Advances in microfluidics for environmental analysis
Jana C.
Jokerst
,
Jason M.
Emory
and
Charles S.
Henry
*
Colorado State University, Department of Chemistry, Fort Collins, CO 80523, USA
First published on 18th October 2011
Abstract
During the past few years, a growing number of groups have recognized the utility of microfluidic devices for environmental analysis. Microfluidic devices offer a number of advantages and in many respects are ideally suited to environmental analyses. Challenges faced in environmental monitoring, including the ability to handle complex and highly variable sample matrices, lead to continued growth and research. Additionally, the need to operate for days to months in the field requires further development of robust, integrated microfluidic systems. This review examines recently published literature on the applications of microfluidic systems for environmental analysis and provides insight in the future direction of the field.
 Jana C. Jokerst | Jana Jokerst received a BS in Chemistry at the University of Arkansas in Fayetteville, AR where she worked on a microchip ELISA platform for the detection of pathogens in drinking water. She is currently a PhD candidate at Colorado State University in Fort Collins, CO. Her interests in monitoring water and food quality led to graduate research projects involving the development of a novel microfluidic device for the determination of perchlorate in drinking water as well as the development of a paper-based biosensor for the detection of foodborne pathogens. |
 Jason M. Emory | Jason M. Emory received a BS and MS in Chemistry from the University of North Carolina at Charlotte where he worked on ultrasensitive detection of proteins in glass microfluidic devices. He went on to earn his Ph.D. in Chemistry at Louisiana State University working on single molecule detection techniques in microfluidic devices for molecular diagnostics and homeland security applications. He is currently a Post-Doc at Colorado State University working to develop new techniques and instrumentation for the determination of ionic pollutants in water. |
 Charles S. Henry | Charles S. Henry received his B.S. in Chemistry from Missouri Southern State College and his Ph.D. in Analytical Chemistry at the University of Arkansas. His Ph.D. research focused ultramicroelectrodes for bioanalytical and environmental studies. Dr Henry completed an NIH Postdoctoral fellowship in Pharmaceutical Chemistry at the University of Kansas. Dr Henry started his academic career at Mississippi State University before moving to Colorado State University. His research group is active in the general areas of microchip electrophoresis and microfluidic biosensors with application to environmental monitoring and clinical diagnostics. |
Introduction
Over the past few decades, tremendous growth in the biochemical, industrial, pharmaceutical, and medical industries has led to a growing list of emerging contaminants and an increase in environmental regulations. As the number of regulated pollutants increases, greater demands are placed on environmental analysis. A number of powerful analytical techniques have been used for environmental monitoring; however, the instrumentation can be complex, leading to time-consuming and costly analysis resulting in incomplete assessment of pollutant distribution. Some researchers have turned to microfluidic devices to realize improvements in environmental analysis. Microfluidic devices offer a number of advantages, including rapid detection and identification of compounds, low sample and reagent consumption, and the potential for field monitoring. While the capabilities of microfluidic systems have made great strides over the years, there remain important features that are still lacking. For example, the ideal microfluidic device for environmental analysis would perform on-chip sample preparation from complex matrices such as ground water in a high-throughput manner with sensitive and selective detection while operating unattended in the field for days to months. Microfluidic devices for biological applications can meet some but not all of these needs. Finally, the devices should be low-cost and portable to allow broad distribution and mobile sensing and/or identification of point source contamination. All of these requirements place a high demand on microfluidic systems in a way that is very different from biological analysis.The high demands on microfluidic ingenuity stem from the complexity and diversity of environmental sample matrices and the broad classifications of contaminants, including endocrine disrupting compounds, polyaromatic hydrocarbons, haloacetic acids and other disinfectant byproducts, pesticides and herbicides, inorganic ions, toxic metals, and microorganisms to name a few.1,2 We believe a review of the current advances in microfluidics for environmental analysis, discussing both the latest developments in the field and the fundamental unresolved issues, is pertinent to the future directions of microfluidic technology. In recent years, similar articles have been published. Chen et al. focused on the broad environmental applications of microchip electrophoresis coupled with electrochemical detection.3 Richardson published a comprehensive review of the most commonly implemented analytical techniques (most of which are based on traditional methods), organized by pollutant type.4 Li et al. discussed current progress in microchip electrophoresis and electrochromatography for environmental applications.5 This review, while far from exhaustive, discusses literature published in the past three years on the advances in sample preparation, novel separation methods, improvements in detection, and developments in integration across all areas of microfluidic analysis for environmental samples.
Sample preparation
Preparative cleanup and preconcentration steps are required with most environmental analyses due to the complexity of sample matrix and the low relative abundance of pollutants (ppm-ppt levels are common).4–6 Various preconcentration strategies have been developed off-line to improve sensitivity; conversely, these methods are time-consuming and run the risk of contamination and/or generation of artifacts. Performing the necessary preparation steps on-chip is ideal; however, integration of pretreatment into a microfluidic network for trace analysis is challenging. On-chip preconcentration methods can be broadly categorized as either static or dynamic.6 Static mechanisms include extraction, surface adsorption, and membrane filtration, while dynamic mechanisms manipulate analyte velocities to achieve selective enrichment and include electrophoretic stacking, focusing, and sweeping. In static methods, the use of a functionalized solid support for sample pretreatment has shown great promise for isolation and concentration of target analytes in a complex sample matrix. The challenge of these methods lies in the difficulty of constructing frits, weirs and other physical features to trap sorbents. Nonetheless, advancements in the area of microfluidics continue in an effort to implement on-chip sample cleanup. Recently, Tennico and Remcho published a solid-phase extraction technique using functionalized, magnetic iron oxide nanoparticles (NPs) as the solid support in a microchip device.7 The particles are directed to an extraction zone in the microfluidic network and held there with the use of a magnet (Fig. 1), making the packing, removal and replenishment of the extraction bed straightforward. In addition to their magnetic properties, iron oxide NPs are stable, biocompatible, and easily modified. The authors synthesized silica-coated iron oxide NPs functionalized with octadecylsilane for extraction of parabens and nonsteroidal anti-inflammatory drugs (NSAIDs) in water and reported 90% analyte recovery.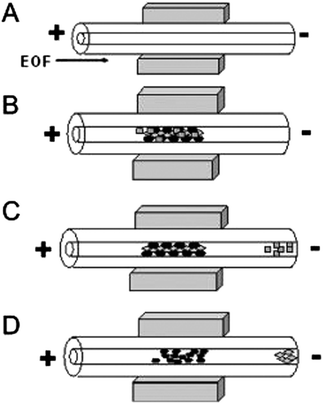 | ||
| Fig. 1 Schematic diagram of in-line magnetic extraction with CE. (A) Conditioning step: capillary is conditioned and NdFeB permanent magnets are placed around the capillary. (B) Sample loading: sample mixture containing magnetic particles is introduced into the capillary and retained by the magnets. (C) Washing step: analytes of interest interact with sorbents, whereas interfering components are eluted. (D) Elution step: retained analytes are eluted by applying a stronger eluent. Reprinted from Tennico and Remcho5 with permission from Wiley InterScience (Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
Monolithic phases have also been used for static preconcentration because they are generally easier to integrate than silica materials.6 Xu et al. developed an in situ polymerized butyl methacrylate monolithic column.8Polymerization of the monolithic material in poly(dimethyl siloxane) microchannels allows for controlled fabrication of an extraction bed within the microfluidic network. The authors were able to achieve a 10-fold improvement in the detection limit of their model analyte, promethazine-luminal-potassium ferricyanide. Similarly, Faure et al. developed a lauryl methacrylate monolith polymerized in cyclic olefin copolymer (COC) chips for reverse-phase electrochromatography with the goal of generating a more portable and disposable analytical device.9 Other recent reports on in situ monolith fabrication in a microfluidic network include Landers,10 Kang,11 and Woolley12 for biological and clinical analytes that have utility for environmental analysis.
In electrophoresis, a number of electric field-based dynamic methods are available for preconcentration. Field-amplified sample stacking (FASS) and field-amplified sample injection (FASI) techniques have been studied for several decades and are commonly used in conventional CE systems as a single-step preconcentration method for achieving high sensitivity.13 In FASS, a sample prepared in low conductivity solution is injected into a microchip channel filled with a higher conductivity background electrolyte. Because the electric field strength is higher in the sample solution, an increase in analyte mobility is observed. Analytes are then stacked at the interface between the sample solution and the buffer. Guan et al. presented a thorough characterization of sample stacking in a PDMS microchip, reporting a 16-fold decrease in the detection limit of a model analyte.14 In the work of Noh et al., FASS and FASI methods are uniquely combined with a background electrolyte modified with gold nanoparticles to enhance preconcentration and separation performance.15 The presence of nanoparticles in the background electrolyte provides additional sites for solute interaction and has the ability to alter the apparent mobility of analytes as well as electroosmotic flow. The optimized microchip device showed a 200-fold increase in sensitivity in the cyclic voltammetric determination of phenolic endocrine disruptors when using FASS, FASI, and a nanoparticle-modified buffer. A schematic of the microfluidic device and resulting electropherograms are shown in Fig. 2, demonstrating the enhanced performance when using stacking. Drinking and surface water samples were tested for the presence of five endocrine disrupting species with detection limits in the fM range.
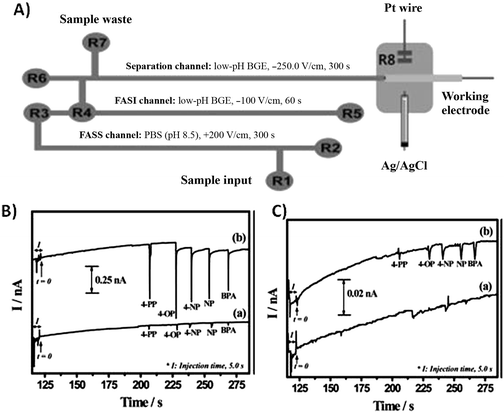 | ||
| Fig. 2 (A) Schematic of microfluidic device (B) Electropherograms for five endocrine disruptors obtained with (a) FASI and (b) both FASI and FASS. (C) Electrpherograms of (a) BGE without gold NPs and (b) BGE with gold NPs. Reprinted from Noh et al.15 with permission from Wiley InterScience (Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
In addition to chemical preconcentration, bacterial enrichment is necessary for selective enumeration of bacterial species and sensitive detection from environmental samples. Enrichment is a time-consuming process in which a species-selective growth medium is inoculated with a sample containing the target species allowing the bacteria to multiply over a period of several hours to days, in some cases. Additionally, the media often contains inhibitors to suppress the growth of competing microorganisms. After enrichment, the target bacterial species population is large enough to be analyzed, while background interferences have been minimized. The work of Dharmasiri et al. demonstrates an enrichment method that is integrated into the microchip device for improved sensitivity and decreased analysis time of E. coli.16 The U.S. Environmental Protection Agency enforces a very stringent maximum contaminant level for E. coli of 0 cfu/mL in drinking water since the minimum infectious dose can be as low as 10 cells.17,18 Recognizably, detecting E. coli at these very low levels requires enrichment of cells as a preconcentration method. Dharmasiri et al. used selective antibodies to immobilize E. coli on the surface of a PMMA microchannel where enrichment takes place. The bacteria were then directed into a PCR microtube for off-chip detection. The authors reported a recovery of 72%, PCR detection limits between 6–10 cfu/mL, and a total process time of 5 h.
Similarly, Beyor et al. developed an immunomagnetic bead-based cell concentration microdevice for pathogen isolation from a dilute sample.19 The device is constructed from PDMS and glass and incorporates on-chip pneumatic pumps for fluid flow. Superparamagnetic polystyrene beads were loaded into the chip and held in the microchannels using an external magnet (Fig. 3), while isolation and preconcentration of E. colicells is achieved through immunological interactions. Following this pre-concentration step, off-chip PCR and capillary electrophoresis (CE) were performed. The authors achieved capture efficiencies of 70% and a limit of detection of 2 cfu/μL.
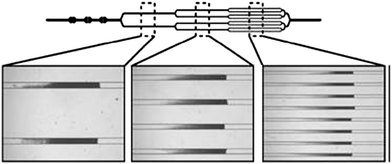 | ||
| Fig. 3 Images of beads loaded in the channels. A multi-step loading process using a sequentially bifurcated design enables equal distribution of bead beds in the multiple channels. Reprinted from Beyor et al. | ||
Detection methods
There are two general trends in detection methodologies for microfluidics: the development of a high-throughput interface coupling the microchip device with conventional instrumentation and the development of stand-alone integrated detection systems.5 Undoubtedly, the latter of the two approaches better lends itself to more compact and less expensive platforms at the expense of performance. Furthermore, while techniques such as mass spectrometry, FTIR, and chemiluminescence are proven to be powerful detection methods for environmental analysis, many are not suitable as portable diagnostic tools. This section will focus on detection methods commonly used for environmental analysis.Electrochemical detection
Electrochemistry is an attractive detection mode because the instrumentation is easily miniaturized and there is minimal loss of performance as size scales are reduced. Electrochemical detection techniques are capable of being universal (conductivity detection), semi-selective (amperometric detection by adjusting the potential) and highly selective by modification of the electrode surface, such as electrochemical immunoassays.20,21 Furthermore, many electrochemical techniques have been adapted to microfluidic devices and show high sensitivity, including amperometry and conductometry.3 As a result, incorporation of microelectrodes into microfluidic devices has become commonplace. Conductivity detection (CD), which can be used in either contact or contactless modes where contact refers to electrodes in galvanic contact with the working solution, is particularly promising for the analysis of small ions. Contactless detection is advantageous because it can be used with a wide range of background electrolytes and can take place at any location along a channel. However, electrical shielding is critical in contactless CD and, therefore, requires an insulating layer between the electrodes and solution. On the other hand, contact CD requires isolation of the electrodes from the high voltage and is subject to electrochemical reactions but utilizes mature conductivity detection instrumentation.22 Gertsch et al. reported a microchip capillary electrophoresis device with contact conductivity detection for the determination of perchlorate in water at the ppb level.23Perchlorate competitively inhibits the uptake of iodide into the thyroid, and long-term exposure has been linked to developmental and neurological disorders in infants and fetuses and also associated with thyroid cancer. Separation of perchlorate from competing anions in drinking water was achieved using a micellar pseudostationary phase containing a zwitterionic surfactant to selectively retain perchlorate. The method is capable of 5.6 ppb detection limits in drinking water with total analysis time of 60 s. A schematic of the microfluidic device as well as electropherograms of perchlorate separation in drinking water are shown in Fig. 4.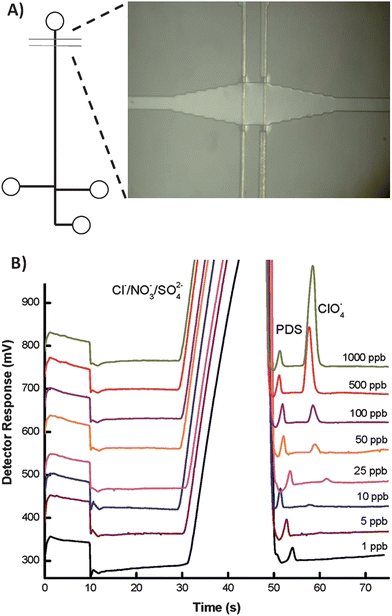 | ||
| Fig. 4 (A) Schematic of the microfluidid device, featuring a bubble cell (image) at the detection zone (B) Electropherograms showing separation of drinking water spiked with 0.12 ppm propane disulfonate (internal standard) and concentrations of perchlorate ranging from 1 to 1000 ppb. Reprinted from Gertsch et al.18 | ||
Recently, Liu et al. developed a microchip CE device with a contactless conductivity detector for separation and detection of inorganic ions and heavy metals.24 The detection circuit is based on a lock-in amplifier and was designed on printed circuit boards for easy miniaturization and integration within the microfluidic network. The device is capable of an average limit of detection of 0.4 μM for inorganic cations in water, a level below that of other contactless detectors. Rogers and Ding reported a microchip CE method for the determination of haloacetic acids using contactless CD.25 Haloacetic acids are a class of disinfectant byproducts generated during the water chlorination treatment process.26,27 These compounds are a major health concern due to their known toxicity and carcinogenicity.28 An off-chip solid-phase extraction step was implemented prior to microchip CE analysis, allowing determination of three HAAs in swimming pool water with limits of detection ranging from 38–500 ppb.
The work of Ha et al. describes a microchip electrophoresis device for monitoring endocrine disrupting species (EDCs) via amperometric detection.29Amperometry (and related pulsed amperometric detection modes) is attractive because it can provide enhanced selectivity relative to conductivity detection. Thin film electrodes fabricated from Prussian Blue-modified indium tin oxide were incorporated into a serpentine microchannel configuration. Separation of four endocrine disruptors was achieved in just over 2 min with detection limits as low as 59 nM. In 2010, Nie et al. reported a carbon disk electrode modified with mesoporous carbon material (CMK-3) for amperometric detection of nitroaromatic compounds (NACs) in water.30NACs are highly toxic, carcinogenic compounds used in explosives, fabric dyes, and agricultural products.31,32NAC contamination of water and soil is a serious concern throughout the US as these compounds are considered dangerous to human health at low (ppb) levels. Chemically modified electrodes can greatly improve sensitivity and selectivity of electrochemical detection.33 The CMK-3-modified working electrode exhibits chemical stability and good electrical properties as well as a highly ordered mesoporous structure with high surface area and large pore volume. Although the separation of four NACs was carried out via a conventional CE instrument rather than the microchip format, using this novel electrode material, limits of detection of 3.0–4.7 ppb were achieved in drinking water, wastewater, and river samples without complex sample pretreatment.
Optical detection
Optical detection techniques comprise some of the most inexpensive, simple, and universal detection modes. In microfluidic devices, however, the limited path length (generally 5 to 50 μm) associated with microchannels greatly hinders sensitivity and detection limits in absorbance measurements and decreases the overall intensity of fluorescent signals.34 Despite this drawback, many groups are working to improve optical detection on microchips. Absorbance has been used for a number of analytes in microfluidic platforms including phenolic chemicals; air pollutants; benzene, toluene, and xylene (BTX) transition metal ions; as well as nitroaromatic and nitramine explosives.35–38 Ohlsson et al. reported a microchip CE device with integrated waveguides for simultaneous absorbance and native UV-excited fluorescence detection.39 Combining the two methods, a wide range of analytes may be analyzed. Additionally, enhanced sensitivity is achieved for compounds that exhibit native fluorescence, for example polycyclic aromatic hydrocarbons40 and naphthalene sulfones.41 This detection scheme could be particularly useful for the identification of analytes and quantification of co-eluting species. Gaspar et al. used a microphotometer coupled to a spectrometer to enhance the cross-sectional area of the irradiation zone, effectively increasing the optical path length in PDMS microchips, diminishing background noise, and improving detection limits.42 Schulze and Belder summarized literature published in the last decade on fluorescence detection in CE and microchip CE.34 The review discusses some of the challenges of fluorescence detection such as derivatization, which is often necessary, but the article also points out the unparalleled sensitivity and the potential for miniaturized optical components.Fluorescence detection is a good choice for detection in microfluidics due to the inherent sensitivity and low detection limits achievable. These advantages overcome the normal short-comings associated with absorbance due to the short path length in a microfluidic device. However, fluorescence detection is not without its own drawbacks, including the lack of native fluorescence for a number of environmental analytes and the large size and complexity of optical instrumentation. The attachment of a fluorophore to environmental analytes requires additional sample preparation steps and increases the cost of the assay. Despite these disadvantages, fluorescence has been used successfully for environmental analysis with microfluidic devices. Walworth et al. demonstrated the analysis of multi-component mixtures representing three different classes of compounds in real sample matrices (Tennessee River and Second Creek).43 A high-performance extraction disk cartridge (HPEDC) was used to preconcentrate analyte mixtures containing several classes of compounds (toxin, pharmaceutical, and endocrine disrupting compounds) in aqueous samples. The HPEDC extracts were analyzed using CD-modified MEKC (CD-MEKC) with a confocal laser induce fluorescence (LIF) detection setup.
Shen et al. developed a microfluidic analytical system for characterization of dissolved organic carbon (DOC) in environmental waters.44 The design was based on a capillary gel electrophoresis (CGE) device with a LIF detector using microchips made from polymethylmethacrylate. The authors demonstrated reproducible peaks for standard organic solutions with analysis times less than 70 s. Additionally, DOC in environmental water from the Biwa Lake and the Hino River was analyzed, showing the content of DOC in the Biwa Lake changed seasonally.45
The major drawback of fluorescence detection is the need to derivatize most analytes with a fluorophore prior to analysis. An alternative to derivatization is the use of native fluorescence. Tolba et al. demonstrated native fluorescence utilizing deep-UV excitation in microchannel electrophoresis. Different analytes such as pollutants, neurotransmitters, and proteins have been successfully detected in more or less complex matrices.46 Organic pollutants can be separated and detected on microfluidic devices with LIF detection. An example is a μCEC device with LIF detection which was applied to the separation and detection of polyaromatic hydrocarbons (PAHs) of anthracene, pyrene, 1,2-benzofluorene, and benzo[a]pyrene. The PAHs were excited at 325 nm by a He-Cd laser, and the fluorescence emitted was detected at 350 nm.47 Benhabib et al. used microchip capillary electrophoresis to identify nine PAH components. The authors demonstrated that benzo[a]pyrene and perylene were distinguishable from a coeluting peak of anthanthrene by spectral analysis.48 One of the disadvantages of deep UV native fluorescence, however, is the high background signal arising from Rayleigh scattering. Another approach to avoid derivatization was presented by Wallenborg and Bailey in which they employed indirect LIF detection with MEKC to determine 14 explosives with a limit of detection of 1 μg mL−1 in natural water.49
Fluorescently labeled antibodies are commonly used in immunoassay platforms, providing sensitive detection of a captured analyte. Ramon-Azcon et al. used microparticles as solid supports to improve immunological detection of herbicides in a microchip device.50 Microparticles increase the surface area and allow faster assay kinetics. Microparticles were used in combination with dielectrophoresis (n-DEP) for the sorting and separating particles and cells of interest using simple electrode components without any moving actuators. The microparticles were functionalized with bovine serum albumin conjugated with atrazine (atrazine-BSA) and were incubated with anti-atrazine IgG antibody and atrazine. The immunocomplex was then injected into the microfluidic device comprised of two caged areas surrounded by n-DEP-generated electric barriers in order to trap the microparticles. The anti-atrazine IgG antibody immobilized on the atrazine-BSA microparticles was retained in the caged area created by n-DEP, whereas antibodies that reacted with free atrazines flowed downstream. A solution containing FITC-labeled anti-rabbit IgG antibody (FITC-anti-IgG) was used to detect the immunocomplex. Detection limits as low as 0.11 ppb were determined for the immunosensing microchip device. The authors proposed simultaneous measurements using a device with different caged areas for multiple analytes.
Som-Aum et al. developed chemiluminescence (CL) detection for the determination of arsenate in water samples based on luminol CL with a heteropoly acid complex.51 The method was based on the complex between luminol and vanadomolybdoarsenate heteropoly acid (VMoAs-HPA) in basic solution. CL was performed by the reaction between the adsorbed ion-pair complex and alkaline luminol. Integration of CL detection was relatively simple and has potential for on-site detection of low levels of contaminants. Advantages of this technique include improved sensitivity, high specificity due to the VMoAs-HPA complex, and the elimination of interference [Co(II), Cu(II) and Fe(II)]. However, an anion exchange resin was necessary to remove interfering ions such as chromate and phosphate. A detection limit of 8.9 mM was achieved for As(V) and a linear calibration range from 1.0 × 10−7 to 5.0 × 10−5 M, which spans the USEPA's maximum contaminant level (MCL) in drinking water.
Recently, surface-enhanced Raman scattering (SERS) techniques have been used for highly sensitive detection in a microfluidic chip.52 The observed enhancement factor of SERS provides a sensitivity that is comparable to fluorescence detection. Additionally, information regarding molecular structure can be obtained, and the detection and identification of non-fluorescent samples is possible using this technique. Raman spectroscopy has been integrated into microfluidics to quantitatively analyze p-aminobenzoic acid (PABA), benzene, toluene, ethylbenzene, o-,m-, p-xylene, pyridine, nicotinic acid, and several pesticides such as carbendazim and metazachlorine.53 Ashok et al. reported a directly embedded split-fiber-probe based on a Raman detection system in a microfluidic chip.54 The embedded split-fiber-probe offered flexibility to modify the collection geometry, minimizing the background signal and allowing an alignment free system. The development of this device affirms the feasibility of using Raman spectroscopy with portable, lab-on-a-chip devices for environmental analytes.
Mass spectrometry
Mass spectrometry (MS) has been widely applied to many different fields of chemical analysis, including environmental analysis. The complex sample pretreatment, large solvent consumption, and complexity of maintenance, however, has restricted the application of on-line environmental sample analysis. Wei et al. developed a microfluidic-based device which combined ESI-Q-TOF-MS with a single particle analysis for the determination of herbicides in water.55 The authors demonstrated a simplified pretreatment process in which analytes are retained on a silicone polymer-coated silica gel modified with C30 alkyl chains within a microfluidic channel. The microchip is coupled to an ESI-Q-TOF-MS for direct detection of herbicides upon desorption from the single particle. In principle, single particle analysis on microfluidic devices coupled with online MS detection could be applied to a variety of environmental samples.Microchip integration
Microfluidic devices offer the possibility of environmental analysis systems with far-reaching advantages such as autonomous on-site monitoring using an instrument that possesses a small footprint, relative to conventional systems.56 Unfortunately, many existing microfluidic systems fail to fully realize this goal because of the peripheral equipment required for signal acquisition and processing, such as the electronics and optics. The peripheral equipment size typically over-shadows the small footprint of the microfluidic chip and can prevent its use for in-field monitoring applications. Thus, continued miniaturization of not only the microfluidic chip but also the peripheral equipment is paramount to the delivery of true point-of-use microfluidic systems.Ramirez-Garcia et al. presented work towards a fully integrated microanalytical instrument with focus on the development of a pump and detector.57 Their pump design was based on conducting polypyrrole (ppy). Ppy was synthesized electrochemically by oxidizing the monomer in the presence of large anions which become trapped in the structure of the polymer. The polymer swells and shrinks with repeated reduction and oxidation due to the hydration and dehydration of the charged backbone, producing mechanical work such as pumping. The swelling of the polypyrrole was used to deform thin walled polyurethane tubes, generating liquid movement in microfluidic channels with very low power consumption. Further integration was achieved by using Light Emitting Diodes (LEDs), which provided low-power consumption, long lifetime and extremely low detection limits.
Alves-Segundo et al. demonstrated the fabrication of miniaturized, continuous flow, analytical microsystems based on photometric detection with optical elements such as light emitting diodes and photodiodes.58Fig. 5 shows the integrated continuous flow microfluidic system. The integration of a glass window eliminates the transparency problem of ceramic material and allows for the bubble-shaped flow cell to increase the area of the light beam. The microsystem was used for the colorimetric determination of Cr(VI) ion in waters based on the diphenylcarbazide reagent. The authors demonstrated a limit of detection of 50 ppb and a linear response range of 0.1 to 20 ppm. The sensitivity was enhanced by the use of the bubble-configuration flow cell coupled to an LED and photodiode. A fully integrated microsystem with the capability of automation, robustness or portability to field applications is still being developed; while the integration of an LED and photodiode for nitrite detection off-chip was shown.
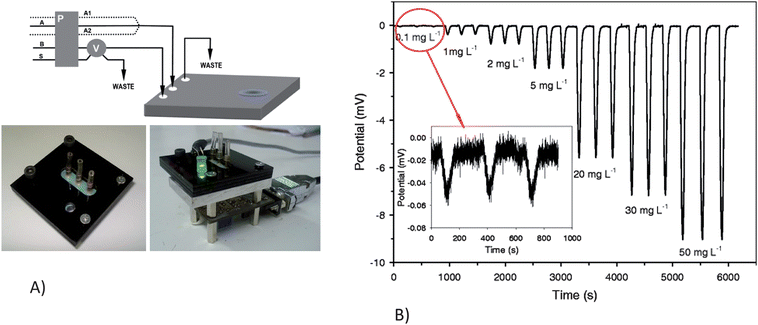 | ||
| Fig. 5 A)Continuous flow microsystem set-up. a) Experimental manifold: A, acidified DPC reagent solution; A1 H2SO4 solution; A2, DPC reagent solution; B, deionized water; S, sample; P, peristaltic pump; V, six-port injection valve. b) Protective PMMA black support for the external optical components. c) Optical detection set-up: (1) LED; (2) PMMA support; (3) Photodetector and associated electronics; (4) DB9 (RS232) computer connector. B) Response signal obtained, by injecting Cr(VI) from 0.1 to 50 mg L−1, using the optimal experimental conditions Reprinted from Alves-Segundo et al. | ||
Sieben et al. demonstrated the use of microfluidic devices which integrate fluid processing and optical detection, enabling the development of a low-cost, miniature, portable and sensitive nitrite sensor based on the Griess assay.59 The sample was mixed with the Griess reagents, forming a colored Azo dye which can be measured by absorbance to determine the nitrite concentration. The design incorporated four separate absorbance cells (2.5 cm path length) separated by three serpentine mixers to monitor the reaction kinetics and mixing efficiency. The on-chip integration of the LED, photodiode, and the absorbance cell are shown in Fig. 6. The detection system had integrated LEDs and photodiodes which yielded a limit of detection of 14 nM and a linear range between 50 nM and 10 mM. The tinted PMMA was used to adsorb non-directional and background light, substantially reducing the background signal. The authors aim to implement an integrated and miniaturized system to continuously measure nitrite concentrations in the environment, generating a profile of the temporal and spatial distribution of chemistry in the oceans.
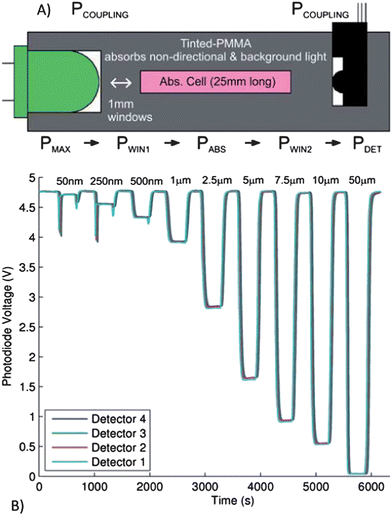 | ||
| Fig. 6 A) Cross-sectional view of the microfluidic absorption cell with optical power budget (see text for details). PMAX is the maximum power that will reach the detector from the LED (determined by the solid angle), PCOUPLING is the power remaining after the lumped losses from coupling the LED and photodetector to the microfluidic device, PWIN1 is the power remaining after the first window, PABS is the power remaining after the adsorption of the Azo dye formed in the presence of nitrite (desired measurement), PWIN2 is the power remaining after the detection window and PDET is the optical power that is detected. B) The photodiode output voltage for all four detectors for a sequence of nitrite samples of varying concentrations from 50 nM to 50 mM, flowing through the microfluidic device. Reprinted from Sieben et al. | ||
Barat et al. designed an integrated optical system for particle analysis on-chip using a combination of scattered light and fluorescence.60 Scattered light is collected at two different angles using optical fibers inserted into microfabricated grooves. The optical fibers are used for both excitation and emission detection, eliminating bulky off-chip optics and the need for a complex, integrated lens. Within the microfluidic device, the fibers act as waveguides to deliver excitation light to a defined location of the device in a well controlled volume, producing high photon irradiances. The illumination volume is controlled by the core diameter of the fiber optics as well as the acceptance angle of the fiber. In working toward a low-cost, disposable platform suitable for environmental applications, simple optical components that may be incorporated into a microfluidic device during the fabrication process are highly advantageous.
In 2009, Beyor et al. further advanced their lab-on-a-chip system for pathogen detection that integrates cell preconcentration, purification, PCR and CE analysis.61 The device is constructed from a four-layer design using glass-glass-PDMS-glass. Micropumps and valves are employed for fluid direction and control throughout the system. Superparamagentic beads are functionalized for immunological cell capture of E. coliO157 and K12 serotypes, and an external magnet is used for retaining beads in capture channels. After this preconcentration step, bead-cell duplexes are hydrodynamically transferred to an on-chip PCR reactor for amplification of the target microorganisms. Finally, the PCR products are injected onto a 5 cm CE column and detected using laser-induced fluorescence. A schematic of the integrated system is shown in Fig. 7. The authors achieved a detection limit of 0.2 cfu/μL for E. coliO157 and confirmed no false positive results even in the presence of a high background (104 cfu/μL) of E. coliK12 cells.
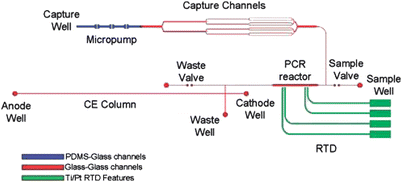 | ||
| Fig. 7 Cell capture-PCR-CE microdevice that integrates a capture structure with PCR and capillary electrophoresis. Immunomagnetic beads are immobilized by an external magnet in the capture channels as sample solution is driven through the bead bed. After capture and washing, the bead-cell duplexes are pumped into the 100 nL PCR reactor, where an external heater drives the reaction. Finally, the PCR amplicons are injected onto the CE column, separated, and detected near the anode using LIF. Channels in blue are enclosed using a PDMS-glass sandwich. Channels in red are enclosed by thermally bonding the etched glass wafer to the RTD wafer. Green features delineate the Ti/Pt resistance temperature detector features. Reprinted from Beyor et al. | ||
Emerging applications and technologies
Lab-on-a-chip technology offers many advantages to environmental analysis by reducing analysis time, improving detection limits, and allowing on-line, real time monitoring. Although the literature discussed thus far focuses primarily on water quality analysis, the field of microfluidics is certainly growing to include air quality and aerosol monitoring. Many of the challenges associated with air quality assessment are common to those of water quality, such as the difficulty in determining low levels of pollutants in a complex sample. Dossi et al. reported a microchip electrophoresis method for the determination of aldehydes in the atmosphere.62 The method involves forcing air samples through a silica gel cartridge coated with 2, 4-dinitrophenylhydrazine for derivatization of aldehydes followed by elution onto the microchip device. Noblitt et al. recently demonstrated an integrated microfluidic device for analysis of ambient aerosol composition.63 The instrumentation involves a water condensation particle collector coupled with an electrophoresis microchip using contact conductivity detection. As aerosol particles were collected onto the microchip, the concentration of inorganic anions was measured every 60 s using the electrophoresis system. In preliminary results, the system ran continuously for 28 h, demonstrating the ability to perform long-term field work in this new area of research.Low-cost, paper-based diagnostic and analytical devices are another emerging technology for environmental analysis. Paper-based analytical devices have several advantages including simplicity, portability, disposability, and cost-effective fabrication. Furthermore, electrochemical sensing provides a versatile and quantitative methodology as demonstrated by the use of microfluidic paper-based electrochemical devices (μPEDs) in the selective analysis of Pb(II) in an aqueous solution containing a mixture of Pb(II) and Zn(II).64 The schematic of the paper-based electrochemical detection device is shown in Fig. 8. The measurement of Pb(II) showed a limit of detection of 1.0 ppb. Apilux et al. presented a novel lab-on-paper device combining electrochemical and colorimetric detection for the rapid screening of Au(III) in the presence of a common interference, Fe(III), in industrial waste solutions.65 They used a colorimetric method to simultaneously detect Fe(III) as a screening tool for the determination of Au(III). According to the authors, Fe(III) is the only metal that affects the electrochemical determination of Au(III) when present above a 2.5-fold excess concentration of that of the Au(III). The calibration curve showed good linearity in the concentration range of 1–200 ppm of Au(III) and a limit of detection of 1 ppm.
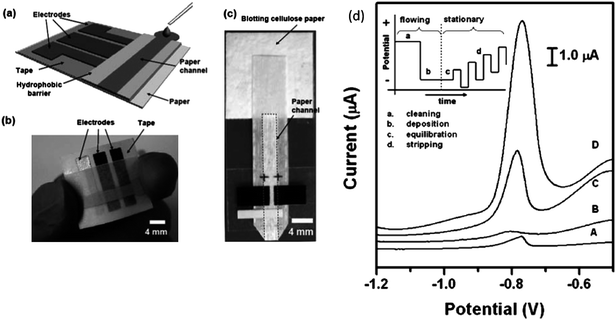 | ||
| Fig. 8 (a) Schematic of a paper-based electrochemical sensing device. The sensor comprises three electrodes printed on a piece of paper substrate (or plastic) and a paper channel. The paper channel was in conformal contact with the electrodes, and was held in place by double sided adhesive tape surrounding the electrodes. A photograph of a paper-based electrochemical sensing device for the analysis of glucose (b), and a hydrodynamic paper-based electrochemical sensing device for the measurement of heavy-metal ions (c). The device consists of two printed carbon d) Square-wave anodic stripping voltammograms for 25 ppb solution of Pb(II) in 0.1 M acetate buffer (pH 4.5) in the presence of 25 ppb Zn(II): (A) a 100 μL solution placed directly on the electrodes; (B) a 100 μL solution added to the stagnant μPEDs (without a pad of blotting paper as sink); (C and D) a solution of analytes continuously wicking the paper channel of the hydrodynamic μPEDs. The deposition time was 120 s (A, B, C) or 360 s (D). The SWASV was performed in the potential range of −1.2 to −0.5 V under optimized conditions: frequency, 20 Hz; amplitude, 25 mV; potential increment, 5 mV; equilibration time, 30 s. Deposition was performed at −1.2 V; ‘cleaning’ was performed at +0.5 V for 60 s. A bismuth(III) concentration of 500 μg L−1 was chosen for the co-deposition of heavy-metal ions. Inset shows the schematic of the four steps of the square-wave anodic stripping voltammetry. Reprinted from Nie et al. | ||
Conclusion
The field of microfluidics has made tremendous strides in recent years with the majority of the effort focused on biological analysis. A growing number of groups, however, have recognized the utility of microfluidic devices for environmental analysis particularly in light of the increasing need to understand the impacts of human activity on the world around us (summary of recent research work is detailed in Table 1). Microfluidic devices are in many respects ideally suited to this task because they can be made at low costs relative to traditional instrumentation and are small and thus potentially quite portable. We believe the future growth of environmental analysis with microfluidics will be focused on two key elements: 1) Production of non-technical instrumentation either through fully integrated systems or elegantly simple paper-based type devices. This would allow for greater application of devices by minimally trained technicians. 2) Portability remains as one of the major selling points of microfluidics over the more conventional techniques. The ability to perform analysis in the field allows for more complete understanding of the complex dynamics of environmental sites with rapid quantitative and qualitative results. As research in this field continues to grow, significant challenges will be faced including the ability to handle complex and highly variable sample matrices, the need to operate for days to months in the field, and the generation of systems that require little to no power. To address these challenges, great strides have been made in on-chip sample preparation and preconcentration techniques such as solid-phase extraction, stacking methods for electrophoretic separations, and integrated bacterial enrichment. Groups have advanced electrochemical, optical, and spectroscopic detection methods on-chip in order to analyze low abundance contaminants in complex environmental samples. Finally, the advent of new device materials, like paper-based analytical devices, allows for simpler fabrication and operation, greater cost efficiency, and enhanced portability.| Authors | Core Technological Component | Class of Pollutant Tested | Limit of Detection | Sample Matrix |
|---|---|---|---|---|
| a N.R. not reported. | ||||
| Tennico et al. | Functionalized magnetic iron oxide nanoparticles as the solid support | Parabens and NSAIDs | N.R. | Standards |
| Xu et al. | Monolithic phase by in situ polymerized butyl methacrylate | Promethazine | 1.6 ng mL−1 | Standards |
| Noh et al. | FASS and FASI | Phenolic endocrine disruptors | 7.1–11.1 fM | Drinking and surface water |
| Dharmasiri et al. | Bacterial cell capture | E. coli | 6–10 cfu/mL | Recreational water |
| Beyor et al. | Bacterial cell capture | E. coli | 0.2 cfu/μL | Standards |
| Gertsch et al. | Contact conductivity | Perchlorate | 5.6 ppb | Drinking water |
| Liu et al. | Contactless conductivity using a lock-in amplifier | Inorganic ions and heavy metals | 0.4 μM | Standards |
| Rogers and Ding | Capacitively coupled contactless conductivity (C4D) | Haloacetic acids | 38–500 ppb | Recreational water |
| Ha et al. | Amperometric detection with thin film electrodes fabricated from Prussian blue | EDC | 59 nM | Water in Styrofoam containers |
| Nie et al. | Carbon disk electrode modified with mesoporous carbon material | Nitroaromatic compounds | 3.0–4.7 ppb | Drinking, ground, cooking waste water |
| Ohlsson et al. | Integrated waveguides for simultaneous absorbance and native UV fluorescence | Polycyclic aromatic hydrocarbons and naphthalene sulfones | 2–4 μM native fluorescence 14–31 μM absorbance | Reference standard |
| Gaspar et al. | Microphotometer coupled to a spectrometer to enhance the cross sectional area of the irradiation zone | Inorganic anions | 0.78 μM | Standards |
| Walworth et al. | High performance extraction disk cartridge | Toxin, pharmaceutical and EDC | N.R. | Surface water |
| Shen et al. | CGE-LIF | Dissolved organic carbon | N.R. | Surface water |
| Tolba et al. | Native fluorescence utilizing deep-UV excitation | pharmaceutical | 250–900 ng mL−1 | Reference standard |
| Banhabib et al. | Used spectral analysis to distinguish coeluting peaks | PAH's | 1 ppb- 400 ppm | Reference standard |
| Wallenborg et al. | Indirect-LIF with MEKC separation | 14 explosive compounds | 1 μg mL−1 | Soil extracts |
| Ramon-Azcon et al. | Microparticle as solid supports in combination with dielectrophoresis | Herbicides | 0.11 ppb | Wine |
| Som-Aum et al. | Chemilumninescence | arsenated | 89 nM | Drinking and mineral water |
| Wei et al. | Combined ESI-Q-TOF mass spectrum with a single particle analysis using a silicone polymer-coated silica gel modified with C30 | Herbicides | 0.11 ppm | Tuber vegetables |
| Ramirez-Garcia et al. | Integrated pump and detector based on polypyrrole | Nitrites | 0.05 ppm | Standards |
| Alves-Segundo et al. | Integration of light emitting diodes and photodiodes for a colorimetric analysis | Cr(VI) | 50 ppb | Surface water |
| Sieben et al. | Integrated fluid processing and optical detection elements with tinted PMMA | Nitrite | 14 nM | Standards |
| Dossi et al. | Forcing air sample through a silica gel cartridge to determine aldehydes in the atmosphere | Aldehydes | 7.2 –9.2 μM | Urban air samples |
| Noblitt et al. | Water condensation particle collector coupled to MCE | Inorganic anions | 19 nM | Aerosol extracts |
| Nie et al. | Electrochemical sensor on a paper-based microfluidic device | Pb(II) | 1.0 ppb1.0 ppb | Standards |
| Apilux et al. | Paper-based electrochemical device for electrochemical and colorimetric detection | Au(III), Fe(III) | 1 ppm AU(III) | Gold-refining waste water |
References
- Z. Y. Xie and R. Ebinghaus, Anal. Chim. Acta, 2008, 610, 156–178 CrossRef CAS.
- J. M. Wu, L. F. Zhang and Z. G. Yang, Crit. Rev. Anal. Chem., 2010, 40, 234–245 CrossRef CAS.
- G. Chen, Y. Lin and J. Wang, Talanta, 2006, 68, 497–503 CrossRef CAS.
- S. D. Richardson, Anal. Chem., 2009, 81, 4645–4677 CrossRef CAS.
- H. F. Li and J. M. Lin, Anal. Bioanal. Chem., 2009, 393, 555–567 CrossRef CAS.
- C. C. Lin, J. L. Hsu and G. B. Lee, Microfluid. Nanofluid., 2011, 10, 481–511 CrossRef.
- Y. H. Tennico and V. T. Remcho, Electrophoresis, 2010, 31, 2548–2557 CrossRef CAS.
- Y. Xu, W. Zhang, P. Zeng and Q. Cao, Sensors, 2009, 9, 3437–3446 CrossRef CAS.
- Y. Ladner, G. Cretier and K. Faure, J. Chromatogr., A, 2010, 1217, 8001–8008 CrossRef CAS.
- C. Cakal, J. P. Ferrance, J. P. Landers and P. Caglar, Anal. Bioanal. Chem., 2010, 398, 1909–1917 CrossRef CAS.
- Q. S. Kang, Y. Li, J. Q. Xu, L. J. Su, Y. T. Li and W. H. Huang, Electrophoresis, 2010, 31, 3028–3034 CrossRef CAS.
- W. C. Yang, M. Yu, X. H. Sun and A. T. Woolley, Lab Chip, 2010, 10, 2527–2533 RSC.
- M. C. Breadmore, M. Dawod and J. P. Quirino, Electrophoresis, 2011, 32, 127–148 CrossRef CAS.
- Q. Guan and C. S. Henry, Electrophoresis, 2009, 30, 3339–3346 CrossRef CAS.
- H. B. Noh, K. S. Lee, B. S. Lim, S. J. Kim and Y. B. Shim, Electrophoresis, 31, pp. 3053–3060 Search PubMed.
- U. Dharmasiri, M. A. Witek, A. A. Adams, J. K. Osiri, M. L. Hupert, T. S. Bianchi, D. L. Roelke and S. A. Soper, Anal. Chem., 2010, 82, 2844–2849 CrossRef CAS.
- W. E. Keene, J. M. McAnulty, F. C. Hoesly, L. P. Williams, Jr., K. Hedberg, G. L. Oxman, T. J. Barrett, M. A. Pfaller and D. W. Fleming, N. Engl. J. Med., 1994, 331, 579–584 CrossRef CAS.
- S. O. Van Poucke and H. J. Nelis, J. Appl. Microbiol., 2000, 89, 390–396 CrossRef CAS.
- N. Beyor, T. Seo, P. Liu and R. Mathies, Biomed. Microdevices, 2008, 10, 909–917 CrossRef CAS.
- H. Dong, C.-M. Li, Y.-F. Zhang, X.-D. Cao and Y. Gan, Lab Chip, 2007, 7, 1752–1758 RSC.
- J.-J. Xu, A.-J. Wang and H.-Y. Chen, TrAC, Trends Anal. Chem., 2007, 26, 125–132 CrossRef CAS.
- S. D. Noblitt and C. S. Henry, Anal. Chem., 2008, 80, 7624–7630 CrossRef CAS.
- J. C. Gertsch, S. D. Noblitt, D. M. Cropek and C. S. Henry, Analytical Chemistry, 82, pp. 3426–3429 Search PubMed.
- B. Liu, Y. Zhang, D. Mayer, H.-J. Krause, Q. Jin, J. Zhao and A. Offenhäusser, Electrophoresis, 2011, 32, 699–704 CrossRef CAS.
- Y. S. Ding and K. Rogers, Electrophoresis, 31, pp. 2602–2607 Search PubMed.
- Y. Qi, C. Shang and I. M. C. Lo, Water Res., 2004, 38, 2375–2383 CrossRef CAS.
- V. Kanokkantapong, T. F. Marhaba, B. Panyapinyophol and P. Pavasant, J. Hazard. Mater., 2006, 136, 188–196 CrossRef CAS.
- M. L. Hanson and K. R. Solomon, Environ. Pollut., 2004, 130, 371–383 CrossRef CAS.
- K. Ha, G. S. Joo, S. K. Jha and Y. S. Kim, Microelectron. Eng., 2009, 86, 1407–1410 CrossRef CAS.
- D. X. Nie, P. Li, D. Zhang, T. S. Zhou, Y. Liang and G. Y. Shi, Electrophoresis, 31, pp. 2981–2988 Search PubMed.
- Y. Bhattacharjee, Science, 2008, 320, 1416–1417 CrossRef CAS.
- J. D. Rodgers and N. J. Bunce, Water Res., 2001, 35, 2101–2111 CrossRef CAS.
- S. Hrapovic, E. Majid, Y. Liu, K. Male and J. H. T. Luong, Anal. Chem., 2006, 78, 5504–5512 CrossRef CAS.
- P. Schulze and D. Belder, Anal. Bioanal. Chem., 2009, 393, 515–525 CrossRef CAS.
- S. Wakida, K. Fujimoto, H. Nagai, T. Miyado, Y. Shibutani and S. Takeda, J. Chromatogr., A, 2006, 1109, 179–182 CrossRef CAS.
- B. C. Giordano, A. Terray and G. E. Collins, Electrophoresis, 2006, 27, 4295–4302 CrossRef CAS.
- Y. Ueno, T. Horiuchi, O. Niwa, H. S. Zhou, T. Yamada and I. Honma, Sens. Actuators, B, 2003, 95, 282–286 CrossRef.
- Q. Lu and G. E. Collins, Analyst, 2001, 126, 429–432 RSC.
- P. D. Ohlsson, O. Ordeig, K. B. Mogensen and J. P. Kutter, Electrophoresis, 2009, 30, 4172–4178 CrossRef CAS.
- J. Kuijt, C. García-Ruiz, G. J. Stroomberg, M. L. Marina, F. Ariese, U. A. T. Brinkman and C. Gooijer, J. Chromatogr., A, 2001, 907, 291–299 CrossRef CAS.
- S. J. Kok, G. P. Hoornweg, T. de Ridder, U. A. T. Brinkman, N. H. Velthorst and C. Gooijer, J. Chromatogr., A, 1998, 806, 355–360 CrossRef CAS.
- A. Gáspár, I. Bácsi, E. Garcia, M. Braun and F. Gomez, Anal. Bioanal. Chem., 2009, 395, 473–478 CrossRef.
- M. J. Walworth, R. M. Connatser and M. J. Sepaniak, J. Sep. Sci., 2009, 32, 2985–2992 CrossRef CAS.
- N. Hudson, A. Baker and D. Reynolds, River Res. Appl., 2007, 23, 631–649 CrossRef.
- S. L. Shen, Y. Li and S. Wakida, Environmental Monitoring and Assessment, 166, pp. 573–580 Search PubMed.
- K. Tolba and D. Belder, Electrophoresis, 2007, 28, 2934–2941 CrossRef CAS.
- B. S. Broyles, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 2003, 75, 2761–2767 CrossRef CAS.
- M. Benhabib, T. N. Chiesl, A. M. Stockton, J. R. Scherer and R. A. Mathies, Anal. Chem., 2010, 82, 2372–2379 CrossRef CAS.
- S. R. Wallenborg and C. G. Bailey, Anal. Chem., 2000, 72, 1872–1878 CrossRef CAS.
- J. Ramon-Azcon, R. Kunikata, F. J. Sanchez, M. P. Marco, H. Shiku, T. Yasukawa and T. Matsue, Biosens. Bioelectron., 2009, 24, 1592–1597 CrossRef CAS.
- W. Som-Aum, H. Li, J. J. Liu and J. M. Lin, Analyst, 2008, 133, 1169–1175 RSC.
- Y. S. Ding, C. D. Garcia and K. R. Rogers, Anal. Lett., 2008, 41, 335–350 CrossRef CAS.
- L. X. Chen and J. B. Choo, Electrophoresis, 2008, 29, 1815–1828 CrossRef CAS.
- P. C. Ashok, G. P. Singh, K. M. Tan and K. Dholakia, Optics Express, 18, pp. 7642–7649 Search PubMed.
- H. B. Wei, H. F. Li and J. M. Lin, J. Chromatogr., A, 2009, 1216, 9134–9142 CrossRef CAS.
- P. S. Dittrich and A. Manz, Anal. Bioanal. Chem., 2005, 382, 1771–1782 CrossRef CAS.
- S. Ramirez-Garcia, M. Baeza, M. O'Toole, Y. Z. Wu, J. Lalor, G. G. Wallace and D. Diamond, Talanta, 2008, 77, 463–467 CrossRef CAS.
- R. Alves-Segundo, N. Ibanez-Garcia, M. Baeza, M. Puyol and J. Alonso-Chamarro, Microchimica Acta, 172, pp. 225–232 Search PubMed.
- V. J. Sieben, C. F. A. Floquet, I. R. G. Ogilvie, M. C. Mowlem and H. Morgan, Analytical Methods, 2, pp. 484–491 Search PubMed.
- D. Barat, G. Benazzi, M. C. Mowlem, J. M. Ruano and H. Morgan, Optics Communications, 283, pp. 1987–1992 Search PubMed.
- N. Beyor, L. N. Yi, T. S. Seo and R. A. Mathies, Anal. Chem., 2009, 81, 3523–3528 CrossRef CAS.
- N. Dossi, S. Susmel, R. Toniolo, A. Pizzariello and G. Bontempelli, Electrophoresis, 2009, 30, 3465–3471 CrossRef CAS.
- S. D. Noblitt, F. M. Schwandner, S. V. Hering, J. L. Collett and C. S. Henry, J. Chromatogr., A, 2009, 1216, 1503–1510 CrossRef CAS.
- Z. H. Nie, C. A. Nijhuis, J. L. Gong, X. Chen, A. Kumachev, A. W. Martinez, M. Narovlyansky and G. M. Whitesides, Lab on a Chip, 10, pp. 477–483 Search PubMed.
- A. Apilux, W. Dungchai, W. Siangproh, N. Praphairaksit, C. S. Henry and O. Chailapakul, Analytical Chemistry, 82, pp. 1727–1732 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |