Catalytic performance and hydrothermal durability of CeO2–V2O5–ZrO2/WO3–TiO2 based NH3-SCR catalysts
Xinquan
Wang
a,
Anju
Shi
a,
Yingfeng
Duan
a,
Jun
Wang
a and
Meiqing
Shen
*ab
aKey Laboratory for Green Chemical Technology of State Education Ministry, School of Chemical Engineering & Technology, Tianjin University, Tianjin 300072, China. E-mail: mqshen@tju.edu.cn; Fax: +86 22 27892301; Tel: +86 22 27892301
bState Key Laboratory of Engines, Tianjin University, Tianjin 300072, China
First published on 29th March 2012
Abstract
Ceria modified V2O5–ZrO2/WO3–TiO2 catalysts with different Ce loading (0, 5, 10 wt%) have been evaluated in the NH3-SCR process before and after hydrothermal aging (750 °C, 10 wt% H2O/air for 12 h). Compared with only Zr containing catalysts, addition of Ce greatly enhances the low temperature activity of the fresh catalyst, but it deactivates obviously after aging. The catalysts were characterized by XRD, XPS, H2-TPR and DRIFTS. The results suggest that not only the enrichment of Ce3+ and the increased redox properties, but also the more active adsorbed nitrates on CeO2 modified catalysts benefit the SCR reaction. The catalyst deactivation after aging is mainly due to the sintering and segregation of CeO2 on the catalysts surface, suggesting the poor hydrothermal stability of the Ce component. However, additional provided NO2 will compensate for the activity loss due to hydrothermal aging and significantly improve the low temperature SCR activity, suggesting a high NO2 sensitivity of the Ce component. Moreover, it was found that a ceria, zirconia containing catalyst exhibits superior SCR activities, with both the fresh and aged 1%V2O5–10%CeO2–10%ZrO2/WO3–TiO2 catalysts showing high NOx conversions (>95%) and selectivity to N2 (>98%) in a wide temperature range of 200–500 °C, at a space velocity of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 in a simulated exhaust containing 500 ppm NOx (NO2/NO = 1) and 5% H2O.
000 h−1 in a simulated exhaust containing 500 ppm NOx (NO2/NO = 1) and 5% H2O.
1. Introduction
The selective catalytic reduction (SCR) of NOx with NH3 or urea has been proven to be the most chemically efficient and cost-effective technology to reduce NOx emission from stationary and lean burn mobile sources.1,2 One commercial catalyst technology is based on the V2O5–WO3–TiO2 system. It is highly efficient in the temperature range of 300–450 °C and has been widely used for industrial applications.3 However, the main drawbacks of these catalysts for mobile application are the insufficient low temperature activity, the poor thermal durability and the potential toxicity that might arise from vanadium during operations.4 In view of these defects, extensive research efforts have been made to modify V2O5 based catalysts with various components, such as SiO2, SO42−, BaO, MnO2 and rare earths.5–13 Recently, Chapman D. M. has found that the loss of vanadia can be significantly reduced by reducing the loss of the surface area of the anatase-TiO2 support during exposure and by minimizing the surface vanadia loading.4 In this aspect, zirconia modified TiO2 based catalysts are found to exhibit superior thermal stability as NH3-SCR catalysts.14,15 This is due to the stabilized anatase phase of the TiO2 support by the formation of ZrxTi1−xO2 solid solutions.15 However, the insufficient low temperatures (<300 °C) activity still exists on these catalysts.Due to the high oxygen storage capacity and excellent redox properties, ceria has been attracting much attention in the NH3-SCR process.13,16–20 Qi and Yang reported a MnOx–CeO2 catalyst that was highly active for the SCR of NO by NH3 at low temperature.16 Xu et al.17 developed the CeO2/TiO2 catalyst, which has high activity in the temperature range of 275–400 °C. Chen et al.13 also found that the doping of ceria could greatly enhance the low temperature NH3-SCR performance of V2O5 based catalysts for diesel application. However, limited information is available in the literature regarding the effect of hydrothermal aging on the reactivity of CeO2 modified catalysts. Thermal durability of SCR catalysts has been much emphasized in vehicle applications,4,11 because the exhaust temperature may reach above 700 °C during the regeneration of the diesel particulate filter (DPF) process.
In this work, we report the effect of harsh hydrothermal aging treatment (750 °C in 10 wt% H2O/air for 12 h) on the activity of ceria modified 1 wt%V2O5–10 wt%ZrO2/WO3–TiO2 catalyst. The performance of NH3-SCR and NH3 oxidation for both the fresh and aged catalysts was evaluated. The effect of H2O and NO2 on the activity of catalysts was also studied to simulate the practical conditions. Moreover, the structural effect of ceria modification on NH3-SCR activity was investigated. DRIFT studies were also performed to investigate the effect of ceria on adsorbed species under reaction conditions. The aim is to probe how ceria would influence the NH3-SCR performance of catalysts before and after aging.
2. Experimental
2.1. Catalysts preparation
The CeO2 (0, 5, 10 wt%) modified V2O5–ZrO2/WO3–TiO2 catalysts with 1 wt% V2O5, 10 wt% ZrO2 loading were prepared by an impregnation method. The catalyst support, 92%TiO2–8%WO3 (WT) with 0.8 wt% of sulfate (from TG analysis), was commercially obtained from Millennium Inorganic Chemicals Inc. The complex of VO(CO2)2 was prepared by reacting V2O5 powder with oxalic acid (1 M) with continuous stirring at 70 °C for 30 min. Calculated amounts of zirconia acetate and cerium nitrate were then added into the solutions. Subsequently, the desired quantity of WO3–TiO2 powder was added into the mixed solutions and stirred for 1 h. The as-synthesized material was dried overnight at 110 °C and then calcined at 500 °C for 3 h to obtain the fresh catalyst. A further hydrothermal aging at 750 °C in 10 wt% H2O/air for 12 h was carried out to obtain the aged catalysts. The fresh catalysts are denoted as V1CexZWT, where x denotes weight percentage of the ceria with respect to the TiO2 support, and the reference catalyst is denoted as V1ZWT. The aged samples are denoted as V1CexZWT-A and V1ZWT-A, respectively.2.2. Catalysts characterization
The powder X-ray diffraction (XRD) experiments were performed on an X’Pert Pro diffractometer employing Cu Kα radiation (λ = 0.15418 nm). The X-ray tube was operated at 40 kV and 40 mA. The X-ray powder diffractogram was recorded at a 0.02° interval in the range of 20° < 2θ < 80°. The identification of the phases was made with the help of JCPDS cards (Joint Committee on Powder Diffraction Standards). The mean crystallite sizes of titania were calculated using the Scherer equation.The surface areas of the samples were measured by N2 physisorption at −196 °C using the Brunauer–Emmett–Teller (BET) method with F-Sorb 3400 volumetric adsorption/desorption apparatus. The samples were degassed in flowing N2 at 150 °C for 3 h before measurement.
The X-ray photoelectron spectroscopy (XPS) experiments were carried out on a PHI-1600 ESCA system. The binding energy was calibrated internally by the carbon deposit C1s binding energy (BE) at 284.8 eV.
H2-TPR experiments were conducted using 100 mg of samples. Samples were pretreated at 500 °C for 1 h in O2 (30 mL min−1) flow and then cooled down to room temperature in N2 flow. The samples were heated in 5% H2/He flow (30 mL min−1) from 50 to 800 °C by 10 °C min−1 while H2 consumption was recorded continuously.
In situ DRIFT spectra of adsorption species were collected on a Nicolet 6700 FTIR spectrometer equipped with a commercial high temperature chamber (Thermofisher) fitted with a ZnSe window. Prior to reactant gas (NH3 or NO) chemisorption, the sample was heated up to 500 °C in a 20% O2/N2 flow (50 mL min−1) in the chamber. After the pre-oxidation treatment at 500 °C for 30 min, the sample was cooled down to the target temperature. The sample was subsequently flushed by 50 mL min−1 N2 for 30 min to remove any physisorbed molecules for background collection. Then, the gas containing 3000 ppm NH3 in N2 (50 mL min−1) passed through the sample at the target temperature for 30 min. The FTIR spectra were collected after purging the weakly adsorbed gas molecules by flowing N2 for 15 min.
In situ FTIR spectra for NO, NH3 and O2 co-adsorption were collected at the similar conditions as the NH3 chemisorptions, where 3000 ppm NO and 3000 ppm NH3 in 4% O2 + N2 were introduced to the system.
2.3. Activity measurement
The catalytic activity measurement for the reduction of NO by NH3 (NH3-SCR) was carried out in a fixed bed reactor made of stainless steel with 0.5 mL catalysts (diluted by silica). The reaction gas mixture consists of 500 ppm NO, 500 ppm NH3, 5% O2 and N2 in balance. The NO conversion was measured from 150 °C to 550 °C at a ramp rate of 5 °C min−1. The total flow of the gas mixture was 1 L min−1 at a gas hourly space velocity (GHSV) of 120000 h−1. The concentrations of NO, NO2, N2O and NH3 were measured by a Thermofisher IS10 FTIR spectrometer equipped with a 2 m path length gas cell.
The N2 concentrations were determined based on nitrogen balance from eqn (1).
| N2out = (1/2) ([NO]in − [NO]out) + (1/2) ([NH3]in − [NH3]out) − (1/2) NO2out − N2Oout | (1) |
And the N2 selectivity for the SCR reaction was calculated from eqn (2).
| N2 selectivity (%) = 2N2out/(2N2out + 2N2Oout + NO2out) | (2) |
NH3 oxidation activities were measured using the same reactor, with 500 ppm NH3 and 5% O2 in N2 as a feed, from 250 to 550 °C. All catalysts were kept at each temperature for 10 min to obtain a stable conversion. N2 selectivity was calculated based on the following equation:
| N2 selectivity (%) = (([NH3]in − [NH3]out) − NOout − NO2out − 2N2Oout)/([NH3]in − [NH3]out) | (3) |
3. Results
3.1. Catalytic performance
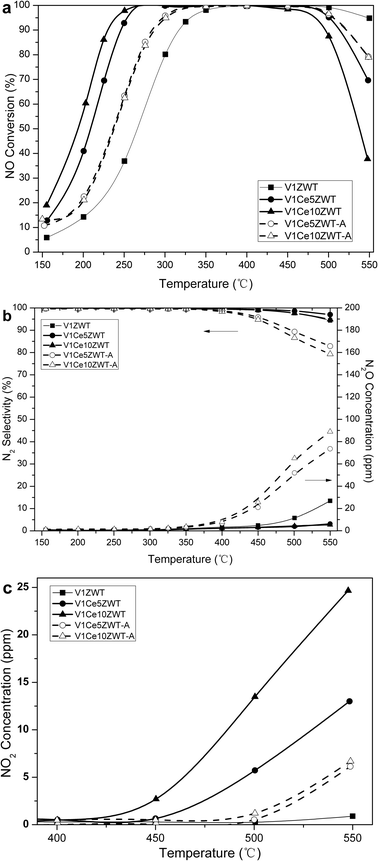 | ||
| Fig. 1 NH3-SCR performance on V1CexZWT and V1CexZWT-A catalysts as a function of temperature. (a) NO conversion. (b) N2O generation and N2 selectivity. (c) NO2 generation. | ||
N2O formation and N2 selectivity during the NH3-SCR reaction are shown in Fig. 1b. On the V1ZWT catalyst, undesired N2O is formed above 500 °C, which is a main side product on vanadia based catalysts.1 Ceria addition strongly inhibits N2O formation on fresh catalysts, while a significant amount of N2O is found on aged ceria modified catalysts, which contributes to the decrease in N2 selectivity of the aged catalysts.
Fig. 1c shows NO2 concentration during the NH3-SCR process. A little NO2 is observed on the V1ZWT sample at 550 °C, while ceria greatly enhances NO2 formation at high temperatures. This is mainly due to the direct NO oxidation by ceria at high temperatures.19 However, NO2 concentration decreases on ceria modified catalysts after aging treatment. The increase of the NO2 product on a fresh catalyst and the decrease on an aged one indicates the decreased oxidation ability of ceria modified catalysts.
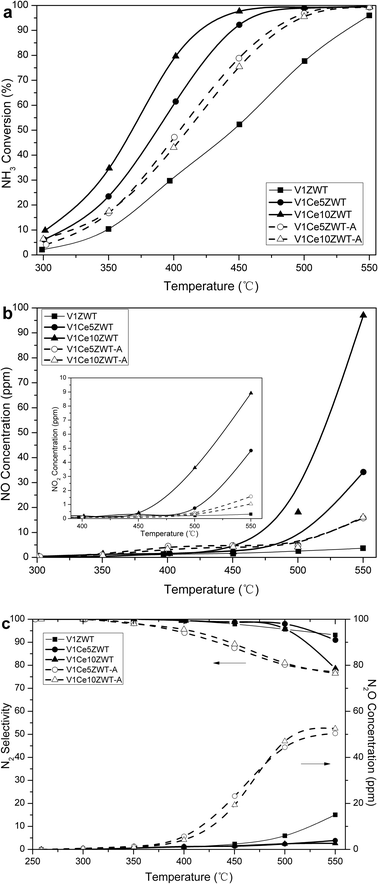 | ||
| Fig. 2 NH3 oxidation on V1CexZWT and V1CexZWT-A catalysts as a function of temperature. (a) NH3 conversion. (b) NO and NO2 generation. (c) N2O generation and N2 selectivity. | ||
Fig. 2b and c show the NO, NO2, N2O formation and N2 selectivity as a function of temperature during the NH3 oxidation process. A small amount of N2O is observed on the V1ZWT catalyst. With the addition of ceria, a significant amount of NO is observed while N2O is inhibited during NH3 oxidation at high temperatures. This high amount of NO side product also explains the drastic decrease in NH3-SCR activity on V1Ce10ZWT at 550 °C. Moreover, NO2 increases with the sharp increase of the NO product on ceria modified catalysts, indicating the strong oxidation capacity of CeO2. After aging, both NO and NO2 products decrease obviously, while N2O increases on the V1Ce10ZWT-A catalyst, which results in the N2 selectivity decrease. The change of side products from NO, NO2 to N2O after aging suggests hydrothermal deactivation of the CeO2 component on the catalyst.
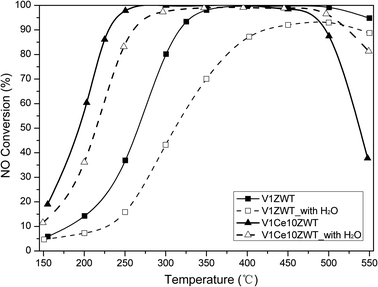 | ||
| Fig. 3 Effect of H2O on NH3 SCR activity of V1ZWT and V1Ce10ZWT catalysts. Reaction conditions: 500 ppm NO, 500 ppm NH3, 5% O2, 5% H2O, N2 balance. | ||
Fig. 4a shows the activity of V1ZWT and V1Ce10ZWT in a NOx feed containing various NO2 concentrations (NOx = 500 ppm, NO2 = 0, 150, 250 ppm) with 500 ppm NH3, 5% O2 and 5% H2O. For the V1ZWT catalyst, the promotional effect of NO2 is limited, with only 35% of NO conversions obtained at 200 °C. Moreover, increasing the NO2 concentration has little effect on low temperature (<300 °C) SCR activity. Comparatively, a clear positive effect of NO2 on V1Ce10ZWT is observed, with the NOx conversion enhanced from 35% (NO alone) to 95% (NO2, NO = 250 ppm) at 200 °C, the overall NOx conversion maintains above 95% from 200 to 500 °C, and the N2O does not exceed 2 ppm at 550 °C. The positive role of NO2 was demonstratively due to the occurrence of fast SCR reactions (2NH3 + NO + NO2 → 2N2 + 3H2O).22–24
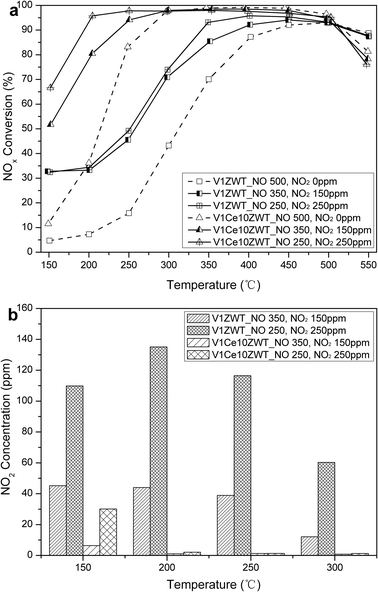 | ||
| Fig. 4 Effect of NO2 on NH3 SCR activity of V1ZWT and V1Ce10ZWT catalysts. (a) NOx conversion. (b) NO2 concentration. Reaction conditions: 500 ppm NOx (NO2 = 0, 150, 250 ppm), 500 ppm NH3, 5% O2, 5% H2O, N2 balance. | ||
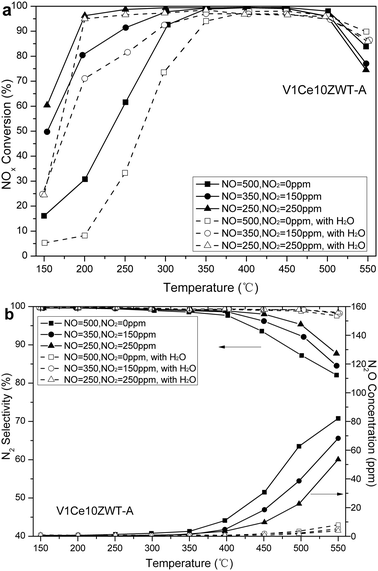
In order to explore the difference in NH3-SCR activities between V1ZWT and V1Ce10ZWT at low temperatures, the outlet NO2 concentrations of the catalysts below 300 °C are shown in Fig. 4b. A large amount of NO2 is observed on the V1ZWT catalyst, and the level of NO2 is almost unchanged (in a feed of 150 ppm NO2) or even increased (in a feed of 250 ppm NO2) from 150 to 250 °C, indicating that less NO2 is participating in the fast SCR reactions. And the increase of NO2 under the conditions of feeding 250 ppm of NO2 could be due to NOx desorption on the V1ZWT catalyst. These results strongly suggest that V1ZWT has rather low sensitivity to NO2 feed, increasing the NO2 concentration or increasing the temperature has little promotional effect on SCR activity. Comparatively, only a little NO2 is observed at 150 °C on the V1Ce10ZWT catalyst, and the level of NO2 is below 2 ppm above 200 °C, indicating that NO2 is more reactive on ceria modified catalysts. The performance of the V1Ce10ZWT-A catalyst with different NO2/NOx ratios is shown in Fig. 5a. The reaction conditions are 500 ppm NOx (NO2 = 0, 150, 250 ppm), 500 ppm NH3, 5% O2 in the absence or presence of 5% H2O. Relatively low activity is observed below 300 °C with only NO added in the absence or presence of 5% H2O. However, the NOx removal efficiency is strikingly improved when NO2 is introduced, with 90% of NOx conversion shifting to low temperature by about 150 °C. Moreover, the presence of both NO2 and H2O can inhibit N2O production, with N2 selectivity significantly improved (Fig. 5b). It is found that the aged V1Ce10ZWT catalyst can still exhibit superior NH3-SCR activity and N2 selectivity in the temperature range 200–550 °C with NO2/NO = 1 in the presence of H2O.
3.2. BET & XRD
The BET surface areas are summarized in Table 1. It is found that CeO2 does not have much effect on the BET surface area of fresh samples but slightly decreases it on aged samples. The X-ray powder diffraction patterns are shown in Fig. 6, and the calculated crystallite sizes of catalysts are summarized in Table 1. On fresh catalysts, all XRD peaks are from the anatase phase of TiO2 and no ceria or zirconia diffractions can be detected, suggesting that CeO2 and ZrO2 are present as amorphous materials. Upon hydrothermal aging, the peaks assigned to anatase-TiO2 on all catalysts become more intense. While the average crystallite sizes of aged catalysts are at the same level, ranging from 28.9 nm to 30.6 nm. This clearly indicates that CeO2 modification does not influence much the structure stability of the TiO2 support. However, WO3 segregation on the V1ZWT-A catalyst is strongly inhibited by CeO2 addition, which can be due to the Ce–W interactions on the catalyst surface.13 Moreover, the peak attributed to tetragonal ZrO2 observed on V1ZWT-A slightly decreases on Ce–Zr modified aged catalysts, with Ce0.75Zr0.25O2 solid solutions forming on the surface.25 The active Ce component would segregate after aging, while the Zr component inclines to incorporate into the CeO2 matrix to form Ce–Zr solid solutions, which is beneficial for the thermal stability of CeO2.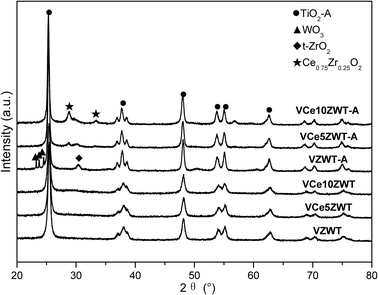 | ||
| Fig. 6 XRD patterns of the catalysts. | ||
| Sample | BET surface area/m2 g−1 | TiO2 crystalline size/nm |
|---|---|---|
| V1ZWT | 82.9 | 16.8 |
| V1Ce5WT | 82.1 | 16.5 |
| V1Ce10WT | 78.5 | 16.4 |
| V1ZWT-A | 21.6 | 28.9 |
| V1Ce5ZWT-A | 17.2 | 30.6 |
| V1Ce10ZWT-A | 16.1 | 29.2 |
3.3. XPS
The XPS spectra of chemical states of Ce over V1Ce10ZWT and V1Ce10ZWT-A catalysts are shown in Fig. 7. The bands labeled u′ and v′ represent the 3d104f1 initial electronic state corresponding to Ce3+, while the peaks labeled u, u′′, u′′′, v, v′′ and v′′′ represent the 3d104f0 state of Ce4+ ions.26 For V1Ce10ZWT, v and u′′′ peaks for Ce4+ were much weaker than v′ and u′ peaks for the Ce3+ state. This result suggests that the Ce component mainly stays in the state of 3+ on the catalyst surface. According to the literature,18,27,28 the presence of the Ce3+ species could create a charge imbalance, and more oxygen vacancies can be generated, which is beneficial for the activation of surface oxygen species in the SCR reaction. This is critical for the improved low temperature activity of ceria modified catalysts. However, after severe hydrothermal aging, the characteristic peaks assigned to Ce3+ and Ce4+ almost disappear. This is probably due to the segregation of CeO2 on the catalyst surface, which has little chance to be accessed due to its large diameters above the detection limit of XPS. And the disappearance of Ce3+ and segregation of the active CeO2 component (as shown in Fig. 6) could contribute to the deactivation of V1Ce10ZWT-A catalysts at low temperature.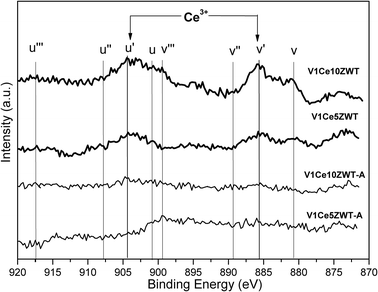 | ||
| Fig. 7 XPS spectra of Ce 3d on the catalysts. | ||
3.4. H2-TPR test
H2-TPR is usually carried out to study the redox property of various catalysts. As shown in Fig. 8, all the catalysts show a broad peak for the temperature range of 400–800 °C. The reduction profile of a commercial 8%WO3–92%TiO2 support (reference) is also shown for comparison. It is found that the TiO2 support exhibits a major peak at around 621 °C. As the reduction of WOx and TiOx is usually at high temperatures above 800 °C,29,30 this peak could be due to reduction of some sulfate species that existed on the commercial TiO2 support, as reported in other literature.8,31 The presence of sulfate is also confirmed by our DRIFT results (not shown here). For the V1ZWT catalyst, a strong reduction peak centered at 557 °C is observed. As pure ZrO2 does not have redox properties,32 this peak on the V1ZWT sample could be due to the reduction of surface V5+ to V3+ and sulfate species. When Ce is introduced into the catalysts, a sharp peak at 608 °C and a novel shoulder peak around 306 °C are found. According to the literature on ceria based catalysts,33 this shoulder peak is attributed to the reduction of surface oxygen of nonstoichiometric ceria of type Ce3+–O–Ce4+.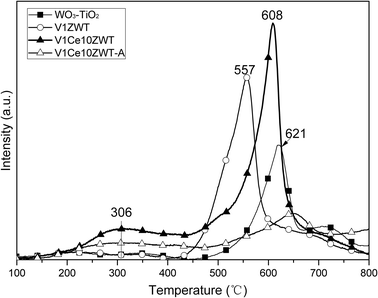 | ||
| Fig. 8 H2-TPR profiles on the catalysts. | ||
On the other hand, the strong peak at 608 °C lines between the reduction peaks of TiO2 and V1ZWT catalysts, but with higher intensity. This is probably due to the interactions between CeO2, V2O5 and sulfates on the TiO2 surface. These results strongly suggest the increased redox properties on CeO2 modified catalysts. However, after hydrothermal aging, the intensity of these peaks decrease, which indicates the decreased redox properties on the V1Ce10ZWT-A catalyst.
3.5. DRIFT
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of sulfate on the catalyst surface.35 These sulfate species are found to be derived from the commercial TiO2 support, confirmed by our FTIR results (not shown here), and contribute to Brønsted acid sites on the surface.36 With an increase in temperature, the intensity of the 1453 cm−1 band decreases noticeably at high temperature, while the 1242 cm−1 band still remains and shifts to a higher wave band at 1255 cm−1. This indicates that NH3 bonded to Lewis acid sites is more stable on the catalyst surface. With CeO2 addition, the peaks assigned to Brønsted acid sites and Lewis acid sites decrease obviously. Moreover, the band assigned to Brønsted acid sites decreases more rapidly with increasing temperature on V1Ce10ZWT. These results suggest that the addition of CeO2 decreases both the amount and strength of Brønsted acid sites on the catalyst surface. Although many research studies have proved the critical role of surface acidity in the SCR reaction,37,38 the promotional effect of CeO2 on NH3-SCR activity could not be explained by the acidity factor in this catalyst system.
O stretching mode of sulfate on the catalyst surface.35 These sulfate species are found to be derived from the commercial TiO2 support, confirmed by our FTIR results (not shown here), and contribute to Brønsted acid sites on the surface.36 With an increase in temperature, the intensity of the 1453 cm−1 band decreases noticeably at high temperature, while the 1242 cm−1 band still remains and shifts to a higher wave band at 1255 cm−1. This indicates that NH3 bonded to Lewis acid sites is more stable on the catalyst surface. With CeO2 addition, the peaks assigned to Brønsted acid sites and Lewis acid sites decrease obviously. Moreover, the band assigned to Brønsted acid sites decreases more rapidly with increasing temperature on V1Ce10ZWT. These results suggest that the addition of CeO2 decreases both the amount and strength of Brønsted acid sites on the catalyst surface. Although many research studies have proved the critical role of surface acidity in the SCR reaction,37,38 the promotional effect of CeO2 on NH3-SCR activity could not be explained by the acidity factor in this catalyst system.
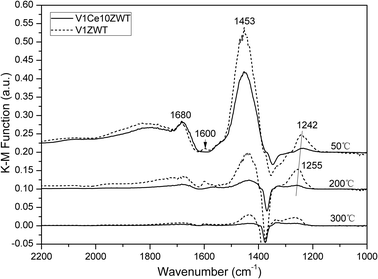 | ||
| Fig. 9 DRIFT spectra of V1ZWT and V1Ce10ZWT catalysts arising from NH3 adsorption at various temperatures. | ||
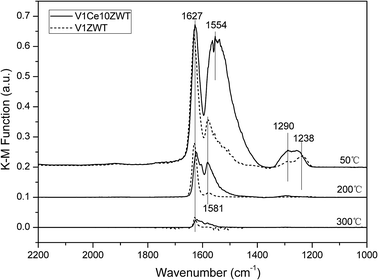 | ||
| Fig. 10 DRIFT spectra of V1ZWT and V1Ce10ZWT catalysts arising from NO + O2 adsorption at various temperatures. | ||
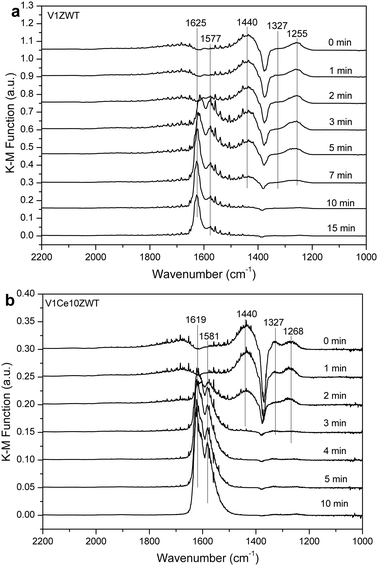
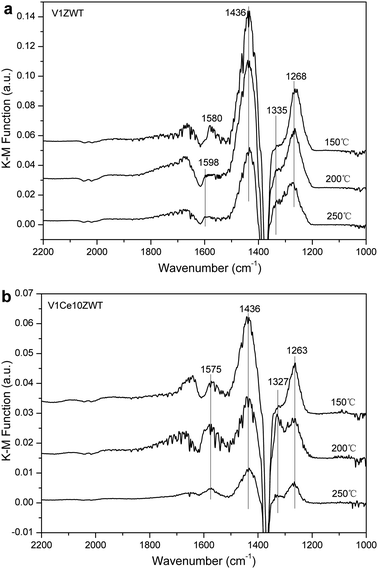
4. Discussion
CeO2 is found to enhance the low temperature NH3-SCR activity of the V2O5–ZrO2/WO3–TiO2 catalyst in the fresh state. NH3 oxidation results suggest that the enhanced activity is directly correlated with the increased oxidation property of the catalysts. According to XRD and XPS results, ceria is highly dispersed on the catalyst surface (Fig. 6), and mainly exists in the form of Ce3+ oxide (Fig. 7). The enrichment of Ce3+ could create a charge imbalance, and more oxygen vacancies are formed. This could facilitate the activation and transportation of active oxygen species, and enhance the NO oxidation to NO2 process, which is beneficial for the SCR reaction.18,44 Furthermore, the H2-TPR result reveals a new active oxygen species (Ce3+–O–Ce4+) with high reducibility by ceria addition (Fig. 8), indicating the enhanced redox property of the catalysts, which also contributes to the increase in SCR activity at low temperatures.In addition, surface acidity of the catalyst is an important factor for vanadia based SCR reactions,37,38 although some different mechanisms were proposed based on Brønsted or Lewis acid sites.43,45–47 Chen L. et al.13 proposed that the promotional effect of Ce on V0.1W6Ti was due to the fact that the Ce additive could provide stronger and more active Brønsted acid sites. However, based on our NH3-DRIFT results shown in Fig. 9, both the Brønsted acid sites (associated with V5+–OH, W6+–OH, S–OH) and Lewis acid sites (associated with Ti4+, Zr4+) decrease obviously on CeO2 modified catalysts.1,15,35,48 It could be suggested that although the presence of ceria could provide additional Brønsted acid sites,13 the coverage effect of CeO2 on these NH3 adsorption sites could be the overwhelming factor for the decrease in acidity in Brønsted and Lewis acid sites. However, the SCR activity is clearly improved by CeO2 addition (Fig. 1). This result indicates that the surface acidity (Brønsted acid sites) is not the determining factor for low temperature activity enhancement on our ceria modified catalysts, while it is the enhancement of redox properties by ceria. In our previous work we have confirmed that the amount of Brønsted acid sites could not predict the SCR reaction rate.15
On the other hand, based on our DRIFT results, noticeable NOx adsorption on V1ZWT and ceria modified catalysts was observed (Fig. 10). The nitrate species adsorbed on zirconia containing catalysts is inactive in reacting with adsorbed NH3 species (Fig. 11a), and even could not be present under simulated SCR reaction conditions above 200 °C (Fig. 12a). Nevertheless, the amount and strength of nitrate species were both increased by CeO2 addition (Fig. 10). And the nitrate on ceria modified catalysts is more active with adsorbed NH3 (Fig. 11b) and could still exist under SCR reaction conditions at 250 °C (Fig. 12b). Compared with only ZrO2 containing catalysts, the adsorbed nitrate plays a more important role in NH3-SCR reaction on ceria modified catalysts.
The NO2/NOx ratio test from Section 3.1.3 indicates higher NO2 sensitivity of ceria modified catalysts. According to the literature,22–24 a VWTi catalyst is sensitive to fast SCR reactions. And the surface nitrates play a key role in reoxidizing the V-related redox sites during the reaction.49 However, in the ZrO2 containing VWTi catalysts, fast SCR reaction is less effective and NO2 can hardly participate in the reaction. ZrO2 is found to be inactive for SCR reactions.50,51 The addition of zirconia may decrease the active VO sites, which inhibits fast SCR reactions. The inhibition effect of ZrO2 on the activity of the VWTi catalyst has been discussed in our previous work.15 With the addition of CeO2, additional redox sites are provided. Moreover, the adsorbed surface nitrates on CeO2 modified catalysts are stronger and more active in SCR reactions. These are beneficial for the enhanced fast SCR reactivity of the ceria modified catalysts.
After hydrothermal aging treatment, a clear decrease in SCR activity was observed on ceria modified catalysts (Fig. 1). This is mainly due to the sintering and segregation of the Ce component (Fig. 6), resulting in the decreased redox property and almost disappearance of Ce3+, and the process of NO oxidation to NO2 is inhibited (as indicated by NO2 formation from Fig. 1 and Fig. 2). CeO2 is found to have poor hydrothermal stability in SCR reactions. However, additional provided NO2 drastically improves the low temperature SCR activity of the V1Ce10ZWT-A catalyst (Fig. 5), indicating the high NO2 sensitivity of ceria modified catalysts. It can be found that hydrothermal aging deactivates the capability of ceria for the NO oxidation process, while additional introduced NO2 will compensate for the activity loss. It is worth noting that both the fresh and aged ceria modified catalysts can exhibit superior NH3-SCR activity and N2 selectivity in the temperature range 200–550 °C with NO2/NO = 1 in the presence of 5 wt% H2O, which is a good candidate catalyst for diesel applications.
5. Conclusions
The CeO2 and ZrO2 co-modified catalysts prepared by an impregnation method were highly active for NH3-SCR reactions in the temperature range 220–500 °C. The higher Ce loading leads to higher NO conversion. The promotional effect of CeO2 is attributed to the increased redox properties and the enrichment of Ce3+ on the surface. Moreover, DRIFTS results indicate that the adsorbed nitrates can be more active on CeO2 modified catalysts, which is beneficial for SCR reactions. However, CeO2 modified catalysts deactivate obviously at low temperature after severe hydrothermal aging at 750 °C, 10 wt% H2O/air for 12 h. This is mainly due to the sintering and segregation of CeO2 on the catalyst surface, suggesting the poor hydrothermal stability of the Ce component. Additional provided NO2 will compensate for the activity loss due to hydrothermal aging and significantly improve the low temperature SCR activity, suggesting a high NO2 sensitivity of the ceria component. It is worth noting that both the fresh and aged 1%V2O5–10%CeO2–10%ZrO2/WO3–TiO2 catalysts exhibit high NOx conversions (>95%) and superior selectivity to N2 (>98%) during 200–500 °C, at a space velocity of 120000 h−1 under conditions of NO2/NO = 1 and 5% H2O, which suggest that it is a good catalyst candidate for diesel applications.
Acknowledgements
The authors are grateful to the financial support from the National High-Tech Research and Development Program of China (No. 2011AA03A405), the Program of the Natural Science Foundation of China (No. 50972104) and the Research Fund for the Doctoral Program of Higher Education of China (No. 20090032110020).Notes and references
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- P. Forzatti, Catal. Today, 2000, 62, 51–65 CrossRef CAS.
- P. Forzatti, Appl. Catal., A, 2001, 222, 221–236 CrossRef CAS.
- D. M. Chapman, Appl. Catal., A, 2011, 392, 143–150 CrossRef CAS.
- M. Kobayashi, R. Kuma, S. Masaki and N. Sugishima, Appl. Catal., B, 2005, 60, 173–179 CrossRef CAS.
- M. Kobayashi and M. Hagi, Appl. Catal., B, 2006, 63, 104–113 CrossRef CAS.
- S. T. Choo, Y. G. Lee, I. S. Nam, S. W. Ham and J. B. Lee, Appl. Catal., A, 2000, 200, 177–188 CrossRef CAS.
- L. Baraket, A. Ghorbel and P. Grange, Appl. Catal., B, 2007, 72, 37–43 CrossRef CAS.
- S. T. Choo, S. D. Yim, I. S. Nam, S. W. Ham and J. B. Lee, Appl. Catal., B, 2003, 44, 237–252 CrossRef CAS.
- J. H. Goo, M. F. Irfan, S. D. Kim and S. C. Hong, Chemosphere, 2007, 67, 718–723 CrossRef CAS.
- M. A. L. Vargas, M. Casanova, A. Trovarelli and G. Busca, Appl. Catal., B, 2007, 75, 303–311 CrossRef CAS.
- M. Casanova, E. Rocchini, A. Trovarelli, K. Schermanz and I. Begsteiger, J. Alloys Compd., 2006, 408–412, 1108–1112 CrossRef CAS.
- L. Chen, J. H. Li and M. F. Ge, J. Phys. Chem. C, 2009, 113, 21177–21184 CAS.
- N. Marcotte, B. Coq, C. Savill-Jovitt, P. Bichon, R. Cavalier and R. Durand, Appl. Catal., B, 2011, 105, 373–376 CrossRef CAS.
- A. J. Shi, X. Q. Wang, T. Yu and M. Q. Shen, Appl. Catal., B, 2011, 106, 359–369 CrossRef CAS.
- G. Qi and R. T. Yang, J. Catal., 2003, 217, 434–441 CAS.
- W. Q. Xu, Y. B. Yu, C. B. Zhang and H. He, Catal. Commun., 2008, 9, 1453–1457 CrossRef CAS.
- T. Gu, Y. Liu, X. L. Weng, H. Q. Wang and Z. B. Wu, Catal. Commun., 2010, 12, 310–313 CrossRef CAS.
- X. Gao, Y. Jiang, Y. Zhong, Z. Y. Luo and K. F. Cen, J. Hazard. Mater., 2010, 174, 734–739 CrossRef CAS.
- S. S. R. Putluru, A. Riisager and R. Fehrmann, Catal. Lett., 2009, 133, 370–375 CrossRef CAS.
- G. Ramis, L. Yi, G. Busca, M. Turco, E. Kotur and R. J. Willey, J. Catal., 1995, 157, 523–535 CrossRef CAS.
- M. Koebel, G. Madia and M. Elsener, Catal. Today, 2002, 73, 239–247 CrossRef CAS.
- C. Ciardellia, I. Nova, E. Tronconi, D. Chatterjee, T. Burkhardt and M. Weibel, Chem. Eng. Sci., 2007, 62, 5001–5006 CrossRef.
- E. Tronconi, I. Nova, C. Ciardelli, D. Chatterjee and M. Weibel, J. Catal., 2007, 245, 1–10 CrossRef CAS.
- B. M. Reddy, P. Lakshmanan and A. Khan, J. Phys. Chem. B, 2004, 108, 16855–16863 CrossRef CAS.
- F. B. Noronha, E. C. Fendley, R. R. Soares, W. E. Alvarez and D. E. Resasco, Chem. Eng. J., 2001, 82, 21–31 CrossRef CAS.
- X. W. Liu, K. B. Zhou, L. Wang, B. Y. Wang and Y. D. Li, J. Am. Chem. Soc., 2009, 131, 3140–3141 CrossRef CAS.
- S. X. Yang, W. P. Zhu, Z. P. Jiang, Z. X. Chen and J. B. Wang, Appl. Surf. Sci., 2006, 252, 8499–8505 CrossRef CAS.
- M. A. Reiche, E. Ortelli and A. Baiker, Appl. Catal., B, 1999, 23, 187–203 CrossRef CAS.
- M. A. Reiche, M. Maciejewski and A. Baiker, Catal. Today, 2000, 56, 347–355 CrossRef CAS.
- S. M. Jung and P. Grange, Appl. Catal., B, 2001, 32, 123–131 CrossRef CAS.
- D. Pietrogiacomi, A. Magliano, P. Ciambelli, D. Sannino, M. C. Campa and V. Indovina, Appl. Catal., B, 2009, 89, 33–40 CrossRef CAS.
- X. Gao, Y. Jiang, Y. C. Fu, Y. Zhong, Z. Y. Luo and K. F. Cen, Catal. Commun., 2010, 11, 465–469 CrossRef CAS.
- R. Q. Long and R. T. Yang, J. Catal., 2000, 196, 73–85 CrossRef CAS.
- R. T. Yang, W. B. Li and N. Chen, Appl. Catal., A, 1998, 169, 215–225 CrossRef CAS.
- J. P. Chen and R. T. Yang, J. Catal., 1993, 139, 277–288 CrossRef CAS.
- J. P. Chen and R. T. Yang, Appl. Catal., A, 1992, 80, 135–148 CrossRef CAS.
- L. Lietti, I. Nova, G. Ramis, L. Dall’Acqua, G. Busca, E. Giamello, P. Forzatti and F. Bregani, J. Catal., 1999, 187, 419–435 CrossRef CAS.
- F. D. Liu and H. He, Catal. Today, 2010, 153, 70–76 CrossRef CAS.
- G. M. Underwood, T. M. Miller and V. H. Grassian, J. Phys. Chem. A, 1999, 103, 6184–6190 CrossRef CAS.
- A. Trovarelli, Catal. Rev., 1996, 38, 439–520 CAS.
- L. Lietti, I. Nova, E. Tronconi and P. Forzatti, Catal. Today, 1998, 45, 85–92 CrossRef CAS.
- G. Ramis, G. Busca, F. Bregani and P. Forzatti, Appl. Catal., 1990, 64, 259–278 CrossRef CAS.
- L. Chen, J. H. Li, M. F. Ge and R. H. Zhu, Catal. Today, 2010, 153, 77–83 CrossRef CAS.
- G. Ramis, G. Busca, V. Lorenzelli and P. Forzatti, Appl. Catal., 1990, 64, 243–257 CrossRef CAS.
- N. Y. Topsøe, H. Topsøe and J. A. Dumesic, J. Catal., 1995, 151, 226–240 CrossRef.
- N. Y. Topsøe, H. Topsøe and J. A. Dumesic, J. Catal., 1995, 151, 241–252 CrossRef.
- X. Guo, C. Bartholomew, W. Hecker and L. L. Baxter, Appl. Catal., B, 2009, 92, 30–40 CrossRef CAS.
- E. Tronconi, I. Nova and M. Colombo, Ind. Eng. Chem. Res., 2010, 49, 10374–10385 CrossRef CAS.
- R. T. Yang, J. P. Chen, E. S. Kikkinides and L. S. Cheng, Ind. Eng. Chem. Res., 1992, 31, 1440–1445 CrossRef CAS.
- D. Pietrogiacomi, A. Magliano, P. Ciambelli, D. Sannino, M. C. Campa and V. Indovina, Appl. Catal., B, 2009, 89, 33–40 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |