 Open Access Article
Open Access ArticleA DNA based five-state switch with programmed reversibility†‡
Jonathan R.
Burns
a,
Søren
Preus
b,
Daniel G.
Singleton
a and
Eugen
Stulz
*a
aSchool of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: est@soton.ac.uk; Web: http://www.soton.ac.uk/~stulz Tel: +44 (0)2380 599 369
bDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen, Denmark
First published on 24th September 2012
Abstract
A programmable switch based on a DNA hairpin loop is functionalised with a rigid or flexible porphyrin or FAM and TAMRA FRET pair, which provides insight into the restructuring of the hairpin as well as porphyrin–porphyrin coupling. The switch contains five discrete states which can be accessed independently and followed by real-time spectroscopy, opening the way to a quinary computing code.
The unique self-recognition properties of DNA have been explored extensively for the formation of new self-assembled nano-architectures over the past decades. By taking the DNA out of its biological context, the emerging field of DNA nanotechnology is becoming increasingly attractive to advance research in drug delivery, autonomous machines or computing.1 Additional functionalities such as redox active metal complexes2 or organic chromophores3 are increasingly being incorporated into DNA, resulting in operational DNA based nano-devices. Still, the incorporation of chemically modified DNA into these nano-architectures is in its infancy, and the exploration of modified DNA as building blocks remains an important aspect to understand their behaviour and suitability for DNA nanotechnology. In particular, organic chromophores4 such as pyrenes5 and porphyrins6 are gaining increasing attention as DNA modifiers e.g. for the creation of photonic wires, or as diagnostic tools since their optical properties (absorption, emission) can vary with the DNA sequence or a change in the environment such as pH, temperature or secondary structure.
Particularly intriguing structures arise when partially self-complementary DNA strands assemble to form intramolecular hairpin loops. The loops can be opened and closed through sequential addition of suitable complementary DNA strands, thus creating moving parts within a DNA nanostructure. The concept has been used in molecular beacons for DNA analysis,7 in switchable DNA nanostructures with optical responses,8 and in autonomous DNA walkers.9 Here, we report a programmable switch based on a molecular beacon, where the DNA is partially self-complementary with repeating ATTA–TAAT sequences (Fig. 1). An additional 13 base sequence allows for specific recognition of complementary strands including various repeats of the complementary ATTA–TAAT box, thus enabling controlled elongation or contraction of the stem region. To demonstrate functionality, FRET pairs were attached to the extremes of the repeat region, giving access to a tuneable energy transfer system with well-defined chromophore distances from the same DNA strand by simply adding the appropriate complementary strand. The porphyrin based FRET system (denoted 1P) comprises of a zinc porphyrin (donor, D) and a free-base porphyrin (acceptor, A), where the porphyrins are attached either via a rigid alkynyl linker or a more flexible propargyl-amide linker.10 The rigid alkynyl linker ensures that the chromophores have a low diffusional mobility and remain in a well oriented environment, particularly in terms of the transition dipole moment, introduced in order to study the influence of the porphyrin distance and angle on the electronic coupling compared to the flexible linker. As a control system, we have also synthesised the analogous FAM and TAMRA labelled switch strand (denoted 1F). Because this FRET pair is tethered via a longer and more flexible linker, the angular dependence on the FRET efficiency should in ideal cases be eliminated (κ2 = 2/3).
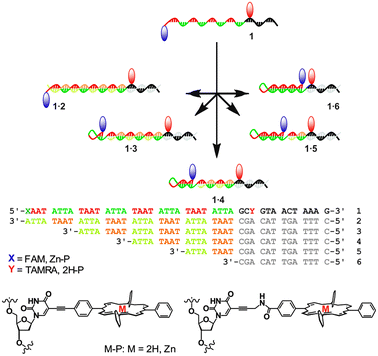 | ||
| Fig. 1 Schematic of the adjustable hairpin loops with DNA sequences. The red and blue markers indicate attachment of FRET pairs. | ||
The Tm values of the two extreme complexes (the full length duplex 1P·2 and the hairpin with the longest possible stem of DNA 1P·6) show a clear difference, where 1P·2 has a Tm value of 54.1 °C, and for 1P·6 the Tm was determined to be 61.2 °C (Table 1). The molecular beacon 1P on its own displays a Tm of 61.7 °C, overall confirming the intramolecular stabilisation effect on the duplex. In the presence of the complementary strands, the other hairpin-duplex combinations show intermediate Tm values in the expected order.
| DNA system | T m | T m | FRET FAM–TAMRA |
|---|---|---|---|
| 1P a/°C | 1F b/°C | Exp. (calcd) | |
| a T m values obtained from UV-melting (260 nm). b T m values obtained from fluorescence melting (λex = 495 nm, λem = 510 nm). | |||
| 1 | 61.7 | 60.0 | 0.8 (0.98) |
| 1·2 | 54.1 | 59.0 | 0.00 (0.01) |
| 1·3 | 50.8 | 59.1 | 0.01 (0.03) |
| 1·4 | 54.4 | 58.6 | 0.17 (0.13) |
| 1·5 | 55.2 | 59.7 | 0.52 (0.57) |
| 1·6 | 61.2 | 60.2 | 0.81 (0.98) |
To determine changes in the direct environment of the modifications, we attempted to record the melting profiles monitoring the porphyrin region. However, the thermal lability of the zinc porphyrin complex interferes with the measurement (loss of metal at higher temperatures),10a thus fluorescence melting was conducted on the FAM and TAMRA system (Fig. 2a). This system displayed clear transitions when monitoring the donor emission of FAM. The initial donor emission intensity at 20 °C corresponds to the programmed donor–acceptor distance, and decreases in the expected order from 1F·2 to 1F·6, resulting from the variable length of the stem. The denaturing profiles, however, are very different to the UV-melting curves,‡ and show an initial decrease in donor emission, indicating a decrease in distance of the chromophores and increase in FRET between the donor and acceptor. Thus the dissociation of the complementary DNA strand is accompanied by a restructuring of the self-complementary DNA to form a longer and more stable stem-loop system as an intermediate structure, which then denatures to give the random coil single strand DNA, though the intermediate full length stem may not be formed in all cases. This is also corroborated by the fact that the fluorescence melting shows the same Tm of around 60 °C for all systems. The final donor fluorescence intensity is equal in all systems and corresponds to the initial intensity of the system 1F·2. This confirms both the complete denaturing at higher temperature as well as formation of the full length duplex in 1·2.
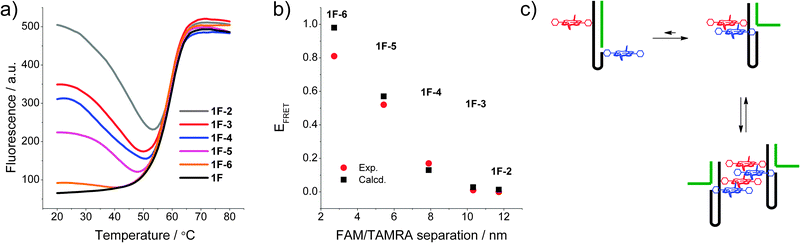 | ||
| Fig. 2 (a) Fluorescence melting curves of the DNA switch by monitoring FAM emission (λex = 495 nm, λem = 510 nm); (b) calculated vs. experimental FRET of the modified system in the FAM and TAMRA system 1F; (c) putative intra- and intermolecular stacking of the porphyrins in the DNA beacon systems. | ||
The FRET efficiencies for both the Zn- and 2H-porphyrin and the FAM and TAMRA system were measured and compared, in order to evaluate the difference between the FRET pairs where the chromophores are attached via a rigid or flexible linker. Since energy transfer efficiencies can vary greatly with probe diffusion and reorientation, the linker moiety plays an important role. The FRET parameters of the two FRET pairs (quantum yield, extinction coefficient and spectral overlap) were identified by steady state fluorescence spectroscopy.‡ The FRET efficiencies (EFRET) were determined by spectral decomposition of the combined donor and acceptor emissions at an excitation wavelength of 426 nm. The quantum yield of Zn–P was determined to be Φ = 0.12 (using quinine sulphate as the standard) which in combination with a spectral overlap integral of J = 2.3 × 1014 M−1 cm−1 nm4 yields a Förster distance of R0 = 28.4 Å. The Förster distance of the FAM and TAMRA pair was determined to be R0 = 57 Å. In addition, the FRET efficiencies were predicted theoretically for all systems using a custom made FRET simulation program (FRET matrix).11 Here, all DNA conformations were simulated as rigid B-form geometries and a full atomistic description of the tethered dyes was used to calculate the theoretical donor–acceptor distances.‡
The experimental FRET values of the 1F·x combinations are in excellent agreement with the theoretically predicted values (Fig. 2b) and show a decreasing FRET efficiency upon increasing the fluorophore distance. Exceptions are the states 1F and 1F·6 showing a smaller measured FRET efficiency compared to that expected from the calculated donor–acceptor distance. This deviation is often observed for closely spaced dyes and is likely to be a result of direct dye–dye interactions interfering with the FRET process.12 Overall, the good correlation between expected and measured FRET efficiencies demonstrates the well-defined, discrete states of the switch.
Compared to the FAM and TAMRA control system, the porphyrin FRET system shows a very different behaviour. Despite that porphyrins attached to duplex DNA show FRET efficiencies which are dependent on the distance (including a control sequence used to determine the quantum yields of the porphyrin–DNA, see ESI‡), the switch sequences all show a FRET efficiency close to 100%. No variation could be detected in either the different conformations or upon changing the linker to the porphyrin. Based on the R0 value, the calculations would predict FRET efficiencies of 43% for 1P·6 and <10% for all other combinations. This indicates that the porphyrins are in close contact at a maximum distance of about 3–10 Å (10–30% of R0 where 100% FRET is expected). The system is therefore disrupted by the hydrophobic interactions of the porphyrins which are probably stacked to give a very efficient energy transfer between the Zn- and 2H-porphyrin (Fig. 2c). At this point we cannot rule out intermolecular interactions as we10a and others6c have found efficient intermolecular stacking of porphyrins and small organic molecules covalently attached to DNA, which can act as a molecular glue between DNA strands.13 Since the association constant for these intermolecular assemblies is unknown, it cannot be estimated to which extent they contribute to the FRET if they do so.
DNA has recently been used to generate molecular machines, where addition of specific complementary strands is used to trigger switching between different structures and states, and up to eleven discrete states have been achieved using a DNA tile actuator.14 Our simple switch equally responds to a change in the system: by extending the complementary strand by eight bases to form an overhang (toehold), the strand can be peeled off, and the system switches to a different state by subsequent addition of a longer or shorter complementary strand. The switching can be monitored using real-time fluorescence spectroscopy (Fig. 3). Starting with the full-length duplex 1F·2, subsequent switching between the various states leads to a system where five different states can easily and reversibly be addressed. The concept is schematically drawn in Fig. 3 for the switching between the states 1F·2 → 1F·6 → 1F·3. In this way, not only a binary open-closed code can be made, but this system could form the basis for a quinary code that could significantly enhance the information density in DNA based computing.15
In summary, a DNA based adjustable strap was obtained by synthesising DNA strands with partially self-complementary boxes, and addition of complementary strands allows for a programmable adjustment of the stem-loop size. Such a system may be used as part of a molecular motor or computer, where a DNA input results in a change in structure thus encouraging motion and yielding a corresponding change in output (e.g. change in energy transfer). The presented programmable switch, containing fluorophores attached by both rigid and flexible linkers, reveals that the formation of a particular DNA system can strongly be influenced by the nature of the modification. The hydrophobic nature of the porphyrins disrupts the stem structure in the beacon, and close contacts between the chromophores are evident, which shows the system's limitations. However, by tuning the properties of the modification specific structures and motion are retained.
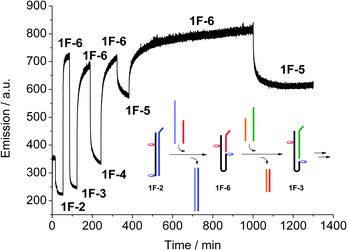 | ||
| Fig. 3 Real-time monitoring of the switching between the five different switching states, recording acceptor emission (λex = 495 nm, λem = 583 nm), and schematic drawing of the first steps. The final switches between 1F·6 and 1F·5 show the reversibility and stability of the system. | ||
We acknowledge generous support by ATDBio (Southampton, UK) and sponsorship by the EPSRC (CASE-DTA to JRB); MS analysis by the EPSRC National Mass Spectrometry Service, Swansea, is greatly acknowledged.
Notes and references
- (a) E. Stulz, Chem.–Eur. J., 2012, 18, 4456–4469 CrossRef CAS; (b) E. Stulz, G. Clever, M. Shionoya and C. Mao, Chem. Soc. Rev., 2011, 40, 5633–5635 RSC; (c) T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. Nguyen and E. Stulz, Chem. Soc. Rev., 2011, 40, 138–148 RSC.
- (a) J. R. Burns, J. Zekonyte, G. Siligardi, R. Hussain and E. Stulz, Molecules, 2011, 16, 4912–4922 CrossRef CAS; (b) S. Bezer, S. Rapireddy, Y. A. Skorik, D. H. Ly and C. Achim, Inorg. Chem., 2011, 50, 11929–11937 CrossRef CAS; (c) S. I. Khan, A. E. Beilstein and M. W. Grinstaff, Inorg. Chem., 1999, 38, 418–419 CrossRef CAS.
- (a) V. L. Malinovskii, D. Wenger and R. Häner, Chem. Soc. Rev., 2010, 39, 410–422 RSC; (b) H. Kashida, T. Takatsu, K. Sekiguchi and H. Asanuma, Chem.–Eur. J., 2010, 16, 2479–2486 CrossRef CAS; (c) M. Nakamura, Y. Murakami, K. Sasa, H. Hayashi and K. Yamana, J. Am. Chem. Soc., 2008, 130, 6904–6905 CrossRef CAS.
- (a) M. M. Rubner, C. Holzhauser, P. R. Bohlander and H. A. Wagenknecht, Chem.–Eur. J., 2012, 18, 1299–1302 CrossRef CAS; (b) A. Ruiz-Carretero, P. G. A. Janssen, A. Kaeser and A. Schenning, Chem. Commun., 2011, 47, 4340–4347 RSC.
- (a) D. Lindegaard, A. S. Madsen, I. V. Astakhova, A. D. Malakhov, B. R. Babu, V. A. Korshun and J. Wengel, Bioorg. Med. Chem., 2008, 16, 94–99 CrossRef CAS; (b) M. E. Ostergaard and P. J. Hrdlicka, Chem. Soc. Rev., 2011, 40, 5771–5788 RSC; (c) C. Boonlua, C. Vilaivan, H. A. Wagenknecht and T. Vilaivan, Chem.–Asian J., 2011, 6, 3251–3259 CrossRef CAS; (d) S. P. Sau and P. J. Hrdlicka, J. Org. Chem., 2012, 77, 5–16 CrossRef CAS.
- (a) T. Nguyen, A. Brewer and E. Stulz, Angew. Chem., Int. Ed., 2009, 48, 1974–1977 CrossRef CAS; (b) K. Borjesson, J. G. Woller, E. Parsa, J. Martensson and B. Albinsson, Chem. Commun., 2012, 48, 1793–1795 RSC; (c) A. Mammana, G. Pescitelli, T. Asakawa, S. Jockusch, A. G. Petrovic, R. R. Monaco, R. Purrello, N. J. Turro, K. Nakanishi, G. A. Ellestad, M. Balaz and N. Berova, Chem.–Eur. J., 2009, 15, 11853–11866 CrossRef CAS; (d) A. W. I. Stephenson, A. C. Partridge and V. V. Filichev, Chem.–Eur. J., 2011, 17, 6227–6238 CrossRef CAS.
- R. T. Ranasinghe and T. Brown, Chem. Commun., 2005, 5487–5502 RSC.
- R. Varghese and H. A. Wagenknecht, Org. Biomol. Chem., 2010, 8, 526–528 CAS.
- R. A. Muscat, J. Bath and A. J. Turberfield, Nano Lett., 2011, 11, 982–987 CrossRef CAS.
- (a) A. Brewer, G. Siligardi, C. Neylon and E. Stulz, Org. Biomol. Chem., 2011, 9, 777–782 RSC; (b) L. A. Fendt, I. Bouamaied, S. Thöni, N. Amiot and E. Stulz, J. Am. Chem. Soc., 2007, 129, 15319–15329 CrossRef CAS.
- S. Preus, K. Kilså, F.-A. Miannay, B. Albinsson and L. M. Wilhelmsson, Nucleic Acids Res., 2012 DOI:10.1093/nar/gks1856.
- N. Di Fiori and A. Meller, Biophys. J., 2010, 98, 2265–2272 CrossRef CAS.
- (a) H. Kashida, T. Hayashi, T. Fujii and H. Asanuma, Chem.–Eur. J., 2011, 17, 2614–2622 CrossRef CAS; (b) D. Baumstark and H. A. Wagenknecht, Angew. Chem., Int. Ed., 2008, 47, 2612–2614 CrossRef CAS.
- Z. Zhang, E. M. Olsen, M. Kryger, N. V. Voigt, T. Torring, E. Gultekin, M. Nielsen, R. Mohammad Zadegan, E. S. Andersen, M. M. Nielsen, J. Kjems, V. Birkedal and K. V. Gothelf, Angew. Chem., Int. Ed., 2011, 50, 3983–3987 CrossRef CAS.
- G. M. Church, Y. Gao and S. Kosuri, Science, 2012, 337, 1628 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Nucleic acids: new life, new materials’ web themed issue. |
| ‡ Electronic supplementary information (ESI) available: Theoretical FRET calculations, synthetic methods and spectroscopic analysis of the DNA strands. See DOI: 10.1039/c2cc35799b |
| This journal is © The Royal Society of Chemistry 2012 |