The size-dependent anode-catalytic activity of nickel-supported palladium nanoparticles for ethanol alkaline fuel cells
Partha Sarathi
Roy
,
Joyeeta
Bagchi
and
Swapan Kumar
Bhattacharya
*
Physical Chemistry Section, Department of Chemistry, Jadavpur University, Kolkata, 700 032, India. E-mail: skbhatt7@yahoo.co.in; Fax: +91 3324146584; Tel: +91 9831699643
First published on 9th July 2012
Abstract
Spherical nanoparticles of palladium with varying particle diameters have been prepared from PdCl2 by wet chemical single pot synthesis using citric acid as reducing agent in the presence of PVA. The size of the nanoparticles has been tuned by changing the duration of reflux. The resulting nanoparticles have been dip-coated on Ni-foil, and evaluated as anode catalysts for oxidation of ethanol under alkaline conditions. The morphology and surface characteristics of the Pd nano-catalyst have been investigated by TEM and FE-SEM in conjugation with EDS. Measurements of catalytic activity by electrochemical methods (cyclic voltammetry, chronopotentiometry and electrochemical impedance spectroscopy) reveal that the majority of the nanoparticle-embedded anodes, despite less Pd0 loading, act as superior electrocatalysts as compared to the nickel-supported Pd electrode constructed electrochemically. In this study, the effects of the different extent of catalyst-loading on the experimental parameters have been rationalized to obtain the size-dependence of the electrocatalytic activity. The experimental results clearly show that there is noticeable development in the intrinsic catalytic activity and poisoning resistance of the anode catalysts. In addition, the intrinsic electrocatalytic activity of the nano-palladium is found to be size-dependent, which increases with decrease in particle size particularly below the diameter of 19 nm.
Introduction
Alcohols of low molar mass are widely proposed as possible fuel for mobile applications such as electric vehicles.1,2 Thus, development of novel catalysts with high electrocatalytic activity for methanol or ethanol oxidation has been receiving much attention.3–5 By comparing the performance of fuel cells operating on some small alcohols, it resulted that ethanol may replace methanol in a direct alcohol fuel cell (DAFC).6,7 Direct ethanol fuel cells (DEFCs) have attracted more and more attention because ethanol is less toxic compared to methanol and can be produced in large quantities by the fermentation of sugar containing raw materials.8–10 So, it is of significant importance to develop new anode electrocatalysts for ethanol electro-oxidation in order to enhance the performance of DEFCs.Electrocatalytic activity of anodic materials is one of the main factors influencing the practical application of DAFCs.11–19 Pt-based catalysts are recognized as the best catalysts for low temperature fuel cells.20,21 However, the limitations of the use of Pt-based electrocatalysts come from their high cost and limited resource of Pt.22 Moreover, Pt gets easily poisoned due to surface coverage by poisonous intermediates like Pt–CO during oxidation of alcohols, as Pt is not enough oxophilic to form a ‘rescuer’ intermediate, Pt–OH, at lower potential. On the other hand, Pd seems to be better than Pt, since it is at least fifty times more abundant than Pt and thus able to make the anodes of fuel cells cost-effective.18 Pd has the ability to reduce proton, store and release hydrogen23 which can be further utilized to remove absorbed Pt–CO formed during electro-oxidation of alcohols. Reportedly, Pd has been used as an excellent catalyst for various chemical and electrochemical reactions.24–27 Zhou et al. reported recently that Pd nanoparticles of diameter 5–7 nm are the most favorable for formic acid electro-oxidation due to the structure induced particle size effect.27 Xu et al. reported that Pd is a good electrocatalyst for ethanol oxidation and showed higher activity than that of Pt in alkaline media.28 So, Pd seems to be a unique substitute to Pt.18,19 Its importance as an electrocatalyst is also found in a recently published comprehensive review for alcohol oxidation in half cells and direct alcohol fuel cells.29 A variety of palladium nanoparticles have been fabricated in order to examine their electrocatalytic activity for alkaline oxidation of alcohols,11 such as nanocrystalline oxide-Pd/C promoted electrocatalysts,30 Pd nanowire arrays,31 Pd nanoparticles-supported on multi-walled carbon nanotubes (MWCNT),32 Pd nanoparticles supported on TiO2 nanotubes.33 Many binary or ternary composite catalysts involving Pd have been developed also in order to enhance the electrocatalytic activity of Pd for alcohol oxidation,29 including Pd–NiO/C,34 Pt–Pd/Ru nanoparticles,35 heterobimetallic Ru/Pd,36 Pd–In2O3/CNTs,37 oxide (CeO2, NiO, Co3O4, Mn3O4)-promoted Pd/C,38 Ni–Pd supported by silicon microchannel plates,39 Pd–Pt/Ti,40 and novel porous palladium electrodes11etc. However, the study of size-dependent electrocatalytic activity of palladium nanoparticles for alkaline oxidation of ethanol is still limited in literature. Notably, besides platinum metals, cheaper nickel nanoparticles are also used as a component of the modified electrodes for alcohol sensing41,42 and fuel cell development43–46 particularly in alkaline medium although electrocatalytic activity of bare Ni foil is insignificant at low potential.19
In view of this backdrop, in this study we have embedded Pd-metal nanoparticles of various average diameters on Ni-foils and tested their electrocatalytic activity in anodic oxidation of ethanol in alkaline medium by cyclic voltammetry, chronopotentiometry, and electrochemical impedance spectroscopy. Interestingly, the findings indicate marked differences in the nanoparticle-size-dependent electrocatalytic activity in ethanol oxidation. The morphology and the diameters of the nanoparticles of Pd adsorbed on the Ni surface were obtained by high resolution field emission scanning electron microscopy (FE-SEM). The atom% of adsorbed Pd0 as obtained from energy dispersive X-ray spectroscopy (EDX) was conducive to understanding the surface characteristics of the constructed electrodes including the roughness factor, θsurface, defined as true area per unit apparent area. The stability of the electrodes against poisoning is measured using the transition time of the chronopotentiometric response. It is important that in our case the embedded Pd nanoparticles are prepared by a single pot synthetic technique46,47 and unlike the nanoparticles of other synthetic methods; these nanoparticles do not require surface treatments like leaching, ignition etc. to bring them in the same surface environment. Further, in spite of bigger size, the synthesized nanoparticles retain their inherent excellent catalytic activity whereas some other nanometer- and even subnanometer-sized particles are known to be catalytically inactive due to surface environmental effects46 and strong particle–stabilizer interaction.46
Experimental
Materials
The metal precursor palladium chloride (99% pure, Arora Matthey Ltd.), citric acid (Merck, India), polyvinyl alcohol (PVA) (number average relative molar mass = 125![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Lab Rasayan Co.), nickel foil (99.9 + % gold level, Aldrich Chem. Company Inc.) of thickness 0.0125 cm and absolute ethanol (M Tedia, Canada) were used as received. All the other reagents like NaOH, HCl were of AR/GR grade and from Merck, India. Triple distilled water was used for all experimental purposes.
000, Lab Rasayan Co.), nickel foil (99.9 + % gold level, Aldrich Chem. Company Inc.) of thickness 0.0125 cm and absolute ethanol (M Tedia, Canada) were used as received. All the other reagents like NaOH, HCl were of AR/GR grade and from Merck, India. Triple distilled water was used for all experimental purposes.
Preparation of palladium nanocatalysts
PVA-protected palladium nanoparticles for electrocatalysis of ethanol oxidation were prepared by our method46,47 developed previously. About 1.0 g of PVA was dissolved in about 80 ml of hot water taken in a three-neck round bottom flask of 250 ml capacity. It was then cooled to room temperature (around 25 °C) and 4 ml of aqueous solution of 2 wt% PdCl2 in 2 N HCl (distilled from constant boiling solution at 381.8 K, 101 kPa) was added followed by dissolution of 0.864 g of solid citric acid. The volume of the solution was made up to 100 ml by adding required quantity of water, as determined by a similar control experiment in a 100 ml volumetric flask. The mixture was then refluxed continuously for about 3 hours and a little amount of resulting Pd suspension was withdrawn at successive intervals of 1, 1.5, 2 and 2.33 hours to get colloidal palladium nanoparticles of different average diameters trapped into the PVA matrix (Pd/PVA). These aliquots contained impurities like unreacted citric acid, minute quantity of PdCl2, byproduct acetone dicarboxylic acid, etc. In each case the dispersed phase consisting of Pd/PVA was separated from the aqueous medium with the expulsion of water soluble molecules and ions by treating the sols with excess ethanol. The purified Pd/PVA nanocomposite collected by centrifugation was re-dispersed in water with further formation of Pd hydrosol which was then used for the construction of the anode. Transmission electron microscopic study was made by using a Hitachi H-600 model microscope operating at 100 kV.Preparation of electrodes
The Ni-foil was used as a substrate for the electrocatalyst.13,18,19 The middle portion of the foil was enwrapped with Teflon tape (Champion) keeping both ends bare. Both chemical and electrochemical deposition of palladium was implemented on one end of the foil and the other end was kept for electrical connection. Out of the five electrodes studied, four were prepared by a “dip and dry” technique of chemical solution deposition as we have adopted in our previous study.46,47 For this purpose one end of the Ni-foil was dipped into the chosen sol, kept for a definite few seconds, and then withdrawn vertically from the sol. This was done consecutively ten times. Then it was dried using a vacuum desiccator. The whole process of dipping and drying was carried out twice to construct each dip-coated electrode. A thin film of a coloured composite was found to adhere onto the Ni-surface. These dip-coated electrodes designated as Ni/Pd(i) (i = 1–4) were prepared from 1, 1.5, 2 and 2.33 h-refluxed sol, respectively, in the whole electrochemical and surface studies. Another electrode, Ni/Pd(e-chem), was also constructed by the galvanostatic deposition of Pd on planar Ni-foil at a current density of 5 mA cm−2 for 10 minutes from a solution containing 0.04 wt% of PdCl2 in 2 M HCl solution.Surface study
The surface morphology and composition of the prepared anodes were investigated with a JEOL JSM-6700F Field Emission Scanning Electron Microscope (FE-SEM) coupled with EDX. The Pd content of each Ni/Pd(i) electrode was determined by measuring the mass difference of each before and after deposition with a Mettler electronic balance AE 240 and using EDX data.Electrochemical measurements
The electrochemical measurements were conducted at 30 ± 2 °C in a two compartment glass-cell fitted with a conventional three-electrode assembly. In all electrochemical measurements, the reference electrode used was Hg/HgO/OH− (1 M) (MMO) having an equilibrium electrode potential of ∼0.1 V with respect to the standard hydrogen electrode (SHE). A large Pt-foil (1 cm × 1 cm) was used as counter electrode and potential data were recorded with respect to MMO.Cyclic voltammetric study was performed using a computer aided Potentiostat/Galvanostat (AEW-2, Munistst, Sycopel Scientific Ltd., UK). Cyclic voltammogram (CV) of each electrode was recorded at the scan rate 50 mV s−1 for several consecutive cycles until a steady profile was obtained. Chronopotentiometry was also performed by applying a current density of 5 mA cm−2 with the help of a constant current charger (DB-300, DB – Electronics) and the potential was recorded with an EC digital multimeter (DM 610 4B) as described before.13 Electrochemical impedance measurements were carried out using an amplitude of 5 mV and the frequency range 30 kHz to 30 mHz at a few steady potentials by a computer controlled potentiostat/galvanostat with PG STAT 12 - FRA modules (Eco Chemie BV, The Netherlands).
Results and discussions
SEM, TEM and EDX study
In order to understand the morphology of the adsorbed species on the surface of Ni/Pd(i) (i = 1–4, e-chem) electrodes, scanning electron microscopic study has been carried out. Fig. 1a shows the low resolution SEM image which reveals the overall morphology of the Ni/Pd(1) electrode, a representative of all the Ni/Pd(i) (i = 1–4) electrodes. In the SEM image, the black lumps or patches of various shapes are associated with relatively smooth white deposits. The white deposit is also present within the black lumps and relatively large black dots or patches. These black and white portions are mainly PVA and Pd, respectively, as confirmed by the EDX study. A representative EDX spectrum, in Fig. 1b corresponding to the SEM image (Fig. 1a), reveals the presence of the elements (palladium, carbon, oxygen) adsorbed on the Ni surface. The SEM-EDX study ensures successful embedding of the PVA–palladium nanoparticle-composite on the planar nickel-surface. The surface composition of the electrodes has been analyzed by EDX study and is summarized in Table 1. Notably the atomic ratio, C:O, for all the electrodes (vide Table 1) is found to be almost 2:1 indicating the co-existence of PVA with the metal nanoparticles adsorbed on the Ni-surface. The SEM image (Fig. 1c) of the electrochemically deposited electrode, Ni/Pd(e-chem), shows black and white slabs of deposits. Some of the white slabs overlap with the other white slabs confirming layer deposition of palladium. Some slabs are scrolled, indicating loose binding18 as Pd–Pd bond strength is low (100 kJ mol−1).48
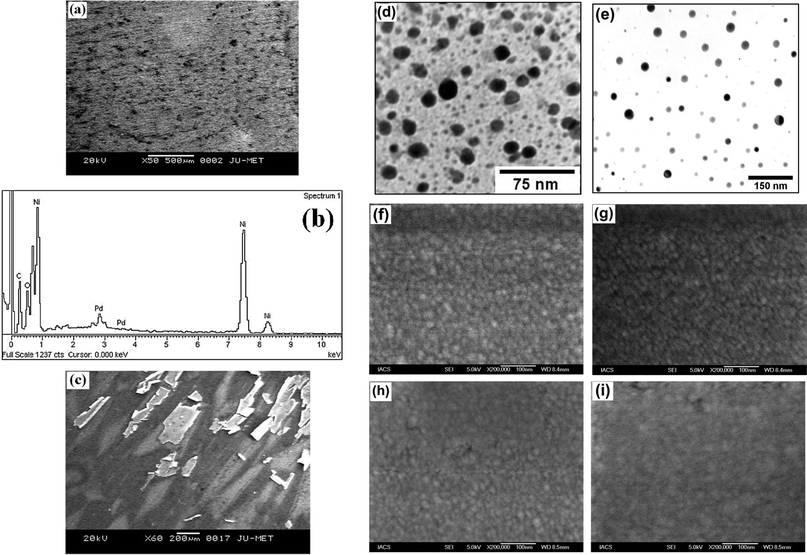 | ||
| Fig. 1 (a) Low magnification SEM image and (b) EDX spectrum of Ni/Pd(1), (c) SEM image of Ni/Pd(e-chem), (d, e) representative TEM images of Pd nanoparticles from 1 h and 2 h-refluxed sol, (f–i) high magnification SEM images of Ni/Pd(i) (i = 1–4) respectively. | ||
| Electrode | Elemental composition (atom%) | d/nm | Mass of deposit per sq cm × 106 (g cm−2)a | Moles of deposited Pd0 per cm2 × 108 (m/A) |
θ
surface
|
|||
|---|---|---|---|---|---|---|---|---|
| C | O | Ni | Pd | |||||
| a For Ni/Pd(i) (i = 1–4) electrodes deposit denotes the Pd–PVA composite whereas it is pure Pd for the Ni/Pd(e-chem) electrode. | ||||||||
| Ni/Pd(1) | 34.88 | 18.46 | 45.83 | 0.83 | 12.0 ± 2.2 | 96.39 | 2.291 | 1.025 |
| Ni/Pd(2) | 50.97 | 26.25 | 22.16 | 0.62 | 14.0 ± 3.7 | 173.49 | 4.483 | 1.718 |
| Ni/Pd(3) | 47.90 | 23.68 | 27.71 | 0.71 | 18.9 ± 3.6 | 200.63 | 5.335 | 1.515 |
| Ni/Pd(4) | 50.84 | 24.65 | 24.04 | 0.47 | 28.8 ± 2.5 | 328.36 | 6.202 | 1.155 |
| Ni/Pd(e-chem) | — | 7.28 | 86.02 | 6.70 | — | 1654.2 | 1554.4 | — |

Fig. 1d and e are the two representative TEM images of Pd nanoparticles present in 1 h and 2 h-refluxed sol. It is evident from the figures that the particles are well separated and globular in shape. The average diameters of the particles are computed to be 11.0 ± 4.6 nm and 21.0 ± 6.2 nm, respectively, which are also the same as that obtained from the other TEM images collected from the different parts of the grid. Fig. 1f–i represent high magnification FE-SEM images of the electrodes, Ni/Pd(i) (i = 1–4), which consist of distinct white spots on their surface, representing Pd nanoparticles as also confirmed from EDX study. Unlike TEM, the particles in FE-SEM images are not well separated probably because of a greater extent of catalyst loading on the electrode surface. The average particle sizes of the nanoparticles obtained from the magnification of FE-SEM images (Fig. 1f–i) are found to be 12.0 ± 2.2, 14.0 ± 3.7, 18.9 ± 3.6 and 28.8 ± 2.5 nm, respectively, for the electrodes Ni/Pd(1–4). It is worth mentioning that these values are in good agreement with those obtained from TEM study.
In order to determine the roughness factor of the constructed electrodes eqn (1) is used.
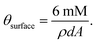 | (1) |
Cyclic voltammetry
Cyclic voltammograms (CVs) of bare Ni and Ni/Pd(i) (i = 1–4, e-chem) electrodes in alkali solution (1 M NaOH) in the presence and absence of ethanol (1 M) are depicted in Fig. 2a and b respectively. Notably, in blank alkali solution the electrodes Ni/Pd(i) (i = 1 and 4) serve as the representative of all the electrodes, Ni/Pd(i) (i = 1–4). The profile for bare Ni shows a pair of quasi-reversible peaks at potentials ca. 0.480 and 0.397 V at zones 1 and 2, respectively, for the conversion of Ni(OH)2 to NiOOH and vice versa, as found in the literature.18,19,49,50 All the Ni/Pd(i) (i = 1–4, e-chem) electrodes also exhibit a similar characteristic CV pattern in zones 1 and 2 as that of bare Ni but they differ in zone 3, where an additional cathodic peak is obtained for all the above-mentioned electrodes during reverse scanning. Moreover, a careful analysis reveals that in the potential region between −0.25 and 0.40 V, there is notable anodic charge for the electrodes over and above that observed for bare Ni, illustrating that on these electrodes an extra anodic process like Pd oxidation occurs along with the existing anodic process on the Ni surface (eqn (2)):| Ni + 2OH− = Ni(OH)2 + 2e− | (2) |
The increment in the anodic peak current density in zone 1 for all the Ni/Pd(i) electrodes in comparison to that for planar Ni foil indicates the synchronous oxidation of Pd(0) to Pd(OH)2 according to eqn (3):
| Pd + 2OH− = Pd(OH)2 + 2e− | (3) |
| Pd(OH)2 + Ni = Ni(OH)2 + Pd | (4) |
Here the additional formation of Ni(OH)2 by the transmetallation reaction helps the generation of more NiOOH in zone 1, according to the well known reaction (eqn (5)):
| Ni(OH)2 + OH− = NiOOH + H2O + e− | (5) |
So the peak current density for the electrodes is greater than that for bare Ni not only at zone 1 but also at zone 2 where the sole reduction process is NiOOH to Ni(OH)2.50 Since the diameter of the palladium nanocrystallites decreases in the order: Ni/Pd(4) > Ni/Pd(3) > Ni/Pd(2) > Ni/Pd(1), all the peak current densities in zones 1, 2 and 3, for all these electrodes, follow the reverse order as shown by the representative electrodes, Ni/Pd(1 and 4), in Fig. 2a. The cathodic peak obtained in zone 3 is due to simultaneous reduction of Pd(OH)2 to Pd and Ni(OH)2 to Ni, as reported in other studies51 but the contribution for the latter reaction is insignificant as it appears in the CV of pure Ni (Fig. 2a).
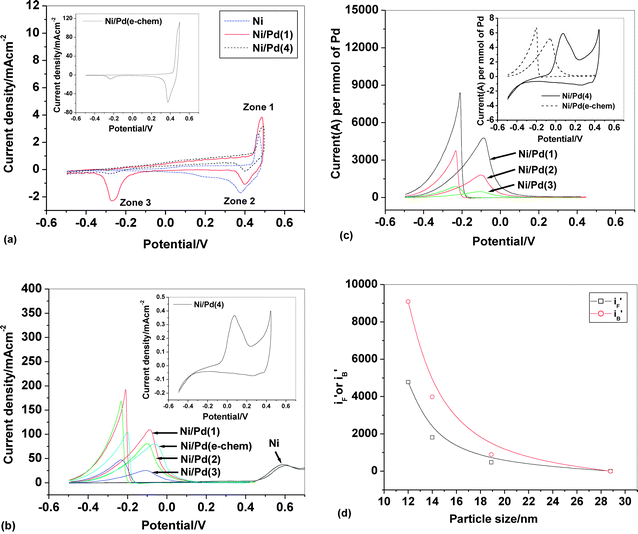 | ||
Fig. 2 (a) Cyclic voltammograms (in mA cm−2) in 1 M NaOH on bare Ni, Ni/Pd(i) (i = 1, 4) and Ni/Pd(e-chem) (inset), (b) cyclic voltammograms (in mA cm−2) of steady cycles of bare Ni, Ni/Pd(i) (i = 1–3, e-chem) and Ni/Pd(4) (inset) for alkaline (1 M) oxidation of ethanol (1 M), (c) cyclic voltammograms (in Am mol−1) of steady cycles of Ni/Pd(i) (i = 1–3), Ni/Pd(4) (inset) and Ni/Pd(e-chem) (inset) for alkaline (1 M) oxidation of ethanol (1 M), (d) a plot of 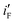 or or 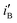 versus average diameter of Pd nanoparticles. versus average diameter of Pd nanoparticles. | ||
Now it is evident from Table 1 that the deposition of the nanocrystallites causes an increase in the true surface area of the electrodes e.g., the maximum increase in true surface area with respect to apparent area is ca. 1.7 times. However the control experiment shows that the peak current density (in zone 3) of the electrode Ni/Pd(1) is found to increase ca. 12.8 times than Ni/Pd(4). It is worth noting that the peak in zone 3 corresponds mainly to the reduction of Pd(OH)2 to Pd0. Therefore it seems reasonable that an increase in the reactivity with decrease in diameter of Pd nanoparticles is mainly due to particle-size-induced decrease in activation energy of the electrodic reaction. It is evident from Fig. 2b that the potential, EF, corresponding to forward peak current density, iF, and the potential, EB, corresponding to backward peak current density, iB, appear at around 0.6 V for Ni foil whereas EF, EB values for the dip-coated electrodes are found in between −0.107 V and 0.074 V, −0.313 V and −0.201 V, respectively. This indicates a large cathodic shift of EF and EB and hence a significant improvement of the electrocatalytic activity for the chemically deposited electrodes. Table 2 illustrates a comparative study of the electrocatalytic activities of the deposited electrodes in terms of iF, iB, EF, EB and iF/iB. The iF and iB values of the electrodes are found to follow the order: Ni/Pd(1) > Ni/Pd(2) > Ni/Pd(e-chem) > Ni/Pd(3) > Ni/Pd(4). This shows that most of the chemically deposited electrodes are electrocatalytically more efficient than the electrochemically deposited electrode, Ni/Pd(e-chem). The loading of Pd0 on Ni-foil is much greater for Ni/Pd(e-chem) (see Table 1) which is electrocatalytically less active. It transpires that superior electrocatalytic activity of the most of the chemically deposited electrodes is plausibly due to smaller diameter and greater dispersion ability of nanocrystallites on the Ni surface.
| Electrode | E F/V | E B/V | i F/mA cm−2 | i B/mA cm−2 | i F/iB |
|
|
|
|
|---|---|---|---|---|---|---|---|---|---|
| Ni/Pd(1) | −0.089 | −0.208 | 109.47 | 208.26 | 0.526 | 4777 | 9089 | 114.85 | 218.5 |
| Ni/Pd(2) | −0.101 | −0.232 | 81.19 | 178.55 | 0.455 | 1811 | 3983 | 47.25 | 103.9 |
| Ni/Pd(3) | −0.107 | −0.231 | 25.58 | 47.67 | 0.537 | 479.5 | 893.5 | 16.89 | 31.47 |
| Ni/Pd(4) | 0.074 | −0.313 | 0.3702 | −0.0507 | − | 5.970 | −0.8180 | 0.32 | −0.0444 |
| Ni/Pd(e-chem) | −0.064 | −0.201 | 80.34 | 107.02 | 0.751 | 5.170 | 6.890 | — | — |




Since loading of the catalyst-composite is different for the electrodes, peak current densities of the CVs for the electrodes are analyzed in two ways. First, peak currents per unit mole of Pd 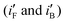 are computed by dividing peak current densities (iF and iB) with mole of Pd (atom) adsorbed per unit area of the surface. Secondly, peak currents per unit true area,
are computed by dividing peak current densities (iF and iB) with mole of Pd (atom) adsorbed per unit area of the surface. Secondly, peak currents per unit true area, 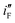 and
and 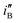 , are obtained by dividing iF and iB, respectively, by θsurface. The
, are obtained by dividing iF and iB, respectively, by θsurface. The 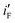 and
and 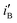 for the electrode Ni/Pd(1) are about 924 (=4777.35/5.17) and 1319 (=9088.61/6.89) fold greater than those of the corresponding values of Ni/Pd(e-chem) as depicted in Fig. 2c. This indicates significant improvement of electrocatalytic activity of the nanoparticle-embedded electrodes compared to Ni/Pd(e-chem). The
for the electrode Ni/Pd(1) are about 924 (=4777.35/5.17) and 1319 (=9088.61/6.89) fold greater than those of the corresponding values of Ni/Pd(e-chem) as depicted in Fig. 2c. This indicates significant improvement of electrocatalytic activity of the nanoparticle-embedded electrodes compared to Ni/Pd(e-chem). The 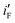 and
and 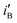 values of the electrodes are found to follow the order (Fig. 2d): Ni/Pd(1) > Ni/Pd(2) > Ni/Pd(3) > Ni/Pd(4). This clearly shows that smaller is the average diameter of the embedded nanocrystallites of Pd, greater is the electrocatalytic activity of the electrodes for anodic oxidation of ethanol. The
values of the electrodes are found to follow the order (Fig. 2d): Ni/Pd(1) > Ni/Pd(2) > Ni/Pd(3) > Ni/Pd(4). This clearly shows that smaller is the average diameter of the embedded nanocrystallites of Pd, greater is the electrocatalytic activity of the electrodes for anodic oxidation of ethanol. The 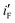 value of Ni/Pd(1) is ca. 800 times greater than that of Ni/Pd(4). This suggests the remarkable impact of the diameter of the nanoparticles on improving the rate of ethanol oxidation. True surface area of Ni/Pd(1) is ca. 0.89 times greater than that of Ni/Pd(4). Therefore, the increase in the electrocatalytic activity of the electrodes due to change of the average diameters of the particles is basically intrinsic in nature. Since
value of Ni/Pd(1) is ca. 800 times greater than that of Ni/Pd(4). This suggests the remarkable impact of the diameter of the nanoparticles on improving the rate of ethanol oxidation. True surface area of Ni/Pd(1) is ca. 0.89 times greater than that of Ni/Pd(4). Therefore, the increase in the electrocatalytic activity of the electrodes due to change of the average diameters of the particles is basically intrinsic in nature. Since 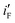 represents the rationalised electrocatalytic activity of the electrodes per millimole of metal electrocatalysts, it bears the compounding effect of both intrinsic electrocatalytic activity and true surface area. So, in order to get the intrinsic catalytic activity of the electrodes by eliminating the effect of surface area of the embedded nanoparticles,
represents the rationalised electrocatalytic activity of the electrodes per millimole of metal electrocatalysts, it bears the compounding effect of both intrinsic electrocatalytic activity and true surface area. So, in order to get the intrinsic catalytic activity of the electrodes by eliminating the effect of surface area of the embedded nanoparticles, 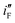 and
and 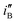 have been determined. The results show that
have been determined. The results show that 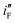 for Ni/Pd(1) is about 359 times greater than that of Ni/Pd(4) while the diameter of nanoparticles is decreased by only about 2.4 (=28.8/12.0) times. Thus the improvement in electrocatalytic activity of the Pd nanoparticle-embedded Ni electrodes is basically intrinsic in nature.
for Ni/Pd(1) is about 359 times greater than that of Ni/Pd(4) while the diameter of nanoparticles is decreased by only about 2.4 (=28.8/12.0) times. Thus the improvement in electrocatalytic activity of the Pd nanoparticle-embedded Ni electrodes is basically intrinsic in nature.
It is to be noted that iB, 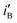 and
and 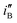 arise due to the secondary effect of the removal of adsorbed poisonous material during reverse scan in cyclic voltammetry. So these have not been considered directly in the relative measurement of catalytic capability. It is known that the tolerance of the anode towards poisonous carbonaceous species is normally measured by the ratio of iF to iB.14 It is here maximum for the Ni/Pd(e-chem) electrode, seemingly due to more symmetric and smaller spacing between the nanocrystalline seeds and also for the greater loading18 of Ni/Pd(e-chem) than the electrodes prepared chemically. This result is in conformation with our earlier studies.13,18,19
arise due to the secondary effect of the removal of adsorbed poisonous material during reverse scan in cyclic voltammetry. So these have not been considered directly in the relative measurement of catalytic capability. It is known that the tolerance of the anode towards poisonous carbonaceous species is normally measured by the ratio of iF to iB.14 It is here maximum for the Ni/Pd(e-chem) electrode, seemingly due to more symmetric and smaller spacing between the nanocrystalline seeds and also for the greater loading18 of Ni/Pd(e-chem) than the electrodes prepared chemically. This result is in conformation with our earlier studies.13,18,19
Chronopotentiometry
The chronopotentiograms obtained by the application of a relatively large constant current density of 5 mA cm−2 are depicted in Fig. 3a. The transition time, τ (presented in minutes within parentheses), increases in the following order: Ni/Pd(4)(6) < Ni/Pd(3)(7) < Ni/Pd(e-chem)(17) < Ni/Pd(2)(40) < Ni/Pd(1)(162). This reflects the order of resistance offered by the different electrodes or the stability of the electrodes against poisoning16 during electro-oxidation of ethanol. Thus it appears that the order of stability against poisoning increases with the decrease in the diameter of the spherical nanocrystallites constituting the electrodes e.g., the Ni/Pd(1) electrode offers the strongest resistance against poisoning among the electrodes. Three regions are distinctly observed in the chronopotentiograms (Fig. 3a) of the electrodes. In the lower potential range, the potential increases very sluggishly with time and then in the second region, a sharp increase occurs leading to a third and steady region at the potential ranging from 0.43 to 0.50 V. The first region signifies the region of potential where mainly dehydrogenation occurs in the process of ethanol oxidation. The second region is attributed to fast coverage of the electrode by carbonaceous species making the dehydrogenation step less important. The third steady region indicates particularly the oxidation of the carbonaceous species, which occurs at relatively high potential for completion of oxidation. In this region, besides the oxidation of carbonaceous species, negligible dehydrogenation also occurs simultaneously due to poor availability of active sites on the poisonous electrode surface. So better the catalytic capability of the electrode, more time it would stay at the relatively low potentials engaging itself with more dehydrogenation than oxidation of carbonaceous species. This suggests that Ni/Pd(1) is the best among the electrodes studied. Fig. 3b shows the dependence of τ on average diameter (d). It is evident from the figure that τ increases markedly after ‘d’ becomes less than ca. 19 nm. Thus the ability to resist poisoning increases sharply below that particle diameter. Again, the linear profiles (Fig. 3c) of the E vs.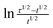 plot conform with the chronopotentiometric relation (eqn (6)) for the reaction system:
plot conform with the chronopotentiometric relation (eqn (6)) for the reaction system: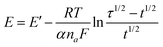 | (6) |
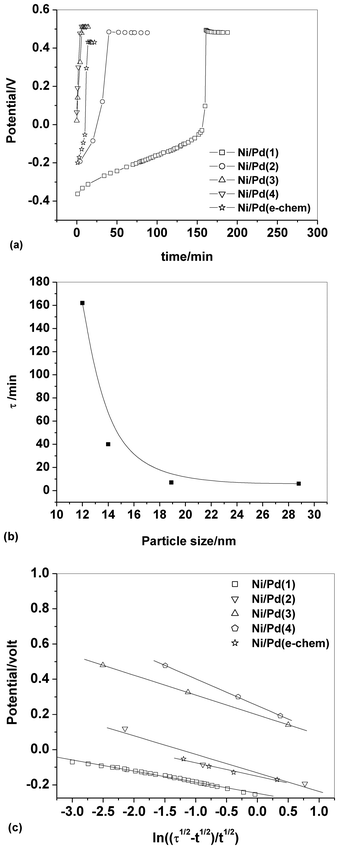 | ||
Fig. 3 (a) Chronopotentiometric profiles for ethanol oxidation on different Ni/Pd(i) electrodes immersed in 1 M ethanolic solution of 1 M NaOH, (b) a plot of τ versus average diameter of Pd nanoparticles, (c) plot of potential versus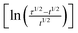 for different Ni/Pd(i) electrodes studied in 1 M ethanolic solution of 1 M NaOH. for different Ni/Pd(i) electrodes studied in 1 M ethanolic solution of 1 M NaOH. | ||
| Electrode | τ/min | −Slope/V | −Intercept/V |
|---|---|---|---|
| Ni/Pd(1) | 162 | 0.0634 | 0.2484 |
| Ni/Pd(2) | 40 | 0.1053 | 0.1319 |
| Ni/Pd(3) | 7 | 0.1127 | −0.1974 |
| Ni/Pd(4) | 6 | 0.1527 | −0.2500 |
| Ni/Pd(e-chem) | 17 | 0.0767 | 0.1512 |
Electrochemical impedance spectroscopy
In impedance spectroscopy, semi-circular Nyquist plots (Fig. 4a–c) are obtained for the electrodes at all the potentials studied except −0.5 V. This indicates very slow kinetics at −0.5 V for the anodic oxidation of ethanol. This observation is consistent with the formal potential data obtained from the chronopotentiometric study. The diameter of the referred semi-circular profiles represents the charge transfer resistance (Rct) for oxidation of alkaline ethanol on the prepared electrodes. At equilibrium, the exchange current, io, is related to the corresponding Rct by eqn (7)15,17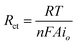 | (7) |
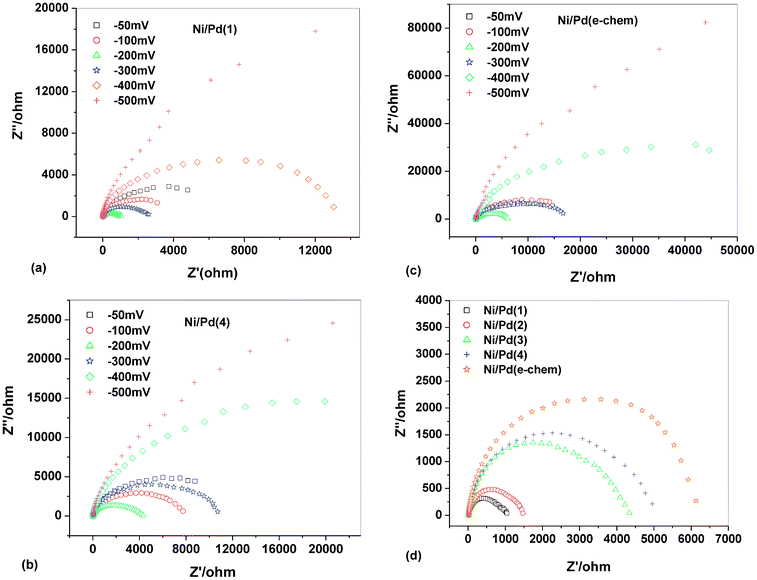 | ||
| Fig. 4 Electrochemical impedance spectra (EIS) in 1 M ethanolic solution of 1 M NaOH at different potentials with respect to MMO on (a) Ni/Pd(1), (b) Ni/Pd(4), (c) Ni/Pd(e-chem), (d) EIS at −0.2 V with respect to MMO on Ni/Pd(i) (i = 1–4, e-chem) electrodes. | ||
Fig. 4a–c are the semicircular Nyquist plots for Ni/Pd(1, 4), the representative electrodes and Ni/Pd(e-chem) electrodes, respectively, at different steady potentials. The profiles altogether indicate that the charge transfer resistance decreases gradually upon increasing the potential (from −0.5 to −0.2 V) and then increases upon further increasing the potential, e.g., at −0.1 and −0.05 V. This illustrates the fact that the reaction kinetics increase gradually with the increase in potential, which is maximum at ca. −0.2 V, the potential which lies in between EF and EB values as obtained from the corresponding cyclic voltammetric study. After applying a potential greater than −0.2 V the rate of charge transfer reaction decreases gradually as evident from the electrochemical impedance spectra (EIS) at −0.1 and −0.05 V. The result may be attributed to the formation of strongly adsorbed intermediates of metal oxides and hydroxides following eqn (2)–(5) beyond −0.2 V. Consequently, the ethanol adsorption (eqn 8) and hence the subsequent poison removal reactions (eqn (11) and (12)) according to Scheme 1 (eqn (8)–(12))12,54–56 gradually decrease and eventually stop at high anodic potential.
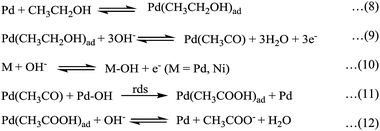 | ||
| Scheme 1 Mechanism of anode catalytic oxidation of ethanol under alkaline conditions. | ||
So impedance study (Fig. 4a–c) shows that the charge transfer resistance is minimal at nearly −0.2 V, indicating the optimum condition for getting the best electrocatalytic activity from the type of electrodes studied. In addition, the promotion of anode catalysis by smaller Pd nanoparticles as observed in the chronopotentiometric study can be explained using Scheme 1 where the formation of the relatively weak Pd–OH bond by these nanoparticles results in a higher reaction rate for the rate determining step (rds) of Scheme 1.
Fig. 4d shows the comparative profiles for all the electrodes at −0.2 V. The charge transfer conductance per unit area (1/ARct) of the electrodes, Ni/Pd(i) (i = 1–4, e-chem), was found to be 0.0407, 0.0352, 0.00974, 0.00937, 0.00756 Ω−1 cm−2 respectively. Hence, the order of the charge transfer conductance per unit area of the electrodes is Ni/Pd(1) > Ni/Pd(2) > Ni/Pd(3) > Ni/Pd(4) > Ni/Pd(e-chem) indicating size-dependence. It is worth mentioning that the relatively depleted Nyquist plots found here signify the complexity of ethanol oxidation reaction and each plot provides a lot of mechanistic kinetic information. For more detailed interpretation of the data we require studies of equivalent circuits based on the mechanism, simulation, etc. which are not included in the perspective of the present study. Hence further rationalization of EIS data directed towards the determination of the mechanism and structural effects is avoided.
Conclusions
Thus the present study leads to the following conclusions:(i) Polyvinyl alcohol-protected spherical Pd nanoparticles of different average diameters, embedded on a planar Ni substrate, are capable of exhibiting size-dependent anode-catalytic activity for the alkaline oxidation of ethanol. Gradual retardation of electrocatalytic activity for this oxidation was found with increase in particle size on the electrode surface.
(ii) The stable nanoparticle on the electrode surface responds considerably to its own electro-oxidation and shows superior catalytic activity for ethanol oxidation as evident from cyclic voltammetric study.
(iii) Despite low Pd0 loading, most of the dip-coated electrodes under similar reaction conditions show much better electrocatalytic activity than the electrochemically constructed electrode, with respect to both reaction rate and poisoning resistance.
(iv) The ascending order of electrocatalytic capability with decrease in the diameter of the adsorbed Pd nanocrystallites on the electrodes is proved by the rationalized peak current per mole in cyclic voltammetric study and charge transfer per mole of the catalyst (before transition) in chronopotentiometric study. The latter study of the electrodes also reveals the sharp increase of poisoning resistance for the nanoparticles lying below the average diameter of 19 nm, suggesting greater extent of Pd–OH formation and weakening of the Pd–poison bond, as shown in Scheme 1.
(v) The smaller size and greater adsorption capability of the smaller nanoparticles are effective in increasing true surface area and the availability of more energized surface Pd atoms comprising the electrode. This results in the enhancement of intrinsic catalytic activity of the electrodes.
Acknowledgements
The authors gratefully acknowledge CSIR (New Delhi) and Jadavpur University for financial support.Notes and references
- R. Ganesan and J. S. Lee, Angew. Chem., Int. Ed., 2005, 44, 6557 CrossRef CAS.
- C. Lamy, E. M. Belgsir and J. M. Leger, J. Appl. Electrochem., 2001, 31, 799 CrossRef CAS.
- Z. Zhang, L. Xin, K. Sun and W. Li, Int. J. Hydrogen Energy, 2011, 36, 12686 CrossRef CAS.
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, Appl. Catal., B, 2006, 63, 137 CrossRef CAS.
- X. Xue, J. Ge, T. Tian, C. Liu, W. Xing and T. Lu, J. Power Sources, 2007, 172, 560 CrossRef CAS.
- X. Ren, T. E. Springer, T. A. Zawodzmski and S. Gottesfeld, J. Electrochem. Soc., 2000, 147, 466 CrossRef CAS.
- A. Heinzel and V. M. Barragan, J. Power Sources, 1999, 84, 70 CrossRef CAS.
- S.-W. Xie, S. Chen, Z.-Q. Liu and C.-W. Xu, Int. J. Electrochem. Sci., 2011, 6, 882 CAS.
- S. Q. Song and P. Tsiakaras, Appl. Catal., B, 2006, 63, 187 CrossRef CAS.
- H. L. Jiang, G. Q. Sun, S. L. Wang, G. X. Wang, Q. Xin, Z. H. Zhou and B. Zhou, Electrochem. Commun., 2005, 7, 663 CrossRef.
- Q. Yi, F. Niu and L. Sun, Fuel, 2011, 90, 2617 CrossRef CAS.
- E. Antolini, J. Power Sources, 2007, 170, 1 CrossRef CAS.
- J. Bagchi and S. K. Bhattacharya, J. Power Sources, 2007, 163, 661 CrossRef CAS.
- Z. Liu, X. Y. Ling, X. Su, J. Y. Lee and L. M. Gan, J. Power Sources, 2005, 149, 1 CrossRef CAS.
- L. Jiang, S. Song, Z. Zhou, S. Yan, H. Li, G. Sun, B. Zhou and Q. Xin, Indian J. Chem., 2005, 44A, 913 CAS.
- T. Iwasita, H. Hoster, A. John-Anaeker, W. F. Lin and W. Vielstich, Langmuir, 2000, 16, 522 CrossRef CAS.
- H. A. Gasteiger, N. Markovic, P. N. Ross Jr. and J. Cairns Elton, J. Phys. Chem., 1994, 98(2), 617 CrossRef CAS.
- J. Bagchi and S. K. Bhattacharya, Transition Met. Chem., 2007, 32(1), 47 CrossRef CAS.
- J. Bagchi and S. K. Bhattacharya, Transition Met. Chem., 2008, 33, 113 CrossRef CAS.
- X. Chen, Z. Lin, T. Jia, Z. Cai, X. Huang, Y. Jiang, X. Chen and G. Chen, Anal. Chim. Acta, 2009, 650, 54 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS.
- F. Benseba, A. A. Farah, D. S. Wang, C. Bock, X. M. Du, J. Kung and Y. L. Page, J. Phys. Chem. B, 2005, 109, 15339 CrossRef.
- J. O'M Bockris, A. K. N. Reddy and S. Khan, Surface Electrochemistry, A Molecular Level Approach, Plenum, New York, 1993 Search PubMed.
- I. L. Finar, Organic Chemistry (roll), Addition Wesley Longman (Singapore) Pvt. Ltd, Indian Branch, Delhi-110092, India, 2001 Search PubMed.
- E. S. Ranganathan, S. K. Bej and L. T. Thomson, Appl. Catal., A, 2005, 289, 153 CrossRef CAS.
- Z. Liu, L. Hong, M. P. Tham and T. H. Lim, J. Power Sources, 2006, 161, 831 CrossRef CAS.
- W. Zhou and J. Y. Lee, J. Phys. Chem. C, 2008, 112, 3789 CAS.
- C. W. Xu, L. Q. Cheng, P. K. Shen and Y. L. Liu, Electrochem. Commun., 2007, 9, 997 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183 CrossRef CAS.
- C. Xu, P. K. Shen and Y. Liu, J. Power Sources, 2007, 164, 527 CrossRef CAS.
- F. Cheng, H. Wang, Z. Sun, M. Ning, Z. Cai and M. Zhang, Electrochem. Commun., 2008, 10, 798 CrossRef CAS.
- V. Bambagioni, C. Bianchini, A. Marchionni, J. Filippi, F. Vizza and J. Teddy, et al. , J. Power Sources, 2009, 190, 241 CrossRef CAS.
- F. Hu, F. Ding, S. Song and P. K. Shen, J. Power Sources, 2006, 163, 415 CrossRef CAS.
- F. Hu, C. Chen, Z. Wang, G. Wei and P. K. Shen, Electrochim. Acta, 2006, 52, 1087 CrossRef CAS.
- A. N. Grace and K. Pandian, Electrochem. Commun., 2006, 8, 1340 CrossRef CAS.
- D. Serra and L. McElwee-White, Inorg. Chim. Acta, 2008, 361, 3237 CrossRef CAS.
- D. Chu, J. Wang, S. Wang, L. Zha, J. He and Y. Hou, et al. , Catal. Commun., 2009, 10, 955 CrossRef CAS.
- C. Xu, Z. Tian, P. K. Shen and S. P. Jiang, Electrochim. Acta, 2008, 53, 2610 CrossRef CAS.
- F. Miao, B. Tao, L. Sun, T. Liu, J. You and L. Wang, et al. , J. Power Sources, 2010, 195, 146 CrossRef CAS.
- J. Lu, S. Lu, D. Wang, M. Yang, Z. Liu and C. Xu, et al. , Electrochim. Acta, 2009, 54, 5486 CrossRef CAS.
- Y. C. Weng and T. C. Chou, J. Electrochem. Soc., 2006, 153(6), H127 CrossRef CAS.
- S. M. A. Shibli, K. S. Beenakumari and N. D. Suma, Biosens. Bioelectron., 2006, 22, 633 CrossRef CAS.
- C. Xu, Y. Hu, J. Rong, S. P. Jiang and Y. Liu, Electrochem. Commun., 2007, 9, 2009 CrossRef CAS.
- N. M. Suleimanov, S. M. Khantimerov, E. F. Kukovitsky and V. L. Matukhin, J. Solid State Electrochem., 2008, 12, 1021 CrossRef CAS.
- V. Bambagioni, C. Bianchini, J. Filippi, W. Oberhauser, A. Marchionni, F. Vizza, R. Psaro, L. Sordelli, M. L. Foresti and M. Innocenti, ChemSusChem, 2009, 2, 99 CrossRef CAS.
- P. S. Roy, J. Bagchi and S. K. Bhattacharya, Colloids Surf., A, 2010, 359, 45 CrossRef CAS.
- P. S. Roy, J. Bagchi and S. K. Bhattacharya, Transition Met. Chem., 2009, 34, 447 CrossRef CAS.
- Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press LLC, 33rd edn, 2002–2003 Search PubMed.
- A. E. Bolzan, J. Electroanal. Chem., 1995, 380, 127 CrossRef.
- M. A. Abdel Rahim, R. M. Abdel Hameed and M. W. Khalil, J. Power Sources, 2004, 134, 160 CrossRef CAS.
- Yy M. Grden, A. Czerwinski, J. Golimowski, E. Bulska, B. Krasnodebska-Ostrega, R. Marassi and S. Zamponi, J. Electroanal. Chem., 1999, 460, 30 CrossRef.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, John Wiley & Sons, 1980, pp. 1–718 Search PubMed.
- N. Hoshi, K. Kida, M. Nakamura, M. Nakada and K. Osada, J. Phys. Chem. B, 2006, 110, 12480 CrossRef CAS.
- Z. X. Liang, T. S. Zhao, J. B. Xu and L. D. Zhu, Electrochim. Acta, 2009, 54, 2203 CrossRef CAS.
- A. V. Tripkovic, K. D. Popovik and J. D. Lovic, Electrochim. Acta, 2001, 46, 3163 CrossRef CAS.
- H. P. Liu, J. P. Ye, C. W. Xu, S. P. Jiang and Y. X. Tong, Electrochem. Commun., 2007, 9, 2334 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |