y. Kobuke and M. Sokabe, Angew. Chem., Int. Ed. Engl., 1995, 34, 693–694 CrossRef CAS.
- D. J. Cram, Science, 1983, 219, 1177–1183 CrossRef CAS.
- N. Yoshino, A. Satake and Y. Kobuke, Angew. Chem., Int. Ed., 2001, 40, 457–459 CrossRef CAS.
- J. de Mendoza, F. Cuevas, P. Prados, E. S. Meadows and G. W. Gokel, Angew. Chem., Int. Ed., 1998, 37, 1534–1537 CrossRef CAS.
- V. Siderov, F. W. Kotch, G. Abdrakhmanova, R. Mizani, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2002, 124, 2267–2278 CrossRef CAS.
- O. Lawal, K. S. J. Iqbal, A. Mohamadi, P. Razavi, H. T. Dodd, M. C. Allen, S. Siddiqui, F. Fucassi and P. J. Cragg, Supramol. Chem., 2009, 21, 55 CrossRef CAS.
- K. S. J. Iqbal, M. C. Allen, F. Fucassi and P. J. Cragg, Chem. Commun., 2007, 3951–3953 RSC.
- M. J. Pregel, L. Jullien, J. Canceill, L. Lacombe and J.-M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1995, 417–426 RSC.
- M. J. Pregel, L. Jullien and J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1992, 31, 1637–1640 CrossRef.
- K. Kim, Y. J. Jeon, H. Kim, S. Jon, N. Selvapalam, D. H. Oh, I. Seo, C.-S. Park, S. R. Jung and D.-S. Koh, J. Am. Chem. Soc., 2004, 126, 15944–15945 CrossRef CAS.
- K. Kim, N. Selvapalam, Y.-H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC.
- I. Tabushi, Y. Kuroda and K. Yokota, Tetrahedron Lett., 1982, 23, 4601–4604 CrossRef CAS.
- P. V. Jog and M. S. Gin, Org. Lett., 2008, 10, 3693–3696 CrossRef CAS.
- N. Madhavan, E. C. Robert and M. S. Gin, Angew. Chem., Int. Ed., 2005, 44, 7584–7587 CrossRef CAS.
- L. Bacri, A. Benkhaled, P. Guegan and L. Auvray, Langmuir, 2005, 21, 5842–5846 CrossRef CAS.
- N. Badi, L. Auvray and P. Guegan, Adv. Mater., 2009, 21, 4054–4057 CrossRef CAS.
- G. W. Gokel and A. Mukhopadhyay, Chem. Soc. Rev., 2001, 30, 274–286 RSC.
- G. W. Gokel, R. Ferdani, J. Liu, R. Pajewski, H. Shabany and P. Uetrecht, Chem.–Eur. J., 2001, 7, 33–39 CrossRef CAS.
- O. Murillo, S. Suzuki, E. Abel, C. L. Murray, E. S. Meadows, T. Jin and G. W. Gokel, J. Am. Chem. Soc., 1997, 119, 5540–5549 CrossRef CAS.
- E. Abel, G. E. M. Maguire, E. S. Meadows, O. Murillo, T. Jin and G. W. Gokel, J. Am. Chem. Soc., 1997, 119, 9061–9062 CrossRef CAS.
- E. Abel, E. S. Meadows, I. Suzuki, T. Jin and G. W. Gokel, Chem. Commun., 1997, 1145–1146 RSC.
- O. Murillo, I. Suzuki, E. Abel and G. W. Gokel, J. Am. Chem. Soc., 1996, 118, 7628–7629 CrossRef CAS.
- C. D. Hall, G. J. Kirkovits and A. C. Hall, Chem. Commun., 1999, 1897–1898 RSC.
- M. Tsikolia, A. C. Hall, C. Suarez, Z. O. Nylander, S. M. Wardlaw, M. E. Gibson, K. L. Valentine, L. N. Onyewadume, D. A. Ahove, M. Woodbury, M. M. Mongare, C. D. Hall, Z. Wang, B. Draghici and A. R. Katritzky, Org. Biomol. Chem., 2009, 7, 3862–3870 RSC.
- J. R. Pfeifer, P. Reiss and U. Koert, Angew. Chem., Int. Ed., 2006, 45, 501–504 CrossRef CAS.
- A. Cazacu, C. Tong, A. van der Lee, T. M. Fyles and M. Barboiu, J. Am. Chem. Soc., 2006, 128, 9541–9548 CrossRef CAS.
- T. M. Fyles, D. Loock, W. F. van Straaten-Nijenhuis and X. Zhou, J. Org. Chem., 1996, 61, 8866–8874 CrossRef CAS.
- T. M. Fyles, D. Loock and X. Zhou, J. Am. Chem. Soc., 1998, 120, 2997–3003 CrossRef CAS.
- L. M. Cameron, T. M. Fyles and C. Hu, J. Org. Chem., 2002, 67, 1548–1553 CrossRef CAS.
- N. Sakai, Y. Kamikawa, M. Nishii, T. Matsuoaka, T. Kato and S. Matile, J. Am. Chem. Soc., 2006, 128, 2218–2219 CrossRef CAS.
- N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2005, 38, 79–87 CrossRef CAS.
- N. Sakai, C. Ni, S. M. Bezrukov and S. Matile, Bioorg. Med. Chem. Lett., 1998, 8, 2743–2746 CrossRef CAS.
- N. Sakai, D. Houdebert and S. Matile, Chem.–Eur. J., 2003, 9, 223–232 CrossRef CAS.
- N. Sakai, N. Sorde, G. Das, P. Perrottet, D. Gerard and S. Matile, Org. Biomol. Chem., 2003, 1, 1226–1231 RSC.
- P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 6528–6529 CrossRef CAS.
- Y. Baudry, D. Pasini, M. Nishihara, N. Sakai and S. Matile, Chem. Commun., 2005, 4798–4800 RSC.
- A. M. Woodhull, J. Gen. Physiol., 1973, 61, 687–708 CrossRef CAS.
- H. Tanaka, G. Bollot, J. Mareda, S. Litvinchuk, D.-H. Tran, N. Sakai and S. Matile, Org. Biomol. Chem., 2007, 5, 1369–1380 RSC.
- J.-C. Meillon and N. Voyer, Angew. Chem., Int. Ed. Engl., 1997, 36, 967–969 CrossRef CAS.
- E. Biron, F. Otis, J.-C. Meillon, J. Lamothe, P. V. Hove, M.-E. Cormier and N. Voyer, Bioorg. Med. Chem., 2004, 12, 1279–1290 CrossRef CAS.
- R. Ferdani and G. W. Gokel, Org. Biomol. Chem., 2006, 4, 3746–3750 RSC.
- G. Jung, T. Redemann, K. Kroll, S. Meder, A. Hirsch and G. Boheim, J. Pept. Sci., 2003, 9, 11–12 Search PubMed.
- L. You, R. Ferdani, R. Li, J. P. Kramer, R. E. K. Winter and G. W. Gokel, Chem.–Eur. J., 2008, 14, 382–396 CrossRef.
- P. K. Eggers, T. M. Fyles, K. D. D. Mitchell and T. Sutherland, J. Org. Chem., 2003, 68, 1050–1058 CrossRef CAS.
- Y. Kobuke, K. Ueda and M. Sokabe, J. Am. Chem. Soc., 1992, 114, 7618–7622 CrossRef CAS.
- Y. Kobuke and K. Morita, Inorg. Chim. Acta, 1998, 283, 167–174 CrossRef CAS.
- Y. Kobuke and A. Ohgoshi, Colloids Surf., 2000, 169, 187–197 Search PubMed.
- T. Renkes, H. J. Schäfer, P. M. Siemens and E. Neumann, Angew. Chem., Int. Ed., 2000, 39, 2512–2516 CrossRef CAS.
- T. M. Fyles, C. Hu and R. Knoy, Org. Lett., 2001, 3, 1335–1337 CrossRef CAS.
- H. Luong and T. M. Fyles, Org. Biomol. Chem., 2009, 7, 733–738 RSC.
- H. Luong and T. M. Fyles, Org. Biomol. Chem., 2009, 7, 725–732 RSC.
- T. M. Fyles, C. Hu and H. Luong, J. Org. Chem., 2006, 71, 8545–8551 CrossRef CAS.
- T. M. Fyles, R. Knoy, K. Müllen and M. Sieffert, Langmuir, 2001, 17, 6669–6674 CrossRef CAS.
- H. L. Luong, University of Victoria, 2008, http://hdl.handle.net/1828/892.
- J. Moszynski and T. M. Fyles, Org. Biomol. Chem., 2010, 8, 5139–5149 RSC.
- N. Illy, L. Bacri, J. Wojno, D. Destouches, B. Brissault, J. Courty, L. Auvray, J. Penelle and V. Barbier, Macromol. Symp., 2010, 287, 60–68 CrossRef CAS.
- W. Wang, R. Li and G. W. Gokel, Chem.–Eur. J., 2009, 15, 10543–10553 CrossRef CAS.
- W. Wang, R. Li and G. W. Gokel, Chem. Commun., 2009, 911–913 RSC.
- G. Deng, T. Dewa and S. L. Regen, J. Am. Chem. Soc., 1996, 118, 8975–8976 CrossRef CAS.
- G. Deng, M. Merritt, K. Yamashita, V. Janout, A. Sadownik and S. L. Regen, J. Am. Chem. Soc., 1996, 118, 3307–3308 CrossRef CAS.
- M. Merritt, M. Lanier, G. Deng and S. L. Regen, J. Am. Chem. Soc., 1998, 120, 8494–8501 CrossRef CAS.
- P. Bandyopadhyay, V. Janout, L. Zhang, J. A. Sawko and S. L. Regen, J. Am. Chem. Soc., 2000, 122, 12888–12889 CrossRef CAS.
- P. Bandyopadhyay, V. Janout, L. Zhang and S. L. Regen, J. Am. Chem. Soc., 2001, 123, 7691–7696 CrossRef CAS.
- Y. Kobuke and T. Nagatani, J. Org. Chem., 2001, 66, 5094–5101 CrossRef CAS.
- M. Yoshii, M. Yamamura, A. Satake and Y. Kobuke, Org. Biomol. Chem., 2004, 2, 2619–2623 RSC.
- C. Goto, M. Yamamura, A. Satake and Y. Kobuke, J. Am. Chem. Soc., 2001, 123, 12152–12159 CrossRef CAS.
- L. Ma, M. Melegari, M. Colombini and J. T. Davis, J. Am. Chem. Soc., 2008, 130, 2938–2939 CrossRef CAS.
- L. Ma, W. A. Harrell and J. T. Davis, Org. Lett., 2009, 11, 1599–1602 CrossRef CAS.
- H. D. Arndt, A. Knoll and U. Koert, ChemBioChem, 2001, 2, 221–223 Search PubMed.
- X. Xie, L. Al-Momani, P. Reiss, C. Griesinger and U. Koert, FEBS J., 2005, 272, 975–986 Search PubMed.
- G. A. Woolley, V. Zunic, J. Karanicolas, A. S. I. Jaikaran and A. V. Starostin, Biophys. J., 1997, 73, 2465–2475 CrossRef CAS.
- P. Reiss, L. Al-Momani and U. Koert, ChemBioChem, 2008, 9, 377–379 CrossRef CAS.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 2nd edn, 1992 Search PubMed.
- X. Li, B. Shen, X.-Q. Yao and D. Yang, J. Am. Chem. Soc., 2007, 129, 7264–7265 CrossRef CAS.
- X. Li, B. Shen, X.-Q. Yao and D. Yang, J. Am. Chem. Soc., 2009, 131, 13676–13680 CrossRef CAS.
- C. R. Yamnitz, S. Negin, I. A. Carasel, R. K. Winter and G. W. Gokel, Chem. Commun., 2010, 46, 2838–2840 RSC.
- W. H. Binder, Angew. Chem., Int. Ed., 2008, 47, 3092–3095 CrossRef CAS.
- J. C. Chung, D. J. Gross, J. L. Thomas, D. A. Tirrell and L. R. Opsahl-Ong, Macromolecules, 1996, 29, 4636–4641 CrossRef CAS.
- F. Vial, A. G. Oukhaled, L. Auvray and C. Tribet, Soft Matter, 2007, 3, 75–78 RSC.
- P. Schlieper and E. De Robertis, Arch. Biochem. Biophys., 1977, 184, 204–208 CrossRef CAS.
- T. K. Rostovtseva, C. L. Bashford, A. A. Lev and C. A. Pasternak, J. Membr. Biol., 1994, 141, 83–90 CAS.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS.
- S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef CAS.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS.
- T. M. Fyles and C. C. Tong, New J. Chem., 2006, 31, 655–661 Search PubMed.
- H. Furukawa, J. Kim, K. E. Plass and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 8398–8399 CrossRef CAS.
- M. Jung, H. Kim, K. Baek and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 5755–5757 CrossRef CAS.
- S. J. Dalgarno, N. P. Power, J. E. Warren and J. L. Atwood, Chem. Commun., 2008, 1539–1541 RSC.
- O. V. Kulikov, R. Li and G. W. Gokel, Angew. Chem., Int. Ed., 2009, 48, 375–377 CrossRef CAS.
- R. Li, O. V. Kulikov and G. W. Gokel, Chem. Commun., 2009, 6092–6094 RSC.
- A. Satake, M. Yamamura, M. Oda and Y. Kobuke, J. Am. Chem. Soc., 2008, 130, 6314 CrossRef CAS.
- A. R. Palmer, Annu. Rev. Ecol. Syst., 2000, 31, 441–480 Search PubMed.
- D. W. Deamer and J. Bramhill, Chem. Phys. Lipids, 1986, 40, 167–188 CrossRef CAS.
- T. Heimberg, Biophys. Chem., 2010, 150, 2–22 CrossRef CAS.
- M. Yafuso, S. J. Kennedy and A. R. Freeman, J. Membr. Biol., 1974, 17, 201–212 CrossRef CAS.
- V. F. Antonov, V. V. Petrov, A. A. Molnar, D. A. Predvoditelev and A. S. Ivanov, Nature, 1980, 283, 585–586 CrossRef CAS.
- K. Kaufmann, W. Hanke and A. Corcica, 1989, http://membranes.nbi.dk/Kaufmann/pdf/1989_Kaufmann_book3_ed.pdf.
- H. Lindsey, N. O. Petersen and S. I. Chan, Biochim. Biophys. Acta, 1979, 555, 147–167 Search PubMed.
- I. R. Mellor and M. S. P. Sansom, Proc. R. Soc. London, Ser. B, 1990, 239, 383–400 Search PubMed.
- A. Blicher, K. Wodzinska, M. Fidorra, M. Winterhalter and T. Heimberg, Biophys. J., 2009, 96, 4581–4591 CrossRef.
- T. Heimberg, Phys. J., 2009, 8, 33–39 Search PubMed.
- B. Wunderlich, C. Leirer, A. Idzko, U. F. Keyser, V. Myles, T. Heimburg and M. Schneider, Biophys. J., 2009, 96, 4592–4597 CrossRef CAS.
- W. M. Leevy, M. E. Weber, M. R. Gokel, G. R. Hughes-Strange, D. D. Daranciang, R. Ferdani and G. W. Gokel, Org. Biomol. Chem., 2005, 3, 1647–1652 RSC.
- T. Zhai, L. Li, Y. Ma, M. Liao, X. Wang, X. Fang, J. Yao, Y. Bando and D. Golberg, Chem. Soc. Rev., 2011, 40, 2986–3004 RSC.
- W. H. Chen, M. Nishikawa, S. D. Tan, M. Yamamura, A. Satake and Y. Kobuke, Chem. Commun., 2004, 872–873 RSC.
Footnotes |
| † A tetracyano derivative of 4a with tridecyl sidechains also shows activity in bilayers. The current is pH dependent and somewhat larger than that reported for 4a. No traces were supplied, so the duration of the openings and the shape trace could not be evaluated.143 |
| ‡ The authors report conductances 1000-fold larger (nS in place of pS), but the scales of the figures and the figure captions indicate this is incorrect. |
| § For as knowledges are now delivered, there is a kind of contract of error between the deliverer and the receiver: for he that delivereth knowledge desireth to deliver it in such a form as may be best believed, and not as may be best examined; and he that receiveth knowledge desireth rather present satisfaction than expectant inquiry; and so rather not to doubt than not to err: glory making the author not to lay open his weakness, and sloth making the disciple not to know his strength. The Advancement of Learning, Francis Bacon, 1605 |
| This journal is © The Royal Society of Chemistry 2012 |