 Open Access Article
Open Access ArticleIce nucleation by particles immersed in supercooled cloud droplets†
B. J.
Murray
*a,
D.
O'Sullivan
a,
J. D.
Atkinson
a and
M. E.
Webb
b
aSchool of Earth and Environment, University of Leeds, Leeds, LS2 9JT, UK. E-mail: b.j.murray@leeds.ac.uk
bSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
First published on 29th August 2012
Abstract
The formation of ice particles in the Earth's atmosphere strongly affects the properties of clouds and their impact on climate. Despite the importance of ice formation in determining the properties of clouds, the Intergovernmental Panel on Climate Change (IPCC, 2007) was unable to assess the impact of atmospheric ice formation in their most recent report because our basic knowledge is insufficient. Part of the problem is the paucity of quantitative information on the ability of various atmospheric aerosol species to initiate ice formation. Here we review and assess the existing quantitative knowledge of ice nucleation by particles immersed within supercooled water droplets. We introduce aerosol species which have been identified in the past as potentially important ice nuclei and address their ice-nucleating ability when immersed in a supercooled droplet. We focus on mineral dusts, biological species (pollen, bacteria, fungal spores and plankton), carbonaceous combustion products and volcanic ash. In order to make a quantitative comparison we first introduce several ways of describing ice nucleation and then summarise the existing information according to the time-independent (singular) approximation. Using this approximation in combination with typical atmospheric loadings, we estimate the importance of ice nucleation by different aerosol types. According to these estimates we find that ice nucleation below about −15 °C is dominated by soot and mineral dusts. Above this temperature the only materials known to nucleate ice are biological, with quantitative data for other materials absent from the literature. We conclude with a summary of the challenges our community faces.
 B. J. Murray | Dr Ben Murray is a Reader in Aerosol Science at the University of Leeds, where he has held an academic post since 2006. He received his PhD from the University of East Anglia (UEA) where he studied the heterogeneous chemistry of mesospheric ice clouds with Professor John Plane. After a short post-doctoral position at UEA he embarked on a post-doctoral fellowship with Professor Allan Bertram at the University of British Columbia. His research focuses on heterogeneous ice nucleation, aerosol properties and the phases of ice in the atmosphere. |
 D. O'Sullivan | Dr Daniel O'Sullivan is a post-doctoral researcher at the University of Leeds, and has worked in Dr Ben Murray's group since 2011. He graduated from University College Cork, Ireland, in 2007 with a BSc (Hons) in Chemistry. He then went on to complete a PhD under the supervision of Prof. John R. Sodeau at University College Cork in 2011. His postgraduate research focused on examining the ice-mediated reactivities of tropospheric gases such as interhalogens, organo-nitrates and sulphur dioxide. Daniel’s current research interests are centred on examining the importance of primary biological aerosol particles as ice nuclei in mixed-phase clouds. |
 J. D. Atkinson | Mr James Atkinson is in his third year of study for a PhD at the University of Leeds under the supervision of Dr Ben Murray and Dr Steven Dobbie. He received his BSc in Meteorology from the University of Leeds in 2009. His research interests focus on cloud microphysical processes which he addresses through laboratory experiments with particular emphasis on ice nucleation by mineral dusts. |
 M. E. Webb | Dr Michael Webb is a Lecturer in Chemical Biology in the School of Chemistry at the University of Leeds. Following a PhD in Biological Chemistry with Professor Chris Abell at the University of Cambridge he undertook postdoctoral work with Professor Stephen Benkovic at the Pennsylvania State University and with Professor Alison Smith in the Department of Plant Sciences at Cambridge before moving to Leeds in 2007. His major research interests are in enzyme mechanism, the regulation of biosynthetic pathways and chemical probes of protein post-translational modification. He has been collaborating with Dr Ben Murray on the role of biological materials in atmospheric ice nucleation since 2010. |
1 Introduction
Clouds in the Earth's atmosphere substantially modify climate.1–3 They interact with both incoming shortwave and outgoing longwave radiation and can strongly influence local and regional wind patterns. Additionally, clouds are an integral part of the hydrological cycle and are responsible for water transport and precipitation, which controls water availability and drives numerous environmental processes. Ice formation in clouds strongly impacts their properties and plays a major role in precipitation formation. However, ice nucleation on solid aerosol particles remains poorly understood and quantified. In this review we assess our current state of knowledge of heterogeneous ice nucleation under conditions relevant for mixed phase (water and ice) clouds between 0 °C and about −37 °C.There are several reviews of ice nucleation in the atmosphere which incorporate findings from field measurements, laboratory studies and modelling work,4–8 as well as a recent article on ice throughout nature.9 These reviews cover multiple modes of ice nucleation for many cloud types including tropospheric mixed phase and ice clouds as well as clouds in the polar stratosphere and mesosphere. In contrast, this review is a detailed assessment of laboratory studies of ice nucleation by particles immersed in supercooled water and how this fundamental information can inform our quantitative understanding of mixed phase clouds.
We start with a general discussion of the importance of ice nucleation in mixed phase clouds and the cloud types in which ice nucleation is important. Our theoretical understanding and methods of describing ice nucleation are then presented. We then go on to examine laboratory studies of mineral dusts, carbonaceous combustion aerosol, biological species and volcanic ash in some detail. The ice nucleation efficiency of these materials is quantified using a time-independent temperature-dependent parameter, which allows comparison between different experimental methods and aerosol species. Using estimated concentrations of these aerosol species in the atmosphere we evaluate the potential concentration of ice particles (or the potential ice nuclei concentration) which they could produce. This highlights which aerosol species are most important for ice nucleation in mixed phase clouds and also where experimental data is lacking.
1.1 Aerosol–cloud interactions and ice formation
The radiative properties and lifetime of clouds are particularly sensitive to aerosol concentration, composition and size.1 This sensitivity arises because the nucleation of droplets and ice crystals directly from the vapour phase is never favourable in the troposphere. Instead, aerosol particles are required to serve as cloud condensation nuclei (CCN) to form liquid cloud droplets or as ice nuclei (IN) to form ice particles. The number of droplets which form in a cloud tends to increase with increasing aerosol number, resulting in clouds containing a greater number of smaller droplets which more effectively scatter light, thus increasing albedo, and also increase cloud lifetime.2 Anthropogenic emissions have increased aerosol concentration in the atmosphere and this is thought to have increased the amount of solar radiation being reflected back to space through increased albedo.1 Ice nucleation causes further changes to clouds which at present are difficult to quantify. While the uncertainties associated with the indirect effects of aerosol (i.e. changes in radiative properties of clouds due to anthropogenic aerosols) on liquid clouds are large, the effect of ice nucleation is even less well understood. In their latest report, the Intergovernmental Panel on Climate Change (IPCC), were unable to estimate the radiative forcing of aerosol on clouds through ice nucleation.1There are two broad categories of tropospheric clouds in which ice is present: cirrus and mixed phase types. Cirrus clouds form in the upper troposphere and typically form from liquid solution droplets which may freeze homogeneously or via heterogeneous mechanisms involving ice nuclei.10–12 While these clouds are important in terms of climate,13 transport of water vapour,14,15 and chemistry,16 for this review we focus on mixed phase clouds.
Mixed phase clouds exist at temperatures between 0 °C and about −37 °C and tend to occur in the low and middle troposphere where clouds have an important impact on climate,3 and are integral to the hydrological cycle.7 This can include clouds which form in very different dynamical conditions and are discussed in more detail in Section 1.3. The upper temperature limit of mixed phase clouds is defined by the melting of ice at 0 °C, but cloud-sized water droplets can persist in a supercooled state to below −37 °C in the absence of particles which can catalyse ice formation.7,17 These clouds can glaciate at any temperature below 0 °C in the presence of the right type of ice nucleating particle, but these particles tend to be rare in comparison with CCN. This has profound implications for cloud properties and precipitation.
1.2 Aerosol and ice nucleating particle concentrations
Aerosol concentrations vary substantially depending on location and season.18–20 In general the aerosol concentration is lower in remote marine locations than continental regions, ranging from <102 cm−3 in clean remote locations to >104 cm−3 in urban locations and 102–103 cm−3 in the free troposphere.18,19 Only a subset of these aerosol particles can serve as IN with typical IN concentrations ranging from 10−4 to 10−1 cm−3,7,21–24 with extreme values in excess of 1 cm−3 within desert dust plumes.25 These measurements indicate that IN are rare in comparison to particles capable of serving as CCN. In an aircraft campaign Rogers et al.23 reported that a fraction of 10−3 to 10−5 of the background aerosol served as IN; they also observed no correlation between IN and CCN concentrations. The fraction of aerosol serving as IN in marine air has been reported to be many orders of magnitude lower.26 More recently it has been shown that in regions not influenced by sea salt aerosol, IN concentrations are correlated to the number of aerosol particles larger than 0.5 μm.21 In addition, observations show the number of particles capable of serving as IN increases dramatically with decreasing temperature.7,22,27 In summary, only a small proportion of aerosol particles can serve as IN at all temperatures and considerable seasonal and spatial variability exists.1.3 Clouds containing supercooled water droplets
Clouds in the Earth's atmosphere have been classified by the meteorological community into types based largely upon visual characteristics and altitude.28 These visual characteristics are determined by a combination of dynamical and microphysical behaviour.Mid-level clouds, forming at altitudes of ∼2–6 km (at mid-latitudes), include altostratus and altocumulus, are usually composed of ice crystals and supercooled water droplets.28 Altostratus are uniform layer clouds which are thin enough to allow the sun to be visible and can extend over thousands of square miles. Altocumulus are patches of cloud in the shape of rounded lumps or flattened globules which are often arranged into groups, lines or waves.28 They form in regions of wide scale ascent with velocities of 5–10 cm s−1 (0.015–0.03 °C min−1; assuming a wet lapse rate of 5 °C km−1),28 although localised updrafts can be substantially faster.29
Low-level clouds, typically below 2 km, include stratocumulus and stratus clouds. Stratus are uniform featureless clouds and form fog when they reach the ground. Arctic stratus mixed phase clouds have received significant attention in recent years due to their large spatial extent and substantial impact on the Arctic climate.30 Stratocumulus are low level clumps or globules of cloud with well-defined bases as low as a few hundred meters and tops at ∼2 km. Vertical velocities in these clouds are typically less than 10 cm s−1 (0.03 °C min−1).28
Other mixed phase cloud types have a much greater vertical extent. Nimbostratus can extend from close to the ground into the mid-troposphere and are associated with rain or snow. These clouds are often caused by regions of large scale slow ascent such as warm fronts. Cumulus clouds are detached dense clouds with heaped tops and flat bottoms and can extend from as low as ∼0.5 km up to ∼6 km. These clouds are associated with areas of fast ascent, with updraft velocities in the range of 1–5 m s−1 (0.3–1.5 °C min−1), caused by convection or cold fronts. If the ascent is strong enough (sometimes referred to as deep convection), they can grow into cumulonimbus clouds and begin to produce precipitation. Such clouds can extend to altitudes of 12 km in the mid-latitudes or even higher in the tropics.28 Cumulonimbus have the greatest updraft speeds of all clouds, with velocities in the 10s of meters per second (10 m s−1, or 36 km h−1, is equivalent to 3 °C min−1) which are driven in part by the release of latent heat from droplet and ice formation.
In addition, clouds can form as air is forced to ascend over a hill or mountain. These orographic clouds include lenticular clouds which are striking lens shaped clouds with sharp edges. Downwind the air may be set into an oscillation with clouds, known as wave clouds, forming in the uplift phase. These clouds have received attention by the cloud community because they provide a well-defined system and have been used as a ‘natural laboratory' in aircraft field experiments.31–33
Given that IN are rare in the Earth's atmosphere it is perhaps not a surprise that many clouds can persist in a supercooled liquid state. In deep convective clouds liquid water has been observed down to −37.5 °C,34 which is close to the limit defined by laboratory experiments for homogeneous freezing. If we consider a cloud of supercooled droplets 10 μm in diameter, 99% of them will freeze within 1 minute at −37.5 °C according to measured nucleation rate co-efficients.17 Significant amounts of supercooled water (not including haze solution particles) are unlikely to persist at lower temperatures and this probably defines the lower limit to supercooled water in the troposphere. In other clouds with weaker updrafts this temperature limit is several degrees warmer due to an increase in time scales.
In many clouds glaciation occurs at much higher temperatures. In general the fraction of clouds which contain supercooled water decreases with decreasing temperature;35–39 this reflects the measurements discussed in Section 1.2 showing that the number of aerosol particles capable of serving as IN increases with decreasing temperature. The fraction of mid-level stratus clouds containing ice in four locations around the globe is shown in Fig. 1; there are some striking differences. For example, the fraction of clouds containing ice is ∼70% at −19 °C above Leipzig (51° N, northern Germany); whereas at a similar latitude in the southern hemisphere (Punta Arenas, 53° S) the same fraction frozen is only achieved below −34 °C.38–41 Kanitz et al.41 suggest that this difference is due to the presence of more extensive sources of IN in continental Europe compared to Punta Arenas which receives air from the Pacific Ocean. At Cape Verde off the west coast of Africa, cloud glaciation only occurs with a frequency of about 5% at −20 °C. This is remarkable considering its location in the path of dust emissions flowing from Africa.25,42 This is a similar finding to that of Ansmann et al.36 who showed that clouds above Morocco remained in a supercooled state down to −18 °C despite being colocated with Saharan dust. In contrast, Sassen et al.43 report that Saharan dust is capable of glaciating altocumulus clouds at temperatures between −5 and −9 °C.
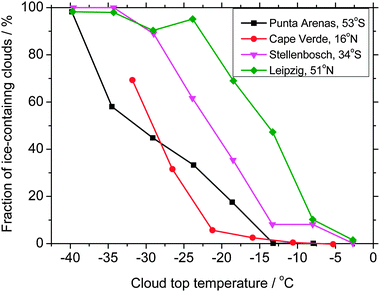 | ||
| Fig. 1 The fraction of mid-level stratus clouds which contained ice determined using polarization lidar in a number of locations. This data is taken from Kanitz et al.41 and is further discussed in other articles.37,39 The clouds examined in this work were mainly within the altitude range 2–8 km and were mostly less than 1 km thick with small optical depths. | ||
The global distribution of the fraction of clouds containing supercooled water has been measured by satellite based LIDAR (a technique comparable to RADAR, but using laser light).35,40 These results highlight the regional and seasonal variability of the occurrence of supercooled water in the atmosphere. Based on this data Choi et al.40 suggest that the presence of dust leads to a significant reduction in cloud albedo, counteracting the direct effect of the dust.
1.4 Consequences of ice nucleation in supercooled clouds
Despite their rarity, IN have a substantial impact on the properties of mixed phase clouds. In part this is because IN are rare in comparison to particles capable of serving as CCN, but it is also related to the fact that a liquid cloud below 0 °C is thermodynamically unstable. In many systems, including clouds, a transition to a more thermodynamically stable state can happen promptly despite the system having previously persisted in a metastable state for a long period of time.44 Ice nucleation in a small fraction of cloud droplets can trigger a transformation in the whole cloud and substantially modify its properties. Since ice is more stable than supercooled water below 0 °C, ice crystals have a lower equilibrium vapour pressure.45 This causes a water vapour concentration gradient to form between the air around ice crystals and the supercooled water droplets, leading to growth of the ice crystals at the expense of the supercooled droplets. This is known as the Bergeron–Findeisen (sometimes referred to as the Wegener–Bergeron–Findeisen) process.7,46 The timescale for glaciation through this process depends on temperature and pressure and in the middle troposphere at −20 °C the timescale is on the order of minutes. This process is thought to be critical in many low- and mid-level clouds, resulting in a cloud containing large ice crystals of a considerably lower concentration than the original liquid droplet concentration. In vigorous convective systems a supersaturation with respect to liquid water may be maintained, even in the presence of ice particles and under these conditions both ice and supercooled droplets will grow.47,48 If sufficiently large ice crystals form, they can collide with supercooled droplets, which freeze on contact in a process referred to as riming. This collision coalescence process is an important mechanism in the formation of rain and hail.Cloud glaciation is further complicated by ice multiplication mechanisms (also termed secondary ice production processes). The mechanism which has received most attention is the Hallett–Mossop process49 in which riming at around −3 to −8 °C (most effective at −5 °C) leads to ice splinters being ejected, yielding ice crystal concentrations that are orders of magnitude higher than IN concentrations.29,50–52 This process requires supercooled water droplets with a diameter greater than 24 μm and relative impact velocities need to be greater than 1.4 m s−1.49,53 Pruppacher and Klett7 present data which suggests ice multiplication mechanisms are important above about −20 °C, which is significantly below the temperature range where the Hallett–Mossop process is known to be active. This is thought to be due to the break-up of fragile dendrites which can form in this temperature range,7,54 but ice multiplication mechanisms remain an area of significant uncertainty. From the point of view of ice nucleation, any IN active in a regime relevant for ice multiplication will have a disproportionate impact on cloud glaciation.
1.5 Modes of ice nucleation in mixed phase clouds
There are a number of ways in which aerosol particles are thought to trigger ice nucleation in the atmosphere. These are: (i) deposition nucleation, which occurs when vapour directly deposits onto a solid surface as ice. (ii) Immersion freezing, which happens when ice nucleates on a solid particle immersed within a supercooled liquid droplet. (iii) Condensation freezing, which occurs when water vapour condenses on a solid particle, possibly due to the presence of some hygroscopic material, and then freezes (this is sometimes referred to as deliquescence freezing55 and is sometimes classed together with immersion freezing31 or deposition freezing56,57). (iv) Contact freezing, in which a solid particle collides with a supercooled liquid droplet, resulting in ice nucleation.7,58 In addition to these standard modes of nucleation, an ‘inside-out’ nucleation process has been identified in which solid particles immersed within a droplet come into contact with the air–water interface and only then initiate freezing.59–61 It has been proposed that this may occur while a droplet evaporates.59In many situations field data and modelling studies indicate that liquid water droplets are a prerequisite for ice formation. This implies that deposition nucleation plays a secondary role under mixed phase cloud conditions. Westbrook and Illingworth62 used radar and lidar measurements to show that 95% of ice particles which formed at temperatures above −20 °C originate within supercooled clouds. In a study of altocumulus clouds over Cape Verde Ansmann et al.37 found that 99% of clouds they probed had a liquid layer at their tops. They concluded from this observation that deposition and condensation ice nucleation are not important mechanisms in these clouds. In the majority of cases above −30 °C at high latitudes de Boer et al.57 showed that for ice to form a liquid cloud was required to form first. Ice supersaturated haze layers are observed in these regions with no apparent ice formation, suggesting that deposition and condensation (below water saturation) nucleation is not important.57 In modelling mountain lee wave clouds Field et al.31 concluded that ice formation required liquid water to form first. Based on the same field study, Twohy et al.33 stated that deposition nucleation in ice-supersaturated, but water-subsaturated, conditions was not observed and that there was no evidence for contact nucleation in the evaporating cloud. In contrast, Sassen and Khvorostyanov63 concluded that ice formed below water saturation in the formation of altocumulus clouds impacted by smoke in Alaska. Nevertheless, most studies conclude that water saturation is a prerequisite for ice formation in low- and mid-level clouds and therefore either contact or immersion freezing dominate. Phillips et al.64 suggest that contact mode nucleation is of secondary importance because thermophoretic effects (force exerted by a temperature gradient) favour contact nucleation only in evaporating droplets, but these droplets tend to disappear before they can freeze.
1.6 What makes an effective ice nucleating particle?
Pruppacher and Klett7 list a number of requirements for a particle to be an effective IN. These are: (i) insolubility, water absorption may cause the substrate to disintegrate; (ii) size, a correlation between the number of ‘larger’ aerosol particles and IN has been reported;7,21 (iii) chemical bond requirement, water must be able to make chemical bonds with the IN surface; and (iv) crystallographic, a good IN should template ice. These requirements might be met either on a particular crystallographic face of a nucleant or at specific active sites such as cracks or defects.These criteria were set out in part to help the community establish which atmospheric materials are likely to serve as IN, but these criteria are perhaps too narrow. For example, it has been shown that crystalline soluble salts such as ammonium sulphate65 and oxalic acid66 nucleate ice in the immersion mode (in saturated solution droplets). These results suggest that the insolubility requirement of IN should be rephrased to be a ‘solid’ requirement, although this would not include ice nucleation by ordered surfactants.67 Hygroscopic solutions in a glassy state,11,68 atmospheric organic aerosol,69 and secondary organic aerosol70 have also been shown to nucleate ice despite their amorphous (non-crystalline) nature which challenges the concept of a lattice match. Soot is also a poorly ordered solid material which is known to catalyse ice formation.56,71,72 In the early literature ice nucleation by the clay mineral kaolinite was ascribed to its lattice match with ice, which is thought to template ice.7 However, more recent computational work indicates that this is not the case, suggesting kaolinite nucleates ice for a different reason, possibly due to defects in the crystal structure.73–75 In summary, these commonly cited guidelines need to be taken with caution since our basic understanding of what makes an effective ice nucleating material remains limited. At present the only way of determining if a particular material is effective at nucleating ice is by quantitative experimentation.
1.7 Impact of ice nucleation in mixed phase clouds on climate: aerosol indirect effects involving ice formation
There have been substantial changes in the amount and composition of aerosol particles in the Earth's atmosphere since pre-industrial times.1 In the present day atmosphere the number of aerosol particles over the continental regions is greatly enhanced due to human activity.1,76 It has been suggested that in pre-industrial times, CCN loading in the continental regions was of the same order of magnitude as in oceanic regions (100–300 cm−3, in the absence of dust storms or fires).20,77,78 Andreae et al.20 state that primary and secondary biogenic sources dominated in the pre-industrial era. In contrast, the present continental aerosol loading is orders of magnitude larger than that over oceans due to proximity to significant anthropogenic sources. The dramatic changes in aerosol loading of the atmosphere from pre-industrial to present day are illustrated in Fig. 2. In the pre-industrial atmosphere aerosol concentrations were much more homogeneous between maritime and continental regions than they are in the present atmosphere, which illustrates the anthropogenic impact on global aerosol concentrations.77 Using modelled global distributions such as this it is possible to estimate the impact humankind has had on CCN concentrations,76,77 but it is not known how the number of IN has changed due to human activity. Hence, assessing the anthropogenic impact on cloud glaciation remains a significant challenge.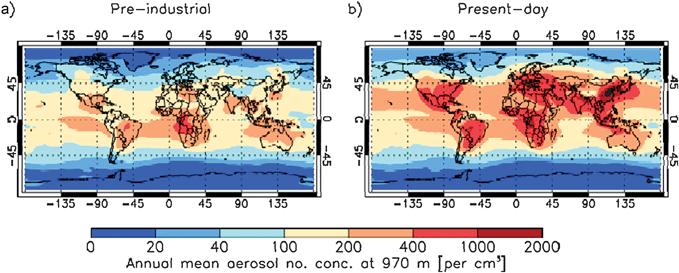 | ||
| Fig. 2 Modelled global distribution of annual mean aerosol particle concentrations at low-cloud altitudes in the pre-industrial and present day atmosphere (personal communication from Anja Schmidt and adapted from Schmidt et al.77). The aerosol particle concentration is equal to the concentration of aerosol particles with a dry diameter larger than 70 nm (including sea salt, sulphate, black carbon, organic carbon and dust) which contain a soluble component and therefore will potentially serve as CCN. Hence, any insoluble material in these droplets will potentially serve as immersion mode ice nuclei. | ||
In this section we discuss a number of potential indirect effects involving ice in mixed phase clouds. Let us start with a description of a cloud forming under ‘clean’ conditions, where ‘clean’ is defined as a natural aerosol loading in the absence of dust storms, fires or volcanic activity. With the relatively few CCN the resulting number of cloud droplets tends to be low and these droplets tend to grow to larger sizes. For example, Rosenfeld et al.79 reported droplet sizes in marine clouds in excess of 40 μm (effective diameter). In such clouds ice-free warm rain processes, in which droplets grow large enough to sediment, are feasible.79 During sedimentation, they can coalesce with more water droplets and with sufficient time precipitation forms. This leads to removal of liquid water from the cloud, which has the effect of removing latent heat that would otherwise be released if ice formation occurred and would lead to a higher and colder cloud top. If IN are present ice crystals may form, and if this occurs in the temperature regime required for the Hallett–Mossop process49,53 even a very small concentration of IN may lead to rapid cloud glaciation.79,80 Clouds are clearly sensitive to the number of ice nuclei, but they are also sensitive to the number of aerosol particles which can serve as CCN as well as the ratio of CCN to IN. We will now consider a range of cases with contrasting ratios of CCN to IN.
(i) Increased CCN and low IN: This might come about if the overall aerosol concentration increases, through an additional source of soluble aerosol particles, but where the number of IN remain constant. In this case a greater number of CCN leads to more, but smaller cloud droplets. In shallow clouds this leads to a decrease in precipitation via the warm rain process.78 In contrast, for convective clouds this inhibits the warm rain process, allowing more water to arrive at the freezing level and more latent heat to be released, leading to a more vigorous updraft. This has a number of effects including increased transport of water, particles and trace gases into the mid and upper troposphere (and sometimes the stratosphere), altering atmospheric dynamics and creating more intense precipitation in convective systems.81–85 The latent heat released during droplet and ice formation is consumed when the hydrometeors evaporate, but when condensed water is removed via precipitation the heat is retained in the atmosphere which energises convection and atmospheric circulation.78 Andreae et al.81 reported that the hail which developed in a smoke-impacted cloud in the Amazon was so large that it dented the nose of an aircraft, which is striking as no hail was observed in clouds not impacted by smoke. Increasing CCN in cumulus clouds has been shown to decrease or eliminate precipitation through reduced droplet size and consequently reduced efficiency of ice multiplication and warm rain processes.48,52 Clouds with an increased number of smaller droplets scatter more incoming solar radiation and it is this effect which is accounted for in the IPCC's assessment of radiative forcing.1 In addition they also tend to have a longer lifetime. Clouds with smaller droplets tend to have a lower glaciation temperature (hence a higher glaciation altitude) since droplets are not large enough for secondary ice multiplication processes (see Section 1.4). Rosenfeld et al.79 reported convective clouds forming in a smoke plume over China with droplet sizes of only 10–15 μm (effective radius) and glaciation only occurring below −33 °C. In this case, the smoke aerosol particles did not serve as an effective IN, but did serve as CCN.
(ii) Increased CCN and IN: DeMott et al.21 reported that the concentration of IN in a number of locations is related to the number of aerosol particles larger than 0.5 μm as well as temperature. Hence, an increase in the number of larger aerosol particles is generally associated with increased IN number densities. As in case (i), increased CCN concentrations suppresses the warm rain process. However, Rosenfeld et al.79 suggested that enhanced IN could then trigger glaciation in convective clouds which will enhance precipitation through the Bergeron–Findeisen process. Hence, suppression of the warm rain process by enhanced CCN may be counteracted by the enhanced precipitation from IN formation.
(iii) Increased IN and low CCN: As discussed in Section 1.2, concentrations of CCN are usually far greater than for IN. If particles which are not only CCN but are also efficient IN are introduced, the total IN number will increase substantially with a minimal increase in CCN number. In this situation one might not expect a dramatic change in cloud droplet size, hence warm rain and also the Hallet–Mossop process may remain active. Increased IN and increased glaciation temperature would be particularly important if the initiation of freezing were shifted into the regime important for secondary ice production. However, Rosenfeld et al.79 suggest that even very small concentrations of IN can effectively glaciate a convective cloud formed with few CCN (due to secondary ice production) and that these clouds may therefore not be particularly sensitive to increased IN.
There have been a limited number of studies at the global scale investigating the impact of ice nucleation in mixed phase clouds. Lohmann and Diehl86 explored the impact of mineral dusts and black carbon as ice nuclei on mixed phase clouds. They show that the lifetime of clouds is reduced due to ice nucleation enhancing precipitation, resulting in a substantial warming due to reduced cloud cover. Their sensitivity study shows that ice nucleation by mineral dust (using ice nucleation parameterisations,87 based on laboratory data lacking any quantification for surface area87–89) had a radiative forcing of up to 2.1 W m−2. This significantly counteracts the impact of increased CCN and similar results have been obtained in other studies.90,91 In a later study, Storelvmo et al.92 assessed the impact of increased ice particle concentrations from increased IN and showed that this created more reflective clouds. This effect largely counteracted the positive radiative forcing from decreased cloud lifetime. In summary, the ice-related aerosol indirect effects are complex and research in this area is still in its infancy. There are many uncertainties in our understanding of clouds and the lack of a quantitative understanding of heterogeneous ice nucleation by atmospheric aerosol is a severe limitation.
2 Classical nucleation theory (CNT)
Classical nucleation theory has been applied in a wide range of fields in which a new phase nucleates from the parent metastable phase.93,94 One of its strengths is that it makes use of readily available macroscopic quantities to estimate the rate at which the new phase nucleates. Although simplifying assumptions are made in CNT, it does a remarkably good job at reproducing observed trends (e.g. see ref. 17, 93, and 95). This theory can be applied to the nucleation of ice from supercooled water or from a supersaturated water vapour as well as to the nucleation of amorphous materials (e.g. liquid) from vapours. The equations set out below are written in a general form that could be applied to any situation in which a new phase nucleates from a metastable phase, but our focus here is on ice nucleating in supercooled water.2.1 Homogeneous classical nucleation theory
In pure water, clusters form and dissipate through the addition and removal of water molecules.| H2O + (H2O)n−1 ⇆ (H2O)n | (1) |
| ΔGcl = ΔGs + ΔGv | (2) |
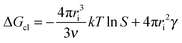 | (3) |
The radius of the critical cluster (rg) is ri where dΔGcl/dri = 0, hence, the critical radius is
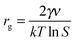 | (4) |
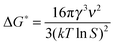 | (5) |
It should be noted that classical nucleation theory has a number of weaknesses. A significant issue is the assumption that parameters such as interfacial energy, density and saturation ratio are the same for a nanometre sized cluster of molecules as they are for a macroscopic well defined material. For example, it is thought that the interfacial energy is size dependent.102 In addition, the assumption that the initial cluster is spherical may be incorrect. Nevertheless, classical theory does provide a theoretical framework with which to understand nucleation and is capable of reproducing experimental observations.
The rate coefficient (J, nucleation events per unit volume per unit time) at which ice crystals appear in supercooled water is related to the Gibbs energy required to form a critical cluster in an Arrhenius form:
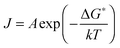 | (6) |
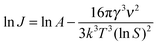 | (7) |
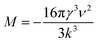 | (8) |
2.2 Heterogeneous classical nucleation theory
A suitable surface in contact with supercooled water can reduce the energy barrier to nucleation and therefore catalyse ice formation. The Arrhenius equation for homogeneous nucleation can be adapted for the heterogeneous nucleation coefficient (Jhet/cm−2 s−1):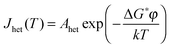 | (9) |
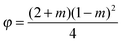 | (10) |
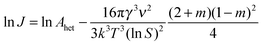 | (11) |
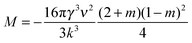 | (12) |
3 The structure of ice which nucleates and grows
In many studies it is implicitly assumed that that stable hexagonal phase of ice nucleates and grows when atmospheric water droplets freeze. This is perhaps reasonable given older literature (e.g. see Hobbs106), which indicate a metastable phase of ice only exists below about −70 °C. However, it is well known that in many systems a metastable phase will nucleate and crystallise in preference to a stable phase and will only later recrystallise to the stable phase at some finite rate; this is known as Ostwald's law of stages.94,96 In a recent study Malkin et al.97 showed that pure water droplets of 0.9 μm diameter exclusively crystallised to a metastable form of ice at about −40 °C, consistent with Ostwald's law of stages. This supports the hypothesis that crystallisation of water always proceeds through a metastable phase.Let us now discuss the metastable phases of ice which might form when water freezes. Out of the 15 crystalline phases of ice that are known only ice I can form at atmospheric pressure.107 Traditionally it was thought that ice I came in two distinct forms: the stable hexagonal phase (ice Ih) and the metastable cubic phase (ice Ic). Both of these phases are made up of water molecules arranged in layers consisting of puckered six-membered rings. In ice Ih each layer is a mirror image of the previous layer, whereas in ice Ic each successive layer is shifted a distance of half of the diameter of the hexagonal ring. However, diffraction data show that ice which forms from pure water droplets is neither of these phases.97 In fact, Malkin et al.97 show that this ice is fully stacking disordered, i.e. it is composed of randomly stacked layers of cubic and hexagonal sequences. This ice is named stacking disordered ice (ice Isd).97 Furthermore, ice which was previously identified as ice Ic was in fact stacking disordered ice.97,108,109
The diffraction work of Malkin et al.97 showed that water droplets crystallised to a stacking disordered ice, but it is important to bear in mind that we do not know from experiments which phase nucleated (i.e. the phase of the critical nucleus). This is important because parameterisations based on classical theory require some knowledge of the phase of the critical nucleus. Usually it is implicitly assumed that ice Ih forms,110,111 but others have suggested the metastable ice Ic may form.17,95,105 Homogeneous nucleation rates are more consistent with a substantially lower interfacial energy of ice Ic,17,95,101 but direct observational evidence of the nucleating phase is lacking. However, computational studies indicate stacking disorder is present to scales approaching that of the critical cluster,97,112–115 and it is possible the critical cluster does not have a well-defined structure. In summary, the crystallisation of an atmospheric droplet is a poorly understood process and it is clear that the assumption that ice Ih nucleates and initially grows may not be correct.
4 Descriptions of experimental ice nucleation data
In order to quantify ice nucleation in models and also to be able to quantitatively state which materials are the most import IN in the atmosphere we need a means of quantifying how efficiently a particular material nucleates ice. In the early days of ice nucleation research it was common to quote the threshold temperature or relative humidity at which ice was observed to form under a particular set of conditions (for example see Mason22 and Pruppacher and Klett7). While a threshold value might be useful when comparing different materials within the context of a single experimental design, it is much less useful when comparing results from different experiments or attempting to estimate the number of ice particles which will form under a given set of atmospheric conditions. For example, on injecting kaolinite aerosol particles into a chamber containing supercooled droplets Mason and Maybank116 reported a threshold temperature of −9 °C, whereas Hoffer88 reported a very different freezing temperature for 100 μm diameter droplets, containing an unspecified quantity of kaolinite, of −32 °C. Given this data alone it is impossible to assess the impact of glaciation by this material in clouds. The variation in freezing temperatures measured in these early experiments most likely stems from differences in both the amount of material in the droplets and the mode of nucleation. In order to quantitatively compare ice nucleation by different materials in laboratory studies it is essential to quantify how much material is available for nucleation and to design the experiment in a manner such that the mode of nucleation is unambiguous.There are two primary motivations for wanting to describe heterogeneous ice nucleation. The first motivation is to attempt to understand and describe ice nucleation from a fundamental perspective, whereas the second is to describe ice nucleation by complex natural materials in a way which captures the dominant ice nucleating properties of natural aerosol in a relatively simple way.
Nucleation is a stochastic process, meaning the probability of nucleation occurring is dependent on time as well as the amount of heterogeneous nucleating material. This is clear in the classical theory described above. In some idealised laboratory experiments the sample being investigated was uniform with each particle having much the same ice nucleating probability as the next.105,117–119 In contrast, natural atmospheric aerosol tend to be much more complex, primarily for the reason that natural aerosol is made up of a wide range of different materials each with its own ice nucleating characteristics. For example, droplets containing relatively pure kaolinite froze over just a few degrees and freezing systematically shifted to higher temperatures as the amount of kaolinite in the droplets was increased or cooling rate was reduced; this data fitted a simple stochastic model.105 In contrast, droplets containing a dust more representative of natural atmospheric mineral dust composed of a range of minerals nucleated ice over a much broader temperature range and required a much more complex model to describe the data.120–122 In order to simplify this complex behaviour it has been suggested that the time dependence, or stochastic behaviour, of ice nucleation can be neglected, which has given rise to the singular description. In the following sections we discuss the stochastic and singular approaches.
4.1 The stochastic description
In this discussion we will consider an array of droplets each containing material which could nucleate ice. The stochastic model is often implicitly assumed to refer to a situation in which each droplet in an array contains exactly the same material and therefore has the same probability of freezing. This case has been referred to as the single component stochastic description, with ‘single component’ referring to the uniformity of droplets, i.e. the freezing of all droplets can be described by a single probability.105,120 The condition of droplet uniformity often does not always hold and droplets may contain different amounts of material or different particle types. This heterogeneity leads to some droplets having a greater probability of freezing at a given temperature and freezing of the whole population of droplets cannot be described by the single component stochastic description. Instead each droplet will have a temperature and time dependent probability of freezing and can then be described by a stochastic model which includes multiple nucleating components to account for the droplet to droplet inhomogeneity. This section begins with a discussion of the single component stochastic description and then expands to describe the how various authors have developed this description to cope with multiple ice nucleating components.The rate (R) at which droplets freeze to form ice particles can be defined as:
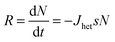 | (13) |
In a time interval from t1 to t2, the number of droplets which are liquid will decrease from N1 to N2:
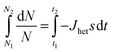 | (14) |
N2 = N1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−JhetsΔt) exp(−JhetsΔt) | (15) |
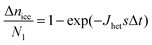 | (16) |
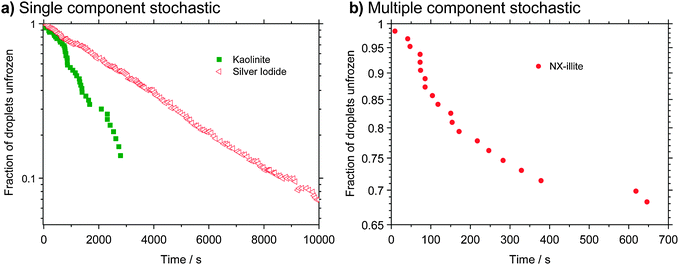 | ||
| Fig. 3 Decay of liquid droplets with time under isothermal conditions. (a) Data are shown for kaolinite105 and also silver iodide;118,119 note that the surface area and temperature were different in the two experiments. The kaolinite experiment was done with multiple kaolinite containing droplets supported on a hydrophobic surface and freezing was monitored over time at −29 °C. The silver iodide experiment was done by repeatedly cooling to −4.9 °C, waiting for freezing, and thawing a droplet containing a silver iodide crystal. Each point represents the time it took for the droplet to freeze in one experiment. Freezing by material in both of these experiments is consistent with a single component stochastic model since the decays are approximately exponential. However, multiple experiments would be needed to assess particle-to-particle variability in the case of the silver iodide particles. (b) Data for an array of droplets held at −30 °C containing a dust which is made up of numerous minerals (NX-illite). Each droplet has a different probability of freezing at this temperature which is consistent with the multiple component stochastic model. Note that the axes displaying the fraction of droplets unfrozen are plotted on a log scale in both panels a and b (although this is difficult to see by eye in panel b). | ||
Some care needs to be taken in justifying the assumption of droplet uniformity required to apply the SCS model. It is not possible to decide if this approximation holds when only a single population of droplets are cooled under a specific set of conditions; other experiments are required.120,124 One approach is to test the time dependence of nucleation under isothermal conditions. A uniform population of liquid droplets will decay exponentially with time as discussed above. The case where the decay is not exponential is discussed in the next section. Another test is to repeat the experiments at different cooling rates. Results from an ice nucleation study by a relatively pure kaolinite particles immersed in a population of droplets were well described by the SCS model for experiments with a wide range of cooling rates.105 Hence, freezing by this kaolinite sample is consistent with the SCS model. A contrasting study is that of Broadley et al.120 who studied an illite rich sample containing many minerals. They show that single data sets (from a single population cooled at a constant rate) can be parameterised with the SCS model. However, when they compared their results for experiments at different cooling rates the SCS model failed, indicating that the assumption of droplet uniformity did not hold for this material. Instead the description of ice nucleation by this complex mixture of minerals must reflect droplet heterogeneity (i.e. different droplets have a different probability of freezing).
There are a number of methods discussed in the literature which we place under the general heading of MCS descriptions.105,120–122,124–128 In general these methods sum the effect of many different ice nucleating particles. Murray et al.105 express the fraction of droplets which activate to ice in a time interval where the droplets have a distribution of ice nucleating abilities as:
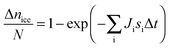 | (17) |
4.2 The singular description
An alternative way of dealing with the complexity of the particle to particle variability is to make the assumption that the time dependence of nucleation is of secondary importance in comparison to the distribution of ice nuclei types. This has given rise to the singular description.7,129,130 A result of this assumption is that nucleation will occur at a particular site at a characteristic set of conditions. Immersion mode freezing by a particular IN type will occur at a characteristic temperature, Tc, above which ice nucleation cannot occur. The temperature at which an IN containing multiple nucleation sites activates to ice is determined by the nucleation site with the highest Tc. A similar statement can be made about the deposition mode, but where there are two variables: temperature and relative humidity.Let us consider the case where there is an array of droplets where the ice-nucleating ability of the particles dispersed through the droplets population is heterogeneous (i.e. different droplets have nucleation sites with different Tc in them). In this case freezing would occur over a range of temperatures, i.e. a spectrum of ice activation temperatures. On cooling, the fraction of droplets that freeze by temperature T, fice(T), can be described by:105,120,131–133
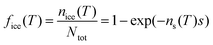 | (18) |
The density of surface sites that become active per unit temperature, k(T), is formally related to ns(T) by:
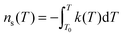 | (19) |
Vali134 defined similar quantities, but rather than expressing the cumulative nucleation sites per unit surface area, it was expressed per unit volume and was termed the cumulative nucleus spectrum K(T):
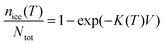 | (20) |
| K = nssspCm | (21) |
| K = nmCm | (22) |
| K = nnCn | (23) |
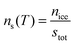 | (24) |
4.3 Modified singular description
In order to test the assumption of time independence, Vali130 conducted a sequence of experiments to study the repeatability of freezing individual water droplets and found that the same droplets tended to freeze within a few degrees on repeated temperature cycling, although differences of up to 5 °C were occasionally observed. According to the standard singular description, if the same droplet is cooled and warmed repeatedly it will always freeze at the same temperature. In comparison with the broad spectrum of freezing temperatures (−5 to −24 °C) this variability on repeated freezing is relatively small, but nevertheless indicates that there is a non-negligible stochastic element to freezing. The time dependent nature of nucleation was illustrated by an observed 0.4 °C shift in freezing temperatures when the cooling rate was increased by a factor of six. In an earlier study Vali and Stansbury137 observed a 0.2 °C change in mean freezing temperature with a factor of two change in cooling rate for the heterogeneous freezing of distilled water droplets. This observation led Vali129 to propose a modification to the singular approximation, presented here in terms of surface area rather than droplet volume:105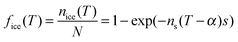 | (25) |
| α = β log(|r|) | (26) |
5 Methodologies for quantifying ice nucleation by particles immersed in supercooled water
The experimental techniques for the investigation of immersion mode ice nucleation can be conveniently classified into two broad categories where droplets are: suspended in gas (e.g. an aerosol of liquid droplets) or immobilised on a surface or in oil.5.1 Droplets suspended in gas
Cloud expansion chambers are large temperature and humidity controlled vessels which are used to quantify deposition, immersion and condensation mode ice nucleation.71,135,138 A typical experiment involves expanding the gas in the chamber via pumping, resulting in a reduction of temperature and enhanced saturation ratio. In order to observe immersion mode freezing in a cloud chamber it is first necessary to increase the humidity to water saturation to activate aerosol particles as liquid droplets. If the sample nucleates ice below water saturation this may not be possible. On continued cooling, freezing can then be quantified by the appearance of ice crystals.Continuous Flow Diffusion Chambers (CFDCs) are commonly employed in the laboratory and field for quantification of the concentration of IN in air samples.21,139–143 In order to determine the IN concentration, aerosol particles are passed into the chamber where the humidity is controlled by two ice coated plates set at different temperatures.143 Particles which nucleate ice in the time the aerosol spends in the supersaturated region are counted as they exit the chamber. A range of relative humidities are accessible in these instruments which allows studies of ice formation below water saturation and also in aerosol particles which have activated to droplets at or above water saturation. Another approach is to activate aerosol to droplets in advance of admitting them to the CFDC, which ensures that only the immersion mode is studied.126
Other techniques: Wind tunnels can be used to suspend single droplets in a flow of gas at controlled temperature and freezing probabilities are established as a function of temperature through observations with many droplets.89,144,145Laminar flow chambers generate a supersaturation by mixing a cool dry flow containing aerosol with warmer more humid air.132,146 The proportion of aerosol which activated to ice under varying conditions is then determined down-stream. Free falling droplet systems have also been used to quantify freezing in micron sized water and solution droplets.147 Streams of droplets are allowed to fall into a well characterised cold chamber and freezing detected using a polarised laser system. Electrodynamic levitation of micron sized droplets has been used to study homogeneous nucleation of water,123 and could also be used for heterogeneous studies. Aerosol flow tubes have been used to study freezing by size selected nanometer scaled solid particles immersed in solution droplets of know composition.148
5.2 Droplets immobilised on a surface or in oil
Supporting droplets on a hydrophobic surface and subsequent cooling is a common method of determining the efficiency with which any suspended particles nucleate ice.149 The droplets used range from microlitre down to picolitre sizes, with advantages at both ends of the range. Microlitre (millimetre sized) droplets are better suited to detection of rare, but highly efficient ice nuclei. A disadvantage is that droplets without any added nucleating material can freeze tens of degrees above the homogeneous limit, probably due to contamination in the droplets or on the supporting surfaces.149 In order to access the full range of atmospheric temperatures, i.e. down to homogeneous freezing, it is necessary to finely divide the droplets which, substantially reduces the probability of finding a contaminating particle in any one droplet. Division of droplets into picolitre volumes (microns in scale – similar to cloud droplets) and working with carefully prepared hydrophobic surfaces has been used to quantify heterogeneous freezing down to ∼−37 °C.17,105Immersing droplets in oil is another method of immobilising droplets for freezing studies. Droplets containing ice nuclei can be supported between two immiscible oil layers,88 or in an oil emulsion with a surfactant to stabilise the suspension.121,150,151 Droplets supported on hydrophobic substrates have also been immersed by oil which blocks mass transfer between supercooled water and ice crystals.105,120 This technique has the advantage that the time dependence of nucleation can be quantified by exposing droplets to a well-defined set of conditions for periods of hours (rather than seconds or minutes which are possible in CFDCs or cloud chambers).105,120 Freezing has commonly been monitored using microscopy,88,120 or differential scanning calorimetry.121,151 Microfluidic techniques have also been applied to heterogeneous freezing in which droplets were carried in a liquid fluorocarbon over a temperature controlled stage, with freezing detected optically.152
6 Classes of ice nucleating aerosol
6.1 Mineral dusts
Large quantities of mineral dust are aerosolised into the atmosphere from a range of sources, most notably from arid regions in Africa, the Middle East and Asia, referred to as the Dust Belt.42 Mineral dusts of varying types were shown to serve as effective ice nuclei decades ago.7,22 Nevertheless, it is only in recent years that quantitative measurements have been made on their ice nucleation efficiency in the immersion mode.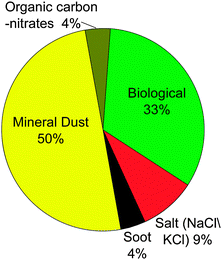 | ||
| Fig. 4 Relative contributions of different particle types in ice residual particles sampled (46 were sampled) in a wave cloud ∼8 km above Wyoming as determined by aerosol time of flight mass spectrometry (ATOFMS). This is an example from a single flight and there was significant variability between different flights. Data taken from Pratt et al.156 | ||
The importance of mineral dusts as ice nuclei is also supported by studies within the modelling community. In 2004, Diehl and Wurzler87 produced parameterisations for illite, kaolinite and montmorillonite based on experiments by Hoffer88 and Pitter and Pruppacher.89 Unfortunately the experimental reports did not contain details of how much solid material was inside the droplets and the resulting parameterisations were therefore independent of the surface area of ice nucleating material. These parameterisations were used by Lohmann and Diehl86 in a general circulation model sensitivity study on mixed phase stratiform clouds and showed that the different mineral parameterisations had a significant effect upon the simulation (these simulations are discussed in Section 1.7). A parameterisation based on observations of ice nuclei using a CFDC has been produced by Phillips et al.,157 who determined that mineral dusts and metallic particles were responsible for a large proportion of IN throughout the atmosphere. Further supporting this conclusion, recent simulations using a global climate model by Hoose et al.110 found that mineral dusts accounted for 77% of IN active between 0 and −38 °C.
Mineral dust concentrations vary greatly on spatial and temporal scales depending on the uplift process and distance of transportation. Smaller scale features, such as dust devils of 1–10 m in size and lasting seconds to minutes,163 are responsible for the uplift of significant amounts of dust.164 Large scale convective systems and atmospheric circulations occur in the hour-month and 0.1–1000 km scales and are able to transport dust in the continental to global scale.165 For example, it has been estimated that 50 Tg of dust is transported annually from Africa to the Amazon basin.166 Surface measurements, which at some locations have been on-going for several decades, show large variations in dust concentration depending upon location and season.159,167
As the nucleating efficiency of individual minerals differs (see Section 6.1.4), knowledge of bulk dust concentration alone is insufficient to understand the dust glaciation effect. Proportional mineralogy of bulk powder samples can be retrieved by X-ray Diffraction (XRD), however the lower limit of detection for most minerals using XRD is typically 0.5–1%.168 Electron microscopy can also be used to determine mineralogy in much smaller masses of material.169Fig. 5 provides a comparison of observations of atmospheric dust mineralogies retrieved via XRD analysis, with the average observed composition shown in Fig. 6. Atmospheric dust is primarily composed of clay minerals (47%), quartz (29%) and feldspar minerals (13%) with other minerals making up the remainder. Dust mineralogy is determined by laboratory analysis of filter samples collected either at the surface or from aircraft. Some details can be retrieved by in situ mass spectrometry,155 but many minerals have chemically similar compositions and mineralogy is not typically reported.
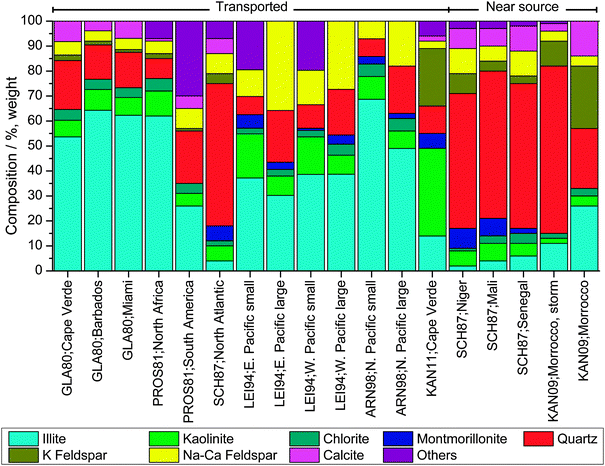 | ||
| Fig. 5 A summary of atmospheric mineral dust compositions from XRD analyses. The data are split into mineral dust which was sampled near the source region (generally continental locations) and at locations a large distance from the source region (generally in marine locations). Dust sampled close to source was richer in materials associated with the coarse fractions, whereas the clay minerals were relatively enhanced in samples in remote locations. ‘Others’ includes materials such as gypsum, goethite, haematite, palygorskite and halite which were also identified in some studies. In addition, for the LEI94 study the ‘other’ category also includes material which could not be identified with X-ray diffraction. From left to right, GLA80: Glaccum and Prospero;331 PROS81: Prospero et al.;332 SCH87: Schütz and Sebert;176 LEI94: Leinen et al.,333 small is particles <2 μm, large between 2 μm and 20 μm; ARN98: Arnold et al.334 sizes as in LEI94; KAN09: Kandler et al.;335 KAN11: Kandler et al.336 | ||
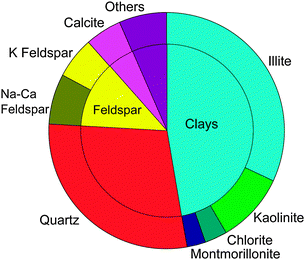 | ||
| Fig. 6 Average of atmospheric dust compositions shown in Fig. 5. Averaging is done without any weighting. Where mineralogy of different particle size bins are provided in a particular study only the smaller size is included. ‘Others’ is defined in the caption for Fig. 5. | ||
Global dust distributions can be retrieved using satellite products (see for example Schepanski et al.,170). Progress has been made towards improving these retrievals to identify individual minerals.171 This would enable the study of how the composition of a dust plume changes with time and, in combination with laboratory studies, would enable a more accurate estimation of global mineral dust IN concentrations.
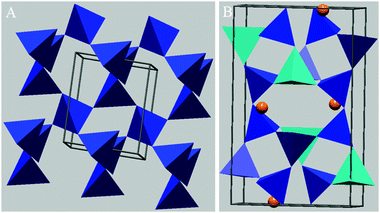
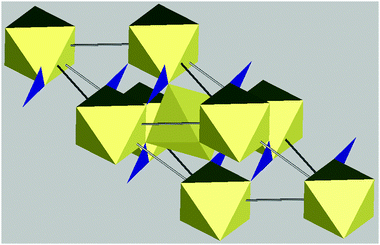
The group of minerals most frequently observed in the atmosphere are the clays. Clays are part of the phyllosilicate group which are secondary minerals with a highly laminated (layered) structure. In the majority of clays, this structure is made up of repeating layers of silicon dioxide tetrahedrals and aluminium oxide octahedrals, with clays categorised by the numbers of each type of layer within the repeating structure. For example, the structure of kaolinite features single silicon and aluminium layers repeating and is referred to as a 1:1 clay mineral (see Fig. 7B). In montmorillonite, the aluminium layer is sandwiched between silicon layers and is referred to as a 2:1 clay mineral (Fig. 8). Within these layers, ion substitutions frequently occur, such as Al3+ for Si4+ and Mg2+ for Al3+. Any resulting charge imbalances caused by these substitutions are balanced by cations located within the inter-layer region. The layered structure of the clay minerals introduces weakness and all have a Mohs hardness of less than 3 (on this scale diamond has a value of 10 and talc has a value of 1).172 Consequentially the clays readily break up into small, thin, plate-like particles with a higher surface to volume ratio and greater asphericity than most other minerals.
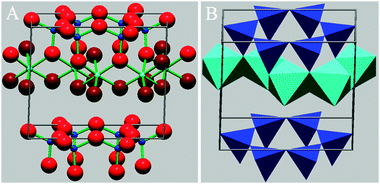 | ||
| Fig. 7 Representations of the structure of kaolinite, a 1:1 clay mineral (see Section 6.1.3). Fig. 7A (left) is a ball and stick structure, explicitly showing the location of all atoms within the structure. Light blue – Al3+, dark blue – Si4+, red (large) – O2−, dark red (large) – OH−. Fig. 7B is a polygonal representation: Light blue octagons represent AlO0.5(OH)2, dark blue tetrahedra representing SiO2. In each polygon, the cation position is in the centre, with the anions on the points. The OH groups, as shown in the spherical representation, occupy the lower side of the Al layer, and the positions on the upper side of the Al layer in the centre of the Si tetrahedral hexagons. Crystal structures are drawn using XtalDraw.337 | ||
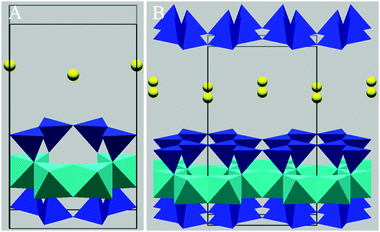 | ||
| Fig. 8 Polygonal representations of montmorillonite. Panel A (left) displays the unit cell, and B (right) displays an expansion of the structure, highlighting the layering and hexagonal arrangements within the structure. As in Fig. 7, light blue octahedrals represent aluminium, specifically AlO(OH), and dark blue tetrahedrals represent SiO2. Yellow spheres represent interlayer Ca2+ cations. All polygon points represent O, except on Al polygons where any point not connecting to a silicon tetrahedral represents an OH group. | ||
6.1.3.1 Clay minerals: kaolinite. Kaolinite has the simplest structure of the clay minerals commonly found in the atmosphere. It is a common component of soil and has a density of approximately 2.65 g cm−3. With repeating aluminium and silicon layers, as shown in Fig. 7, kaolinite is a 1
:1 clay mineral. The chemical composition of kaolinite is Al4Si4(OH)4O10. Ion substitutions within kaolinite are infrequent, with hydrogen bonds providing inter-layer attraction. The relatively high strength of these inter-layer attractions prevents ions or molecules such as water from entering this region.172
6.1.3.2 Clay minerals: montmorillonite. Montmorillonite is a common mineral of the smectite group, with an average density of 2.35 g cm−3. It has a 2
:1 layered structure (see Fig. 8), with frequent substitutions of Mg2+ for Al3+ within the octahedral layer. The resulting charge imbalance is usually countered by the presence of Ca2+ or Na+ ions within the interlayer region and the chemical composition of montmorillonite is (Na,Ca)0.7(Al,Mg)4Si8O20(OH)4·n(H2O).172 The size of these ions combined with their distance from the negatively charged inner layer results in weakly bound layers.173 The consequence of this is the easy infiltration of the interlayer regions by ions and polar molecules (especially water) and as a result montmorillonite is a swelling clay with a high capacity for ion exchange.173,174 This swelling characteristic can make identification of montmorillonite difficult via XRD as the layer separation, and therefore the diffraction pattern, varies with humidity.172
6.1.3.3 Clay Minerals: illite. Illite is the prevalent mineral found in the atmosphere (see Fig. 6) and has an average density of 2.75 g cm−3. It has a 2
:1 structure similar to that of montmorillonite, with frequent ion substitutions especially of aluminium for silicon (see Fig. 9A).172 This results in a higher layer charge than in montmorillonite and consequently stronger interlayer bonding and a non-swelling structure.173 This layer charge is compensated by the addition of potassium and hydronium ions between the repeating layers, resulting in a general chemical composition of K1.5Al4(Si,Al)8O20(OH)4.172
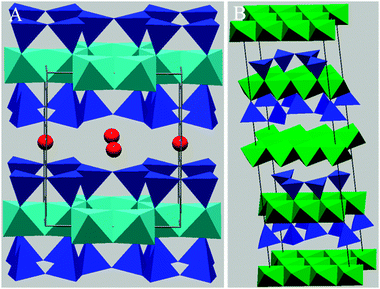 | ||
| Fig. 9 Polygonal representations of illite (panel A) and chlorite (panel B). Panel A includes the unit cell of illite and has the same colour coding as montmorillonite (Fig. 8), but with red spheres representing K. Note the much smaller separation of illite layers vs. montmorillonite layers. Panel B is the unit cell for chlorite with green octahedra represent metal ions, typically Mg2+, Fe2+ or Al3+, blue tetrahedra represent SiO2. The majority of octahedral polygon points in chlorite represent OH− groups, with those constituting part of Si tetrahedrals representing O2−. | ||
6.1.3.4 Clay Minerals: chlorite. While chlorite is not always classified as a true clay,172 it is often treated as a clay since it is a phyllosilicate (layered structure) and can exist in the same size range as typical clays (i.e. typically less than 2 μm).173,175,176 Chlorite refers to a group of minerals with a layer structure which is similar to a 2
:1 clay, but with some important differences. Ion substitutions are similar to illite, but are generally more frequent. Also, unlike illite the interlayer cations are replaced by a layer of hydroxide octahedrals resulting in a 2:1:1 structure (see Fig. 9B). This structure is strongly bound and non-swelling. Members of the chlorite group are classified by the primary non-aluminium or silicon ion present, such as clinochlore (magnesium, (Mg10Al2)(Al2Si6)O20(OH)16) and chamosite (iron(II), (Fe2+10Al2)(Al2Si6)O20(OH)16), with densities in the range of 2.6–3.3 g cm−3.172,173
6.1.3.5 Quartz. Quartz is a primary mineral which is present in most igneous rocks. Unlike the clay minerals, the structure of quartz is formed by a framework exclusively of silicon dioxide tetrahedrals, creating a compact arrangement of six membered loops (see Fig. 10A). This results in a very strong (Mohs hardness of 7), chemically resistant crystal structure which has a density of 2.65 g cm−3 and a tendency to break up into granular particles with a lower surface area to volume ratio than the clays. Also in contrast to the softer clay minerals, this hardness results in a resistance to erosion; quartz is found in most sedimentary rocks, including clay formations.172
 | ||
| Fig. 10 Polygonal representations of quartz (panel A) and feldspar (panel B). Panel A shows quartz which consists of SiO2 tetrahedra arranged in six membered loops. Panel B shows the feldspar albite, with dark blue tetrahedra representing SiO2 and light blue tetrahedral representing AlO2−, arranged in loops of 4. Orange spheres represent Na+, which balances the AlO2− charge. All tetrahedral points are occupied by O2−. | ||
6.1.3.6 Feldspar. The feldspar group of minerals are the most common crustal minerals. They are primary minerals usually of igneous origin, found in most volcanic ashes, and constitute a large proportion of common rocks such as granite. Similar to quartz, the feldspars are composed of a framework of tetrahedrals, except with the replacement of some silicon by aluminium with the resulting negative charges balanced by the addition of a cation (potassium and sodium feldspars have Al
:Si ratios of 1:3, for calcium feldspar this is 1:1). The structure of the sodium feldspar albite is shown in Fig. 10B. The hardness of the feldspars (6–6.5 on the Mohs scale) means they are resistant to physical erosion.172 Plagioclase feldspars (those containing a mixture of calcium and sodium) form kaolinite when weathered by acids, whereas potassium feldspar tends to form illite as well as other clays.172,173 The feldspar minerals all have densities around 2.5–2.7 g cm−3. The chemical compositions of the feldspars are CaAl2Si2O8 (anorthite plagioclase), NaAlSi3O8 (albite plagioclase) and KAlSi3O8 (orthoclase/microcline). The plagioclase feldspars form a series of minerals composed of a mixture of anorthite and albite.172
6.1.3.7 Calcite. Calcite (CaCO3) is a common carbonate mineral and is the principle component of a number of common sedimentary rocks including limestone and chalk. Its structure is similar to ionic substances such as NaCl (see Fig. 11), and due to its weakness (Mohs hardness of 3) easily breaks up into cubic grains.172 It reacts readily with acids, and via reactions with atmospheric sulphuric acid can be processed into gypsum.177 It has a density of 2.7 g cm−3.172
 | ||
| Fig. 11 Calcite, with yellow octahedrals representing Ca2+ and blue triangles representing CO32−. All polygon points are occupied by O2−. | ||
6.1.4.1 Ice nucleation by natural mineral dusts. The active site densities determined for natural minerals dusts from several sources are shown in Fig. 12. The natural mineral dust samples that have been analysed are typically dusts collected from desert surfaces. Dust from non-arid regions contains significant components of biological material which can dominate ice nucleation,178 but we focus here on mineral dusts relevant for arid source regions which dominate mineral dust loadings.42 Connolly et al.131 acquired and tested dust samples from Asia and the Sahara at the AIDA expansion chamber in Karlsruhe. In these experiments dust was aerosolised and then exposed to a supersaturation sufficient for the condensation of water onto the particles. On expansive cooling the mineral dust particles were observed to act as IN and the fraction of aerosol which catalysed freezing was determined. Combining this information with the size distribution of the mineral dust aerosol they were able to determine the cumulative ice nucleus spectrum in the form of the active site densities (ns(T)). The ns values reported by Connolly et al.131 have since been adjusted down by one order of magnitude.135 In a later study, Niemand et al.135 expanded the AIDA study to dusts from Asia, the Sahara, the Canary Isles and Israel. A striking result from the AIDA studies is that mineral dusts from all sources appear to have similar ice nucleating efficiency and can be fitted with a single parameterisation (see Fig. 12). It seems that there is a common component of all these studied natural mineral dusts which causes it to nucleate ice, but unfortunately no mineralogical information was supplied by the authors.
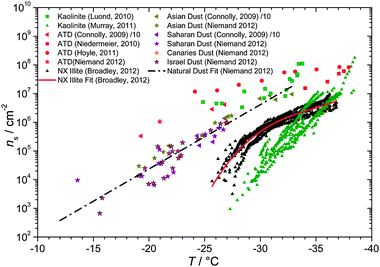 | ||
| Fig. 12 A summary of mineral dust ns values from Connolly et al.,131 Niemand et al.,135 Murray et al.,105 Lüönd et al.,126 Hoyle et al.,181 Niedermeier et al.,132 and Broadley et al.120 Values from Connolly et al. have been adjusted down by a factor of 10, as per Niemand et al.135 in order to correct an earlier error in the data presented by Connolly et al.131 Details of included parameterisations can be found in Table 2. | ||
In a study using emulsified droplets containing mineral dusts, Pinti et al.151 used differential scanning calorimetry (DSC) to study ice nucleation by dust from the Hoggar Mountains in the Sahara. Although they reported a freezing onset temperature of around −26 °C, it is non-trivial to estimate ns values from the data provided. In order to estimate ns values the fraction of droplets frozen as a function of temperature is required; unfortunately this information is not readily available from DSC data.
6.1.4.2 Ice nucleation by dusts composed of single minerals. There are very few ice nucleation studies of single minerals where the composition of the mineral dust was specified. Natural mineral powders sold as single minerals invariably contain finite quantities of other minerals and it is therefore never completely unambiguous which component causes ice nucleation. Natural minerals are never pure because secondary minerals, such as clays and carbonates, usually originate from sedimentary rock formations and frequently include quantities of other minerals which might survive weathering processes, such as quartz and feldspar.172 Due to the similar densities of all the aluminosilicate minerals, it is often impractical to remove such impurities. This must be borne in mind when interpreting ice nucleation results reporting to be for a particular mineral. In this section we review immersion mode ice nucleation studies in which it was reported that a single mineral was introduced to the droplets.
Illite is the most common mineral type in the atmosphere (Fig. 6), but immersion mode ice nucleation by this mineral has only been studied by Hoffer.88 In this study droplets were supported by an oil surface on a cold stage and a median freezing temperature of −24 °C for an unknown concentration of illite was reported (ns values are not determined here because the amount of material per droplet is unknown). Details of the exact mineral composition were not provided, which is a concern since illite samples can contain substantial impurities. For example, Broadley et al. report ice nucleation by a mineral dust marketed as NX-illite, but showed it was made of a mixture of many minerals of which illite was one component (Broadley et al.120 suggest that this mineral dust is a good proxy for natural dusts which have been subject to long range transport, which is supported in this study – see Fig. 19).
Kaolinite, despite being a smaller component of natural atmospheric dust (Fig. 6), has been studied more intensively. Early work by Hoffer88 and Pitter and Pruppacher89 (using a wind tunnel based system) found median freezing temperatures of −33 and −25 °C, respectively. Unfortunately, the amount of material per droplet was not reported in either of these studies and we therefore cannot estimate ns values.
More recently, Lüönd et al.126 used a continuous flow diffusion chamber to investigate particles of 200 to 800 nm diameter of a commercial kaolinite (from Fluka; no more details, such as composition, were provided). They reported median freezing temperatures of −35 to −33 °C. A different kaolinite, of known mineralogy, was investigated by Murray et al.105 using a cold stage with droplets supported on a hydrophobic surface. This resulted in median freezing temperatures in close agreement with Lüönd et al.126 although the surface site densities (ns, see Fig. 12) determined for these two studies are substantially different, with the results from Murray et al.105 up to three orders of magnitude smaller at −27 °C. Lüönd et al.126 report mobility diameters from which we determined a surface area assuming the particles were spherical. In combination with the reported fraction of droplets frozen we determine temperature dependent ns values. In contrast Murray et al.105 used the N2 gas adsorption method to determine surface area. For the 800 nm particles used by Lüönd et al.126 we estimate that the gas adsorption surface area would produce an ns value about four times smaller. This suggests that the discrepancies between Murray et al. and Lüönd et al.126 cannot be simply put down to differences in the method of determining surface area. It may be that there is an intrinsic difference between the kaolinite used in the two studies, since they are from two different sources. Alternatively, ice nucleation in one or both studies may have been dominated by a different mineral present in the samples. The clay mineral society kaolinite (KGa-1b) is a well characterised and documented material with 4% impurity of anatase (titanium dioxide) and crandallite (hydrated phosphate) with traces of dickite (another polymorph of kaolinite) and quartz.179 Unfortunately, the mineralogical make up of Fluke kaolinite used by Lüönd et al.126 is unknown. The enhanced ability of Fluka kaolinite over KGa-1b to nucleate ice was also recently confirmed by Pinti et al.151 in a calorimetry study.
Montmorillonite has been studied by several groups. Hoffer88 and Pitter and Pruppacher89 reported median freezing temperatures of −24 and −21 °C respectively. Details of sample mineralogy and concentrations were not reported. Conen et al.178 performed droplet freezing experiments using a commercial montmorillonite in 50 μL droplets, with freezing beginning at −10 °C. Whilst Conen et al. provided values of nm (ice nucleation sites per mass), they did not provide sufficient information to estimate ns. Pinti et al.151 provided DSC data for four different montmorillonites, of which two were sourced from the Clay Mineral Society (SWy-2 and STx-1b) and two commercially (two acid treated samples from Sigma Aldrich). Significant differences were evident between the samples, with three samples initiating freezing in a small proportion of droplets up to 10 °C higher due to a second, more effective, minor component. All four samples exhibited a freezing onset at −34 to −37 °C. None of the discussed montmorillonite experiments provided sufficient information to enable the inclusion of ns values in Fig. 12.
6.1.4.3 Ice nucleation by proxies for natural atmospheric mineral dust. Using laboratory proxies for natural mineral dust provides the benefits of allowing different groups with different instrumentation to benchmark their equipment and also explore a wide range of conditions with a well characterised material. A good proxy would have a well characterised particle size, surface area, and also mineralogy. To date, two commercial dusts have been suggested as proxies: Arizona Test Dust (ATD) and NX illite.
ATD is a dust supplied by Powder Technology Inc., USA, and has been the subject of numerous studies. It is made of material which has been milled to produce particles with a specific range of sizes and is sold on a commercial basis for testing the efficiency of filters. It is attractive for ice nucleation experiments because it has a well-defined particle size and is available in large quantities. As well as elemental compositions provided by Vlasenko et al.,180 mineralogical composition has been provided by Broadley et al.120 In Fig. 12 we summarise and compare ice active site densities for a number of experiments performed in the immersion mode with ATD. Connolly et al.131 explored ice nucleation by ATD in the AIDA chamber during experiments similar to those described above for natural dusts. Their results are in good agreement with the later study by Niemand et al.135 which also made use of the AIDA chamber (also discussed above). Niedermeier et al.132 report ns values for ATD determined with the LACIS (Leipzig Aerosol Cloud Interaction Simulator) chamber which is a temperature controlled laminar flow tube, allowing the activated fraction of aerosol particles to be determined under specific conditions. In addition, Hoyle et al.181 activated individual size-selected ATD particles to droplets and then measured the ice nucleating fraction using a continuous flow diffusion chamber. We have estimated the surface area based on a spherical assumption in conjunction with the reported fraction frozen data in order to derive ns values. Ice nucleation in the immersion mode was also studied by Marcolli et al.121 using a DSC with emulsified micrometer sized droplets, but it is non-trivial to determine ns values from this data since the fraction of droplets frozen was not reported. The ns values for all four of the studies in which values were derived or reported are consistent with one another (see Fig. 12), which indicates ATD is a useful material for benchmarking and validating instrumentation.
The values of ns for ATD are higher than those for the natural dusts shown in Fig. 12, which indicates that ATD is significantly more efficient at nucleating ice than natural dusts. This is also consistent with differential calorimetry studies which show ATD nucleates ice at higher temperatures than natural samples.121,151 The greater efficiency of ATD may be related to the milling process that ATD is subjected to in its manufacture, but it could also be related to its mineralogy which is significantly different to natural dusts (see Fig. 13).
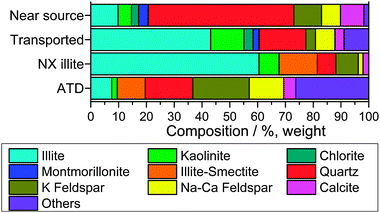 | ||
| Fig. 13 Relative compositions of proxies of atmospheric mineral dust (NX-illite and Arizona Test Dust), in comparison with mineral dust sampled in the atmosphere. The natural atmospheric dust is classified as those near source regions (i.e. continental) and those transported some distance from the source (sampled over the oceans) – see Fig. 5 for classification of individual samples. The illite-smectite group is a mixed layer clay which has been distinguished in the X-ray diffraction patterns of bulk samples of the two proxy dusts. The illite-smectite component is identified on the basis of a weak feature in the diffraction pattern, hence it is not routinely reported in natural samples where signal to noise is limited by small sample sizes. The ‘other’ category for the natural material is defined in the caption for Fig. 5. For ATD the ‘other’ category also includes a substantial proportion of unusual mixed layer clays.120 Mineralogy of ATD and NX-illite are from Broadley et al.120 | ||
A second dust called NX illite, produced by B + M Nottenkaempe, Germany, has been proposed as a suitable proxy for natural dust.120 The efficiency of ice nucleation has been quantified for this material using a cold stage with micron sized droplets containing NX-illite supported on a hydrophobic surface.120 The justification for adopting this dust as a proxy for natural dust which have been subject to long range transport (i.e. dust captured over oceans) is on the basis of its mineralogy. A comparison of the mineralogy of the proxy dusts is provided in Fig. 13 and shows that the mineralogical make up of NX illite and ATD are very different.
On a mineralogical basis, NX-illite is a closer match to transported natural dusts than ATD, which is greatly enhanced in minerals generally associated with the coarse mode (i.e. quartz and feldspars). However, the ice nucleating efficiency, expressed as ns values, for NX illite is substantially smaller than the natural dusts (Fig. 12), but there is a caveat which needs to be considered when comparing these results. The methodologies used to derive surface area across these studies differed. For the natural dusts tested in the AIDA chamber experiments, surface areas were estimated assuming the particles were spheres of radii determined using mobility measurements.131,135 In contrast, the surface area of NX illite particles was based upon gas adsorption measurements in combination with the mass of dust per droplet.120
In a gas adsorption measurement the number of gas molecules, usually N2, required to make a monolayer coverage on the surface is determined and therefore a specific surface area (surface area per mass of solid) can be estimated if an assumption is made about how much space each molecule takes up on the surface.182 Surface areas for various kaolinites measured with atomic force microscopy and gas adsorption techniques agree to within 3%,105,183 which increases confidence in the gas adsorption technique. Electron microscopy120,169,184 and atomic force microscopy183 of dust grains reveals that particles with geometric sizes of greater than 100s of nanometers are often made up of many grains of only 10s of nanometers in size. Hence, mineral dust grains tend to have a true surface area significantly greater than the surface area of a sphere of equivalent size.
As a consequence of the different methods of estimating surface area, the way in which ns is calculated in a particular experiment needs to be taken into consideration when using this data in estimating the number of ice crystals which could nucleate in the atmosphere. It seems sensible that if a spherical approximation has been made in the laboratory, then this assumption should also be made for atmospheric aerosol if they are of a similar size; whereas if a specific surface area has been used then a similar estimate should be made for the atmospheric counterpart. This is illustrated later in this review (Section 6 and Fig. 19) where it is shown that the prediction of concentration of potential ice nuclei using ns values from the natural dusts and NX-illite are consistent with one another in the temperature range where they overlap. This further indicates that NX-illite is a good proxy for natural dusts which have been subject to long range transport.
6.2 Biological material
Although it has been known for more than 40 years that biological particles can act as efficient ice nuclei, their role in ice formation within supercooled clouds remains poorly understood. Primary biological aerosol particles, or PBAPs, encompass all airborne solid particles that are, or were derived from, living organisms.185 Aerosolisation of biological particles occurs as a result of both active and passive processes.186–188 A diverse array of biological entities are found in atmospheric aerosol including fragments of plants and animals, pollen, fungal spores, bacteria and virions (virus particles).185,189,190 Discussions on the potential impacts of biological aerosols upon ice formation processes in the atmosphere have featured in several previous reviews.5,8,157,187,190–192 In the next section, a brief summary of recent evidence from field observations and model studies implicating PBAPs as potentially important atmospheric IN will be introduced. Following this overview, a more detailed discussion on the characteristics of biological ice nuclei derived from laboratory studies is presented.Although the extent to which PBAPs act as atmospheric IN has yet to be quantified, evidence has emerged to suggest that they are significant. In a study examining the nature of IN in precipitation collected from locations around the globe, Christner et al.200 found that for IN active at temperatures ≥ −9 °C, on average 95% were deactivated by heat treatment (95 °C for 10 min) which is consistent with the disruption of the membrane-bound proteins bacteria rely on for their IN activity. The authors also examined the effect of lysozyme (which catalyzes the hydrolysis of peptidoglycan in bacterial cells walls, and thereby disrupts the membranes) on the freezing behaviour and found that on average, 42% of the high temperature ice nuclei were inactivated across all the samples. Recently, Pratt and co-workers156 have also presented evidence that biological materials play a significant role in atmospheric ice nucleation processes. During an investigation into the composition of ice crystal residues sampled from a wave cloud above Wyoming, they used Aerosol Time of Flight Mass Spectrometry (ATOFMS) to show that biological particles and mineral dusts accounted for 33% and 50% respectively of the residues sampled in one particular cloud (Fig. 4).156 The potential importance of biological IN on a regional scale has also been highlighted by Prenni et al.201 during a field campaign investigating IN in aerosols sampled above the canopy at a site in the Amazon basin. By employing transmission electron microscopy and energy-dispersive X-ray diffraction, IN-active aerosol particles were shown to be primarily composed of mineral dust and carbonaceous materials. On the basis of ultraviolet aerodynamic particle sizer (UV-APS) measurements (which provides size resolved number concentrations of total and biological aerosol particles) the authors were able to infer that the carbonaceous fraction of IN was dominated by biological particles.
Bacteria, a single constituent of the biological aerosol, have gained considerable attention from atmospheric researchers interested in ice nucleation. Certain bacteria, designated Ice+, are amongst the most effective ice nucleators currently known, possessing the ability to nucleate ice at temperatures as high as −2 °C.202 Airborne bacteria are ubiquitous over land, with cell densities typically exceeding 10−2 cm−3.203 On a global scale, inferring the proportions of airborne bacteria which possess the Ice+ phenotype is a difficult task given available data. An early study published by Maki and Willoughby,204 found that in snow 15.4% of the bacteria isolated exhibited the Ice+ phenotype, while of the limited number (only five) of isolates in rainwater, none were similarly active. During the 1980s, Lindemann et al.205 sampled air from above a variety of canopies over plants including corn, wheat, alfalfa and pea and found that, at most, 3.9% of the bacterial colony forming units (CFUs) were active as IN above −10 °C. In a recent study of cloud water collected at two mountain sites in the Hebrides (UK), Ahern et al.206 showed that none of the Pseudomonad isolates, which dominated the samples, were Ice+ strains. Of late, questions have been raised over how representative bacterial communities in freshly fallen precipitation or aerosol particles sampled in close proximity to the Earth's surface are of those typically found in mixed-phase clouds.207,208 Clearly, the bacterial community composition in cloud water is a topic which will require further study before firm conclusions can be drawn on the prevalence of Ice+ bacteria in the atmosphere.
While field studies suggest that biological IN may be involved in atmospheric ice formation at least on a regional/seasonal scale, model studies have demonstrated mixed results on the net impacts of PBAPs in atmospheric ice formation processes. Diehl and Wurzler209 have examined the effects of bacterial IN in the immersion mode compared with other known ice nucleating particles such as mineral dust and soot using air parcel model simulations of a convective cloud. They used freezing parameterisations developed in previous studies.87,209,210 The authors found that the impacts of bacterial IN on ice formation in convective clouds were substantially less than that of mineral dusts. However, they assumed that 20 to 25% of the cloud droplets contained mineral dust particles, which seems high when mineral dust typically only makes up a small proportion of atmospheric aerosol.155 In comparison, the assumed fraction of cloud droplets containing bacterial IN was only 1 × 10−8 to 0.01%, hence it is not surprising that bacteria played a minor role in their modelling study. Phillips et al.211 examined the potential impacts from elevated bioaerosol concentrations on continental cloud ensembles, using a cloud-system resolving model. Ice nucleation by biological participles (termed ‘insoluble organic’ by Phillips et al.211) was parameterised using a scheme based on published laboratory data.157 During the simulations, concentrations were raised up to 100 times those typically seen at continental regions; a plausible factor given past observations of the PBAP variability.205 Cloud micro- and macrophysical properties, including average numbers and sizes of ice crystals and droplets in the clouds, horizontal cloud coverage and precipitation were demonstrated by the authors to be sensitive to the allowed variations in PBAP concentrations. In contrast, Hoose and co-workers110,212 found that the average contributions of bacteria, pollen and fungal spores to the global ice nucleation rate were minimal. Using immersion freezing parameterisations based on classical nucleation theory and laboratory measurements, less than 10−5% of heterogeneous ice nucleation in mixed-phase clouds was found to be due to the PBAPs included in the simulations. In contrast, 77% of the simulated heterogeneous nucleation was found to be initiated by mineral dust particles and 23% by soot. However, as noted by the authors the assumed IN active fraction of biological materials in the atmosphere remains uncertain, and further research is necessary to better constrain current estimates.
6.2.2.1 Bacterial ice nuclei. Owing in part to their roles in the frost injury of plants, bacteria are the most intensively studied biological ice nucleators. Early indications that biological materials may exhibit ice nucleation activity (INA) emerged during the early 1970's when Schnell and Vali213,214 discovered that decaying leaves contained a source of highly active ice nuclei. Within a short period of time the causative agent was identified as Pseudomonas syringae,215 an epiphytic (leaf dwelling) bacterium. Although P. syringae may be the most abundant and widely distributed Ice+ bacteria identified to date,205,216,217 numerous other species of highly IN active bacteria have been identified, most of which have been isolated from the phyllosphere (leaf surfaces) and are Gram-negative (Gram staining is a technique to differentiate bacteria into one of two major groups, which differ in their cell wall structure). These include other pseudomonads such as strains of P. fluorescens,218P. viridiflava219 and P. antarctica,220 along with a number of other Gram-negative bacteria including Pantoea agglomerans, Pantoea ananas221 and Xanthomonas campestris pv. translucens.222
Laboratory studies quantifying the ice-nucleating activity of Ice+ bacteria have required careful consideration of both the nature and source of the bacteria under examination, along with the characteristics of the nucleating sites. The ability of certain bacteria to nucleate ice at exceptionally small supercoolings has been localised to a membrane bound protein around 120–180 kDa in size which is proposed to provide an epitaxial fit to ice.223–225 Disruption of the cell membrane, by either physical or chemical means, can lead to a reduction in the activity of Ice+ bacteria.204,215,224,226 Conversely, bacteria do not necessarily need to be viable (able divide and form offspring) in order to express IN activity. For example, Maki and Galyan215 demonstrated that P. syringae remained IN active even after the cells were killed with the antibiotic polymyxin B. For some bacteria, IN activity is not necessarily constrained to the cells themselves; certain strains of P. agglomerans have been observed to shed membrane vesicles 50–200 nm in diameter which retain the nucleating capabilities of the parent cell.227 Within a given population of bacteria, distinct sub-populations of bacteria with varying IN activities exist. While for the most active cells (type I), ice nucleation activity is expressed at temperatures between −2 to −5 °C, other cells termed type II and III are only active at lower temperatures (−5 to −7 °C and −7 to −10 °C respectively; using a microliter drop freezing assay).228 Differences between the nucleating sites are believed to be related to the degree of post-translational glycosylation in the N- and C-terminal regions of the protein and the degree of multimerisation of proteins in the outer membrane.229–231
The extent to which INA is exhibited by individual bacterial cells has been found to be dependent on a wide variety of factors. When grown in vitro for laboratory studies, expression of the Ice+ phenotype is found to be dependent on culture conditions, such as the medium composition,232,233 water activity,234 and temperature235,236 along with the growth stage during which ice nucleation is assayed (an assay is an analysis done to determine a property of a system and is a term common in the biological ice nucleation community).228 Amongst strains of P. syringae the number of cells active as ice nuclei at −5 °C ranges from most cells of some strains, to less than 1 in 107 for others (for an illustration of the range of IN activities exhibited by bacteria see Fig. 14).237 In nature, the frequency of occurrence for IN active P. syringae strains varies with the environment of origin. In a study examining the abundance of P. syringae across a wide variety of potential habitats, Morris et al.217 found that while all strains isolated from snow samples were IN active, the proportion of Ice+ strains found on wild plants and epilithic biofilms (on the surface of rocks) were significantly lower. In addition O’Brien and Lindow have found that the IN activities of P. syringae strains are generally higher when grown on plants than in vitro and under these conditions, ice nucleation activity is controlled by complex and strongly interacting factors such as the host plant species along with environmental variables such as light intensity and relative humidity.238
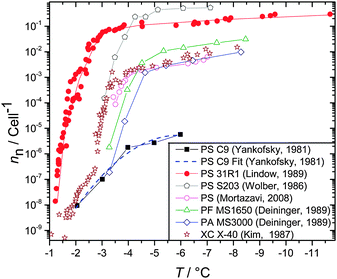 | ||
| Fig. 14 Ice nucleating efficiency for a range of bacteria expressed as nucleation sites per cell (nn). Included are data on various strains of Pseudomonas syringae (PS),224,225,244,338 along with Pseudomonas fluorescens (PF),233Pantoea agglomerans (PA)233 and Xanthomonas campestris (XC).339 See Section 6.2.2.1 for a discussion on the factors underlying the observed variability in activity for ice nucleating bacteria. | ||
When present in cloud waters, the effects of relevant environmental conditions on the INA of Ice+ bacteria must also be considered. Recently, Attard et al.239 have examined how acidic pH levels, exposure to UV-A and to reactive gases such as NO2 and O3 impact the INA of Ice+ bacteria. In agreement with previous reports,231,240,241 the authors demonstrated that increasing acidity had a deleterious effect on the INA of Ice+Pseudomonads. Exposure to UV-A was shown to lead to a significant loss in cell viability, but only minor decreases in INA were observed. Variable results were found on exposure to the reactive pollutants NO2 and O3; while exposure did not significantly reduce INA for two strains of P. syringae isolated from cloud water, the effect on a third strain isolated from leaf surfaces varied between replicates, indicating that as-yet unidentified parameters were causing differences between the tests.
Many of the past studies on the freezing behaviour of bacterial suspensions have involved variants of the drop-freezing assay where a sample is split into many drops. These subunits are cooled and freezing is usually detected by visual inspection or by thermoanalytical techniques such as differential scanning calorimetry.149 By assuming that the time-dependence of nucleation is a second order effect, results from the drop-freezing assay can be interpreted in terms of the cumulative nucleus spectrum K(T), which describes the number of nuclei active per unit volume at temperatures higher than T (see Section 4.2).149 Expressed per unit volume of water, the cumulative nucleus spectrum can readily be determined from the fraction of droplets frozen, fice(T), at a given temperature using the formula:134,242
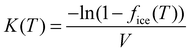 | (27) |
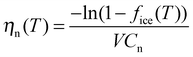 | (28) |
To date, relatively few experiments have examined the activity of bacteria using techniques validated to be free from potential interference such as nucleation by substrates (i.e. using techniques where homogeneous nucleation is achievable in bacteria free droplets). Using droplets in free-fall, Wood et al.147 examined the IN activity of Snowmax™, a freeze-dried and sterilized bionucleant manufactured from P. syringae strain R31 grown under proprietary conditions.245 Given the conditions under which this material is produced, its atmospheric relevance is questionable. In this technique, nanolitre sized falling droplets are frozen under the influence of a vertical temperature gradient maintained within a hollow cylinder. Freezing is detected by illuminating the falling droplets with a linear polarized laser and monitoring the depolarization of the backscattered light. With this setup, the authors were able to successfully demonstrate both homogeneous freezing of water droplets (Tfreeze ∼ −37 °C, ddroplet < 70 μm), and show that Snowmax™ nucleated ice at temperatures (−10 < Tfreeze < −6 °C, ddroplet < 70 μm) similar to those obtained previously by Maki et al.,215 for unfiltered, sonically disrupted cells of P. syringae. In contrast, Junge and Swanson246 employed the same technique to determine the ice nucleating properties of Polar sea ice bacteria. Of the 15 isolates investigated, freezing temperatures for all samples were found to be within two degrees of those observed for the homogeneous freezing of the artificial seawater medium (−42.2 ± 0.3 °C).
Other laboratory techniques used to investigate the IN activity of Ice+ bacteria have included the use of wind tunnels247 and cloud chambers.245,248 During the early 1980s, Levin and Yankovsky247 examined the applicability of desiccated and pulverised Ice+ bacteria isolated from citrus as an artificial IN for weather modification using a vertical wind tunnel. The dry bacterial powder used during the experiments (termed bacterium M1 by the authors) was later identified as P. agglomerans.249 Immersion mode measurements were made by suspending individual bacteria-containing droplets of 440–720 μm diameter in a wind tunnel.247 The work confirmed the conclusions of past researchers that disrupted bacteria can still act as high temperature IN, thereby demonstrating their potential as cloud seeding agents. Studies on the potential of bacterial IN as cloud seeding agents were also conducted by Ward and DeMott248 using a dynamic cloud chamber. By expansion-cooling a dispersion of Snowmax™ powder in air, both the CCN and IN activities of the bacterial particles were investigated. The propensity of IN activities to be centred at narrow temperature ranges was noted by the authors, consistent with the activation of distinct nucleating sites. During isothermal chamber experiments, the authors further demonstrated that the yields of ice crystals per gram of Snowmax™ nucleant added exceeded that of silver iodide at temperatures greater than −5 °C.
Recently, Möhler et al.245 have examined ice nucleation by several live Ice+ bacteria including strains of P. viridiflava, P. syringae and P. agglomerans in both the immersion and condensation modes. Bacterial suspensions were aerosolised and introduced into the cloud chamber where two styles of experiment were conducted. Standard expansion experiments were performed where the supersaturation was controlled by reducing pressure in the chamber in a controlled manner. In the other series of experiments, ice nucleation was probed as the aqueous suspension was nebulised into the chamber. In these so-called “spray experiments” droplets cooled to the surrounding conditions before all the liquid water evaporated and immersion mode ice nucleation was observed. From the number of ice particles, measured using optical particle counters, the authors calculated the fraction of IN active cells from the ratio of the number density of ice particles to that of bacterial cells. Interestingly, when Möhler et al.245 compared the results taken from the cloud chamber to those determined using a popular variant of the drop freezing assay introduced by Lindow,242 lower than expected IN active fractions were found for the latter. While the authors had insufficient evidence to definitively conclude that a systematic difference between the techniques existed, the result highlights the need for further inter-comparison studies on the techniques employed to quantify bacterial IN activity.
Not all bacterial ice nucleators induce freezing at the higher temperatures characteristic of Ice+ bacteria. During recent investigations into the IN activity of bacteria isolated from snow samples collected at ground level, Mortazavi et al.244 found that an intermediate range of activity within bacteria was also possible. While none of the bacteria isolated belonged to the Pseudomonas or Pantoea genera, most of the isolates nucleated ice at temperatures below −16 °C. Similarly, Ponder et al.250 have presented evidence suggesting that a Gram-positive bacterium may also have limited activity as an ice nuclei. These observations raise the fascinating possibility that bacterial ice nucleation may not be restricted to a limited number of Ice+ bacteria, but may in fact be a more widespread phenomenon at lower temperatures.
Despite the large number of lab-based studies which have characterised Ice+ bacteria, questions on the efficacy of bacteria as atmospheric ice nuclei remain. Translating the current body of knowledge to an atmospheric context represents a considerable task. Determining how environmental conditions affects the expression of INA, both before and after bacteria become airborne, will necessitate carefully controlled studies only possible in the laboratory.
6.2.2.2 Fungal and lichen ice nuclei. Although Ice+ bacteria have been the most intensively studied biological ice nucleators to date, similar IN activities have been documented in both lichens (symbiotic organisms composed of a fungus and a photosynthetic partner, either an alga or cyanobacterium) and in certain free-living fungi.251–254 Despite these observations, it is currently difficult to establish whether these biological IN play a relevant role in atmospheric ice formation processes. Estimates of lichen biomass, and consequently concentrations of lichen-derived aerosol particles are difficult.190,243 Margulis255 has estimated that biomass on rock surfaces alone is in the region of 1017 kg globally, while Henderson-Begg et al.243 have suggested that the canopy lichen biomass in temperate forests is similar to leaf biomass and that if lichen particles become airborne, an impact on cloud glaciation is at least plausible. Similarly, the amount of hyphal fragments (the filamentous structures of fungi which collectively constitute the mycelium, or vegatative body) in the atmospheric biological aerosol has yet to be firmly established, but number concentrations of up to 10−3 cm−3 in air over vegetated regions have been reported near the surface.256
INA in lichens was first reported by Kieft251 in 1988, who examined 15 different lichen partnerships collected from a variety of different substrates (rocks, plants and soil) using a drop freezing assay. Of the 15 lichens examined, onset freezing temperatures varied from between −8 °C for the least active lichen partnership to −2.3 °C for the lichen Rhizoplaca chrysoleuca, with threshold temperatures for freezing generally higher in lichens collected from the surface of rocks. Kieft and Ahmadjian252 further examined the INA of pure cultures of lichen fungi (mycobionts) along with lichen algae and cyanobacteria (photobionts) and found that only the fungi could nucleate ice at T ≥ −5 °C.
Several authors have reported warm temperature INA associated with the mycelia of non-lichenised fungi, all of which belonging to the genus Fusarium.253,254,257 Pouleur et al.253 examined the INA of mycelial mats from fungi spanning 20 different genera, and found that strains of F. acuminatum and F. avenaceum had cumulative nucleus spectra similar to those found for Ice+ bacteria on a per unit mass basis.
While the nucleating sites in lichen and Fusarium IN have been identified as proteinaceous, they exhibit several key differences to those found in their Ice+ bacterial counterparts. Activity in both lichen and Fusariam ice nucleators are found to be insensitive to pH; the freezing behaviour of extracts prepared by grinding samples of either Rhizoplaca chrysoleuca or F. avenaceum remained constant throughout the pH range 1–12.253,258 Similarly, while in Ice+ bacteria INA is lost after incubation above 40 °C,204 lichen and Fusarium derived IN maintain their activity after incubation at or above 60 °C. Perhaps the most striking difference between bacterial and lichen/fungal IN is the ability of the latter to pass through 0.22 μm filters, indicating the presence of IN in the absence of cells.253,258
In addition to fungal fragments, spores from fungi also have the potential to affect atmospheric ice formation processes. Fungal spores alone are thought to account for 23% by mass of the total primary emissions of organic aerosol,259 with typical number concentrations of 10−3−10−2 cm−3 observed in the continental boundary layer.259–261 These reproductive units of fungi, which are ejected either passively or actively, have typical diameters which lie in the coarse mode of atmospheric aerosol; spore diameters are most frequently found to be in the range 2–10 μm.260–262 Despite having number densities similar to those found for bacteria, currently few studies have examined the IN activities of fungal spores.
During their survey of biogenic IN sampled from an aircraft above the Arctic, Jayaweera and Flanagan263 found that certain fungal spores exhibited varying ice nucleating efficiencies in drop freezing assays. The IN activity of the fungal spores, reported by the authors as the temperature required to observe an active fraction of 0.03, varied from up to −10 °C for spores of Penicillium digitatum, down to −23 °C for spores of Rhizopus stolonifera. Recently, Iannone et al.264 examined the immersion mode IN activity of spores from Cladosporium species, one of the most abundant types of fungal spore found in the atmosphere. Using a temperature-controlled flow cell coupled to an optical microscope, the authors demonstrated the spores to be relatively inefficient IN, with only 10−3 spores demonstrating IN activity by −28 °C.
6.2.2.3 Pollen ice nuclei. Number densities of pollen in the atmosphere are influenced by a variety of factors including meteorology, location, season and even time of day.186,190 Estimates of typical annual average number densities of pollen over continents are in the range 10−6 to 10−5 cm−3,110 although episodic pollen counts of up to 10−3 cm−3 can occur.265 Typical diameters of pollen grains range from 10–100 μm, leading to short atmospheric residence times. However, elevated pollen number densities observed in inversion layers266 have prompted suggestions that pollen may be temporarily suspended in the atmosphere at concentrations comparable to those of other atmospheric ice nuclei.190,267
Laboratory experiments examining the IN activity of pollen in the immersion mode have shown that pollen grains can induce freezing of droplets at temperatures as high as −9 °C. Using a vertical wind tunnel, Diehl et al.145 and von Blohn et al.268 examined the freezing behaviour of freely suspended droplets (d = 500–760 μm) containing pollen grains. Throughout these experiments, the authors surveyed the IN activities of eight types of pollen including four deciduous tree pollen, three grass pollen types and one conifer pollen. Pollens with small grain diameters (<30 μm), and hence lower settling velocities, were chosen for the studies. Of the eight pollen types investigated, all exhibited ice nucleating abilities at temperatures of −15 °C or higher, with some, such as alder (Alnus incana) pollen demonstrating active fractions approaching 1.0 by −20 °C (Fig. 15).
 | ||
| Fig. 15 Ice nucleating efficiency for pollens expressed as nucleation sites per pollen grain (nn). Calculated from Diehl et al.,145 von Blohn et al.,268 and Pummer et al.,150 assuming pollen grains are spherical and have a density of 0.8 g ml−1. | ||
Recently, Pummer et al.150 examined ice nucleation induced by a range of birch and conifer pollens in water droplets (Vmedian ∼ 22 pL) in an oil and surfactant matrix. Median freezing temperatures ranging from −33 to −19 °C were reported by the authors, largely in agreement with previous reports.145,268 The authors went on to show that when the pollen grains were separated from the suspension, ice nucleation in the remaining water proceeded as efficiently as in the suspensions containing the grains. In contrast to the proteinaceous active sites in bacterial and fungal ice nucleators, the extractible IN from pollen were found to be stable to compounds which degrade proteins, such as proteases and 6 M guanidinium chloride. Ice activity was only lost after exposure to 5 M sulfuric acid, leading the authors to propose that the responsible species may be a polysaccharide. As evident from Fig. 15, the cumulative nucleus spectrum for the more active pollens, such as birch, indicate that the extractible IN from one pollen grain can potentially nucleate ice in more than one water drop. On the basis of their results, the authors argued that the macromolecular IN associated with pollen could be distributed independently in the atmosphere from the grains themselves, perhaps released via the bursting of pollen grains by rain; a process which can discharge allergens and sugars from the bulk.269,270
6.2.2.4 Plankton ice nuclei. The majority of past research examining biological ice nuclei has focused on those with continental sources, with few studies examining potential marine sources of biological ice nuclei. As recently discussed by Burrows et al.,193 evidence from in situ, ship-based measurements over the past 40 years suggests that in remote, biologically active regions of the ocean, background atmospheric IN levels are linked to sea spray production and local marine biological activity. In early laboratory studies examining laboratory cultured marine phytoplankton, Schnell271 demonstrated that cultures of the marine dinoflagellate Heterocapsa niei exhibited remarkably high IN activities. However, a decade later, Fall and Schnell272 identified the IN active species in the mixed algal cultures (which also contained bacteria) as a bacterium, phenotypically similar to Pseudomonas fluorescens biotype G.
Recently, IN activity has been explicitly identified by Knopf and co-workers273–275 in two microalgae, Thalassiosira pseudonana and Nannochloris atomus. IN activity of the two phytoplankters, which were grown in unialgal, axenic (i.e. free of other organisms) cultures was investigated as a function of water activity in aqueous sodium chloride droplets which were conditioned in a humidity-controlled chamber. These two microalgae possess remarkably different cell wall structures; the diatom T. pseudonana has a silaceous cell wall, while N. atomus, a chlorophyte, has only an organic layered cell wall composed of cellulose and other polysaccharides.276 At water activities approaching unity, freezing in droplets containing T. pseudonana or N. atomus (aw = 0.98 and 0.99 respectively) proceeded at median temperatures ∼10 °C above the homogeneous freezing limit.277 In contrast, Emiliania huxleyi, a coccolithophore with an outer wall of calcitic plates was shown by the authors to have negligible IN activity. While future laboratory studies employing techniques which can assess IN activity down to the homogeneous limit will be required to properly constrain source strengths of planktonic ice nuclei, these studies highlight that biogenic particles of marine origin may also play a role in atmospheric ice production.
6.3 Carbonaceous combustion aerosol
There have been dramatic increases in the amount of carbonaceous combustion aerosol released into the atmosphere due to human activity.1,278 Ito and Penner278 estimate that the amount of black carbon emitted globally has increased from 2.1 Tg per year in 1870 to 8.2 Tg per year in 2000. Much of the increase comes from fossil fuel combustion. Combustion aerosol can account for a significant fraction of atmospheric aerosol; for example, one study concluded that 33% of particles sampled in the North American free troposphere were identified as biomass burning particles.279 This material may have a significant impact on cloud properties. In a global model study Spracklen et al.76 estimate that carbonaceous combustion aerosol account for more than half of global CCN and the majority of CCN in polluted regions. The presence of carbonaceous combustion aerosol in cloud droplets supports Lohmann's280 hypothesis of a ‘glaciation indirect effect’, a phrase coined to distinguish the ice nucleation potential of anthropogenic aerosol from the well-known liquid cloud indirect effects.Another major class of particles observed in biomass burning plumes are spherical amorphous carbonaceous particles from 30 to 500 nm, which have been termed tar balls and are thought to form via the condensation of uncombusted low volatility organic compounds.288–290 Tar balls may be a class of amorphous solid aerosol,291–294 and amorphous organic aerosol have been shown to catalyse ice formation.11,68,69
6.3.3.1 Ice nucleation by soot particles. In a study using a controlled expansion cloud chamber, DeMott71 measured the ice nucleating ability of soot produced using an oxygen-deficient acetylene burner. Size-selected soot particles were first exposed to a supersaturation with respect to water in order to activate the majority of soot particles as CCN. On continued cooling, soot particles catalysed freezing below −24 °C. In experiments with the same system, very dilute solution droplets froze homogeneously below −34 °C,71 indicating that soot nucleates ice in the immersion mode. DeMott71 investigated the ice nucleating ability of soot particles with electrical mobility diameters of 0.08 and 0.15 μm and found that almost double the amount of ice crystals were nucleated for the latter. In addition DeMott changed the cooling rate by a factor of two with no significant shift in freezing temperatures, indicating that time dependence was not a significant factor for these conditions.
Diehl and Mitra56 bubbled exhaust from a kerosene burner through water and tested the freezing temperature of droplets made from this suspension using a wind tunnel apparatus. This study was focused on the impact of soot from jet aircraft and they used aviation kerosene to generate soot. The water droplets were between 344 and 818 μm in diameter and were held in the wind tunnel at a specific temperature for about one minute. They showed that the droplets contaminated with exhaust fumes froze at higher temperatures than uncontaminated water droplets, with the probability of freezing increasing with droplet size. In the largest droplet size category 4% of droplets froze at −18 °C and this increased to 70% at −28 °C, clearly indicating that there is some potential for soot to catalyse droplet freezing.
Since soot properties are highly dependent on the fuel and combustion conditions it is perhaps not a surprise to find soot of dissimilar origins can have different ice nucleating abilities. Gorbunov et al.307 studied ice nucleation by soot generated in two separate ways, which produced hydrophilic soot rich in surface hydrogen bonding sites and hydrophobic soot which was deficient in these sites. They show that both soot types could nucleate ice at −10 and −20 °C, but the hydrophilic soot was 3–4 orders of magnitude more efficient at producing ice. However, the mode of ice nucleation in their experiments is unclear. During the experiments, the authors produced a cloud of supercooled droplets within a chamber and then introduced aerosol to this chamber. The chamber was held constant at a temperature for an unspecified period of time. The number of ice particles nucleated was counted by allowing ice crystals to settle onto a coated glass slide and then counting crystals with an optical microscope.308 Although the mode of nucleation in these experiments has been interpreted as contact nucleation in the past,280 it is not clear that this is the case. Given the experimental description, contact, deposition and immersion freezing may have taken place, hence we have not included the Gorbunov et al. data in our assessment of immersion freezing. However, their study does indicate soot can nucleate ice as high as −10 °C and also that the ice nucleating ability of soot strongly depends on the soot particle production conditions.
Popovicheva et al.309 also present evidence that soot particles of different origin nucleate ice with different efficiencies. They study the freezing of millimetre sized droplets contaminated with a range of soots. Unfortunately their uncontaminated water droplets froze at between −7 and −16 °C (with a median of −11.5 ± 2.1 °C), which strongly overlapped with their measured heterogeneous freezing temperatures. However, the median freezing temperatures for several soots were greater than that for uncontaminated water indicating that they may catalyse freezing at temperatures as high as about −7 °C.
In order to compare the efficiencies of ice nucleation by soot particles in the literature we have estimated ns(T) values from the studies of DeMott71 and Diehl and Mitra.56 We selected these studies for this assessment because they provided frozen fractions as a function of temperature together with information on the size and concentration of soot particles. In the case of DeMott’s data it is assumed that each droplet contained a single size selected soot particle. We then determined the surface area of the soot particles assuming they were spheres. Values of ns were then determined using the fraction frozen data. A similar calculation was performed for the data presented by Diehl and Mitra,56 but where the total surface area per droplet was estimated from the measured soot particle size spectrum provided.
The resulting temperature dependent ns values are plotted in Fig. 16. Despite the different sources of soot and the different experimental techniques employed there is consistency between the results from the two experiments. In the temperature range between −24 and −28 °C the two data sets produce similar values of ns in the temperature range in which they overlap. Unfortunately, neither set of authors characterised the soot they used in terms of elemental to organic carbon content or hygroscopicity. Clearly, further experiments are needed to quantify the ice nucleating ability of well characterised soot particles in order to make a more thorough assessment of soot's ability to serve as an IN in the atmosphere.
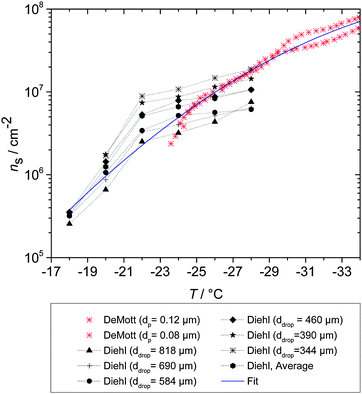 | ||
| Fig. 16 Ice nucleation efficiency expressed as nucleation sites per unit area of soot particles immersed in water (ns). Values of ns have been estimated based on data from DeMott71 and Diehl and Mitra.56 See Section 6.3.3.1 for details. | ||
6.3.3.2 Ice nucleation by carbonaceous particles from biomass burning. Combustion of biomass in nature is likely to produce many aerosol species in addition to soot particles, hence measurements of ice nucleation by biomass combustion aerosol are also needed. At present we are only aware of a single laboratory study on the ice nucleating ability (at water saturation) of particles produced during biomass combustion rather than combustion of liquid fuels. Petters et al.310 used a large combustion chamber facility to generate smoke form 21 biomass fuels from Asia and the United States. A diffusion chamber was used to measure the number of ice nuclei at −30 °C and at water saturation. Only nine of these fuels produced smokes in which there was a detectable fraction of particles (more than 1 in 104 particles) capable of nucleating ice in the condensation or immersion mode. Furthermore, for these nine fuels, not all samples of each fuel produced detectable IN. Petters et al.310 performed a statistical analysis designed to test whether certain factors were correlated to IN emissions. The authors were able to show that organic carbon fractions were significantly lower in smoke containing IN. Petters et al.310 suggest that organic coatings may inhibit ice nucleation in much the same way as has been observed for deposition mode nucleation in a number of studies.302,311 The organic carbon content of the aerosol increased during the smouldering phase of burns and was lowest in the flaming combustion. They also report that aerosols with greater hygroscopicities or those containing water soluble inorganic ions were correlated with increased ice nucleation. Interestingly, the presence of elemental carbon was not found to be correlated with the number of IN, suggesting some other component of the biomass smokes catalysed ice nucleation.
6.4 Volcanic ash
Sulphur emissions from volcanoes are well known to episodically impact human health,312 as well as climate through direct313 and indirect radiative forcings.77 Less well understood is the role volcanic emissions play in cloud glaciation. Historically, there has been some debate in the literature as to the role of volcanic activity in producing atmospheric IN. For example Isono et al.314 reported that IN were enhanced by a factor of 40 in maritime air and linked this to activity at a volcano 140 km from their measurement site in Tokyo. Similarly, Hobbs et al.315 observed around a 10-fold increase in IN concentrations which they linked to volcanic eruptions. They coined the term ‘ice nucleus storms’ to emphasise the sporadic nature of these events. Similarly, Prenni et al.316 showed that IN were enhanced in arctic air passing close to an active volcano. In contrast Schnell and Delany317 found no significant enhancement of ice nuclei near an active volcano in Alaska in addition to evidence suggesting that effluent gases may deactivate natural IN.The recent eruption of Eyjafjallajökull (E15) in Iceland during 2010 provided an opportunity to study the impact of volcanic ash on clouds. Lidar measurements of clouds over Germany reveal a greater propensity for clouds to glaciate within air containing volcanic ash.38 It was reported that all clouds were glaciated below −15 °C in ash influenced clouds, whereas under clear air conditions all clouds were not glaciated until below −25 °C. Bingemer et al.318 reported that volcanic ash made up 53–68% of the particles which served as IN below water saturation. They reported that the total ice nucleus number densities (in the deposition/condensation mode below water saturation) over Germany were much greater in ash influenced air than during intense Saharan dust storms. In addition, IN concentrations were enhanced in Tel Aviv (Israel), some 5000 km from Iceland. Furthermore, Bingemer et al.318 presented evidence that volcanic ash particles subjected to long range transport were substantially more efficient IN in the deposition/condensation mode than freshly emitted material. This is an intriguing finding, but at present there is no adequate explanation for these results.
While it is clear that volcanic ash can glaciate clouds, this effect is episodic and the implications for the planet’s climate are limited to these sporadic events.319 A more direct and perhaps important implication of volcanic ash’s ability to catalyse ice formation is its influence on volcanic clouds (i.e. the aerosol and gas produced by explosive volcanic activity320). Durant et al.320 suggest that latent heat release from water condensation and subsequent glaciation are key drivers in the dynamics of volcanic clouds. Furthermore, the Bergeron–Findeisen process is inhibited since volcanic clouds are so rich in IN and they suggest that most water freezes and subsequently the ice crystals remain small and more stable against sedimentation. Hence, ice nucleation is key in determining the dynamics of the volcanic clouds, important for factors such as injection of sulphate and water into the stratosphere as well as the dispersal of volcanic material.
An important mechanism of ash production is through bubble bursting as lava depressurises and degasses.319,321 The mechanism is analogous to aerosol production by bubble bursting at the surface of oceans.322 This sort of eruption where ash is produced by the degassing of magma and subsequent disintegration of the ‘froth’ is known as a magmatic eruption.321 The morphology of ash particles depends on the eruption type and also the viscosity of the lava. In magmatic eruptions highly viscous lavas tend to produce angular fragments whereas low viscosity magmas can produce spherical ash particles.321 Fine ash production can be enhanced if water is present during the eruption which causes violent steam eruptions in events known as phreatomagmatic eruptions.321 In addition to ash produced from magma, the surrounding rock can also be broken down by explosive forces and form part of the volcanic ash. The properties of this lithic material are dependent on the nature of the rock broken up during the eruption.
Magma is composed of a silica-rich melt with varying amounts of alumina and other metal compounds as well as some crystalline material known as phenocryst. Consequently, volcanic ash tends to a mixture of crystalline material (minerals) and a silicate rich volcanic glass. The presence of glassy (or vitreous) material in volcanic ash distinguishes it from mineral dusts which tend to have a similar mass spectrometry signature.316 The minerals in volcanic ash include some which are also present in natural mineral dust such as silicas (quartz, cristobalite and tridymite), feldspars and micas. In addition, olivines ((Mg,Fe)2SiO4), pyroxenes (typical composition (Mg,Fe,Ca)2Si2O6) and amphiboles (complex alumino-silicates with varying cations) are also present.319,323 Magmas have varying chemical composition and tend to be characterised by how much silica, iron and magnesium they contain; consequently the ashes produced vary substantially.321 Mafic magmas contain a higher proportion of magnesium and iron and this tends to be anticorrelated with the silica component. The silica component increases in the order: basaltic (45–52% SiO2), andesitic (56–59% SiO2) and rhyolitic (63–75% SiO2) magmas.321
The ice nucleating ability of large individual particles (100s μm) of volcanic ash has been investigated when immersed in water droplets.59–61,320 In these studies a single droplet was repeatedly frozen to quantify freezing probabilities as a function of temperature. Despite volcanic ash samples from a range of locations and compositions the median freezing temperature was consistently around −20 °C when the ash particle was immersed in a water droplet.59–61,320 Fornea et al.61 give the temperature dependent probability of freezing as well as a measure of particle size (for Mount St Helens ash) which allows us to estimate the ice active site density (see Fig. 17). A striking conclusion from this style of experiment is that when the particle comes into contact with the interface, the droplets freeze at much greater temperatures.59–61,320 This inside-out contact freezing mechanism is consistent with theoretical studies which suggest heterogeneous freezing is more efficient when a particle is in contact with the droplet surface i.e. where three phases meet.325 However, Gurganus et al.326 recently showed that there was no preference for nucleation at the point where three phases meet in a simplified system. Experiments to determine the importance of inside-out contact freezing with particles of atmospherically relevant size are clearly required.
 | ||
| Fig. 17 Ice nucleating efficiency for volcanic ash samples expressed as nucleation sites per unit area. Values of ns have been estimated based on data from Steinke et al.,136 Hoyle et al.181 and Fornea et al.61 See Section 6.4 for details. | ||
Immersion mode ice nucleation from identical volcanic ash samples from the recent E15 volcanic eruption have recently been investigated by two separate groups using two different techniques. Hoyle et al.181 activated individual size-selected ash particles to droplets and then measured the ice nucleating fraction using a thermal gradient diffusion chamber set at a range of temperatures. They report a temperature dependent probability of freezing and in combination with the size distribution below 3 μm diameter we estimate the active site density (see Fig. 17). It should be noted that all particles smaller than 1.8 μm were placed in one size bin and this may lead to an overestimate in surface area. Steinke et al.136 used a large cloud chamber to quantify ice formation in the immersion and deposition mode. In expansions starting at and above −20 °C no deposition mode nucleation was observed and at water saturation the ash particles activated to water droplets. On continued cooling, the ash particles catalysed freezing between about −23 and −29 °C. Using their measurement of droplet and ice number densities as a function of time together with the size distribution of volcanic ash in the chamber Steinke et al.136 estimated the temperature dependent active site density (plotted in Fig. 17).
Comparison of the active site densities derived from the three sets of experiments discussed above (Fig. 17) yields a surprisingly self-consistent picture of ice nucleation by volcanic ash. The large surface area of the ash particles (250–300 μm diameter) used by Fornea et al.61 means that they were sensitive to smaller active site densities than either of the aerosol based studies. Similarly, in their single particle aerosol system Hoyle et al.181 worked with a much smaller number of droplets than used in the cloud chamber experiments,136 and correspondingly Hoyle et al. observed the lowest freezing temperatures. When the nucleation events are normalised to immersed surface area (i.e. active site density) the data from all three studies falls on a curve (r2 = 0.96) spanning more than seven orders of magnitude. This consistency is despite the variability in experimental technique, implying that there is a characteristic ice nucleating ability of volcanic ash. However, we have only been able to characterise ash from two sources in terms of active site density. Quantitative measurements from more sources and compositions are needed in order to assess the hypothesis of Durant et al.320 that all volcanic ashes have similar ice nucleating ability.
7 Summary and discussion of ice nucleation efficiency of heterogeneous ice nuclei
In order to make a meaningful comparison of the ice nucleating efficiency of different materials we have estimated the cumulative ice active site density (ns) for mineral dust, volcanic ash, soot, fungal spores, pollen grains and bacteria. Our estimates, based on the literature data discussed in the preceding sections, are presented in Fig. 18. In order to estimate ns values we have had to make assumptions about surface areas of materials such as pollen and bacteria, due to which our estimates are prone to errors on the order of a factor of 10. However, the ns values presented here extend over nine orders of magnitude and hence even with these large uncertainties a comparison is still valid. Although there are caveats in the interpretation (see below), Fig. 18 provides a benchmark with which to compare various materials and also serves to highlight potential future research directions. | ||
| Fig. 18 Summary plot of ns values based on literature data. The surface area of a bacteria is assumed to be 5 μm2.239 For birch pollen, a surface area of 1520 μm2 is assumed (d = 22 μm). Note that the data of Murray et al.105 and Broadley et al.120 were determined using a gas adsorption surface area which results in a shift to smaller ns values compared to the other mineral dust results where a spherical approximation was made (see discussion in Section 7). | ||
As discussed in Section 4.2, the singular approximation used here treats the time dependence of nucleation as a second order effect. We justify this approach on the basis that it provides a convenient first order approximation of the efficiency with which a material nucleates ice. However, it should be borne in mind that time dependence of nucleation may be important in some cloud types,29,105,120,128,327 and future studies examining the IN activities of substances should aim to quantify the importance of the stochastic nature of ice nucleation.
A further important point regarding the calculation of ns values can be made in relation to the normalization by surface area. Surface area is quantified in different ways in different experiments. For example, some experiments use gas adsorption surface areas (which are quoted as specific surface areas, surface area per unit mass) and provide a total surface area of all the grains and other small scale features.105,120 Gas adsorption measurements for kaolinite samples are in excellent agreement with surface areas determined from atomic force microscopy measurements,105,183 which suggests that this is an accurate way of determining surface area. This approach is well suited to experiments in which a bulk suspension of solid in water is generated and subsequently finely divided. Another approach is to determine the surface area using the size of aerosolised particulates given by aerosol instrumentation such as the mobility diameter (see for example ref. 131, 132 and 184). Basing surface area on mobility size measurements is clearly a sensible approximation, but it should be borne in mind that dust particles tend to be agglomerates of many smaller particles.120,184 Hence, this assumption may lead to a substantial under-estimate of particle surface area. Broadley et al.120,184 estimated that a 500 nm diameter particle of NX-illite would have a surface area more than a factor of 20 times greater if the gas adsorption surface area were used instead of a spherical approximation. This may help us understand the differences between the measurements of Niemand et al.,135 of natural desert dusts, and Broadley et al.,120 who used a dust with similar mineralogy to natural desert dusts. One would expect these data sets to be consistent with one another, but Niemand et al. report ns values about one order of magnitude above that of Broadley et al.120 The difference may be that Broadley et al. used a gas adsorption surface area whereas Niemand et al. used a spherical approximation. When using these data sets to predict the number of IN in the atmosphere, it is important to consider how the surface area was determined experimentally and apply similar assumptions when determining the surface area of natural aerosol samples.
In order to estimate which materials are most important as IN in the atmosphere, ns values are insufficient on their own, and the amount of each material present in the atmosphere needs to be accounted for. Our estimate of the concentration of potential IN (Nice) for various classes of aerosol particle are presented in Fig. 19. These estimates were produced using the ns values presented in Fig. 18 (or their related nn values) and assuming that each nucleating particle is singly immersed within one water droplet. We also compare this data to thermal gradient diffusion chamber measurements of IN densities from a range of field campaigns reported by DeMott et al.21
 | ||
| Fig. 19 Potential immersion mode ice nuclei concentrations as a function of temperature for a range of atmospheric aerosol species. Calculations performed using concentrations of different aerosol particle sypes listed in Table 1. Also provided are ice crystal number concentrations from DeMott et al.21 for comparison, which were taken using a continuous flow diffusion chamber at water saturation within a 500 m altitude layer. Note that the ice crystal numbers produced for all materials are estimated using global averages of IN numbers, except for volcanic ash where the concentrations are event-based (see Table 1). For bacterial IN, it is assumed that 1% of the total number of airborne bacteria are IN active in line with Phillips et al.211 | ||
The determination of Nice values in Fig. 19 requires some explanation. These calculations were done assuming one particle per droplet. The typical upper and lower concentration of the various particle types used in this calculation are listed in Table 1 and this defines the upper and lower bounds of the estimate in Fig. 19. In the literature, the ice nucleating activities of biological materials are typically presented in the form of nn values (i.e. active sites per particle), which allows the estimation of Nicevia a combination of eqn (20) and (23):
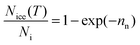 | (29) |
| Material | Upper limit | Lower limit | Units | Ref. |
|---|---|---|---|---|
| Bacteria | 10−2 | 10−5 | cm−3 | Hoose (2010),110 Sesartic (2011)340 |
| Pollen | 10−6 | 10−9 | cm−3 | Hoose (2010)110 |
| Fungal spores | 10−3 | 10−6 | cm−3 | Hoose (2010)110 |
| Soot (d = 0.1 μm) | 100 | 1 | cm−3 | Hoose (2010)110 |
| Dust (d = 1 μm) | 50 | 0.1 | cm−3 | Hoose (2010)110 |
| Volcanic ash | 150 | 30 | μm2 cm−3 | Schumann (2011)328 |
In the case of mineral dust and soot we use the parameterised fits of ns to the data in Fig. 12 and 16, which are listed in Table 2. These ns values are used in conjunction with the mean size (0.1 and 1.0 μm for soot and dust, respectively) and number concentrations listed in Table 1. As discussed in Section 6.1.4.3 if a spherical assumption was made in determining ns a spherical assumption should be made when determining the surface area of atmospheric dust, whereas if a specific surface area was used to determine ns a similar assumption should be made when determining Nice. The appropriate calculation has been done in determining Nice for the proxy of transported natural dust, NX illite, (i.e. specific surface area) and also for the natural mineral dusts (i.e. spherical assumption). The agreement between the Nice predicted for both the natural mineral dusts and the proxy for natural dust is very good in the temperature range over which they overlap.
| Material | n s (T) Parameterisation (cm−2) | Data source |
|---|---|---|
| Dust | 10−4 × exp(−0.517T + 8.934) | 135 |
| NX illite | exp(6.53043 × 104 − 8.215309 × 102 (T − 273.15) + 3.446885376 (T − 273.15)2 − 4.822268 × 10−3 (T − 273.15)3) | 120 |
| Soot | exp(−0.0101T2 − 0.8525T + 0.7667) | 56, 341 |
| Volcanic ash | exp(−0.02287T2 − 1.87203T − 22.05302) | 61, 136, 181 |
| Birch pollen | exp(−0.0309T3 − 1.9893T2 − 42.938T − 297.26) | 150 |
| Cladosporidium spores | exp(−0.02934T3 − 2.91415T2 − 96.59842T − 1056.63326) | 264 |
| P. syringae (low activity) | exp(−0.4325T2 − 5.1067T − 10.399) | 338 |
| P. syringae (high activity) | — | 224 |
The Nice values for volcanic ash are representative of the recent Icelandic E15 eruptions in 2010. In this case Schumann et al.328 report surface areas of ash per volume of atmosphere and we use this information directly in our calculations of Nice rather than assuming a number concentration and size.
From the Nice values in Fig. 19 we can draw a number of conclusions and also draw attention to areas where our knowledge is deficient. One thing to bear in mind is that these values are based on global annual averages of aerosol particle concentrations and substantial deviations will undoubtedly occur episodically on smaller spatial and temporal scales. It should also be noted that there are significant uncertainties in the experimental data as well as approximations made in determining Nice that will affect its accuracy. However, these uncertainties are likely to be relatively small in comparison to the 16 orders of magnitude over which Nice is plotted in Fig. 19, and several conclusions can therefore be drawn from this plot.
The estimates presented in Fig. 19 suggest that cloud glaciation above about −10 °C remains difficult to explain without the presence of IN active bacteria. However, it is also apparent that many of our community’s experimental techniques for ice nucleation on mineral dust and soot may not be sufficiently sensitive to access atmospherically important ns values at temperatures above −15 °C; this should be addressed. Crawford et al.80 studied a mildly supercooled glaciating cumulus cloud and found that 10−3 to 10−4 cm−3 ice nucleus was sufficient at around −7 °C to induce cloud glaciation via the Hallet–Mossop process. Even taking the most efficient bacteria and higher number densities for bacteria it is still difficult to explain glaciation at this warm temperature. We echo the comments of Crawford et al.80 and suggest experimental studies of ice nucleation at warm temperatures are needed.
In their studies of global ice nucleation by various species, Hoose and co-workers110,192,212 conclude that biological particles can only produce a small fraction of primary ice crystals in the Earth’s atmosphere and that soot and mineral dust dominate. This is broadly consistent with our findings, however this does not exclude bacteria as being important in mildly supercooled clouds where ice multiplication may amplify their effects. The parameterisation of Phillips et al.157,211 which is based on thermal gradient diffusion chamber and laboratory studies suggest that biological aerosol (insoluble organic) are significantly more important. Clearly, more experimental work is required to constrain ice nucleation by biological particles and also to improve our understanding of how much biological material is present in the atmosphere.
In making these conclusions we are making the assumption that the materials used in experimental studies are good proxies for aerosol in the atmosphere. Given the relatively small number of studies which we are able to draw on for some materials, we recommend that more experimental efforts are made to quantify ice nucleation by a greater range of materials within these broad categories. We should also remain open to the possibility that there may be as yet uncategorised important classes of IN in the atmosphere. For example, recent work suggests secondary organic aerosol,69,70 and various aerosol species related to anthropogenic activities329,330 may be important ice nuclei. Nevertheless, we present here an up to date picture of the current paradigm of ice nucleation in the atmosphere.
8 Conclusions
Understanding ice nucleation by atmospheric aerosol particles is an integral part of our community’s goal of quantifying the impact of natural and anthropogenic aerosol particles on clouds and climate. There are of course also many other challenges to achieving this goal including improving our understanding of atmospheric dynamics, aerosol composition and distribution, liquid cloud processes as well as model development. Nevertheless, our understanding of the fundamentals of ice nucleation by atmospheric aerosol particles remains a key limitation in understanding aerosol–cloud interactions. In this final section, we conclude by highlighting the key areas requiring future attention. Several of these points have been discussed in depth in previous sections, but for convenience these will be drawn together and highlighted here.While our estimates of the potential IN concentrations presented in Fig. 19 highlight which aerosol species are important, it also highlights areas where our knowledge is lacking. It is clear that aerosol types such as soot and mineral dust are potentially important ice nuclei below about −15 °C, however their importance at higher temperatures remains unclear. The instrumentation which has been used to probe ice nucleation by these materials is insensitive to the potentially small nucleation probabilities which may still be important at higher temperatures. At present, bacteria are the only atmospheric aerosol class which are known to be active in these warm clouds, but it is still under debate if there are sufficient bacteria in the atmosphere to have a significant impact.211,212 It should also be borne in mind that our community has so far restricted its efforts to only a small number of atmospherically relevant materials. For example, soot is a highly variable material, but data suitable for our assessment was only available for two soot types. In addition, it is conceivable that we have so far neglected an important source of ice nucleating aerosol such as SOA or various anthropogenic aerosol species.69,70,329,330
Another major challenge is how to describe ice nucleation by atmospheric aerosol. We have taken a pragmatic approach here and used a time-independent description in order to compare ice nucleation efficiencies of a range of aerosol types. However, there is evidence that the inherent time dependence of nucleation may be important in some cloud types and instruments which operate on short time scales may not always record the presence of all IN which might activate on a longer, but atmospherically relevant time scale.29,105,128,327 Further work is required to characterise time dependence of ice nucleation by the various IN types. In addition, cloud modelling studies are required to test the significance of time dependence.
Differences between experimental studies which may lead to systematic differences in measured ice nucleation efficiencies need to be explored and understood. The sources of variability discussed during this review, such as inconsistent test sample compositions between different studies and different methods of surface area determination or varying experimental conditions require further investigation.
While cooperative work within the laboratory research community will be necessary to improve our current understandings of atmospheric ice nucleation processes, interdisciplinary collaboration involving researchers in the lab, field and modelling communities is required to quantify the impact of ice nucleating aerosol particles on clouds and climate. Unravelling ever more intricate phenomena such as the impacts of environmental processing on mineral dusts, soot or biological materials will only be possible by continuing synergistic efforts across numerous disciplines.
List of symbols used
Notation
| A | Arrhenius pre-exponential factor |
| C m | Mass concentration |
| C n | Number of particles per unit volume of water |
| f ice(T) | Fraction of droplets which freeze on cooling by temperature T |
| ΔGcl | Gibbs free energy of cluster formation |
| ΔGs | Surface excess free energy |
| ΔGv | Volume excess free energy |
| J het | Heterogeneous nucleation rate coefficient |
| J i | Heterogeneous nucleation rate coefficient of ith nucleus |
| k | Boltzmann constant |
| k(T) | Density of surface sites per volume that become active per unit temperature interval |
| K(T) | Cumulative number of nucleation sites per unit volume (cumulative nucleus spectrum) |
| m | Heterogeneous ice nucleating efficiency parameter |
| N tot | Total number of droplets (or number of liquid droplets at t = 0 s) |
| N | Number of liquid droplets at the beginning of a time interval |
| N i | Number of liquid droplets containing potential ice nuclei species i |
| N ice | Concentration of potential ice nuclei (per cm3 of air) |
| n ice(T) | Cumulative number of frozen droplets by temperature T |
| n m | Cumulative number of nucleation sites per unit mass |
| n n | Cumulative number of nucleation sites per unit particle |
| n s | Cumulative number of nucleation sites per unit surface area (active site density) |
| P ice | Vapour pressure of ice |
| P l | Vapor pressure of liquid water |
| R | Cooling nucleation rate (nucleation events per unit volume per unit time) |
| r | Cooling rate |
| r g | Radius of critical cluster |
| r i | Radius of cluster containing i molecules |
| S | Saturation ratio |
| s | Nucleant surface area per droplet |
| s i | Surface area of ith nucleus per droplet |
| s sp | Specific surface area |
| s tot | Total surface area available for nucleation |
| t | Time |
| T c | Characteristic nucleation temperature |
| V | Droplet volume |
| Δnice | Number of droplets which freeze within a time interval |
| α | Modified singular temperature offset |
| β | Modified singular empirical factor |
| γ | Interfacial free energy |
| Φ | Heterogeneous enhancement factor |
| θ | Contact angle |
| ν | Molecular volume of condensed phase |
Acknowledgements
We thank Anja Schmidt for modifying and providing a figure and gratefully acknowledge the European Research Council (FP7, 240449 ICE), and the Natural Environment Research Council (NE/I013466/1) for funding.References
-
P. Forster, V. Ramaswamy, P. Artaxo, T. Berntsen, R. Betts, D. W. Fahey, J. Haywood, J. Lean, D. C. Lowe, G. Myhre, J. Nganga, R. Prinn, G. Raga, M. Schulz and R. Van Dorland, in Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth assessment Report of the Intergovernmental Panel on Climate Change, ed. S. Solomon, D. M. Qin, Z. Manning, M. Chen, K. B. Marquis, M. T. Averyt and H. L. Miller, Cambridge University Press, Cambridge, 2007 Search PubMed
.
- U. Lohmann and J. Feichter, Atmos. Chem. Phys., 2005, 5, 715–737 CrossRef CAS
- D. L. Hartmann, M. E. Ockertbell and M. L. Michelsen, J. Clim., 1992, 5, 1281–1304 CrossRef
- W. Cantrell and A. Heymsfield, Bull. Am. Meteorol. Soc., 2005, 795–807 CrossRef
- C. Hoose and O. Möhler, Atmos. Chem. Phys. Discuss., 2012, 12, 12531–12621 CrossRef
- S. Martin, Chem. Rev., 2000, 100, 3403–3453 CrossRef CAS
-
H. R. Pruppacher and J. D. Klett, Microphysics of Clouds and Precipitation, Kluwer, Dordrecht, 1997 Search PubMed
- W. Szyrmer and I. Zawadzki, Bull. Am. Meteorol. Soc., 1997, 78, 209–228 CrossRef
- T. Bartels-Rausch, V. Bergeron, J. H. E. Cartwright, R. Escribano, J. L. Finney, H. Grothe, P. J. Gutierrez, J. Haapala, W. F. Kuhs, J. B. C. Pettersson, S. D. Price, C. I. Sainz-Diaz, D. J. Stokes, G. Strazzulla, E. S. Thomson, H. Trinks and N. Uras-Aytemiz, Rev. Mod. Phys., 2012, 84, 885–944 CrossRef
-
P. J. DeMott, in Cirrus, ed. D. K. Lynch, K. Sassen, D. C. Starr and G. Stephens, Oxford University Press, Oxford, 2002, pp. 102–135 Search PubMed
- B. J. Murray, T. W. Wilson, S. Dobbie, Z. Q. Cui, S. Al-Jumur, O. Möhler, M. Schnaiter, R. Wagner, S. Benz, M. Niemand, H. Saathoff, V. Ebert, S. Wagner and B. Kärcher, Nat. Geosci., 2010, 3, 233–237 CrossRef CAS
- T. Koop, Z. Physiol. Chem., 2004, 218, 1231–1258 CrossRef CAS
- K.-T. Liou, Mon. Weather Rev., 1986, 114, 1167–1199 CrossRef
- J. R. Holton and A. Gettleman, Geophys. Res. Lett., 2001, 28, 2799–2802 CrossRef CAS
- A. Gettelman and P. M. D. Forster, J. Meteorol. Soc. Jpn., 2002, 80, 911–924 CrossRef
- J. P. D. Abbatt, Chem. Rev., 2003, 103, 4783–4800 CrossRef CAS
- B. J. Murray, S. L. Broadley, T. W. Wilson, S. J. Bull, R. H. Wills, H. K. Christenson and E. J. Murray, Phys. Chem. Chem. Phys., 2010, 12, 10380–10387 RSC
-
J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics, John Wiley and Sons, New York, 1998 Search PubMed
- D. V. Spracklen, K. S. Carslaw, J. Merikanto, G. W. Mann, C. L. Reddington, S. Pickering, J. A. Ogren, E. Andrews, U. Baltensperger, E. Weingartner, M. Boy, M. Kulmala, L. Laakso, H. Lihavainen, N. Kivekas, M. Komppula, N. Mihalopoulos, G. Kouvarakis, S. G. Jennings, C. O'Dowd, W. Birmili, A. Wiedensohler, R. Weller, J. Gras, P. Laj, K. Sellegri, B. Bonn, R. Krejci, A. Laaksonen, A. Hamed, A. Minikin, R. M. Harrison, R. Talbot and J. Sun, Atmos. Chem. Phys., 2010, 10, 4775–4793 CrossRef CAS
- M. O. Andreae and D. Rosenfeld, Earth–Sci. Rev., 2008, 89, 13–41 CrossRef
- P. J. DeMott, A. J. Prenni, X. Liu, S. M. Kreidenweis, M. D. Petters, C. H. Twohy, M. S. Richardson, T. Eidhammer and D. C. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11217–11222 CrossRef CAS
-
B. J. Mason, The Physics of Clouds, Clarendon Press, Oxford, 1971 Search PubMed
- D. C. Rogers, P. J. DeMott, S. M. Kreidenweis and Y. L. Chen, Geophys. Res. Lett., 1998, 25, 1383–1386 CrossRef CAS
- T. Eidhammer, P. J. DeMott and S. M. Kreidenweis, J. Geophys. Res., [Atmos.], 2009, 114, D06202 CrossRef
- P. J. DeMott, K. Sassen, M. R. Poellot, D. Baumgardner, D. C. Rogers, S. D. Brooks, A. J. Prenni and S. M. Kreidenweis, Geophys. Res. Lett., 2003, 30, 1732 CrossRef
- J. Rosinski and G. M. Morgan, J. Aerosol Sci., 1988, 19, 531–538 CrossRef
- M. P. Meyers, P. J. DeMott and W. R. Cotton, J. Appl. Meteorol., 1992, 31, 708–721 CrossRef
-
B. J. Mason, Clouds, rain and rainmaking, Cambridge University Press, Cambridge, 2nd edn, 1975 Search PubMed
- J. Crosier, K. N. Bower, T. W. Choularton, C. D. Westbrook, P. J. Connolly, Z. Q. Cui, I. P. Crawford, G. L. Capes, H. Coe, J. R. Dorsey, P. I. Williams, A. J. Illingworth, M. W. Gallagher and A. M. Blyth, Atmos. Chem. Phys., 2011, 11, 257–273 CrossRef CAS
- H. Morrison, G. de Boer, G. Feingold, J. Harrington, M. D. Shupe and K. Sulia, Nat. Geosci., 2012, 5, 11–17 CrossRef CAS
- P. R. Field, A. J. Heymsfield, B. J. Shipway, P. J. DeMott, K. A. Pratt, D. C. Rogers, J. Stith and K. A. Prather, J. Atmos. Sci., 2012, 69, 1066–1079 CrossRef
- A. J. Heymsfield and L. M. Miloshevich, J. Atmos. Sci., 1993, 50, 2335–2353 CrossRef
- C. H. Twohy, P. J. DeMott, K. A. Pratt, R. Subramanian, G. L. Kok, S. M. Murphy, T. Lersch, A. J. Heymsfield, Z. Wang, K. A. Prather and J. H. Seinfeld, J. Atmos. Sci., 2010, 67, 2437–2450 CrossRef
- D. Rosenfeld and W. L. Woodley, Nature, 2000, 405, 440–442 CrossRef CAS
- R. J. Hogan, M. D. Behera, E. J. O'Connor and A. J. Illingworth, Geophys. Res. Lett., 2004, 31, L05106 CrossRef
- A. Ansmann, M. Tesche, D. Althausen, D. Müller, P. Seifert, V. Freudenthaler, B. Heese, M. Wiegner, G. Pisani, P. Knippertz and O. Dubovik, J. Geophys. Res., [Atmos.], 2008, 113, D04210 CrossRef
- A. Ansmann, M. Tesche, P. Seifert, D. Althausen, R. Engelmann, J. Fruntke, U. Wandinger, I. Mattis and D. Müller, J. Geophys. Res., [Atmos.], 2009, 114, D17208 CrossRef
- P. Seifert, A. Ansmann, S. Gross, V. Freudenthaler, B. Heinold, A. Hiebsch, I. Mattis, J. Schmidt, F. Schnell, M. Tesche, U. Wandinger and M. Wiegner, J. Geophys. Res., [Atmos.], 2011, 116, D00U04 CrossRef
- P. Seifert, A. Ansmann, I. Mattis, U. Wandinger, M. Tesche, R. Engelmann, D. Müller, C. Perez and K. Haustein, J. Geophys. Res., [Atmos.], 2010, 115, D20201 CrossRef
- Y. S. Choi, R. S. Lindzen, C. H. Ho and J. Kim, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11211–11216 CrossRef CAS
- T. Kanitz, P. Seifert, A. Ansmann, R. Engelmann, D. Althausen, C. Casiccia and E. G. Rohwer, Geophys. Res. Lett., 2011, 38, L17802 CrossRef
- J. M. Prospero, P. Ginoux, O. Torres, S. E. Nicholson and T. E. Gill, Rev. Geophys., 2002, 40, 1002 CrossRef
- K. Sassen, P. J. DeMott, J. M. Prospero and M. R. Poellot, Geophys. Res. Lett., 2003, 30, 1633 CrossRef
- E. J. Murray, B. J. Murray and V. Sivakumar, Adv. Geotech.–Eng. Tunnelling, 2008, 553–558 Search PubMed
- D. M. Murphy and T. Koop, Q. J. R. Meteorol. Soc., 2005, 131, 1539–1565 CrossRef
- A. Korolev and G. Isaac, Q. J. R. Meteorol. Soc., 2003, 129, 19–38 CrossRef
- Z. Q. Cui, K. S. Carslaw, Y. Yin and S. Davies, J. Geophys. Res., 2006, 111, D05201 CrossRef
- V. T. J. Phillips, T. W. Choularton, A. M. Blyth and J. Latham, Q. J. R. Meteorol. Soc., 2002, 128, 951–971 CrossRef
- J. Hallett and S. C. Mossop, Nature, 1974, 249, 26–28 CrossRef CAS
- Y. Huang, A. M. Blyth, P. R. A. Brown, T. W. Choularton, P. Connolly, A. M. Gadian, H. Jones, J. Latham, Z. Cui and K. Carslaw, New J. Phys., 2008, 10, 105021 CrossRef
- Y. Huang, A. M. Blyth, P. R. A. Brown, R. Cotton, J. Crosier, K. N. Bower, M. W. Gallagher, H. Jones, A. M. Gadian, T. W. Choularton, J. Cardwell, H. Coe, S. D. Mobbs and M. Hagen, Q. J. R. Meteorol. Soc., 2011, 137, 275–286 CrossRef
- V. T. J. Phillips, A. M. Blyth, P. R. A. Brown, T. W. Choularton and J. Latham, Q. J. R. Meteorol. Soc., 2001, 127, 1513–1534 CrossRef
- S. C. Mossop, Q. J. R. Meteorol. Soc., 1976, 102, 45–57 CrossRef
- J. I. Yano and V. T. J. Phillips, J. Atmos. Sci., 2011, 68, 322–333 CrossRef
- V. I. Khvorostyanov and J. A. Curry, J. Atmos. Sci., 2004, 61, 2676–2691 CrossRef
- K. Diehl and S. K. Mitra, Atmos. Environ., 1998, 32, 3145–3151 CrossRef CAS
- G. de Boer, H. Morrison, M. D. Shupe and R. Hildner, Geophys. Res. Lett., 2011, 38, L01803 CrossRef
- G. Vali, Bull. Am. Meteorol. Soc., 1985, 66, 1426 Search PubMed
- A. J. Durant and R. A. Shaw, Geophys. Res. Lett., 2005, 32, L20814 CrossRef
- R. A. Shaw, A. J. Durant and Y. Mi, J. Phys. Chem. B, 2005, 109, 9865–9868 CrossRef CAS
- A. P. Fornea, S. D. Brooks, J. B. Dooley and A. Saha, J. Geophys. Res., 2009, 114, D13201 CrossRef
- C. D. Westbrook and A. J. Illingworth, Geophys. Res. Lett., 2011, 38, L14808 CrossRef
- K. Sassen and V. I. Khvorostyanov, Environ. Res. Lett., 2008, 3, 025006 CrossRef
- V. T. J. Phillips, L. J. Donner and S. T. Garner, J. Atmos. Sci., 2007, 64, 738–761 CrossRef
- B. Zuberi, A. K. Bertram, T. Koop, L. T. Molina and M. J. Molina, J. Phys. Chem. A, 2001, 105, 6458–6464 CrossRef CAS
- R. Wagner, O. Möhler, H. Saathoff, M. Schnaiter and T. Leisner, Atmos. Chem. Phys., 2011, 11, 2083–2110 CrossRef CAS
- D. A. Knopf and S. M. Forrester, J. Phys. Chem. A, 2011, 115, 5579–5591 CrossRef CAS
- T. W. Wilson, B. J. Murray, R. Wagner, O. Möhler, H. Saathoff, M. Schnaiter, J. Skrotzki, H. C. Price, T. L. Malkin, S. Dobbie and S. M. R. K. Al-Jumur, Atmos. Chem. Phys. Discuss., 2012, 12, 8979–9033 CrossRef
- D. A. Knopf, B. Wang, A. Laskin, R. C. Moffet and M. K. Gilles, Geophys. Res. Lett., 2010, 37, L11803 CrossRef
- B. Wang, A. T. Lambe, P. Massoli, T. B. Onasch, P. Davidovits, D. R. Worsnop and D. A. Knopf, J. Geophys. Res., 2012 DOI:10.1029/2012JD018063
- P. J. DeMott, J. Appl. Meteorol., 1990, 29, 1072–1079 CrossRef
- P. J. DeMott, Y. Chen, S. M. Kreidenweis, D. C. Rogers and D. E. Sherman, Geophys. Res. Lett., 1999, 26, 2429–2432 CrossRef CAS
- T. Croteau, A. K. Bertram and G. N. Patey, J. Phys. Chem. A, 2010, 114, 2171–2178 CrossRef CAS
- X. L. Hu and A. Michaelides, Surf. Sci., 2010, 604, 111–117 CrossRef CAS
- X. L. Hu and A. Michaelides, Surf. Sci., 2007, 601, 5378–5381 CrossRef CAS
- D. V. Spracklen, K. S. Carslaw, U. Poeschl, A. Rap and P. M. Forster, Atmos. Chem. Phys., 2011, 11, 9067–9087 CrossRef CAS
- A. Schmidt, K. S. Carslaw, G. W. Mann, A. Rap, K. J. Pringle, D. V. Spracklen, M. Wilson and P. M. Forster, Atmos. Chem. Phys., 2012, 12, 7321–7339 CrossRef
- D. Rosenfeld, U. Lohmann, G. B. Raga, C. D. O'Dowd, M. Kulmala, S. Fuzzi, A. Reissell and M. O. Andreae, Science, 2008, 321, 1309–1313 CrossRef CAS
- D. Rosenfeld, X. Yu, G. Liu, X. Xu, Y. Zhu, Z. Yue, J. Dai, Z. Dong, Y. Dong and Y. Peng, Geophys. Res. Lett., 2011, 38, L21804 CrossRef
- I. Crawford, K. N. Bower, T. W. Choularton, C. Dearden, J. Crosier, C. Westbrook, G. Capes, H. Coe, P. J. Connolly, J. R. Dorsey, M. W. Gallagher, P. Williams, J. Trembath, Z. Cui and A. Blyth, Atmos. Chem. Phys., 2012, 12, 4963–4985 CrossRef
- M. O. Andreae, D. Rosenfeld, P. Artaxo, A. A. Costa, G. P. Frank, K. M. Longo and M. A. F. Silva-Dias, Science, 2004, 303, 1337–1342 CrossRef CAS
- Y. Yin, K. S. Carslaw and G. Feingold, Q. J. R. Meteorol. Soc., 2005, 131, 221–245 CrossRef
- Y. Yin, K. S. Carslaw and D. J. Parker, Atmos. Chem. Phys., 2002, 2, 293–306 CrossRef CAS
- S. S. Lee, L. J. Donner and V. T. J. Phillips, Atmos. Chem. Phys., 2009, 9, 2555–2575 CrossRef CAS
- S. S. Lee, L. J. Donner, V. T. J. Phillips and Y. Ming, Q. J. R. Meteorol. Soc., 2008, 134, 1201–1220 CrossRef
- U. Lohmann and K. Diehl, J. Atmos. Sci., 2006, 63, 968–981 CrossRef
- K. Diehl and S. Wurzler, J. Atmos. Sci., 2004, 61, 2063–2072 CrossRef
- T. E. Hoffer, J. Meteorol., 1961, 18, 766–778 CrossRef
- R. L. Pitter and H. R. Pruppacher, Q. J. R. Meteorol. Soc., 1973, 99, 540–550 CrossRef
- T. Storelvmo, J. E. Kristjansson and U. Lohmann, J. Atmos. Sci., 2008, 65, 3214–3230 CrossRef
- C. Hoose, U. Lohmann, R. Erdin and I. Tegen, Environ. Res. Lett., 2008, 3, 025003 CrossRef
- T. Storelvmo, C. Hoose and P. Eriksson, J. Geophys. Res., [Atmos.], 2011, 116, D05207 CrossRef
-
D. Kashchiev, Nucleation: Basic Theory with Applications, Butterworth-Heinemann, Oxford, 2000 Search PubMed
-
J. W. Mullin, Crystallization, Elsevier Butterworth-Heinemann, Oxford, Forth edn, 2001 Search PubMed
- J. F. Huang and L. S. Bartell, J. Phys. Chem., 1995, 99, 3924–3931 CrossRef CAS
- W. Ostwald, Z. Phys. Chem., 1897, 22, 289 CAS
- T. L. Malkin, B. J. Murray, A. V. Brukhno, J. Anwar and C. G. Salzmann, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1041–1045 CrossRef CAS
- J. Wolk and R. Strey, J. Phys. Chem. B, 2001, 105, 11683–11701 CrossRef
- R. C. Miller, R. J. Anderson, J. L. Kassner and D. E. Hagen, J. Chem. Phys., 1983, 78, 3204–3211 CrossRef CAS
- B. J. Murray and E. J. Jensen, J. Atmos. Sol.–Terr. Phys., 2010, 72, 51–61 CrossRef CAS
- A. Manka, H. Pathak, S. Tanimura, J. Wolk, R. Strey and B. E. Wyslouzil, Phys. Chem. Chem. Phys., 2012, 14, 4505–4516 RSC
- A. Bogdan, J. Chem. Phys., 1997, 106, 1921–1929 CrossRef CAS
- A. Pant, M. T. Parsons and A. K. Bertram, J. Phys. Chem. A, 2006, 110, 8701–8709 CrossRef CAS
- M. T. Parsons, J. L. Riffell and A. K. Bertram, J. Phys. Chem. A, 2006, 110, 8108–8115 CrossRef CAS
- B. J. Murray, S. L. Broadley, T. W. Wilson, J. D. Atkinson and R. H. Wills, Atmos. Chem. Phys., 2011, 11, 4191–4207 CrossRef CAS
-
P. V. Hobbs, Ice Physics, Clarendon Press, Oxford, 1974 Search PubMed
- C. G. Salzmann, P. G. Radaelli, B. Slater and J. L. Finney, Phys. Chem. Chem. Phys., 2011, 13, 18468–18480 RSC
-
T. C. Hansen, A. Falenty and W. F. Kuhs, in Physics and Chemistry of Ice, ed. W. F. Kuhs, The Royal Society of Chemistry, Cambridge, 2007, pp. 201–208 Search PubMed
- E. B. Moore and V. Molinero, Phys. Chem. Chem. Phys., 2011, 13, 20008–20016 RSC
- C. Hoose, J. E. Kristjánsson, J. P. Chen and A. Hazra, J. Atmos. Sci., 2010, 67, 2483–2503 CrossRef
- J.-P. Chen, A. Hazra and Z. Levin, Atmos. Chem. Phys., 2008, 8, 7431–7449 CrossRef CAS
- J. C. Johnston and V. Molinero, J. Am. Chem. Soc., 2012, 134, 6650–6659 CrossRef CAS
- M. A. Carignano, J. Chem. Phys. C, 2006, 111, 501–504 CrossRef
- E. Pluhařov, L. Vrbka and P. Jungwirth, J. Chem. Phys. C, 2010, 114, 7831–7838 CrossRef
- P. Pirzadeh and P. G. Kusalik, J. Am. Chem. Soc., 2011, 133, 704–707 CrossRef CAS
- B. J. Mason and J. Maybank, Q. J. R. Meteorol. Soc., 1958, 84, 235–241 CrossRef CAS
- B. Vonnegut and M. Baldwin, J. Appl. Meteorol., 1984, 23, 486–490 CrossRef
- A. F. Heneghan, P. W. Wilson and A. D. J. Haymet, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9631–9634 CrossRef CAS
- A. F. Heneghan, P. W. Wilson, G. Wang and A. D. J. Haymet, J. Chem. Phys., 2001, 115, 7599–7608 CrossRef CAS
- S. L. Broadley, B. J. Murray, R. J. Herbert, J. D. Atkinson, S. Dobbie, T. L. Malkin, E. Condliffe and L. Neve, Atmos. Chem. Phys., 2012, 12, 287–307 CrossRef CAS
- S. Marcolli, S. Gedamke, T. Peter and B. Zobrist, Atmos. Chem. Phys., 2007, 7, 5081–5091 CrossRef
- D. Niedermeier, R. A. Shaw, S. Hartmann, H. Wex, T. Clauss, J. Voigtländer and F. Stratmann, Atmos. Chem. Phys., 2011, 11, 8767–8775 CrossRef CAS
- B. Krämer, O. Hubner, H. Vortisch, L. Woste, T. Leisner, M. Schwell, E. Ruhl and H. Baumgartel, J. Chem. Phys., 1999, 111, 6521–6527 CrossRef
- D. Barahona, Atmos. Chem. Phys., 2012, 12, 3733–3752 CrossRef
- V. Stoyanova, D. Kashchiev and T. Kupenova, J. Aerosol Sci., 1994, 25, 867–877 CrossRef CAS
- F. Lüönd, O. Stetzer, A. Welti and U. Lohmann, J. Geophys. Res., 2010, 115, D14201 CrossRef
- M. J. Wheeler and A. K. Bertram, Atmos. Chem. Phys., 2012, 12, 1189–1201 CrossRef CAS
- A. Welti, F. Lüönd, Z. A. Kanji, O. Stetzer and U. Lohmann, Atmos. Chem. Phys. Discuss., 2012, 12, 12623–12662 CrossRef
- G. Vali, J. Atmos. Sci., 1994, 51, 1843–1856 CrossRef
- G. Vali, Atmos. Chem. Phys., 2008, 8, 5017–5031 CrossRef CAS
- P. J. Connolly, O. Möhler, P. R. Field, H. Saathoff, R. Burgess, T. Choularton and M. Gallagher, Atmos. Chem. Phys., 2009, 9, 2805–2824 CrossRef CAS
- D. Niedermeier, S. Hartmann, R. A. Shaw, D. Covert, T. F. Mentel, J. Schneider, L. Poulain, P. Reitz, C. Spindler, T. Clauss, A. Kiselev, E. Hallbauer, H. Wex, K. Mildenberger and F. Stratmann, Atmos. Chem. Phys., 2010, 10, 3601–3614 CrossRef CAS
- P. J. DeMott, Atmos. Res., 1995, 38, 63–99 CrossRef CAS
- G. Vali, J. Atmos. Sci., 1971, 28, 402–409 CrossRef
- M. Niemand, O. Möhler, B. Vogel, H. Vogel, C. Hoose, P. Connolly, H. Klein, H. Bingemer, P. DeMott, J. Skrotzki and T. Leisner, J. Atmos. Sci., 2012 DOI:10.1175/JAS-D-11-0249.1
- I. Steinke, O. Möhler, A. Kiselev, M. Niemand, H. Saathoff, M. Schnaiter, J. Skrotzki, C. Hoose and T. Leisner, Atmos. Chem. Phys., 2011, 11, 12945–12958 CrossRef CAS
- G. Vali and E. J. Stansbury, Can. J. Phys., 1966, 44, 477–502 CrossRef CAS
- P. J. Connolly, O. Möhler, P. Field, H. Saathoff, R. Burgess, T. Choularton and M. Gallagher, Atmos. Chem. Phys., 2009, 2805–2824 CrossRef CAS
- O. Stetzer, B. Baschek, F. Lüönd and U. Lohmann, Aerosol Sci. Technol., 2008, 42, 64–74 CrossRef CAS
- Z. A. Kanji and J. P. D. Abbatt, Aerosol Sci. Technol., 2009, 43, 730–738 CrossRef CAS
- H. M. Jones, M. J. Flynn, P. J. DeMott and O. Möhler, Atmos. Chem. Phys., 2011, 11, 53–65 CrossRef CAS
- B. Friedman, G. Kulkarni, J. Beranek, A. Zelenyuk, J. A. Thornton and D. J. Cziczo, J. Geophys. Res., [Atmos.], 2011, 116, D17203 CrossRef
- D. C. Rogers, Atmos. Res., 1988, 22, 149–181 CrossRef
- K. V. Beard and H. R. Pruppacher, J. Atmos. Sci., 1969, 26, 1066–1072 CrossRef
- K. Diehl, S. Matthias-Maser, R. Jaenicke and S. K. Mitra, Atmos. Res., 2002, 61, 125–133 CrossRef
- U. Bundke, B. Nillius, R. Jaenicke, T. Wetter, H. Klein and H. Bingemer, Atmos. Res., 2008, 90, 180–186 CrossRef
- S. E. Wood, M. B. Baker and B. D. Swanson, Rev. Sci. Instrum., 2002, 73, 3988–3996 CrossRef CAS
- H. M. Hung, A. Malinowski and S. T. Martin, J. Phys. Chem. A, 2003, 107, 1296–1306 CrossRef CAS
-
G. Vali, in Biological Ice Nucleation and Its Applications, ed. R. E. Lee, G. Warren and L. Gusta, APS press, St. Paul, Minnesota, 1995 Search PubMed
- B. Pummer, H. Bauer, J. Bernardi, S. Bleicher and H. Grothe, Atmos. Chem. Phys., 2012, 12, 2541–2550 CrossRef CAS
- V. Pinti, C. Marcolli, B. Zobrist, C. R. Hoyle and T. Peter, Atmos. Chem. Phys., 2012, 12, 5859–5878 CrossRef
- C. A. Stan, G. F. Schneider, S. S. Shevkoplyas, M. Hashimoto, M. Ibabescu, B. J. Wiley and G. M. Whitesides, Lab on a Chip, 2009, 9, 2293–2305 RSC
- M. Kumai, J. Meteorol., 1961, 18, 139–150 CrossRef
- D. J. Cziczo, P. J. DeMott, C. Brock, P. K. Hudson, B. Jesse, S. M. Kreidenweis, A. J. Prenni, J. Schreiner, D. S. Thomson and D. M. Murphy, Aerosol Sci. Technol., 2003, 37, 460–470 CrossRef CAS
- P. J. DeMott, D. J. Cziczo, A. J. Prenni, D. M. Murphy, S. M. Kreidenweis, D. S. Thomson, R. Borys and D. C. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14655–14660 CrossRef CAS
- K. A. Pratt, P. J. DeMott, J. R. French, Z. Wang, D. L. Westphal, A. J. Heymsfield, C. H. Twohy, A. J. Prenni and K. A. Prather, Nat. Geosci., 2009, 2, 397–400 CrossRef
- V. T. J. Phillips, P. J. DeMott and C. Andronache, J. Atmos. Sci., 2008, 65, 2757–2783 CrossRef
- C. S. Zender, D. Newman and O. Torres, J. Geophys. Res., 2003, 108, 4543 CrossRef
- J. M. Prospero and P. J. Lamb, Science, 2003, 302, 1024–1027 CrossRef CAS
- C. S. Zender, R. L. Miller and I. Tegen, Eos Trans. AGU, 2004, 85, 509 CrossRef
- I. Tegen and I. Fung, J. Geophys. Res., 1995, 100, 18707–18726 CrossRef
- I. N. Sokolik and O. B. Toon, Nature, 1996, 381, 681–683 CrossRef CAS
- P. C. Sinclair, J. Appl. Meteorol., 1969, 8, 32–45 CrossRef
- D. A. Gillette and P. C. Sinclair, Atmos. Environ., Part A, 1990, 24, 1135–1142 CrossRef
- P. Knippertz and M. C. Todd, Rev. Geophys., 2012, 50, 28 CrossRef
- I. Koren, Y. Kaufman, J. R. Washington, M. Todd, C. Y. Rudich, J. V. Martins and D. Rosenfeld, Environ. Res. Lett., 2006, 1, 014005 CrossRef
- J. M. Prospero, J. E. Bullard and R. Hodgkins, Science, 2012, 335, 1078–1082 CrossRef CAS
-
S. Hillier, Clay Mineral Cements in Sandstones, Blackwell Publishing Ltd., 1999, pp. 213–251 Search PubMed
- K. Kandler, N. Benker, U. Bundke, E. Cuevas, M. Ebert, P. Knippertz, S. Rodriguez, L. Schuetz and S. Weinbruch, Atmos. Environ., 2007, 41, 8058–8074 CrossRef CAS
- K. Schepanski, I. Tegen, B. Laurent, B. Heinold and A. Macke, Geophys. Res. Lett., 2007, 34, L18803 CrossRef
- L. Klüser, P. Kleiber, T. Holzer-Popp and V. H. Grassian, Atmos. Environ., 2012, 54, 419–427 CrossRef
-
W. A. Deer, R. A. Howie and J. Zussman, An introduction to the rock forming minerals, Addison Wesley Longman, Harlow, UK, 2nd edn, 1992 Search PubMed
-
A. Meunier, Clays, Springer, Berlin, Germany, 2005 Search PubMed
- D. Borden and R. F. Giese, Clay Clay Miner., 2001, 49, 444–445 CAS
-
T. Zoltai and J. H. Stout, Mineralogy concepts and principles, Burgess, Minneapolis, 1985 Search PubMed
- L. Schütz and M. Sebert, J. Aerosol Sci., 1987, 18, 1–10 CrossRef
- F. Bouchelaghem, Applied Mathematical Modelling, 2010, 34, 467–480 CrossRef
- F. Conen, C. E. Morris, J. Leifeld, M. V. Yakutin and C. Alewell, Atmos. Chem. Phys., 2011, 11, 9643–9648 CrossRef CAS
- S. J. Chipera and D. L. Bish, Clay Clay Miner., 2001, 49, 398–409 CAS
- A. Vlasenko, S. Sjogren, E. Weingartner, H. W. Gaggeler and M. Ammann, Aerosol Sci. Technol., 2005, 39, 452–460 CrossRef CAS
- C. R. Hoyle, V. Pinti, A. Welti, B. Zobrist, C. Marcolli, B. Luo, Á. Höskuldsson, H. B. Mattsson, O. Stetzer, T. Thorsteinsson, G. Larsen and T. Peter, Atmos. Chem. Phys., 2011, 11, 9911–9926 CrossRef CAS
-
S. L. Gregg and K. S. W. Sing, Adsorption surface area and porosity, Academic Press, London, 1982 Search PubMed
- B. R. Bickmore, K. L. Nagy, P. E. Sandlin and T. S. Crater, Am. Mineral., 2002, 87, 780–783 CAS
- A. Welti, F. Lüönd, O. Stetzer and U. Lohmann, Atmos. Chem. Phys., 2009, 9, 6705–6715 CrossRef CAS
- S. Matthias-Maser and R. Jaenicke, Atmos. Res., 1995, 39, 279–286 CrossRef
- A. M. Jones and R. M. Harrison, Sci. Total Environ., 2004, 326, 151–180 CrossRef CAS
- A.-M. Delort, M. Vaïtilingom, P. Amato, M. Sancelme, M. Parazols, G. Mailhot, P. Laj and L. Deguillaume, Atmos. Res., 2010, 98, 249–260 CrossRef CAS
- S. M. Burrows, T. Butler, P. Jockel, H. Tost, A. Kerkweg, U. Pöschl and M. G. Lawrence, Atmos. Chem. Phys., 2009, 9, 9281–9297 CrossRef CAS
- L. Deguillaume, M. Leriche, P. Amato, P. A. Ariya, A. M. Delort, U. Pöschl, N. Chaumerliac, H. Bauer, A. I. Flossmann and C. E. Morris, Biogeosciences, 2008, 5, 1073–1084 CrossRef CAS
- V. R. Després, J. A. Huffman, S. M. Burrows, C. Hoose, A. S. Safatov, G. Buryak, J. Fröhlich-Nowoisky, W. Elbert, M. O. Andreae and U. Pöschl, Tellus, Ser. B, 2012, 64, 15598 CrossRef
- O. Möhler, P. DeMott, G. Vali and Z. Levin, Biogeosciences Discuss., 2007, 4, 2559–2591 CrossRef
- C. Hoose, Nat. Geosci., 2009, 2, 385–386 CrossRef CAS
- S. M. Burrows, C. Hoose, U. Pöschl and M. G. Lawrence, Atmos. Chem. Phys. Discuss., 2012, 12, 4373–4416 CrossRef
- R. Jaenicke, Science, 2005, 308, 73 CrossRef CAS
- R. Jaenicke, S. Matthias-Maser and S. Gruber, Environ. Chem., 2007, 4, 217–220 CrossRef CAS
- S. Gruber, S. Matthias-Maser and R. Jaenicke, J. Aerosol Sci., 1999, 30(Suppl. 1), S9–S10 CrossRef
- C. C. White, C. M. Kenny and S. G. Jennings, J. Aerosol Sci., 1999, 30(Suppl. 1), S809–S810 CrossRef
- B. Graham, P. Guyon, W. Maenhaut, P. E. Taylor, M. Ebert, S. Matthias-Maser, O. L. Mayol-Bracero, R. H. M. Godoi, P. Artaxo and F. X. Meixner, J. Geophys. Res., 2003, 108, 4765 CrossRef
- C. E. Morris, D. C. Sands, M. Bardin, R. Jaenicke, B. Vogel, C. Leyronas, P. A. Ariya and R. Psenner, Biogeosciences, 2011, 8, 17–25 CrossRef CAS
- B. C. Christner, R. Cai, C. E. Morris, K. S. McCarter, C. M. Foreman, M. L. Skidmore, S. N. Montross and D. C. Sands, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18854 CrossRef CAS
- A. J. Prenni, M. D. Petters, S. M. Kreidenweis, C. L. Heald, S. T. Martin, P. Artaxo, R. M. Garland, A. G. Wollny and U. Pöschl, Nat. Geosci., 2009, 2, 401–404 CrossRef
- R. Lundheim, Philos. Trans. R. Soc., B, 2002, 357, 937–943 CrossRef CAS
- S. M. Burrows, W. Elbert, M. G. Lawrence and U. Pöschl, Atmos. Chem. Phys., 2009, 9, 9263–9280 CrossRef CAS
- L. Maki and K. Willoughby, J. Appl. Meteorol., 1978, 17, 1049–1053 CrossRef
- J. Lindemann, H. A. Constantinidou, W. R. Barchet and C. D. Upper, Appl. Environ. Microbiol., 1982, 44, 1059–1063 CAS
- H. E. Ahern, K. A. Walsh, T. C. J. Hill and B. F. Moffett, Biogeosciences, 2007, 4, 115–124 CrossRef CAS
- R. M. Bowers, C. L. Lauber, C. Wiedinmyer, M. Hamady, A. G. Hallar, R. Fall, R. Knight and N. Fierer, Appl. Environ. Microbiol., 2009, 75, 5121 CrossRef CAS
- P. S. Kourtev, K. A. Hill, P. B. Shepson and A. Konopka, Atmos. Environ., 2011, 45, 5399–5405 CrossRef CAS
- K. Diehl and S. Wurzler, Atmos. Environ., 2010, 44, 4622–4628 CrossRef CAS
- K. Diehl, M. Simmel and S. Wurzler, J. Geophys. Res., 2006, 111, D07202 CrossRef
- V. T. J. Phillips, C. Andronache, B. Christner, C. E. Morris, D. C. Sands, A. Bansemer, A. Lauer, C. McNaughton and C. Seman, Biogeosciences, 2009, 6, 987–1014 CrossRef
- C. Hoose, J. Kristjánsson and S. Burrows, Environ. Res. Lett., 2010, 5, 024009 CrossRef
- R. Schnell and G. Vali, Nature, 1973, 246, 212–213 CrossRef
- R. Schnell and G. Vali, Nature, 1972, 236, 163–165 CrossRef
- L. R. Maki, E. L. Galyan, M. M. Chang-Chien and D. R. Caldwell, Appl. Environ. Microbiol., 1974, 28, 456–459 CAS
- H. Constantinidou, S. Hirano, L. Baker and C. Upper, Phytopathology, 1990, 80, 934–937 CrossRef
- C. E. Morris, D. C. Sands, B. A. Vinatzer, C. Glaux, C. Guilbaud, A. Buffiere, S. C. Yan, H. Dominguez and B. M. Thompson, ISME J., 2008, 2, 321–334 CrossRef CAS
- H. Obata, Y. Saeki, J. Tanishita, T. Tokuyama, H. Hori and Y. Higashi, Agric. Biol. Chem., 1987, 51, 1761–1766 CrossRef CAS
- H. Obata, T. Nakai, J. Tanishita and T. Tokuyama, J. Ferment. Bioeng., 1989, 67, 143–147 CrossRef CAS
- H. Obata, N. Muryoi, H. Kawahara, K. Yamade and J. Nishikawa, Cryobiology, 1999, 38, 131–139 CrossRef CAS
- S. Lindow, D. Arny and C. Upper, Phytopathology, 1978, 68, 523–527 CrossRef
- H. Azad and N. W. Schaad, Phytopathology, 1988, 78, 95–100 CrossRef
- L. Corotto, P. Wolber and G. Warren, EMBO J., 1986, 5, 231 CAS
- S. Lindow, E. Lahue, A. Govindarajan, N. Panopoulos and D. Gies, Mol. Plant–Microbe Interact., 1989, 2, 262–272 CrossRef CAS
- P. K. Wolber, C. A. Deininger, M. W. Southworth, J. Vandekerckhove, M. Van Montagu and G. J. Warren, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 7256 CrossRef CAS
- A. G. Govindarajan and S. Lindow, J. Biol. Chem., 1988, 263, 9333–9338 CAS
- P. Phelps, T. H. Giddings, M. Prochoda and R. Fall, J. Bacteriol., 1986, 167, 496 CAS
- N. Cochet and P. Widehem, Appl. Microbiol. Biotechnol., 2000, 54, 153–161 CrossRef CAS
- J. A. Ruggles, M. Nemecek-Marshall and R. Fall, J. Bacteriol., 1993, 175, 7216–7221 CAS
- L. Kozloff, M. Turner and F. Arellano, J. Bacteriol., 1991, 173, 6528–6536 CAS
- M. Turner, F. Arellano and L. Kozloff, J. Bacteriol., 1990, 172, 2521 CAS
- L. Pooley and T. A. Brown, FEMS Microbiol. Lett., 1991, 77, 229–232 CrossRef CAS
- C. A. Deininger, G. M. Mueller and P. K. Wolber, J. Bacteriol., 1988, 170, 669–675 CAS
- A. Blondeaux, J. F. Hamel, P. Widehem and N. Cochet, J. Ind. Microbiol. Biotechnol., 1999, 23, 514–519 CrossRef CAS
- J. Rogers, R. Stall and M. Burke, Cryobiology, 1987, 24, 270–279 CrossRef
- M. Nemecek-Marshall, R. Laduca and R. Fall, J. Bacteriol., 1993, 175, 4062–4070 CAS
- D. C. Gross, Y. S. Cody, E. L. Proebsting Jr, G. K. Radamaker and R. A. Spotts, Appl. Environ. Microbiol., 1983, 46, 1370–1379 CAS
- R. D. O'Brien and S. E. Lindow, Appl. Environ. Microbiol., 1988, 54, 2281–2286 CAS
- E. Attard, H. Yang, A. M. Delort, P. Amato, U. Pöschl, C. Glaux, T. Koop and C. E. Morris, Atmos. Chem. Phys. Discuss., 2012, 12, 9491–9516 CrossRef
- H. Kawahara, Y. Tanaka and H. Obata, Biosci., Biotechnol., Biochem., 1995, 59, 1528–1532 CrossRef CAS
- L. Kozloff, M. Schofield and M. Lute, J. Bacteriol., 1983, 153, 222 CAS
- S. E. Lindow, S. S. Hirano, W. R. Barchet, D. C. Arny and C. D. Upper, Plant Physiol., 1982, 70, 1090 CrossRef CAS
- S. K. Henderson-Begg, T. Hill, R. Thyrhaug, M. Khan and B. F. Moffett, Atmos. Sci. Lett., 2009, 10, 215–219 Search PubMed
- R. Mortazavi, C. T. Hayes and P. A. Ariya, Environ. Chem., 2008, 5, 373–381 CrossRef CAS
- O. Möhler, D. G. Georgakopoulos, C. E. Morris, S. Benz, V. Ebert, S. Hunsmann, H. Saathoff, M. Schnaiter and R. Wagner, Biogeosciences, 2008, 5, 1425–1435 CrossRef
- K. Junge and B. D. Swanson, Biogeosciences, 2008, 5, 865–873 CrossRef
- Z. Levin and S. A. Yankofsky, J. Clim., 1983, 1964–1966 Search PubMed
- P. Ward and P. DeMott, J. Wea. Mod, 1989, 21, 9–13 Search PubMed
- A. L. Fall and R. Fall, Curr. Microbiol., 1998, 36, 370–376 CrossRef CAS
- M. A. Ponder, S. J. Gilmour, P. W. Bergholz, C. A. Mindock, R. Hollingsworth, M. F. Thomashow and J. M. Tiedje, FEMS Microbiol. Ecol., 2005, 53, 103–115 CrossRef CAS
- T. L. Kieft, Appl. Environ. Microbiol., 1988, 54, 1678–1681 CAS
- T. L. Kieft and V. Ahmadjian, Lichenologist, 1989, 21, 355–362 CrossRef
- S. Pouleur, C. Richard, J. G. Martin and H. Antoun, Appl. Environ. Microbiol., 1992, 58, 2960–2964 CAS
- C. Richard, J.-G. Martin and S. Pouleur, Phytoprotection, 1996, 77, 83–92 CrossRef
-
L. Margulis, Symbiotic planet: a new look at evolution, Basic Books (AZ), 1999 Search PubMed
- S. Pady and P. Gregory, Trans. Br. Mycol. Soc., 1963, 46, 609–613 CrossRef
- H. Tsumuki, H. Konno, T. Maeda and Y. Okamoto, J. Insect Physiol., 1992, 38, 119–125 CrossRef
- T. L. Kieft and T. Ruscetti, J. Bacteriol., 1990, 172, 3519–3523 CAS
- C. L. Heald and D. V. Spracklen, Geophys. Res. Lett., 2009, 36, L09806 CrossRef
- W. Elbert, P. E. Taylor, M. O. Andreae and U. Pöschl, Atmos. Chem. Phys., 2007, 7, 4569–4588 CrossRef CAS
- J. Fröhlich-Nowoisky, D. A. Pickersgill, V. R. Després and U. Pöschl, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12814–12819 CrossRef
- J. Huffman, B. Treutlein and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 3215–3233 CrossRef CAS
- K. Jayaweera and P. Flanagan, Geophys. Res. Lett., 1982, 9, 94–97 CrossRef
- R. Iannone, D. Chernoff, A. Pringle, S. Martin and A. Bertram, Atmos. Chem. Phys., 2011, 11, 1191–1201 CrossRef CAS
- M. Sofiev, P. Siljamo, H. Ranta and A. Rantio-Lehtimäki, Int. J. Biometeorol., 2006, 50, 392–402 CrossRef CAS
- H. Linskens and W. Jorde, Allergologie, 1986, 9, 55–58 Search PubMed
- K. Diehl, C. Quick, S. Matthias-Maser, S. Mitra and R. Jaenicke, Atmos. Res., 2001, 58, 75–87 CrossRef
- N. von Blohn, S. K. Mitra, K. Diehl and S. Borrmann, Atmos. Res., 2005, 78, 182–189 CrossRef
- G. Schappi, P. E. Taylor, M. Pain, P. Cameron, A. Dent, I. A. Staff and C. Suphioglu, Clin. Exp. Allergy, 1999, 29, 633–641 CrossRef CAS
- K. E. Yttri, C. Dye and G. Kiss, Atmos. Chem. Phys., 2007, 7, 4267–4279 CrossRef CAS
- R. Schnell, Geophys. Res. Lett., 1975, 2, 500–502 CrossRef
- R. Fall and R. C. Schnell, J. Mar. Res., 1985, 43, 257–265 CrossRef
- P. A. Alpert, J. Y. Aller and D. A. Knopf, Phys. Chem. Chem. Phys., 2011, 13, 19882–19894 RSC
- P. A. Alpert, J. Y. Aller and D. A. Knopf, Atmos. Chem. Phys., 2011, 11, 5539–5555 CrossRef CAS
- D. A. Knopf, P. A. Alpert, B. Wang and J. Y. Aller, Nat. Geosci., 2011, 4, 88–90 CrossRef CAS
-
M. T. Madigan, J. M. Martinko, D. A. Stahl and D. P. Clark, Brock Biology Of Microorganisms, Pearson Education, Inc., San Francisco, 13th edn, 2012 Search PubMed
- T. Koop, B. P. Luo, A. Tsias and T. Peter, Nature, 2000, 406, 611–614 CrossRef CAS
- A. Ito and J. E. Penner, Global Biogeochem. Cycles, 2005, 19, GB2028 CrossRef
- P. K. Hudson, D. M. Murphy, D. J. Cziczo, D. S. Thomson, J. A. de Gouw, C. Warneke, J. Holloway, J. R. Jost and G. Hubler, J. Geophys. Res., [Atmos.], 2004, 109, 11 CrossRef
- U. Lohmann, Geophys. Res. Lett., 2002, 29, 1052 CrossRef
- M. O. Andreae and A. Gelencsér, Atmos. Chem. Phys., 2006, 6, 3131–3148 CrossRef CAS
- K. A. Koehler, P. J. DeMott, S. M. Kreidenweis, O. B. Popovicheva, M. D. Petters, C. M. Carrico, E. D. Kireeva, T. D. Khokhlova and N. K. Shonija, Phys. Chem. Chem. Phys., 2009, 11, 7906–7920 RSC
- V. Tishkova, B. Demirdjian, D. Ferry and M. Johnson, Phys. Chem. Chem. Phys., 2011, 13, 20729–20735 RSC
- O. B. Popovicheva, N. M. Persiantseva, E. E. Lukhovitskaya, N. K. Shonija, N. A. Zubareva, B. Demirdjian, D. Ferry and J. Suzanne, Geophys. Res. Lett., 2004, 31, L11104 CrossRef
- M. Posfai, J. R. Anderson, P. R. Buseck and H. Sievering, J. Geophys. Res., [Atmos.], 1999, 104, 21685–21693 CrossRef CAS
- M. T. Baeza-Romero, J. M. Wilson, E. M. Fitzpatrick, J. M. Jones and A. Williams, Energy Fuels, 2010, 24, 439–445 CrossRef CAS
- P. R. Buseck and M. Posfai, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3372–3379 CrossRef CAS
- M. Posfai, R. Simonics, J. Li, P. V. Hobbs and P. R. Buseck, J. Geophys. Res., [Atmos.], 2003, 108, 8483 CrossRef
- M. Posfai, A. Gelencser, R. Simonics, K. Arato, J. Li, P. V. Hobbs and P. R. Buseck, J. Geophys. Res., [Atmos.], 2004, 109, D06213 CrossRef
- K. Adachi and P. R. Buseck, J. Geophys. Res., [Atmos.], 2011, 116, D05204 CrossRef
- B. Zobrist, C. Marcolli, T. Peter and T. Koop, J. Phys. Chem. A, 2008, 112, 3965–3975 CrossRef CAS
- T. Koop, J. Bookhold, M. Shiraiwa and U. Poeschl, Phys. Chem. Chem. Phys., 2011, 13, 19238–19255 RSC
- B. J. Murray, Atmos. Chem. Phys., 2008, 8, 5423–5433 CrossRef CAS
- A. Virtanen, J. Joutsensaari, T. Koop, J. Kannosto, P. Yli-Pirila, J. Leskinen, J. M. Makela, J. K. Holopainen, U. Poeschl, M. Kulmala, D. R. Worsnop and A. Laaksonen, Nature, 2010, 467, 824–827 CrossRef CAS
- C. H. Twohy, A. D. Clarke, S. G. Warren, L. F. Radke and R. J. Charlson, J. Geophys. Res., [Atmos.], 1989, 94, 8623–8631 CrossRef
- K. A. Pratt, C. H. Twohy, S. M. Murphy, R. C. Moffet, A. J. Heymsfield, C. J. Gaston, P. J. DeMott, P. R. Field, T. R. Henn, D. C. Rogers, M. K. Gilles, J. H. Seinfeld and K. A. Prather, J. Geophys. Res., [Atmos.], 2010, 115, 17 Search PubMed
- M. Kamphus, M. Ettner-Mahl, T. Klimach, F. Drewnick, L. Keller, D. J. Cziczo, S. Mertes, S. Borrmann and J. Curtius, Atmos. Chem. Phys., 2010, 10, 8077–8095 CrossRef CAS
- J. P. Schwarz, R. S. Gao, D. W. Fahey, D. S. Thomson, L. A. Watts, J. C. Wilson, J. M. Reeves, M. Darbeheshti, D. G. Baumgardner, G. L. Kok, S. H. Chung, M. Schulz, J. Hendricks, A. Lauer, B. Kärcher, J. G. Slowik, K. H. Rosenlof, T. L. Thompson, A. O. Langford, M. Loewenstein and K. C. Aikin, J. Geophys. Res., 2006, 111, D16207 CrossRef
- S. Mertes, B. Verheggen, S. Walter, P. Connolly, M. Ebert, J. Schneider, K. N. Bower, J. Cozic, S. Weinbruch, U. Baltensperger and E. Weingartner, Aerosol Sci. Technol., 2007, 41, 848–864 CrossRef CAS
- J. Cozic, S. Mertes, B. Verheggen, D. J. Cziczo, S. J. Gallavardin, S. Walter, U. Baltensperger and E. Weingartner, J. Geophys. Res., [Atmos.], 2008, 113, D15209 CrossRef
- M. Dymarska, B. J. Murray, L. M. Sun, M. L. Eastwood, D. A. Knopf and A. K. Bertram, J. Geophys. Res., 2006, 111, D04204 CrossRef
- O. Möhler, C. Linke, H. Saathoff, M. Schnaiter, R. Wagner, A. Mangold, M. Krämer and U. Schurath, Meteorol. Z., 2005, 14, 477–484 CrossRef
- I. Crawford, O. Möhler, M. Schnaiter, H. Saathoff, D. Liu, G. McMeeking, C. Linke, M. Flynn, K. N. Bower, P. J. Connolly, M. W. Gallagher and H. Coe, Atmos. Chem. Phys., 2011, 11, 9549–9561 CrossRef CAS
- O. Möhler, S. Buttner, C. Linke, M. Schnaiter, H. Saathoff, O. Stetzer, R. Wagner, M. Krämer, A. Mangold, V. Ebert and U. Schurath, J. Geophys. Res., [Atmos.], 2005, 110, D11210 CrossRef
- Z. A. Kanji and J. P. D. Abbatt, J. Geophys. Res., [Atmos.], 2006, 111, 31–41 CrossRef
- Z. A. Kanji, P. J. DeMott, O. Möhler and J. P. D. Abbatt, Atmos. Chem. Phys., 2011, 11, 31–41 CrossRef CAS
- B. Gorbunov, A. Baklanov, N. Kakutkina, H. L. Windsor and R. Toumi, J. Aerosol Sci., 2001, 32, 199–215 CrossRef CAS
- A. E. Pashenko and B. Z. Gorbunov, J. Aerosol Sci., 1987, 18, 269–275 CrossRef CAS
- O. Popovicheva, E. Kireeva, N. Persiantseva, T. Khokhlova, N. Shonija, V. Tishkova and B. Demirdjian, Atmos. Res., 2008, 90, 326–337 CrossRef CAS
- M. D. Petters, M. T. Parsons, A. J. Prenni, P. J. DeMott, S. M. Kreidenweis, C. M. Carrico, A. P. Sullivan, G. R. McMeeking, E. Levin, C. E. Wold, J. L. Collett and H. Moosmuller, J. Geophys. Res., [Atmos.], 2009, 114, 10 Search PubMed
- O. Möhler, S. Benz, H. Saathoff, M. Schnaiter, R. Wagner, J. Schneider, S. Walter, V. Ebert and S. Wagner, Environ. Res. Lett., 2008, 3, 025007 CrossRef
- A. Schmidt, B. Ostro, K. S. Carslaw, M. Wilson, T. Thordarson, G. W. Mann and A. J. Simmons, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15710–15715 CrossRef CAS
- A. Robock, Rev. Geophys., 2000, 38, 191–219 CrossRef CAS
- K. Isono, M. Komabayasi and A. Ono, Nature, 1959, 183, 317–318 CrossRef
- P. V. Hobbs, C. M. Fullerton and G. C. Bluhm, Nature Phys. Sci. (London), 1971, 230, 90–91 Search PubMed
- A. J. Prenni, P. J. DeMott, D. C. Rogers, S. M. Kreidenweis, G. M. McFarquhar, G. Zhang and M. R. Poellot, Tellus, Ser. B, 2009, 61, 436–448 CrossRef
- R. C. Schnell and A. C. Delany, Nature, 1976, 264, 535–536 CrossRef CAS
- H. Bingemer, H. Klein, M. Ebert, W. Haunold, U. Bundke, T. Herrmann, K. Kandler, D. Müller-Ebert, S. Weinbruch, A. Judt, A. Wéber, B. Nillius, K. Ardon-Dryer, Z. Levin and J. Curtius, Atmos. Chem. Phys., 2012, 12, 857–867 CrossRef CAS
- A. J. Durant, C. Bonadonna and C. J. Horwell, Elements, 2010, 6, 235–240 CrossRef CAS
- A. J. Durant, R. A. Shaw, W. I. Rose, Y. Mi and G. G. J. Ernst, J. Geophys. Res., 2008, 113, D09206 CrossRef
- G. Heiken, Geol. Soc. Am. Bull., 1972, 83, 1961–1988 CrossRef CAS
- S. J. Norris, I. M. Brooks, G. de Leeuw, A. Sirevaag, C. Leck, B. J. Brooks, C. E. Birch and M. Tjernström, Ocean Sci., 2011, 7, 129–139 CrossRef
- C. J. Horwell and P. J. Baxter, Bull. Volcanol. (Heidelberg), 2006, 69, 1–24 CrossRef
- T. L. Lathem, P. Kumar, A. Nenes, J. Dufek, I. N. Sokolik, M. Trail and A. Russell, Geophys. Res. Lett., 2011, 38, L11802 CrossRef
- Y. S. Djikaev and E. Ruckenstein, J. Phys. Chem. A, 2008, 112, 11677–11687 CrossRef CAS
- C. Gurganus, A. B. Kostinski and R. A. Shaw, J. Phys. Chem. Lett., 2011, 2, 1449–1454 CrossRef CAS
- B. Vonnegut, J. Clim. Appl. Meteorol., 1987, 26, 322 CrossRef
- U. Schumann, B. Weinzierl, O. Reitebuch, H. Schlager, A. Minikin, C. Forster, R. Baumann, T. Sailer, K. Graf, H. Mannstein, C. Voigt, S. Rahm, R. Simmet, M. Scheibe, M. Lichtenstern, P. Stock, H. Rüba, D. Schäuble, A. Tafferner, M. Rautenhaus, T. Gerz, H. Ziereis, M. Krautstrunk, C. Mallaun, J. F. Gayet, K. Lieke, K. Kandler, M. Ebert, S. Weinbruch, A. Stohl, J. Gasteiger, S. Groß, V. Freudenthaler, M. Wiegner, A. Ansmann, M. Tesche, H. Olafsson and K. Sturm, Atmos. Chem. Phys., 2011, 11, 2245–2279 CrossRef CAS
- M. Ebert, A. Worringen, N. Benker, S. Mertes, E. Weingartner and S. Weinbruch, Atmos. Chem. Phys., 2011, 11, 2805–2816 CrossRef CAS
- D. J. Cziczo, O. Stetzer, A. Worringen, M. Ebert, S. Weinbruch, M. Kamphus, S. J. Gallavardin, J. Curtius, S. Borrmann, K. D. Froyd, S. Mertes, O. Möhler and U. Lohmann, Nat. Geosci., 2009, 2, 333–336 CrossRef CAS
- R. A. Glaccum and J. M. Prospero, Mar. Geol., 1980, 37, 295–321 CrossRef CAS
- J. M. Prospero, R. A. Glaccum and R. T. Nees, Nature, 1981, 289, 570–572 CrossRef CAS
- M. Leinen, J. M. Prospero, E. Arnold and M. Blank, J. Geophys. Res., [Atmos.], 1994, 99, 21017–21023 CrossRef
- E. Arnold, J. Merrill, M. Leinen and J. King, Global Planet. Change, 1998, 18, 137–159 CrossRef
- K. Kandler, L. Schütz, C. Deutscher, M. Ebert, H. Hofmann, S. Jäckel, R. Jaenicke, P. Knippertz, K. Lieke, A. Massling, A. Petzold, A. Schladitz, B. Weinzierl, A. Wiedensohler, S. Zorn and S. Weinbruch, Tellus, Ser. B, 2009, 61, 32–50 CrossRef
- K. Kandler, L. Schütz, S. Jäckel, K. Lieke, C. Emmel, D. Muller-Ebert, M. Ebert, D. Scheuvens, A. Schladitz, B. Segvic, A. Wiedensohler and S. Weinbruch, Tellus, Ser. B, 2011, 63, 459–474 CrossRef CAS
- R. T. Downs and M. Hall-Wallace, Am. Mineral., 2003, 88, 247–250 CAS
- S. Yankofsky, Z. Levin, T. Bertold and N. Sandlerman, J. Appl. Meteorol., 1981, 20, 1013–1019 CrossRef
- H. K. Kim, C. Orser, S. E. Lindow and D. Sands, Plant Dis., 1987, 71, 994–996 CrossRef
- A. Sesartic, U. Lohmann and T. Storelvmo, Atmos. Chem. Phys. Discuss., 2011, 11, 1457–1488 CrossRef
- P. J. DeMott and D. C. Rogers, J. Atmos. Sci., 1990, 47, 1056–1064 CrossRef
Footnote |
| † Part of the atmospheric chemistry themed issue. |
| This journal is © The Royal Society of Chemistry 2012 |