Chemistry and physics of a single atomic layer: strategies and challenges for functionalization of graphene and graphene-based materials
Liang
Yan
ab,
Yue Bing
Zheng
c,
Feng
Zhao
ab,
Shoujian
Li
d,
Xingfa
Gao
ab,
Bingqian
Xu
e,
Paul S.
Weiss
*c and
Yuliang
Zhao
*abd
aChinese Academy of Sciences Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology of China, Beijing 100190, China. E-mail: zhaoyuliang@ihep.ac.cn
bInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
cCalifornia NanoSystems Institute, and Departments of Chemistry & Biochemistry and Materials Science & Engineering, University of California, Los Angeles, Los Angeles, California 90095, USA. E-mail: psw@cnsi.ucla.edu
dThe College of Chemistry, Sichuan University, Chengdu 610064, China
eFaculty of Engineering & Nanoscale Science and Engineering Center, University of Georgia, Athens, Georgia 30602, USA
First published on 16th November 2011
Abstract
Graphene has attracted great interest for its superior physical, chemical, mechanical, and electrical properties that enable a wide range of applications from electronics to nanoelectromechanical systems. Functionalization is among the significant vectors that drive graphene towards technological applications. While the physical properties of graphene have been at the center of attention, we still lack the knowledge framework for targeted graphene functionalization. In this critical review, we describe some of the important chemical and physical processes for graphene functionalization. We also identify six major challenges in graphene research and give perspectives and practical strategies for both fundamental studies and applications of graphene (315 references).
Liang Yan is a graduate student of Chinese Academy of Sciences Key Laboratory for Biomedical Effects of Nanomaterials & Nanosafety. He received his BSc in Chemistry from Sichuan University in 2009. He is now studying on the chemical functionalization of graphenes as well their biomedical effects. |
Yue Bing Zheng is a postdoctoral scholar with Prof. Weiss at the University of California, Los Angeles. He received his PhD in Engineering Science and Mechanics from The Pennsylvania State University in 2010, his MS in Physics from the National University of Singapore in 2004, and his BS in Physics from Nankai University, China, in 2001. From 2004 to 2006, he was a Research Fellow in the Institute of Materials Research and Engineering, Singapore. His research centers on designing, measuring, and controlling molecules and light at the nanoscale. |
Feng Zhao is currently a research fellow at the Chinese Academy of Sciences Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences. She received her BSc in Chemistry from the College of Chemical Engineering, Tsinghua University in 2002, and MSc in Biology from the College of Biology, Tsinghua University in 2005. Her current research interests include nanotoxicology, and studies on nanoparticles and their interactions with cells or biomacromolecules. |
Shoujian Li received his PhD in Radiochemistry at Sichuan University. He is the head of the research group Functional Materials for Radionuclides Separation of the Chemistry College of Sichuan University. His research interests include the design and synthesis of new functional carbonaceous materials, advanced materials and technologies for spent fuel reprocessing, and treatment and disposal of nuclear wastes, including the use of carbon nanomaterials. |
Xingfa Gao received his PhD from the Institute of High Energy Physics, Chinese Academy of Sciences in 2006. After working as a postdoctoral fellow (2006–2011) at the Institute for Molecular Science (Japan) and Rensselaer Polytechnic Institute (USA), he returned to the Institute of High Energy Physics as a professor. His research interests lie in applying computational methods to π-conjugated systems including fullerenes, carbon nanotubes, graphene and their hybrids. He has great interest in developing efficient theoretical methods to investigate novel π-conjugated molecules and reactions through close interplay between theoretical predictions and experimental tests. |
Bingqian Xu received his PhD degree in Materials Science and Engineering from Arizona State University (ASU), Tempe, in 2004. Then, he was with the Electrical Engineering Department, ASU, as a Faculty Research Associate in molecular electronics. In 2006, he moved to the University of Georgia (UGA), Athens, and now is an Associate Professor, directing the Molecular Nanoelectronics Laboratory. His research interests include molecular electronics and single-molecule studies of biomolecular assemblies and systems. Dr Xu is a member of the American Chemical Society, Materials Research Society, and American Physical Society. |
Paul S. Weiss received his PhD in Chemistry in 1986 from UC Berkeley. He was a postdoctoral fellow at AT&T Bell Laboratories and IBM Almaden Research Center. He began his academic career at Penn State, becoming Distinguished Professor of Chemistry and Physics before moving to UCLA in 2009. At UCLA, he is the Fred Kavli Chair in NanoSystems Sciences, the Director of the California NanoSystems Institute, and Distinguished Professor of Chemistry & Biochemistry and Materials Science & Engineering. He is the founding Editor-in-Chief of ACS Nano. His research interests are in single-molecule/assembly function, chemical patterning, self-assembly, and nanoscale analyses. |
Yuliang Zhao's research interests include Nanotoxicological Chemistry (nanotoxicology, cancer nanotechnology, and nanochemistry), Nanobioanalytical Sciences, and Molecular Dynamics Simulations of biochemical processes at the nano/bio interface. He serves as editorial board member for eight international journals in the United States and Europe. He is Professor and Director, Chinese Academy of Sciences Key Laboratory for Biomedical Effects of Nanomaterials & Nanosafety, and also serves as Deputy Director-General of National Center for Nanoscience and Technology of China, and a member of National Steering Council for Nanosciences and Technology of China. |
1. Introduction
Since its conception,1–18graphene has attracted enormous interest due to its unique physical properties, such as novel magneto transport,2 electromechanical modulation,5 extremely high carrier mobility,6,7 tunable band gap,8 quantum Hall effect,2,9–11,19 Klein tunneling nature,12 and electron confinement effects.14 These properties make graphene a promising candidate for a broad range of applications in next-generation nanotechnologies where current materials are limited in functionality.For example, current chemical separation materials cannot efficiently remove residual actinide fuels from radioactive fission fragments.20 This costs more than $2 billion per year for treating the large amount of radioactive wastes generated from nuclear power facilities.21–23Graphene exhibits superior properties as separation materials due to its selective adsorption of heavy metals (e.g., lanthanides and actinides) with super loading and in situ rebirth capacity. For example, we may use graphene-nanoparticle multilayers for separation column materials in nuclear fuel recycling. Other fields in which graphene may have potential applications include electronics, photonics, optoelectronics, and mechanics (Fig. 1).1–12,24,25
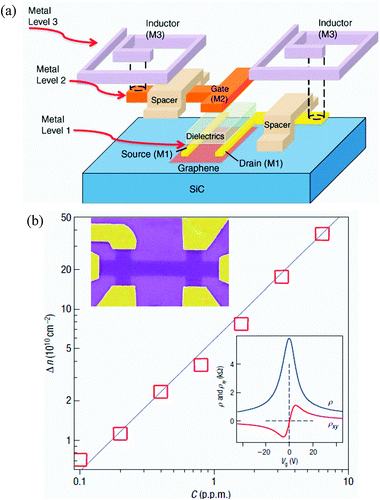 | ||
| Fig. 1 Application examples of functionalized graphene in electronics and single-molecule gas sensors: (a) schematic exploded illustration of a graphene mixer circuit. The critical design aspects include a top-gated graphene transistor and two inductors connected to the gate and the drain of the graphene field-effect transistor (GFET).24 (b) Concentration, Δn, of chemically induced charge carriers in a single-layer graphene exposed to different concentrations, C, of NO2. Upper inset: scanning electron micrograph of this device. Lower inset: characterization of the graphene device by using the electric-field effect.28 Reproduced from ref. 24. Copyright 2011 American Association for the Advancement of Science. Reproduced from ref. 28. Copyright 2007 Nature Publishing Group. | ||
Despite the wide range of possible applications, there are still many challenges for graphene to reach its full potential. For example, graphene has an intrinsic zero band-gap energy. We have to manage to open up the band gap for semiconductor applications, possibly even as a successor of silicon in the post-Moore's law electronics era,26,27 as a single-molecule gas or bio-sensor,28–30 and as a stretchable transparent electrode or electron devices.31–38 In addition, graphene is insoluble in organic solvents and susceptible to aggregation in aqueous solutions. Even for relatively simple uses of graphene, e.g., filler in polymer matrices, the surface properties of graphene sheets must be altered through further functionalization to obtain stable dispersions in solvents. Most recently, it was found that a supercapacitor fabricated by chemical activation of graphene with KOH possessed high values of gravimetric capacitance and energy density,39 higher than the supercapacitor produced by the chemically reduced graphene.40 Therefore, functionalization approaches that can modify the structural, electronic, and chemical properties of graphene are critical for applications.41–47
In principle, graphene can be functionalized at two classes of locations: the basal plane and the edges. On the basal plane, sp2 hybridization of carbon leads to a strong covalent bonding, as well as to delocalization of the π electrons. The interaction of the basal plane with guest atoms or molecules leads to modification of the π–π conjugation and thus the electron density distribution and the physical and chemical properties. The dangling bonds at edge sites of graphene are highly reactive to guest atoms or molecules.
Typically, functionalization approaches used for fullerenes and carbon nanotubes (CNTs) can be applied to graphene. There are significant chemical differences due to the single atomic layer of graphene sheets. Much of this chemistry remains to be explored. The rehybridization from sp2 to sp3via covalent reaction occurs both at the edges and on the basal plane. Before discussing functionalization methods, we summarize six critical issues to consider regarding functionalization of graphene and related materials.
1.1. Multiple methods for graphene production
So far, several methods have been successfully established for graphene preparation, such as peeling-off graphite,1 liquid-phase exfoliation,48–50chemical vapor deposition,39,51–53graphitization of silicon carbide,26,54 templating,55,56reduction of graphene oxide,57,58 unzipping carbon nanotubes,59,60 organic synthesis,61,62 and anodic bonding.63 These different methods produce graphene with different size, shape, chemical composition, and environment (Fig. 2), all of which have different requirements for functionalization. | ||
| Fig. 2 Graphene prepared by different methods: (a) Large graphene crystal prepared on an oxidized Si wafer by the scotch-tape technique. (b) Left panel: suspension of microcrystals obtained by ultrasound cleavage of graphite in chloroform. Right panel: such suspensions can be printed on various substrates. (c) The first graphene wafers are now available as polycrystalline one- to five-layer films grown on Ni and transferred onto a Si wafer. (d) State-of-the-art SiC wafer with atomic terraces covered by a graphitic monolayer (indicated by “1”). Double and triple layers (“2” and “3”) grow at the steps.7 (e) Representation of the gradual unzipping of one wall of a carbon nanotube to form a nanoribbon.60 Reproduced from ref. 7. Copyright 2009 American Association for the Advancement of Science. Reproduced from ref. 60. Copyright 2009 Nature Publishing Group. | ||
1.2. Defects
All current methods cannot produce structurally and morphologically perfect graphene sheets because of the defects created unintentionally and unavoidably.64,65 These defects, including structural imperfections and chemical impurities, complicate graphene functionalization.1.3. Edges
Combining the two basic types of edge configurations (i.e., armchair and zigzag) leads to a variety of edges in graphene. This makes it difficult to characterize the structural and electronic properties and thus to control the functionalization processes at the edges.1.4. Stoichiometry
The functionalized graphene is non-stoichiometric in its chemical composition. This makes it difficult to control its properties.1.5. Low reactivity
Graphene has a much lower chemical reactivity than fullerenes and CNTs.66,67 The low reactivity limits the effective approaches for functionalizing graphene.411.6. Low solubility
Graphene has low solubility in both aqueous and organic solutions. This makes it difficult to manipulate graphene in solutions in which many functionalization processes occur. Thus, pre-treatment is sometimes required to increase the solubility of graphene for further functionalization.The electronic structure of graphene underlies its chemical properties. Ideal graphene is an infinite-scale two-dimensional sheet without edges and basal plane fluctuations. Composed of sp2carbon, ideal graphene is chemically unsaturated (Fig. 3A). Carbon atoms can form an extra covalent bond, which converts sp2 to sp3 hybridization and makes carbon reach its saturated state. Thus, this “unsaturation” is regarded as the origin of graphene's reactivity in covalent addition reactions. However, graphene is chemically inert (or stable) because all its pz atomic orbitals are strongly coupled and stabilized in a giant, delocalized π bonding system (Fig. 3A). On one hand, this π system usually precludes graphene from covalent addition. On the other hand, as a kind of π ligand, it renders versatile complexation reactions for graphene, e.g., with organic compounds and transition metals through π–π, H–π and metal–π interactions. The associated anti-bonding π* molecular orbitals can accommodate electrons, which facilitates favorable adsorption between graphene and electron-rich particles such as ions and alkali metals.
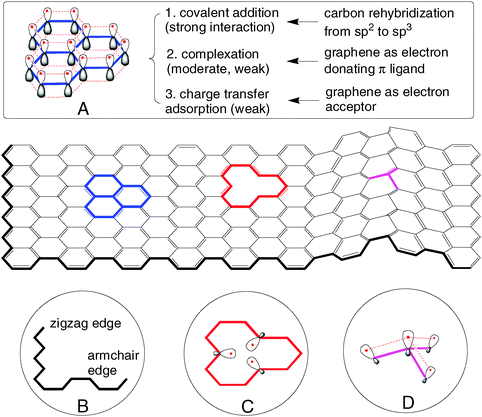 | ||
| Fig. 3 Origin of chemical reactivity of graphene. (A) Intrinsic reactivity arising from the delocalized π-bonding system. (B) Zigzag and armchair edges. (C) Monovacancy. (D) Local structure of a curved graphene sheet. In (A) and (D), the pz atomic orbitals are shown; the dashed lines represent overlap between pz orbitals. In (C), the dangling σ bonds are shown. | ||
In contrast to ideal graphene, practical graphene unavoidably contains edges, basal plane fluctuations, vacancies, and other chemical impurities. By altering the electronic structure, these structural “imperfections” can alter the chemical properties and reactions of graphene. During covalent addition, inner carbons that are strictly constrained in the basal plane have to protrude outward from the plane to adopt a tetrahedral sp3 geometry, causing strain in the plane. Edge carbon atoms are usually terminated by chemical groups such as hydrogen. Unlike inner carbon atoms, edge carbon atoms can adopt tetrahedral geometries more freely without causing extra strain. Therefore, edge carbons are preferred in covalent addition. Because frontier molecular orbitals are mainly localized at zigzag edges,68,69 zigzag edges are particularly reactive in graphene (Fig. 3B).67 For the same reason, vacancies like edges created inside the graphene basal plane, are also very reactive (Fig. 3C). Basal plane fluctuations cause curvature of graphene sheets. The curvature reduces the overlap of the pz atomic orbital of one carbon with pz orbitals of the three surrounding carbons (Fig. 3D). Thus, the curvature can lead to localized states with higher energies, which enhances the reactivity of the carbon.70
This paper reviews recent progress on graphene functionalization based on the different types of reactivity discussed above. We discuss the functionalization of graphene by basal plane covalent addition (Section 2). Because edges are particularly reactive sites in graphene, the covalent functionalization of graphene edges is discussed separately (Section 3). Unlike covalent addition, both complexation and charge-transfer adsorption do not destroy graphene's π bonding network and are discussed in Section 4. Finally, we review perspectives and challenges for graphene functionalization (Section 5). It is worth noting that practical reactions of graphene are usually complicated. Different types of reactivity may be involved simultaneously or at different stages of reactions. It is usually difficult to attribute a reaction solely to one or two origins of reactivity. Therefore, the above classification of the origins of reactivity, as well as the settings of Sections 2 through 4, are only attempted to emphasize the main driving force for the reactions.
2. Functionalization of graphene targeting basal plane covalent addition
Composed of sp2carbon, graphene is chemically unsaturated. Intrinsically, it is possible to undergo covalent addition to change the carbons from sp2 to sp3 hydridization. However, carbon atoms in the graphene basal plane are protected by their π-conjugation system, whose motion is constrained by surrounding carbon atoms. Therefore, basal plane covalent addition usually encounters large energy barriers, and reactive chemical groups, such as atomic hydrogen, fluorine, and precursors of other chemical radicals, are usually needed as the reactants. So far, the chemical modification of graphene cannot be fully controlled. Therefore, most of the reactions discussed in this section can also take place on graphene edges.2.1. Hydrogenation and dehydrogenation
Hydrogenation of the free-standing graphene and of graphene located on top of oxidized Si substrates has been investigated both experimentally and theoretically.66,71–75 The supported graphene displays different structures and electronic properties before and after hydrogenation. Hydrogenation changes the hybridization of carbon atoms from sp2 to sp3, resulting in elongated C–C bonds in the H-modified graphene. Hydrogen atoms tend to react with both surfaces of the plane of pristine graphene (Fig. 4). If only one side is hydrogenated, it can then be rolled to form CNTs because of the unbalanced external stress.76 The semi-hydrogenated graphene possesses ferromagnetic semiconductor properties because the partial hydrogenation can destroy the delocalized π bonding network of graphene.77 The fully hydrogenated graphene is called “graphane”, which is prepared under hydrogen plasma atmosphere and has also been the subject of a number of studies.75 In addition, it is worth mentioning that plasma activation supplies an alternative way to prepare the precursors of graphene derivatives, such as graphenol, graphenoic acid, and graphenamine.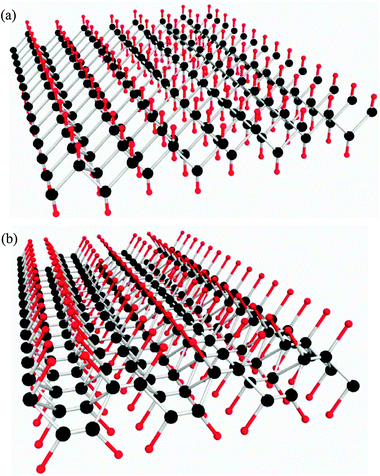 | ||
| Fig. 4 The most favorable conformations of graphene after hydrogenation, the chair (a) and boat (b) conformations. Carbon is shown as black and hydrogen is shown as red. In functionalization, hydrogen tends to attach to both sides of the graphene plane. | ||
Alternatively, several research groups also studied the chemisorptions of hydrogen on the graphene surface, in which case only small subareas of the graphene basal plane are hydrogenated unintentionally.78–81 There are four types of configurations that hydrogen pairs form on the basal plane from thermal graphitization of SiC (0001) after exposing to a 1600 K D-atom beam.79 The adsorbate forms and morphologies depend on various conditions, such as temperature, pressure, and hydrogen coverage. And hydrogens can be desorbed by annealing without influencing the electronic properties of graphene. Moreover, an energy bandgap can be induced via patterned hydrogen chemisorption.78
The dehydrogenation of graphene can proceed via an annealing process through which the properties of graphene can be partially restored.82 The reactivity of annealed graphene is higher than that of pristine graphene66 largely because defects form during annealing. Hydrogenation and dehydrogenation are useful processes for graphene functionalization, particularly by which ferromagnetism can be introduced, the band gap can be opened, and the π-conjugated skeleton of graphene can be altered. Additionally, the dehydrogenated graphene displays higher chemical reactivity than pristine graphene. This opens up new means for generating multifunctional graphene-based materials. Hydrogenation also offers a practical means to control electronic structure, which is the basis for fabricating novel devices. In addition, graphane, (CH)n, if produced, may be an excellent new material for hydrogen storage.72
2.2. Fluorination reactions
Ab initio pseudopotential calculations indicate that the electronic structure of fluorinated graphene sheets has a periodic structure in charge distribution near the Fermi energy.83 It is known that fluorine on the sidewall of fluoro-CNTs can be replaced by alkyl groups in the presence of alkyl lithium,84 alkylidene amino,85 or Grignard reagents.86 The replacement reactions can be extended to fluoro-graphene functionalization. The fluorination of graphene is also useful for further substitution and functionalization. For example, fluorinated graphene sheets successfully reacted in situ with butylamine87 to obtain the alkylated graphene. Alkylated graphenes are important graphene-based materials because they are readily dispersed in common organic solvents such as dichlorobenzene, dichloromethane, and THF. Moreover, they can be completely dealkylated by annealing to recover the original properties of pristine graphene sheets.88
periodic structure in charge distribution near the Fermi energy.83 It is known that fluorine on the sidewall of fluoro-CNTs can be replaced by alkyl groups in the presence of alkyl lithium,84 alkylidene amino,85 or Grignard reagents.86 The replacement reactions can be extended to fluoro-graphene functionalization. The fluorination of graphene is also useful for further substitution and functionalization. For example, fluorinated graphene sheets successfully reacted in situ with butylamine87 to obtain the alkylated graphene. Alkylated graphenes are important graphene-based materials because they are readily dispersed in common organic solvents such as dichlorobenzene, dichloromethane, and THF. Moreover, they can be completely dealkylated by annealing to recover the original properties of pristine graphene sheets.88
The fluorinated graphene has been prepared by plasma treatment of chemically converted graphene (CCG) at room temperature followed by subsequent reaction with butylamine.87 If the initial graphene sheets are prepared by thermally exfoliating graphene oxide or CCG, they have some residual oxygen-containing species on the basal plane. The presence of oxygen-containing species may prevent fluorine atoms from attaching to the graphene sheet in the fluorination process, which undoubtedly reduces the degree of fluorination.
2.3. Oxidation reactions
The oxidized graphene sheet is one of the most important forms of graphene, in which graphene is heavily oxygenated with a wide variety of oxygen species such as carbonyl, carboxyl, and hydroxyl groups.89 Because of the existence of oxygen-containing functional groups, graphene oxides react easily with soluble moieties. This enables changing the hydrophilicity, hydrophobicity, or organophilicity of graphene, as required for many applications. For example, many modified graphene sheets are readily dispersed in organic solvents for further functionalization, or for mixing with organic matrices to form new nanocomposite materials. Oxidation can also generate a monotype of oxygen-containing functional group, such as graphene with only hydroxyls (graphenol), or only carboxyls (graphenic acid) on the basal plane.Three experimental chemical routes have been developed for graphene oxidation. The first is a one-step process, and is achieved through direct oxidization of graphene with strong oxidants such as concentrated sulfuric acid, concentrated nitric acid, or potassium permanganate.90 The second is a two-step process, in which graphite is oxidized through Hummers',91Brodies',92 Staudenmaiers',93 or modified Hummer's methods,94 or electrochemic oxidation,95,96 followed by exfoliating or thermally expanding the graphene oxide obtained.97,98 The third is a physicochemical process: graphene oxide nanoribbons are created through lengthwise cutting and unraveling the side walls of multiwalled carbon nanotubes (MWCNTs) by oxidative processes.59,60 During the oxidation process, graphitic structures break down into smaller fragments. The formation mechanism, electronic properties, and conformations of these fragments were studied both experimentally and theoretically.99–102 It is worth noting that direct oxidization is not usually used to prepare graphene oxide but has the potential to control the size, shape, number of layers, and electronic properties of graphene. For example, experimentally, thermal oxidation of few-layer graphene with O2 can remove 2–3 layers of graphene and eventually yield a single layer, half-metallic graphene.103
2.4. Diazotization
Graphene has an electron-rich surface due to its π electrons. When electron-accepting moieties such as aryl diazonium salts react with graphene, electrons can transfer from the basal plane to the reactant. Because of the increase of pyramidalization of the deformed plane, diazonium salts easily react with graphene. Diazotization could be used to regulate electrical conductivity of the graphene since it can modulate the surface potential of graphenevia regiofunctionalization.104–108Both CCG and epitaxial graphene (EG) have been successfully modified by diazotization at room temperature.98,109 When nitrophenyl groups were attached to the EG surface, the resultant diazonium-functionalized graphene sheets were easily dispersed in polar aprotic solvents such as dimethylformamide (DMF), dimethylacetamide (DMAc), and N-methylpyrrolidone (NMP). Nitro groups on the surface of diazotized graphene can be reduced further to amine. The amine groups make subsequent graphene functionalization possible because amine can react with many other groups, such as hydroxyl radicals, carboxyl groups, and acyl chlorides. Interestingly, nonvolatile memory devices were fabricated by attachment of gold nanoparticles (AuNPs)-4-mercapto-benzenediazonium tetrafluoroborate salt (MBDT) conjugates (AuNPs-MBDTs) through diazotization reactions onto the π-conjugated skeleton of CCG.110
Different graphene preparation methods lead to graphene with different properties after diazotization. For EG on a substrate, there is a preferential adjustment of the density distribution of electrons in response to the regiofunctionalization of the surface. The diazotization process can form a defect-free and oxygen-free surface for EG that can be used to fabricate sensors, detectors, and other electronic devices. For CCG, both surfaces of the basal plane can be diazotized, but with defects and oxygenated groups. As a result, the conductivity of the diazotized CCG is significantly lower than that of the diazotized EG.
2.5. Other cycloaddition reactions
The thermal functionalization of fullerenes111 and carbon nanotubes112,113 by nitrenes via [2+1] cycloaddition reactions has been studied extensively. These reactions have also been applied to graphene to form aziridine adducts and to immobilize graphene on silicon wafers.114 The first step of the reaction protocol is the thermal or photochemical decomposition of perfluorophenylazides (PFPA) vianitrogen elimination, giving rise to the highly reactive single perfluorophenylnitrene. The second step consists of [2+1] cycloaddition of the nitrene to the surface of graphene. Alkyne-terminated molecules have been coupled to graphene sheets via “click” reactions to form stable heterocyclic linkages under mild reaction conditions.115Cycloaddition reactions of fullerenes and CNTs such as the Bingel [2+1] cyclopropanation reaction,116 can also be applied to graphene functionalization. And 1,3-dipolar cycloaddition has been successfully carried out not just at the edge but also at the internal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of graphene.117Claisen rearrangement was also used to functionalize graphene oxide, yielding a water-soluble derivative.118 The modular zwitterion-mediated transformations that have been used to functionalize CNTs and fullerenes119 can also be applied to graphene. These processes introduce a variety of functional groups for further reaction of graphene to expand the library of graphene-based materials for practical applications. For example, if graphene is coupled with chelating ligands, it can complex metal ions, and hence can be used to detect and to remove toxic heavy metal ions from the environment, or can be used as a novel filler to fabricate the columns for the efficient separation/treatment of radioactive wastes of nuclear power stations.
C bonds of graphene.117Claisen rearrangement was also used to functionalize graphene oxide, yielding a water-soluble derivative.118 The modular zwitterion-mediated transformations that have been used to functionalize CNTs and fullerenes119 can also be applied to graphene. These processes introduce a variety of functional groups for further reaction of graphene to expand the library of graphene-based materials for practical applications. For example, if graphene is coupled with chelating ligands, it can complex metal ions, and hence can be used to detect and to remove toxic heavy metal ions from the environment, or can be used as a novel filler to fabricate the columns for the efficient separation/treatment of radioactive wastes of nuclear power stations.
2.6. Reverse reactions of covalent addition
Chemical and thermal reduction of graphene oxide provide alternative approaches for the production of CCG120,121 and graphene-based composites.122 The oxygen-containing functional groups on the basal plane of graphene can be removed by thermal treatment or chemical reaction with reductants such as hydroquinone, reducing sugar, L-glutathione, N2H4, and their derivatives.57–58,121–134 Heating graphene oxide in ultrahigh vacuum is effective in controlling the percentage of oxygen-containing groups being removed.124 Solvothermal reduction of graphene oxide occurs at a much lower reaction temperature than pyrolysis and provides a means to reduce the defects in graphene.135–137 Photothermal heating has also been used to reduce graphene oxide. In contrast to chemical and thermal treatments, a photothermal heating process is rapid and does not require other reactants. In addition, heated atomic force microscope (AFM) tips can be used to reduce graphene oxide locally.138 However, none of these methods are capable of achieving complete reduction of graphene oxide. As a result, the reduced graphene oxide exhibits non-metallic behavior due to defects related to residual oxygen functional groups.57,58,124,129,130A combination of several different reduction methods may enhance the degree of reduction and improve the quality of graphene.139 For example, hydrogen reduction, combined with thermal treatment, can remove oxygen species effectively with little contamination on the basal plane. Nonetheless, the reduction process makes graphene hydrophobic and easy to aggregate.140 One way to prevent aggregation is to add polymers into the graphene oxide solution during the reduction process. However, the polymers also introduce extra impurities into the graphene, which are undesirable for some applications because the impurities can modify both surface and electronic properties. An alternative solution to the aggregation issue is to graft a charged unit such as sulfonate (–SO3H) to the graphene plane so that it can maintain the stability of graphene suspension via repulsive electrostatic forces.139,141,142 In addition, it is worth noting that controlled reduction is significant to adjust the optical properties of graphene oxide,143 opening up a way to explore their optical applications.
2.7. Graphene architectures by basal plane covalent addition
Many nanostructures such as fullerenes and CNTs can be attached covalently to the basal plane of graphene. They convert the two-dimensional (2D) graphene into three-dimensional (3D) complex networks. The networks result in new materials that exhibit special electronic and structural properties.144–147 According to theoretical studies, in pillared graphene, tunable electronic transport between graphene layers is achieved by controlling the lengths of the connecting CNTs.148 Based on atomic orbital density-functional theory, C60 molecules can be attached covalently to the basal plane of graphene, forming periodic graphene nanobuds.144 These nanobuds are thermally stable and are analogous to carbon nanobuds with C60 attached to carbon nanotubes.149 With unique structural properties, these materials could be used for gas or liquid storage133 and for functional sensors. However, it is difficult to make these 3D network nanostructures and the structure–property relationships remain to be measured. Fortunately, the Cheng group has achieved three-dimensional foam-like graphene macrostructures with outstanding electrical and mechanical properties by template-directed chemical vapour deposition. This method provides an approach to fabricating graphene architectures.1503. Functionalization of graphene based on edge covalent addition
Unlike inner carbons, edge carbons can adopt tetrahedral geometries more freely without causing extra strain. Therefore, edge carbon atoms are preferred in covalent addition reactions. Zigzag edges are particularly reactive in graphene because of the localized frontier molecular orbitals.The presence of edge defects in graphene facilitates chemical or physical reactions and provides opportunities for functionalization.121,151–155 There are several types of defect sites for functionalization in graphene: edge sites with dangling bonds, vacancies, and defect sites decorated with functional groups such as carbonyl, epoxy, and carboxylic groups.
3.1. Functionalization of graphene edge sites with benzyne- and carbene-like reactions
The π-electron structure at the edge is different from that on the basal plane for graphene with termination-free edge sites and the resulting dangling bonds.67,68,156–162 Depending on the π-electron structure, the carbon atoms at the edge form two types of configurations (i.e., armchair and zigzag) that lead to graphene of different properties. The different configurations can be tracked with an integrated transmission electron microscope (TEM)-scanning tunneling microscope (STM)163 (Fig. 5) and have been controlled with chemical methods.164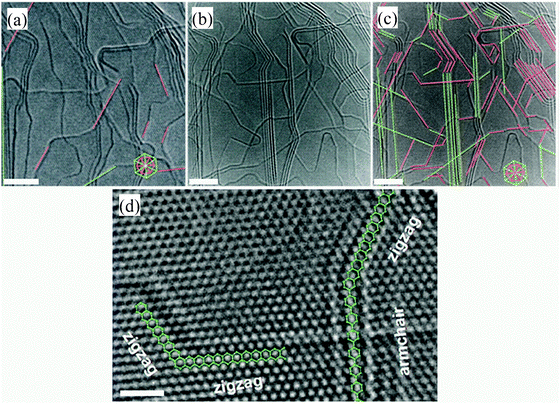 | ||
| Fig. 5 Joule-heating control and track of the armchair and zigzag configurations in graphitic nanoribbons with an integrated TEM-STM system: (a) ribbon sample before Joule heating, showing a few zigzag (pink lines) and armchair edges (green lines). (b) The same ribbon sample after Joule heating (for 10 min at 1.6 V), in which most of the edges seen are either zigzag or armchair edges, as indicated in (c). The inset hexagons indicate the zigzag- or armchair-edge orientations associated with the lattice patterns in (a) and (c). (d) High-magnification image of the annealed sample showing that well-defined zigzag-armchair and zigzag-zigzag edges are formed. The green hexagons in (c) help with the identification of the atomic structure at the armchair and zigzag edges. Scale bars in (a), (b) and (c), 4 nm; in (d), 1 nm.163 Reproduced from ref. 163. Copyright 2009 American Association for the Advancement of Science. | ||
Under ambient conditions, the termination-free zigzag or armchair graphene edges quickly react with absorbed molecules. Therefore, physical or chemical modification of the edge sites is significant for producing graphene with specific edge configurations and functionality. The electronic structure is such that the armchair configuration resembles a singlet benzyne and the zigzag configuration resembles a triplet carbene.161 As a result, the zigzag edge has much higher reactivity than the armchair edge, because the former has an edge state closer to the Fermi level.67 Moreover, the electrons at zigzag sites are unpaired π electrons that distribute around the edge with on average 0.14 electrons per atom.67 The zigzag sites are thus also regarded as partial radicals,67,165 and enable many carbene-like reactions such as cycloaddition and insertion. Still, reactions such as pericyclic and insertion can also be applied to armchair-edged graphene nanoribbons (GNR) for functionalization despite the lower activity of benzyne-like edges due to the delocalized π electrons.67 Recently, Diels–Alder chemistry has been carried out on graphene, and becomes a powerful strategy to adjust the electronic properties of graphene under mild conditions.166
At high temperature, the edge atoms can form loop structures.167 Loop formation saturates dangling bonds and reduces the reactivity of edges. Therefore, high-temperature processes should be avoided in functionalization of graphene edges unless loop structures are desired.
3.2. Adventitious functionalization of graphene edge sites
The edge sites of graphene can also be decorated adventitiously with moieties such as carbonyl, carboxylic, and epoxy groups. With these groups, one can further functionalize graphene to make it hydrophilic or organophilic.168,169 For example, this functionalization strategy was applied to synthesize graphene–polymer composites by managing the mixture of graphene with organic polymers.122The oxygen in graphene oxide is often used for further functionalization. For example, graphene that is dispersible in polar aprotic solvents was produced by coupling isocyanate to graphene oxide via the formation of amides and carbamate esters.122,170 The organophilic graphene that is highly soluble in organic solvents such as THF, DMF, and ethanol, was successfully produced by functionalization of graphene oxide with octadecylamine followed by reduction with hydroquinone or with tetrabutylammonium bromide.126,171 Similarly, the hydrophobic graphene was also synthesized via the functionalization of graphene oxide with octadecylamine.172High-resolution transmission electron microscopy (HRTEM) analysis revealed that the functionalized graphene retained its original crystal structure. This indicates that the chemical reactions occurred only at the edges and thus graphene functionalized in this way retains its electronic properties. Water-soluble poly(N-isopropylacrylamide)-graphene sheets were also synthesized via “click” chemistry, which has great potential as a novel drug-delivery vehicle.173Graphene modified by poly(ethylene glycol) (PEG) is biocompatible and used widely in biomedical studies.174,175
In a similar way, the functionalized graphene oxide can be further modified with additional functional groups such as allylamine and phenylisocyanate using solvothermal synthesis methods.176 The hydrophobic phenylisocyanate-functionalized graphene materials are soluble in many organic solvents, such as DMF, n-hexane, and THF. Graphene-based materials could be produced by controlling reactions of residual groups with molecules of selected functionality.177–179 For example, graphene-based materials for optical limiting were prepared by functionalizing graphene with oligothiophene viaamide bonds.180 For graphene-based fluorescent materials, fluorescence intensity can be controlled by adjusting the distance between graphene and the fluorescence moiety during the functionalization process. Such fluorescent materials have potential applications in devices such as bio-detectors, bio-sensors, and substrates of suppressed fluorescence.181–183
The edge sites of graphene oxide can also be functionalized by bi-functional moieties, one example of which is toluene diisocyanate (TDI).170 The TDI-functionalized graphene could react further with both hydrophilic and hydrophobic organic moieties in the same sample.163 This approach is useful for achieving the amphiphilic graphene that is dispersible in both organic and aqueous solutions.
The edge sites of graphene can be functionalized with various biomolecules via formation of acyl linkages such as amino acids and nucleic acids.184 This biofunctionalization process opens up new ways to construct graphene-based biomedical sensors, DNA transistors, and biomolecule cargo.182–185
3.3. Functionalization of graphene by creating edges
Graphene can effectively be functionalized by cutting it with metallic particles (Fig. 6). When catalytically active Ni,186,187Fe,188 or Ag189 particles are brought into proximity, they can cut a graphene sheet into smaller pieces. The cutting process has been demonstrated as a simple way to achieve GNR and other semiconductor graphene nanostructures.186–189 In the cutting experiments, the catalytic nanoparticles at the graphene edges can catalyze oxidation or hydrogenation of the carbon atoms to create a trench in the graphene sheet. Simultaneously, carbon atoms at the newly cut edges are saturated by hydrogen or oxygen atoms. | ||
| Fig. 6 Graphene tailoring. Cutting graphene from the edge is more like a physical process (scissors) using metallic particles (lower left).187 More advanced techniques with STM lithography can cut graphene sheets with a desired geometry, and tailor graphene into different nano-architectures (lower right). Reproduced from ref. 187, Copyright 2009 American Chemical Society. Reproduced from ref. 197, Copyright 2008 Nature Publishing Group. | ||
Alternatively, size-controllable nanopores can be cut into the basal plane of graphene by ion or electron etching through pre-patterned masks.190 These nanopores can be further functionalized with atoms and molecules, producing graphene with new properties.191 The nanoporous graphene has been used as filters for separating ions, atoms, or molecules, and as sensors for detecting DNA.190,192–194 One of the potential applications is that the filters can even be used for hydrogen atoms and standard gases including helium since they cannot diffuse through intact graphene planes.80,195
In pursuit of capturing single-layer graphene nanostructures, Tour and co-workers have developed single-atomic-layer lithography.196 This lithography enables layer-by-layer removal of graphene by sputtering zinc and then subsequent acid treatment. When used with predesigned zinc patterns, this method can pattern graphene with single-atomic-layer resolution for the fabrication of devices.
So far, most of the cutting processes proceed in a random manner. It is difficult to control the shape and crystallographic direction of the newly cut edges. One exception is STM lithography.197 It has been demonstrated that STM lithography can cut graphene sheets into desired geometries by adjusting lithography parameters such as bias voltage, tunneling current and scanning direction. This technique offers a means for fabricating graphene-based nanoelectronic integrated circuits (Fig. 7a).
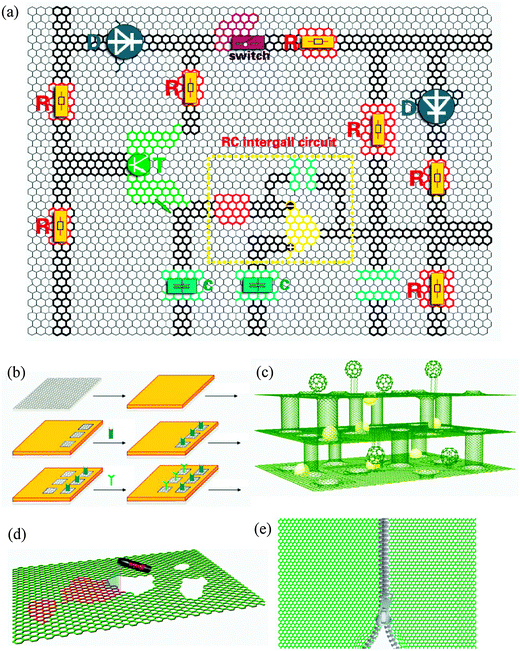 | ||
| Fig. 7 (a) Basal plane functionalization could lead to graphene-based integrated circuits. All electronic devices such as resistors (R), capacitors (C), triodes (T), diode (D), and molecular switches can be fabricated by passivation of the graphene surface via functionalization processes described in the text. The gray areas are insulating in the nano-circuit, possibly formed via regiofunctionalization with hydrogen. (b) Regiofunctionalization of graphene towards multifunctional and all-purpose devices. In the example, different functional probes can be bonded selectively to the graphene substrate. (c) The superior performance of graphene-based materials. One example may be superior catalysts. We can first load transitional metal nanoparticles onto graphene sheets and then package them into the multi-layered architecture of 3D network materials as superior catalysts. It is possible to load different nanoparticles/catalysts onto different layers to enhance performance, or even to regenerate the catalystin situ.147 (d) Mending graphene defects. To repair defects in graphene sheets is of great significance, and may be possible by making use of noncovalent functionalization such as basal plane loading of conjugated molecules. (e) Suture of graphene sheets. If the long edges of two graphene sheets can be stitched together, we can first functionalize different graphene sheets and then attach the functionalized sheets together to create multifunctional materials. For example, one approach for construction of GNRs can be applied for the graphene suture.62 | ||
4. Functionalization of graphene based on complexation and charge transfer adsorption
Transition metals, ions, molecules, and other atomic clusters can be loaded onto the basal plane of graphene through complexation reactions or charge-transfer adsorption (Fig. 8). Since these interactions do not convert carbon from sp2 to sp3, the π bonding system of graphene remains. Therefore, graphene decorated with these methods can be highly conductive. It can be used as an electrode material for manufacturing novel sensors for molecular detection28,198 and a substrate material for catalysis.199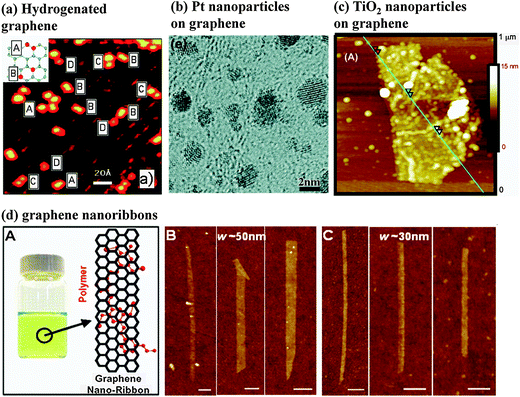 | ||
| Fig. 8 Functionalization of graphene based on noncovalent interactions: (a) scanning tunneling microscopy image of the hydrogenated graphene. The protrusions visible in the image are atomic hydrogen adsorbate structures identified as A = ortho-dimers, B = para-dimers, C = elongated dimers, and D = monomers (imaging parameters: Vt = −0.245 V, It = −0.26 nA). Inset in (a): schematic of the A ortho- and B para-dimer configuration on the graphene lattice.79 (b) High-resolution transmission electron microscopy image of Pt nanoparticles on graphene.225 (c) Atomic force microscopy (AFM) image of TiO2 nanoparticles on graphene. The cross-sections show individual particle heights as great as 7 nm.239 (d) (A) Photograph of a polymer PmPV/DCE solution with graphene nanoribbons (GNRs) stably suspended in solution, and schematic drawing of a GNR with two units of a PmPV polymer chain adsorbed on top of the graphenevia π stacking. (B and C) AFM images of selected GNRs with widths of 50 and 30 nm, respectively.270 Reproduced from ref. 79. Copyright 2009 American Chemical Society. Reproduced from ref. 225. Copyright 2009 American Chemical Society. Reproduced from ref. 239. Copyright 2008 American Chemical Society. Reproduced from ref. 270. Copyright 2008 American Association for the Advancement of Science. | ||
4.1. Graphene basal plane decorated with atoms and ions
Due to the potential applications in catalysis and sensors, the adsorption behavior of atoms (especially transition metal atoms) has been widely investigated.200–203 Based on density functional theory, the most stable configuration of a single Au atom sits above a carbon atom, while for Au dimers, it is perpendicular to the graphene plane above the bridge site.204 The adsorption behavior of Cu is the same as that of Au.205 However, Xe atoms preferentially occupy the hollow sites on the graphene surface.206 Additively, using Raman spectra, Pollak et al. found that the adsorption behavior of Li+ on single- and few-layer graphene was different.2074.2. Graphene basal plane decorated with molecules
The delocalized π electrons of graphene are heavily affected by adsorbates. As a result, molecules adsorbed on a graphene surface can change the local carrier concentration and also open the band gap, due to the charge carrier transfer between the two components.198,208–210 Based on the charge of the electroconductivity after adsorbing molecules, vapor sensors have been fabricated, with high levels of sensitivity and easy manipulation.28,29,211,212 The gap of graphene can be tuned up to ∼0.206 eV via the controlled adsorption of water molecules to the surface of graphene.213 This paves an avenue to adjusting the graphene gap without disturbing the π-skeleton structure.4.3. Graphene basal plane decorated with transition-metal nanoparticles
Transition-metal nanoparticles can also be adsorbed and distributed evenly on the basal plane of graphene.214–224 The size and shape of the nanoparticles on graphene can be adjusted by controlling solvents or reducing agents. In some cases, nanocomposites made of transition metals and graphene have shown superior catalytic performance to conventional transition-metal catalysts or transition-metal carbides.199,225–229 For example, Pt nanoparticles can adhere strongly to the defect-free graphene due to electron transfer from graphene to the nanoparticles.230,231 These transition-metal nanoparticles, in combination with the superior conductivity and large surface area of graphene, have the potential applications in catalysis and sensors.232,233 Much remains to be explored in these promising areas.4.4. Graphene basal plane decorated with metal oxides
Graphene–metal-oxide composites exhibit superior electronic properties for applications in energy harvesting, conversion, and storage.123,234–241Graphene–SnO2 nanocomposites have been prepared by reassembling graphene nanosheets in SnO2 nanoparticles237 or by in situ chemical synthesis.123 The graphene sheets could be homogeneously distributed among the loosely packed SnO2 nanoparticles, which enhanced the cyclability of graphene–SnO2-based electrodes. TiO2 nanoparticles have been attached to graphene sheets by the UV-assisted reduction of graphene oxide in TiO2/ethanol suspensions. The reduced graphene sheets remained well dispersed in solvent due to their interactions with TiO2 nanoparticles.239 The electrical resistance of the TiO2–graphene nanocomposites was reduced by an order of magnitude. Using graphene–TiO2 nanocomposites to form an interfacial layer in dye-sensitized solar cells can greatly enhance the photoconversion efficiency.242LiFePO4–graphene nanocomposites possess all the advantages of pristine LiFePO4, such as low cost, environmental compatibility, and cycling stability, while their conductivity, delivery capacity and cycle performance are much better than conventional LiFePO4.240 Thus, LiFePO4–graphene composites are candidate materials for lithium secondary batteries.243,244ZnO nanorod–graphene hybrid architectures are composed of arrays of ZnO nanorods on graphene films. Being transparent, conductive, and flexible, the ZnO–graphene composites are applicable for next-generation electronic and optoelectronic systems.245 The graphene/Fe3O4 hybrid paper, which exhibits actuation strain as large as 0.1% at −1 V applied potential, can be used to fabricate electromechanical actuators.246 In addition, when graphene oxide and Na0.44MnO2 nanowires were co-assembled, they formed aligned nanowires that were successfully transferred to a Si substrate viasolvent evaporation.247 The co-assembly process transferred the Na0.44MnO2 nanowires from superhydrophobicity into hydrophilicity with contact angles of up to 50°. This result suggests that graphene oxide can serve as a surfactant to improve the surface properties of hydrophobic materials.4.5. Graphene basal plane decorated with conjugated compounds
Graphene aggregates easily in solution due to its hydrophobicity. This limits its potential chemical functionalization and applications. Decorating graphene with conjugated compounds can be used to obtain stable suspensions of individual graphene sheets.248–255 The conjugated compounds can be attached to graphene sheets via noncovalent interactions such as π–π stacking or hydrogen bonding. Graphene dispersions can be prepared via exfoliation of graphite in N-methyl-pyrrolidone (NMP),48 where the energy required to exfoliate graphite is partially compensated by the strong interaction between graphene and NMP. A similar process, noncovalent functionalization of graphene oxide with 1-pyrenebutyrate followed by reduction of the functionalized graphene oxide with hydrazine monohydrate,256,257 can also be used to produce stable aqueous dispersions of graphene sheets. 5,10,15,20-Tetrakis(1-methyl-4-pyridinio)porphyrin (TMPyP) was successfully loaded on CCG sheets through electrostatic and π–π stacking cooperative interactions.258TMPyP-CCG complexes are used as optical probes for rapid and selective detection of Cd2+ ions.258 Conjugated molecules on graphene can also affect the absorption of metal nanoparticles in two different ways. First, the molecules weaken the interactions between the metal nanoparticles and graphene. Second, the conjugated molecules provide new reactive sites for the deposition of the nanoparticles.259,260Surfactants and biomolecules such as cetyltrimethylammonium bromide (CTAB),261sodium cholate (NaC),251sodium dodecyl benzenesulfonate (SDBS)49 and tryptophan262 have also been used to disperse graphene in different solvents. These surfactants adsorb onto the electron-rich surfaces of graphene sheets. In large-scale production processes of graphene by electrolytic exfoliation of graphite in a poly(sodium-4-styrenesulfonate) (PPS) electrolyte, the π–π interactions between the aromatic rings of PPS and the graphene plane play major roles in its dispersion.263
Alternatively, due to the π-skeleton structure and large specific surface area, graphene/graphene oxide has the potential to be used as a drug carrier. Drug molecules (mainly aromatic) can adsorb onto the surface of graphene oxide via π–π interactions, improving their water solubility. When mixing the PEGylated nanographene oxide with 7-ethyl-10-hydroxy-camptothecin (SN38) and doxorubicin (DOX) (water-insoluble cancer drugs), both the drug molecules are readily adsorbed onto the graphene surface with high loading potency and strong chemical attachment.264,265 During this process, the high potency of the free drug molecules was retained. Moreover, the graphene oxide-based carriers displayed no cytotoxicity by themselves.
4.6. Graphene basal plane decorated with polymers
The noncovalent functionalization of graphene with polymers is another important approach to graphene modification in solvents. The polymer chains can enhance the dispersion of graphene in diverse solvents, yielding polymer–graphene composites for future reactant applications. Strategically, we may further divide the processes for the noncovalent attachment of polymers to graphene into two methods: “loading from” and “loading to” (Fig. 9).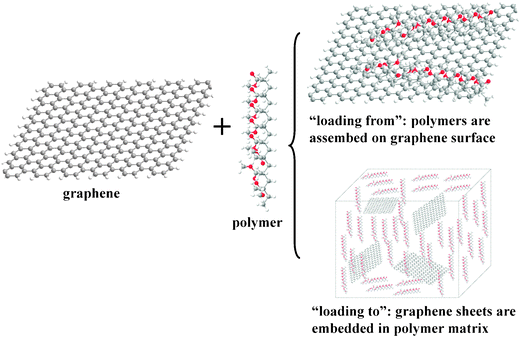 | ||
| Fig. 9 Schematic of processes for the noncovalent attachment of polymers to graphene: “loading from”; “loading to”. | ||
“Loading from” describes all processes in which graphene serves as the matrix for loading polymers onto the graphene surface. This method has been widely applied in graphene preparation mainly to regulate the solubility of graphene so that it can be manipulated in solvent,140,266 or to adjust the electronic structure.267,268 For example, Dai and co-workers successfully loaded 1,2-distearoyl-sn-glycero-3-phospho-ethanolamine-N-[methoxy(polyethyleneglycol)-5000] (DSPE-mPEG) and poly(m-phenylenevinylene-co-2,5-dioctoxy-p-phenylenevinylene) (PmPV) onto graphene to form homogeneous suspensions that have been used to obtain Langmuir–Blodgett films and ultra-smooth nanoribbon semiconductors.269,270 The Dai group also noncovalently functionalized the reduced graphene oxide with an amphiphilic PEGylated polymer, rendering them stable in biological solutions.271 Loading perylene bisimide-containing poly(glyceryl acrylate) (PBIPGA) onto the reduced graphene oxide resulted in improved dispersions. Moreover, the conjugate exhibits low cytotoxicity toward 3T3 fibroblasts.272 Polyoxyethylene sorbitan laurate and poly(norepinephrine) were used to improve the dispersion of the reduced graphene oxide in water.273,274 Cationic poly(ethyleneimine) (PEI)-modified graphene sheets could be co-assembled with acid-oxidized multiwalled carbon nanotubes to form hybrid carbon films. The interconnected networks of carbon structures with well-defined nanopores in these hybrid films are expected to be useful in the fabrication of supercapacitor electrodes.275 Using the tight binding model, Chen et al. reported that the bandgap can be effectively opened after loading pyrene molecules on both sides of graphene.276
“Loading to” represents all processes in which the graphene serves as nanofiller to be loaded into a polymer matrix. This method usually forms graphene–polymer composites through mixing graphene/graphene derivatives with a polymer suspension.94,122,277–282 The prepared graphene–polymer composites have shown enhanced and even new properties.257,283,284 For example, a graphene–PMMA composite showed strong interfacial interactions between PMMA and graphene, which dramatically enhanced many important properties of the composite such as its Young's modulus and thermal stability.277Graphene–polyaniline composites showed a red-shift in absorption wavelength due to π–π interactions and hydrogen bonding between graphene and the polymer skeleton.285 In addition, graphene–polyaniline composites have increased conductivity and specific capacitance.285,286 Loading graphene oxide coated with Fe3O4 into polystyrene led to a novel magnetic composite with high flexibility and optical transparency.287 When graphene was loaded into epoxy compounds, the composites were lightweight and showed effective electromagnetic interference (EMI) shielding. The EMI shielding makes graphene–epoxy composites excellent candidates for stealth materials in military airplanes.288Graphene-conjugated polymer composites of poly(3-hexylthiophene-2,5-diyl) (P3HT) and poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] (MEH-PPV) show strong fluorescence quenching and are suitable for organic hetero-junction solar cells.289
4.7. Assembly and co-assembly of graphene
The assembly and co-assembly of nanostructures present a path to multifunctional materials. For graphene, noncovalent forces are driving forces, although other external forces, such as Langmuir–Blodgett compression, microfluidic flow, and stick-slip motion, can also be important in facilitating assembly and co-assembly processes. The covalent assembly of graphene for large-production stable conjugated nanomaterials has also been proposed.290There are methods, such as rapid freezing by spraying,58dip coating,291 drop casting,57,120 and filtering,256,292,293 that have been used to achieve mono- or multilayer sheets of graphene from graphene oxide suspensions. Vacuum filtration has been successfully used to produce uniform films with controllable numbers of layers over large areas.32 These thin films can be used for transparent conducting materials in solar cells31,291 and thin-film transistors.44 A graphene oxide paper of macroscale thickness with strong mechanical properties can also be produced by filtering graphene oxide colloids.294Pulsed laser deposition (PLD)295 and matrix-assisted laser desorption/ionization (MALDI)296 have been used in the deposition of graphene sections (like poly-aromatic hydrocarbons, PAHs) onto substrates without decomposition. Layer-by-layer (LBL) approaches are suitable for preparing metal-doped graphene films.297,298Graphene can also be deposited on a defined surface to produce structured graphene sheets. For example, sheets of graphene or graphene oxide can be assembled onto patterned and functionalized Au surfaces.299 A new type of carbon nanotube was also produced by carbonizing disc-like graphene assemblies in porous alumina membrane templates.300 The self-assembly of graphene oxide sheets can take place outside and inside CNTs to create new tube-in-tube nanostructures.301 The sheets were also loaded onto the surfaces of poly(ethylene glycol)-coated submicrometre sulfur particles to render the sulfur particles electrically conducting.302 Chuvilin et al. reported that sulfur-terminated graphene nanoribbons can spontaneously self-assemble within SWNTs, achieving a conjugate with novel properties.303 In addition, delaminating and co-stacking were developed for co-assembling graphene with other materials.304
Langmuir–Blodgett deposition has been used to assemble both monolayer and multilayer films of graphene and graphene oxide, which were transferable to solid substrates.269,305 These transparent films have high conductivity and may enable utilization in high-performance solar cell electrodes.306,307 The transparency of the films was inversely proportional to film thickness, and the electrical conductivity was independent of the thickness.
5. Perspectives and challenges
Adoption of graphene-based technologies requires rational and robust functionalization of graphene.308 This, in turn, requires fundamental understanding of the chemistry and physics of graphene and related materials.309 A number of key challenges associated with graphene, including its production, defects, control of edges, reactivity, control of stoichiometry, and solubility, complicate the implementation of functionalization methods. We have described some of the driving forces that can be exploited in graphene to construct a systematic framework for the chemical and physical modification of graphene. Next, we provide some perspectives on the major challenges for the future of graphene functionalization and application.Graphene functionalization occurs randomly: there exist few methods to exert precise control over how and where chemical reactions occur. For example, defects exist both at edges and on the basal plane. Defect-induced chemical reactions occur at both positions simultaneously. Thus, it is a singular challenge to establish techniques for the control of graphene functionalization such that it occurs either at the edge or on the basal plane, but not both. Regiochemistry, in which covalent or noncovalent functionalization proceeds in a selected region of graphene, may provide the solution.310,311 Furthermore, if atoms or molecules can react selectively with a specific region of graphene, it may become possible to build integrated circuits (Fig. 7a), multifunctional sensors (Fig. 7b), and devices (Fig. 1).
The second major challenge is in developing graphene-based materials with superior performance to those currently available. For example, graphene decorated with transition metal nanoparticles may only function as a superior catalyst if there are large specific surface areas and/or high electron mobilities. This requires substantial understanding of the interactions between graphene and metal nanoparticles (one suggested catalyst structure is shown in Fig. 7c). In Fig. 7c, graphene layers are linked by fullerenes and nanotubes of different diameters. Fullerenes can be linked directly or indirectly to graphene layers. With direct linkages, the bond energy at the high curvature positions is larger and results in more reactive sites. With indirect linkages, organic moieties lead to increased surface areas. Recently, we found that carbon nanotubes decorated with blood proteins show reduced cytotoxicity.312 This finding may pave the way toward new applications of graphene in biomedicine.
The third major challenge is defect mending in graphene. Eliminating defects from graphene would open doors to promising new applications for graphene and graphene-based materials. It may be possible to repair defects based on noncovalent functionalization that loads the basal plane of graphene with conjugated molecules (Fig. 7d).
The fourth challenge is developing graphene sutura. Developing techniques for ‘sewing’ the edges of two small graphene pieces would enable an additional dimension of graphene functionalization (Fig. 7e). By combining sutura and regiofunctionalization techniques, we could functionalize specific areas of small pieces of graphene sheets and sew them together into the integrated circuits (Fig. 7a). Some approaches used for the construction of GNRs might be applicable for graphene sutura.62
Structural uniformity is another important goal. Graphene prepared by various methods exhibits different properties due to structure and morphology differences. This limits the practical utility of graphene in devices. Thus, it is necessary to establish manufacturing methods that can prepare graphene with uniform structures.313,314
Lastly, as in all areas of nanoscience, graphene research requires new experimental tools. To explore these intriguing areas discussed above, we need to build advanced experimental tools with new capabilities in manipulation, explicit characterization, and precise analyses of graphene-based materials and products.315
Acknowledgements
The authors are grateful for the support of MOST 973 program (2011CB933403, 2012CB934001, 2010CB934000), NSF (NSF-CHE-1041943), and the Kavli Foundation. We thank Heidi Bednar, Sarawut Cheunkar, Shelley Claridge, Nate Hohman, Brian Kiraly, John Thomas, and Amit Vaish for helpful discussions.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- Y. Zhang, Y.-W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS.
- M. S. Dresselhaus, ACS Nano, 2010, 4, 4344–4349 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- J. S. Bunch, A. M. van der Zande, S. S. Verbridge, I. W. Frank, D. M. Tanenbaum, J. M. Parpia, H. G. Craighead and P. L. McEuen, Science, 2007, 315, 490–493 CrossRef CAS.
- M. Orlita, C. Faugeras, P. Plochocka, P. Neugebauer, G. Martinez, D. K. Maude, A.-L. Barra, M. Sprinkle, C. Berger, W. A. de Heer and M. Potemski, Phys. Rev. Lett., 2008, 101, 267601 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- M. Y. Han, B. Öezyilmaz, Y. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS.
- K. S. Novoselov, E. McCann, S. V. Morozov, V. I. Fal'ko, M. I. Katsnelson, U. Zeitler, D. Jiang, F. Schedin and A. K. Geim, Nat. Phys., 2006, 2, 177–180 CrossRef.
- K. S. Novoselov, Z. Jiang, Y. Zhang, S. V. Morozov, H. L. Stormer, U. Zeitler, J. C. Maan, G. S. Boebinger, P. Kim and A. K. Geim, Science, 2007, 315, 1379 CrossRef CAS.
- N. Stander, B. Huard and D. Goldhaber-Gordon, Phys. Rev. Lett., 2009, 102, 026807 CrossRef CAS.
- A. B. Kaiser and V. Skákalová, Chem. Soc. Rev., 2011, 40, 3786–3801 RSC.
- S.-H. Phark, J. Borme, A. L. Vanegas, M. Corbetta, D. Sander and J. Kirschner, ACS Nano, 2011, 5, 8162–8166 CrossRef CAS.
- M. G. Sung, H. Lee, K. Heo, K.-E. Byun, T. Kim, D. H. Seo, S. Seo and S. Hong, ACS Nano, 2011, 5 DOI:10.1021/nn202135g.
- A. T. Valota, I. A. Kinloch, K. S. Novoselov, C. Casiraghi, A. Eckmann, E. W. Hill and R. A. W. Dryfe, ACS Nano, 2011, 5 DOI:10.1021/nn202878f.
- A. K. Geim, Angew. Chem., Int. Ed., 2011, 50, 6966–6985 CrossRef CAS.
- K. S. Novoselov, Angew. Chem., Int. Ed., 2011, 50, 6986–7002 CrossRef CAS.
- C. R. Dean, A. F. Young, P. Cadden-Zimansky, L. Wang, H. Ren, K. Watanabe, T. Taniguchi, P. Kim, J. Hone and K. L. Shepard, Nat. Phys., 2011, 7, 693–696 CrossRef CAS.
- Basic research needs for advanced nuclear energy systems, Panel 3 report: separations science, current status, DOE Report, p. 31, October 2006 (The full report is available at http://www.sc.doe.gov/bes/reports/files/ANES_rpt.pdf).
- The economics of the nuclear fuel cycle, OECD Report, Nuclear Energy Agency, ISBN: 9264127143 (pbk), OECD Publications and Information Center, Paris (1994).
- D. Butler, Nature, 2004, 429, 238–240 CrossRef CAS.
- F. N. von Hippel, Science, 2001, 293, 2397–2398 CrossRef CAS.
- Y. M. Lin, A. Valdes-Garcia, S. J. Han, D. B. Farmer, I. Meric, Y. Sun, Y. Wu, C. Dimitrakopoulos, A. Grill, P. Avouris and K. A. Jenkins, Science, 2011, 332, 1294–1297 CrossRef CAS.
- S. Guo and S. Dong, Chem. Soc. Rev., 2011, 40, 2644–2672 RSC.
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912–19916 CrossRef CAS.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1196 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- V. Dua, S. P. Surwade, S. Ammu, S. R. Agnihotra, S. Jain, K. E. Roberts, S. Park, R. S. Ruoff and S. K. Manohar, Angew. Chem., Int. Ed., 2010, 49, 2154–2157 CrossRef CAS.
- H. Wang, Q. Zhang, X. Chu, T. Chen, J. Ge and R. Yu, Angew. Chem., Int. Ed., 2011, 50, 7065–7069 CrossRef CAS.
- X. Wang, L. Zhi, N. Tsao, Ž. Tomović, J. Li and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 2990–2992 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7778 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- D. A. Areshkin and C. T. White, Nano Lett., 2007, 7, 3253–3259 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS.
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, ACS Nano, 2011, 5 DOI:10.1021/nn202710s.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- L. Yan, F. Zhao, S. Li, Z. Hu and Y. Zhao, Nanoscale, 2011, 3, 362–382 RSC.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS.
- Y. Gogotsi, J. Phys. Chem. Lett., 2011, 2, 2509–2510 CrossRef CAS.
- S. Niyogi, E. Bekyarova, J. Hong, S. Khizroev, C. Berger, W. de Heer and R. C. Haddon, J. Phys. Chem. Lett., 2011, 2, 2487–2498 CrossRef CAS.
- Z. Sun, D. K. James and J. M. Tour, J. Phys. Chem. Lett., 2011, 2, 2425–2432 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS.
- A. Mukherjee, J. Kang, O. Kuznetsov, Y. Sun, R. Thaner, A. S. Bratt, J. R. Lomeda, K. F. Kelly and W. E. Billups, Chem. Mater., 2010, 23, 9–13 CrossRef.
- T. A. Land, T. Michely, R. J. Behm, J. C. Hemminger and G. Comsa, J. Chem. Phys., 1992, 97, 6774–6783 CrossRef CAS.
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 30–35 CrossRef CAS.
- S. Bae, H. Kim, Y. Lee, X. Xu, J.-S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. Ri Kim, Y. Song, Y.-J. Kim, K. S. Kim, B. Özyilmaz, J.-H. Ahn, B. H. Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS.
- K. V. Emtsev, A. Bostwick, K. Horn, J. Jobst, G. L. Kellogg, L. Ley, J. L. McChesney, T. Ohta, S. A. Reshanov, J. Röhrl, E. Rotenberg, A. K. Schmid, D. Waldmann, H. B. Weber and T. Seyller, Nat. Mater., 2009, 8, 203–207 CrossRef CAS.
- D. Wei, Y. Liu, H. Zhang, L. Huang, B. Wu, J. Chen and G. Yu, J. Am. Chem. Soc., 2009, 131, 11147–11154 CrossRef CAS.
- W. Zhang, J. Cui, C. A. Tao, Y. Wu, Z. Li, L. Ma, Y. Wen and G. Li, Angew. Chem., Int. Ed., 2009, 48, 5864–5868 CrossRef CAS.
- C. Gómez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499–3503 CrossRef.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394–3398 CrossRef CAS.
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877–880 CrossRef CAS.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS.
- X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS.
- H. Qian, F. Negri, C. Wang and Z. Wang, J. Am. Chem. Soc., 2008, 130, 17970–17976 CrossRef CAS.
- T. Moldt, A. Eckmann, P. Klar, S. V. Morozov, A. A. Zhukov, K. S. Novoselov and C. Casiraghi, ACS Nano, 2011, 5, 7700–7706 CrossRef CAS.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS.
- J. Stadler, T. Schmid and R. Zenobi, ACS Nano, 2011, 5, 8442–8448 CrossRef CAS.
- S. Ryu, M. Y. Han, J. Maultzsch, T. F. Heinz, P. Kim, M. L. Steigerwald and L. E. Brus, Nano Lett., 2008, 8, 4597–4602 CrossRef CAS.
- D.-E. Jiang, B. G. Sumpter and S. Dai, J. Chem. Phys., 2007, 126, 134701 CrossRef.
- K. Nakada, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954–17961 CrossRef CAS.
- T. Akasaka, F. Wudl and S. Nagase, in Chemistry of Nanocarbons, ed. D.-E. Jiang, X. Gao, S. Nagase, Z. Chen, D.-E. Jiang, X. Gao, S. Nagase and Z. Chen, Wiley, ch.18, p. 433–465 Search PubMed.
- X. Gao, Y. Wang, X. Liu, T.-L. Chan, S. Irle, Y. Zhao and S. B. Zhang, Phys. Chem. Chem. Phys., 2011, 13 10.1039/C1CP22491C.
- A. Savchenko, Science, 2009, 323, 589–590 CrossRef CAS.
- J. O. Sofo, A. S. Chaudhari and G. D. Barber, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153401 CrossRef.
- A. K. Singh, E. S. Penev and B. I. Yakobson, ACS Nano, 2010, 4, 3510–3514 CrossRef CAS.
- T. Wassmann, A. P. Seitsonen, A. M. Saitta, M. Lazzeri and F. Mauri, Phys. Rev. Lett., 2008, 101, 096402 CrossRef.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610–613 CrossRef CAS.
- D. Yu and F. Liu, Nano Lett., 2007, 7, 3046–3050 CrossRef CAS.
- J. Zhou, Q. Wang, Q. Sun, X. S. Chen, Y. Kawazoe and P. Jena, Nano Lett., 2009, 9, 3867–3870 CrossRef CAS.
- R. Balog, B. Jorgensen, L. Nilsson, M. Andersen, E. Rienks, M. Bianchi, M. Fanetti, E. Laegsgaard, A. Baraldi, S. Lizzit, Z. Sljivancanin, F. Besenbacher, B. Hammer, T. G. Pedersen, P. Hofmann and L. Hornekaer, Nat. Mater., 2010, 9, 315–319 CrossRef CAS.
- R. Balog, B. Jørgensen, J. Wells, E. Lægsgaard, P. Hofmann, F. Besenbacher and L. Hornekær, J. Am. Chem. Soc., 2009, 131, 8744–8745 CrossRef CAS.
- N. P. Guisinger, G. M. Rutter, J. N. Crain, P. N. First and J. A. Stroscio, Nano Lett., 2009, 9, 1462–1466 CrossRef CAS.
- T. Roman, W. A. Diño, H. Nakanishi, H. Kasai, T. Sugimoto and K. Tange, Carbon, 2007, 45, 218–220 CrossRef CAS.
- Y. Wang, X. Xu, J. Lu, M. Lin, Q. Bao, B. Özyilmaz and K. P. Loh, ACS Nano, 2010, 4, 6146–6152 CrossRef CAS.
- M. Igami, S. Okada and K. Nakada, Synth. Met., 2001, 121, 1233–1234 CrossRef CAS.
- R. K. Saini, I. W. Chiang, H. Peng, R. E. Smalley, W. E. Billups, R. H. Hauge and J. L. Margrave, J. Am. Chem. Soc., 2003, 125, 3617–3621 CrossRef CAS.
- J. L. Stevens, A. Y. Huang, H. Peng, I. W. Chiang, V. N. Khabashesku and J. L. Margrave, Nano Lett., 2003, 3, 331–336 CrossRef CAS.
- P. J. Boul, J. Liu, E. T. Mickelson, C. B. Huffman, L. M. Ericson, I. W. Chiang, K. A. Smith, D. T. Colbert, R. H. Hauge, J. L. Margrave and R. E. Smalley, Chem. Phys. Lett., 1999, 310, 367–372 CrossRef CAS.
- S. B. Bon, L. Valentini, R. Verdejo, J. L. Garcia Fierro, L. Peponi, M. A. Lopez-Manchado and J. M. Kenny, Chem. Mater., 2009, 21, 3433–3438 CrossRef.
- K. A. Worsley, P. Ramesh, S. K. Mandal, S. Niyogi, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2007, 445, 51–56 CrossRef CAS.
- J. Chattopadhyay, A. Mukherjee, C. E. Hamilton, J. Kang, S. Chakraborty, W. Guo, K. F. Kelly, A. R. Barron and W. E. Billups, J. Am. Chem. Soc., 2008, 130, 5414–5415 CrossRef CAS.
- K. S. Subrahmanyam, S. R. C. Vivekchand, A. Govindaraj and C. N. R. Rao, J. Mater. Chem., 2008, 18, 1517–1523 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. C. Brodie, Ann. Chim. Phys., 1860, 59, 466–471 Search PubMed.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS.
- L. J. Cote, R. Cruz-Silva and J. Huang, J. Am. Chem. Soc., 2009, 131, 11027–11032 CrossRef CAS.
- M. J. Hudson, F. R. Hunter-Fujita, J. W. Peckett and P. M. Smith, J. Mater. Chem., 1997, 7, 301–305 RSC.
- J. W. Peckett, P. Trens, R. D. Gougeon, A. Pöppl, R. K. Harris and M. J. Hudson, Carbon, 2000, 38, 345–353 CrossRef CAS.
- Z. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS.
- Z. Li, W. Zhang, Y. Luo, J. Yang and J. G. Hou, J. Am. Chem. Soc., 2009, 131, 6320–6321 CrossRef CAS.
- O. Hod, V. Barone, J. E. Peralta and G. E. Scuseria, Nano Lett., 2007, 7, 2295–2299 CrossRef CAS.
- X. Gao, L. Wang, Y. Ohtsuka, D.-E. Jiang, Y. Zhao, S. Nagase and Z. Chen, J. Am. Chem. Soc., 2009, 131, 9663–9669 CrossRef CAS.
- Q. Liaoa, H. J. Zhanga, K. Wua, H. Y. Lia, S. N. Baoa and P. He, Appl. Surf. Sci., 2010, 257, 82–86 CrossRef.
- Z. Osváth, A. Darabont, P. Nemes-Incze, E. Horváth, Z. E. Horváth and L. P. Biró, Carbon, 2007, 45, 3022–3026 CrossRef.
- R. Sharma, J. H. Baik, C. J. Perera and M. S. Strano, Nano Lett., 2010, 10, 398–405 CrossRef CAS.
- A. Sinitskii, A. Dimiev, D. A. Corley, A. A. Fursina, D. V. Kosynkin and J. M. Tour, ACS Nano, 2010, 4, 1949–1954 CrossRef CAS.
- H. Liu, S. Ryu, Z. Chen, M. L. Steigerwald, C. Nuckolls and L. E. Brus, J. Am. Chem. Soc., 2009, 131, 17099–17101 CrossRef CAS.
- F. Koehler, N. Luechinger, D. Ziegler, E. Athanassiou, R. Grass, A. Rossi, C. Hierold, A. Stemmer and W. Stark, Angew. Chem., Int. Ed., 2009, 48, 224–227 CrossRef CAS.
- P. Huang, H. Zhu, L. Jing, Y. Zhao and X. Gao, ACS Nano, 2011, 5, 7945–7949 CrossRef CAS.
- E. Bekyarova, M. E. Itkis, P. Ramesh, C. Berger, M. Sprinkle, W. A. de Heer and R. C. Haddon, J. Am. Chem. Soc., 2009, 131, 1336–1337 CrossRef CAS.
- P. Cui, S. Seo, J. Lee, L. Wang, E. Lee, M. Min and H. Lee, ACS Nano, 2011, 5, 6826–6833 CrossRef CAS.
- M. Yan, S. X. Cai and J. F. W. Keana, J. Org. Chem., 1994, 59, 5951–5954 CrossRef CAS.
- M. Holzinger, J. Abraham, P. Whelan, R. Graupner, L. Ley, F. Hennrich, M. Kappes and A. Hirsch, J. Am. Chem. Soc., 2003, 125, 8566–8580 CrossRef CAS.
- A. Hirsch and O. Vostrowsky, in Functional Molecular Nanostructures, ed. A. D. Schlüter, Springer, Berlin/Heidelberg, 2005, vol. 245, pp. 193–237 Search PubMed.
- L.-H. Liu and M. Yan, Nano Lett., 2009, 9, 3375–3378 CrossRef CAS.
- A. Devadoss and C. E. D. Chidsey, J. Am. Chem. Soc., 2007, 129, 5370–5371 CrossRef CAS.
- K. S. Coleman, S. R. Bailey, S. Fogden and M. L. H. Green, J. Am. Chem. Soc., 2003, 125, 8722–8723 CrossRef CAS.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS.
- W. R. Collins, W. Lewandowski, E. Schmois, J. Walish and T. M. Swager, Angew. Chem., Int. Ed., 2011, 50, 8848–8852 CrossRef CAS.
- W. Zhang, J. K. Sprafke, M. Ma, E. Y. Tsui, S. A. Sydlik, G. C. Rutledge and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 8446–8454 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- I. Jung, D. A. Dikin, R. D. Piner and R. S. Ruoff, Nano Lett., 2008, 8, 4283–4287 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- J. Yao, X. Shen, B. Wang, H. Liu and G. Wang, Electrochem. Commun., 2009, 11, 1849–1852 CrossRef CAS.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice Jr and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CAS.
- G. Wang, X. Shen, B. Wang, J. Yao and J. Park, Carbon, 2009, 47, 1359–1364 CrossRef CAS.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2008, 4, 25–29 CrossRef.
- S. Park, J. An, R. D. Piner, I. Jung, D. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170–1171 CrossRef CAS.
- R. Arsat, M. Breedon, M. Shafiei, P. G. Spizziri, S. Gilje, R. B. Kaner, K. Kalantar-zadeh and W. Wlodarski, Chem. Phys. Lett., 2009, 467, 344–347 CrossRef CAS.
- O. Akhavan, Carbon, 2010, 48, 509–519 CrossRef CAS.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- T. A. Phama, J. S. Kimb, J. S. Kima and Y. T. Jeong, Colloids Surf., A, 2011, 384, 543–548 CrossRef.
- H. Wang, J. T. Robinson, X. Li and H. Dai, J. Am. Chem. Soc., 2009, 131, 9910–9911 CrossRef CAS.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS.
- T. H. Han, Y.-K. Huang, A. T. L. Tan, V. P. Dravid and J. Huang, J. Am. Chem. Soc., 2011, 133, 15264–15267 CrossRef CAS.
- Z. Wei, D. Wang, S. Kim, S.-Y. Kim, Y. Hu, M. K. Yakes, A. R. Laracuente, Z. Dai, S. R. Marder, C. Berger, W. P. King, W. A. de Heer, P. E. Sheehan and E. Riedo, Science, 2010, 328, 1373–1376 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- G. Zhao, L. Jiang, Y. He, J. Li, H. Dong, X. Wang and W. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS.
- P. Johari and V. B. Shenoy, ACS Nano, 2011, 5, 7640–7647 CrossRef CAS.
- X. Wu and X. C. Zeng, Nano Lett., 2008, 9, 250–256 CrossRef.
- Y. Okamoto, Chem. Phys. Lett., 2006, 420, 382–386 CrossRef CAS.
- C. H. Hu, S. Q. Wu, Y. H. Wen, Y. Yang and Z. Z. Zhu, J. Phys. Chem. C, 2010, 114, 19673–19677 CAS.
- G. K. Dimitrakakis, E. Tylianakis and G. E. Froudakis, Nano Lett., 2008, 8, 3166–3170 CrossRef CAS.
- F. D. Novaes, R. Rurali and P. Ordejón, ACS Nano, 2010, 4, 7596–7602 CrossRef CAS.
- A. G. Nasibulin, P. V. Pikhitsa, H. Jiang, D. P. Brown, A. V. Krasheninnikov, A. S. Anisimov, P. Queipo, A. Moisala, D. Gonzalez, G. Lientschnig, A. Hassanien, S. D. Shandakov, G. Lolli, D. E. Resasco, M. Choi, D. Tomanek and E. I. Kauppinen, Nat. Nanotechnol., 2007, 2, 156–161 CrossRef CAS.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS.
- X. Wang, X. Li, L. Zhang, Y. Yoon, P. K. Weber, H. Wang, J. Guo and H. Dai, Science, 2009, 324, 768–771 CrossRef CAS.
- X. Gao, L. Liu, S. Irle and S. Nagase, Angew. Chem., Int. Ed., 2010, 49, 3200–3202 CrossRef CAS.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- F. Cervantes-Sodi, G. Csányi, S. Piscanec and A. C. Ferrari, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 165427 CrossRef.
- B. Biel, F. Triozon, X. Blase and S. Roche, Nano Lett., 2009, 9, 2725–2729 CrossRef CAS.
- T. Enoki, Y. Kobayashi and K.-I. Fukui, Int. Rev. Phys. Chem., 2007, 26, 609–645 CrossRef CAS.
- X. Gao, Z. Zhou, Y. Zhao, S. Nagase, S. B. Zhang and Z. Chen, J. Phys. Chem. C, 2008, 112, 12677–12682 CAS.
- C. Ö. Girit, J. C. Meyer, R. Erni, M. D. Rossell, C. Kisielowski, L. Yang, C.-H. Park, M. F. Crommie, M. L. Cohen, S. G. Louie and A. Zettl, Science, 2009, 323, 1705–1708 CrossRef CAS.
- H. Nagai, M. Nakano, K. Yoneda, H. Fukui, T. Minami, S. Bonness, R. Kishi, H. Takahashi, T. Kubo, K. Kamada, K. Ohta, B. Champagne and E. Botek, Chem. Phys. Lett., 2009, 477, 355–359 CrossRef CAS.
- Y. Niimi, T. Matsui, H. Kambara, K. Tagami, M. Tsukada and H. Fukuyama, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 085421 CrossRef.
- L. R. Radovic and B. Bockrath, J. Am. Chem. Soc., 2005, 127, 5917–5927 CrossRef CAS.
- Y.-W. Son, M. L. Cohen and S. G. Louie, Nature, 2006, 444, 347–349 CrossRef CAS.
- X. Jia, M. Hofmann, V. Meunier, B. G. Sumpter, J. Campos-Delgado, J. M. Romo-Herrera, H. Son, Y.-P. Hsieh, A. Reina, J. Kong, M. Terrones and M. S. Dresselhaus, Science, 2009, 323, 1701–1705 CrossRef CAS.
- Z. Wang, Ž. Tomović, M. Kastler, R. Pretsch, F. Negri, V. Enkelmann and K. Müllen, J. Am. Chem. Soc., 2004, 126, 7794–7795 CrossRef CAS.
- E. F. Sheka and L. A. Chernozatonskii, Int. J. Quantum Chem., 2010, 110, 1938–1946 CAS.
- S. Sarkar, E. Bekyarova, S. Niyogi and C. Haddon, J. Am. Chem. Soc., 2011, 133, 3324–3327 CrossRef CAS.
- J. Campos-Delgado, Y. A. Kim, T. Hayashi, A. Morelos-Gómez, M. Hofmann, H. Muramatsu, M. Endo, H. Terrones, R. D. Shull, M. S. Dresselhaus and M. Terrones, Chem. Phys. Lett., 2009, 469, 177–182 CrossRef CAS.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720–7721 CrossRef CAS.
- T. A. Phama, N. A. Kumara and Y. T. Jeong, Synth. Met., 2010, 160, 2028–2036 CrossRef.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- J. Shen, N. Li, M. Shi, Y. Hu and M. Ye, J. Colloid Interface Sci., 2010, 348, 377–383 CrossRef CAS.
- W. Li, X.-Z. Tang, H.-B. Zhang, Z.-G. Jiang, Z.-Z. Yu, X.-S. Du and Y.-W. Mai, Carbon, 2011, 49, 4724–4730 CrossRef CAS.
- Y. Pan, H. Bao, N. G. Sahoo, T. Wu and L. Li, Adv. Funct. Mater., 2011, 21, 2754–2763 CrossRef CAS.
- K. Yang, S. Zhang, G. Zhang, X. Sun, S.-T. Lee and Z. Liu, Nano Lett., 2010, 10, 3318–3323 CrossRef CAS.
- K. Yang, J. Wan, S. Zhang, Y. Zhang, S.-T. Lee and Z. Liu, ACS Nano, 2011, 5, 516–522 CrossRef CAS.
- G. Wang, B. Wang, J. Park, J. Yang, X. Shen and J. Yao, Carbon, 2009, 47, 68–72 CrossRef CAS.
- X. Zhang, Y. Huang, Y. Wang, Y. Ma, Z. Liu and Y. Chen, Carbon, 2009, 47, 334–337 CrossRef CAS.
- J. L. Delgado, P. de la Cruz, A. Urbina, J. T. López Navarrete, J. Casado and F. Langa, Carbon, 2007, 45, 2250–2252 CrossRef CAS.
- N. Karousis, S. P. Economopoulos, E. Sarantopoulou and N. Tagmatarchis, Carbon, 2010, 48, 854–860 CrossRef CAS.
- Y. Liu, J. Zhou, X. Zhang, Z. Liu, X. Wan, J. Tian, T. Wang and Y. Chen, Carbon, 2009, 47, 3113–3121 CrossRef CAS.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS.
- N. Mohanty and V. Berry, Nano Lett., 2008, 8, 4469–4476 CrossRef CAS.
- L. Xie, X. Ling, Y. Fang, J. Zhang and Z. Liu, J. Am. Chem. Soc., 2009, 131, 9890–9891 CrossRef CAS.
- Z. Liu, L. Jiang, F. Galli, I. Nederlof, R. C. L. Olsthoorn, G. E. M. Lamers, T. H. Oosterkamp and J. P. Abrahams, Adv. Funct. Mater., 2010, 20, 2857–2865 CrossRef CAS.
- J. H. Jung, D. S. Cheon, F. Liu, K. B. Lee and T. S. Seo, Angew. Chem., Int. Ed., 2010, 49, 5708–5711 CrossRef CAS.
- L. Ci, Z. Xu, L. Wang, W. Gao, F. Ding, K. F. Kelly, B. I. Yakobson and P. M. Ajayan, Nano Res., 2008, 1, 116–122 CrossRef CAS.
- L. C. Campos, V. R. Manfrinato, J. D. Sanchez-Yamagishi, J. Kong and P. Jarillo-Herrero, Nano Lett., 2009, 9, 2600–2604 CrossRef CAS.
- S. S. Datta, D. R. Strachan, S. M. Khamis and A. T. C. Johnson, Nano Lett., 2008, 8, 1912–1915 CrossRef CAS.
- N. Severin, S. Kirstein, I. M. Sokolov and J. P. Rabe, Nano Lett., 2008, 9, 457–461 CrossRef.
- G. F. Schneider, S. W. Kowalczyk, V. E. Calado, G. Pandraud, H. W. Zandbergen, L. M. K. Vandersypen and C. Dekker, Nano Lett., 2010, 10, 3163–3167 CrossRef CAS.
- K. Sint, B. Wang and P. Král, J. Am. Chem. Soc., 2008, 130, 16448–16449 CrossRef CAS.
- T. Nelson, B. Zhang and O. V. Prezhdo, Nano Lett., 2010, 10, 3237–3242 CrossRef CAS.
- C. A. Merchant, K. Healy, M. Wanunu, V. Ray, N. Peterman, J. Bartel, M. D. Fischbein, K. Venta, Z. Luo, A. T. C. Johnson and M. Drndic, Nano Lett., 2010, 10, 2915–2921 CrossRef CAS.
- C. Sathe, X. Zou, J.-P. Leburton and K. Schulten, ACS Nano, 2011, 5 DOI:10.1021/nn202989w.
- J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. van der Zande, J. M. Parpia, H. G. Craighead and P. L. McEuen, Nano. Lett., 2008, 8, 2458–2462 CrossRef CAS.
- A. Dimiev, D. V. Kosynkin, A. Sinitskii, A. Slesarev, Z. Sun and J. M. Tour, Science, 2011, 331, 1168–1172 CrossRef CAS.
- L. Tapasztó, G. Dobrik, P. Lambin and L. P. Biró, Nat. Nanotechnol., 2008, 3, 397–401 CrossRef.
- R. K. Joshi, H. Gomez, F. Alvi and A. Kumar, J. Phys. Chem. C, 2010, 114, 6610–6613 CAS.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS.
- K. T. Chan, J. B. Neaton and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235430 CrossRef.
- H. Sevinçli, M. Topsakal, E. Durgun and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195434 CrossRef.
- A. I. Shii, M. Yamamoto, H. Asano and K. Fujiwara, J. Phys.: Conf. Ser., 2008, 100, 052087 CrossRef.
- Z. L. Zhang, Y. P. Chen, Y. E. Xie, M. Zhang and J. X. Zhong, J. Phys. D: Appl. Phys., 2011, 44, 215403 CrossRef.
- R. Varns and P. Strange, J. Phys.: Condens. Matter., 2008, 20, 225005 CrossRef.
- M. Wu, E.-Z. Liu, M. Y. Ge and J. Z. Jiang, Appl. Phys. Lett., 2009, 94, 102505 CrossRef.
- L. Sheng, Y. Ono and T. Taketsugu, J. Phys. Chem. C, 2010, 114, 3544–3548 CAS.
- E. Pollak, B. Geng, K.-J. Jeon, I. T. Lucas, T. J. Richardson, F. Wang and R. Kostecki, Nano Lett., 2010, 10, 3386–3388 CrossRef CAS.
- T. O. Wehling, K. S. Novoselov, S. V. Morozov, E. E. Vdovin, M. I. Katsnelson, A. K. Geim and A. I. Lichtenstein, Nano Lett., 2008, 8, 173–177 CrossRef CAS.
- W. Zhang, C. Lin, K.-K. Liu, T. Tite, C.-Y. Su, C.-H. Chang, Y.-H. Lee, C.-W. Chu, K.-H. Wei, J.-L. Kuo and L.-J. Li, ACS Nano, 2011, 5, 7517–7524 CrossRef CAS.
- N. Jung, N. Kim, S. Jockusch, N. J. Turro, P. Kim and L. Brus, Nano Lett., 2009, 9, 4133–4137 CrossRef CAS.
- B. Huang, Z. Li, Z. Liu, G. Zhou, S. Hao, J. Wu, B.-L. Gu and W. Duan, J. Phys. Chem. C, 2008, 8, 13442–13446 Search PubMed.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137–3140 CrossRef CAS.
- F. Yavari, C. Kritzinger, C. Gaire, L. Song, H. Gullapalli, T. Borca-Tasciuc, Pu. M. Ajayan and N. Koratkar, Small, 2010, 6, 2535–2538 CrossRef CAS.
- M. Machida, T. Mochimaru and H. Tatsumoto, Carbon, 2006, 44, 2681–2688 CrossRef CAS.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263–5266 CAS.
- Z. Jin, D. Nackashi, W. Lu, C. Kittrell and J. M. Tour, Chem. Mater., 2010, 22, 5695–5699 CrossRef CAS.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577–583 CrossRef CAS.
- N. R. Gall, E. V. Rut'kov and A. Y. Tontegode, Carbon, 2000, 38, 663–667 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 520–527 CrossRef CAS.
- K. Vinodgopal, B. Neppolian, I. V. Lightcap, F. Grieser, M. Ashokkumar and P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 1987–1993 CrossRef CAS.
- K. Jasuja, J. Linn, S. Melton and V. Berry, J. Phys. Chem. Lett., 2010, 1, 1853–1860 CrossRef CAS.
- X. Zhou, X. Huang, X. Qi, S. Wu, C. Xue, F. Y. C. Boey, Q. Yan, P. Chen and H. Zhang, J. Phys. Chem. C, 2009, 113, 10842–10846 CAS.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990–7995 CAS.
- W. Hong, H. Bai, Y. Xu, Z. Yao, Z. Gu and G. Shi, J. Phys. Chem. C, 2010, 114, 1822–1826 CAS.
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS.
- Y. Li, L. Tang and J. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- R. Kou, Y. Shao, D. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. Wang, Y. Lin, Y. Wang, I. A. Aksay and J. Liu, Electrochem. Commun., 2009, 11, 954–957 CrossRef CAS.
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2011, 5, 7426–7435 CrossRef CAS.
- A. Rochefort, D. Q. Yang and E. Sacher, Carbon, 2009, 47, 2233–2238 CrossRef CAS.
- S. Guo, D. Wen, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 3959–3968 CrossRef CAS.
- H. Wang, J. T. Robinson, G. Diankov and H. Dai, J. Am. Chem. Soc., 2010, 132, 3270–3271 CrossRef CAS.
- J. L. Johnson, A. Behnam, S. J. Pearton and A. Ural, Adv. Mater., 2010, 22, 4877–4880 CrossRef CAS.
- D. Zhan, L. Sun, Z. H. Ni, L. Liu, X. F. Fan, Y. Wang, T. Yu, Y. M. Lam, W. Huang and Z. X. Shen, Adv. Funct. Mater., 2010, 20, 3504–3509 CrossRef CAS.
- Z.-S. Wu, D.-W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H.-M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2008, 9, 72–75 CrossRef.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS.
- Y. Ding, Y. Jiang, F. Xu, J. Yin, H. Ren, Q. Zhuo, Z. Long and P. Zhang, Electrochem. Commun., 2010, 12, 10–13 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 242–251 CrossRef CAS.
- S. R. Kim, M. K. Parvez and M. Chhowalla, Chem. Phys. Lett., 2009, 483, 124–127 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS.
- V. C. Tung, L.-M. Chen, M. J. Allen, J. K. Wassei, K. Nelson, R. B. Kaner and Y. Yang, Nano Lett., 2009, 9, 1949–1955 CrossRef CAS.
- J. M. Lee, Y. B. Pyun, J. Yi, J. W. Choung and W. I. Park, J. Phys. Chem. C, 2009, 113, 19134–19138 CAS.
- J. Liang, Y. Huang, J. Oh, M. Kozlov, D. Sui, S. Fang, R. H. Baughman, Y. Ma and Y. Chen, Adv. Funct. Mater., 2011, 21, 3778–3784 CrossRef CAS.
- Y. Li and Y. Wu, J. Am. Chem. Soc., 2009, 131, 5851–5857 CrossRef CAS.
- Y. Liang, D. Wu, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 1679–1683 CrossRef CAS.
- X. An, T. Simmons, R. Shah, C. Wolfe, K. M. Lewis, M. Washington, S. K. Nayak, S. Talapatra and S. Kar, Nano Lett., 2010, 10, 4295–4301 CrossRef CAS.
- J. Geng and H.-T. Jung, J. Phys. Chem. C, 2010, 114, 8227–8234 CAS.
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155–3162 CrossRef CAS.
- Q. Yang, X. Pan, F. Huang and K. Li, J. Phys. Chem. C, 2010, 114, 3811–3816 CAS.
- W. Chen, S. Chen, D. C. Qi, X. Y. Gao and A. T. S. Wee, J. Am. Chem. Soc., 2007, 129, 10418–10422 CrossRef CAS.
- H. Huang, S. Chen, X. Gao, W. Chen and A. T. S. Wee, ACS Nano, 2009, 3, 3431–3436 CrossRef CAS.
- X. Tian, J. Xu and X. Wang, J. Phys. Chem. B, 2010, 114, 11377–11381 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS.
- W. Hong, Y. Xu, G. Lu, C. Li and G. Shi, Electrochem. Commun., 2008, 10, 1555–1558 CrossRef CAS.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490–13497 CrossRef CAS.
- X. Wang, S. M. Tabakman and H. Dai, J. Am. Chem. Soc., 2008, 130, 8152–8153 CrossRef CAS.
- J. D. Emery, Q. H. Wang, M. Zarrouati, P. Fenter, M. C. Hersam and M. J. Bedzyk, Surf. Sci., 2011, 605, 1685–1693 CrossRef CAS.
- S. Vadukumpully, J. Paul and S. Valiyaveettil, Carbon, 2009, 47, 3288–3294 CrossRef CAS.
- J. Guo, L. Ren, R. Wang, C. Zhang, Y. Yang and T. Liu, Composites: Part B, 2011, 42, 2130–2135 CrossRef.
- G. Wang, B. Wang, J. Park, Y. Wang, B. Sun and J. Yao, Carbon, 2009, 47, 3242–3246 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS.
- Y. T. Liang and M. C. Hersam, J. Am. Chem. Soc., 2010, 132, 17661–17663 CrossRef CAS.
- A. Nduwimana and X.-Q. Wang, ACS Nano, 2009, 3, 1995–1999 CrossRef CAS.
- H. Lee, S. Yang, J. Choi, Y. Park and S. Kim, J. Phys. Chem. C, 2011, 115, 18736–18739 CAS.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538–542 CrossRef CAS.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. S. Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS.
- L. Q. Xu, L. Wang, B. Zhang, C. H. Lim, Y. Chen, K.-G. Neoh, E.-T. Kang and G. D. Fu, Polymer, 2011, 52, 2376–2383 CrossRef CAS.
- S. Park, N. Mohanty, J. W. Suk, A. Nagaraja, J. An, R. D. Piner, W. Cai, D. R. Dreyer, V. Berry and R. S. Ruoff, Adv. Mater., 2010, 22, 1736–1740 CrossRef CAS.
- S. M. Kang, S. Park, D. Kim, S. Y. Park, R. S. Ruoff and H. Lee, Adv. Funct. Mater., 2011, 21, 108–112 CrossRef CAS.
- D. Yu and L. Dai, J. Phys. Chem. Lett., 2010, 1, 467–470 CrossRef CAS.
- D.-M. Chen, P. M. Shenai and Y. Zhao, Phys. Chem. Chem. Phys., 2011, 13, 1515–1520 RSC.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS.
- Y.-G. Yang, C.-M. Chen, Y.-F. Wen, Q.-H. Yang and M.-Z. Wang, New Carbon Mater., 2008, 23, 193–200 CAS.
- T. Wei, G. Luo, Z. Fan, C. Zheng, J. Yan, C. Yao, W. Li and C. Zhang, Carbon, 2009, 47, 2296–2299 CrossRef CAS.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012 10.1039/C1CS15078B.
- J. R. Pottsa, D. R. Dreyerb, C. W. Bielawskib and R. S. Ruoff, Polymer, 2011, 52, 5–25 CrossRef.
- T. Kuillaa, S. Bhadrab, D. Yaoa, N. H. Kimc, S. Bosed and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef.
- Y. Xu, W. Hong, H. Bai, C. Li and G. Shi, Carbon, 2009, 47, 3538–3543 CrossRef CAS.
- J. Li, S. Guo, Y. Zhai and E. Wang, Electrochem. Commun., 2009, 11, 1085–1088 CrossRef CAS.
- H. Wang, Q. Hao, X. Yang, L. Lu and X. Wang, Electrochem. Commun., 2009, 11, 1158–1161 CrossRef CAS.
- J. Yan, T. Wei, B. Shao, Z. Fan, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 487–493 CrossRef CAS.
- M. Z. Kassaee, E. Motamedi and M. Majdi, Chem. Eng. J., 2011, 172, 540–549 CrossRef CAS.
- J. Liang, Y. Wang, Y. Huang, Y. Ma, Z. Liu, J. Cai, C. Zhang, H. Gao and Y. Chen, Carbon, 2009, 47, 922–925 CrossRef CAS.
- Y. Wang, D. Kurunthu, G. W. Scott and C. J. Bardeen, J. Phys. Chem. C, 2010, 114, 4153–4159 CAS.
- X. Gao, S. B. Zhang, Y. Zhao and S. Nagase, Angew. Chem., Int. Ed., 2010, 49, 6764–6767 CrossRef CAS.
- X. Wang, L. Zhi and K. Mullen, Nano Lett., 2007, 8, 323–327 CrossRef.
- C.-M. Chen, Y.-G. Yang, Y.-F. Wen, Q.-H. Yang and M.-Z. Wang, New Carbon Mater., 2008, 23, 345–350 CAS.
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z.-F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325–7328 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS.
- A. Rouhanipour, M. Roy, X. Feng, H. Räder and K. Müllen, Angew. Chem., Int. Ed., 2009, 48, 4602–4604 CrossRef CAS.
- H. J. Räder, A. Rouhanipour, A. M. Talarico, V. Palermo, P. Samorì and K. Müllen, Nat. Mater., 2006, 5, 276–280 CrossRef.
- F. Günes, H.-J. Shin, C. Biswas, G. H. Han, E. S. Kim, S. J. Chae, J.-Y. Choi and Y. H. Lee, ACS Nano, 2010, 4, 4595–4600 CrossRef.
- Q. Ji, I. Honma, S.-M. Paek, M. Akada, J. P. Hill, A. Vinu and K. Ariga, Angew. Chem., Int. Ed., 2010, 49, 9737–9739 CrossRef CAS.
- Z. Wei, D. E. Barlow and P. E. Sheehan, Nano Lett., 2008, 8, 3141–3145 CrossRef CAS.
- L. Zhi, J. Wu, J. Li, U. Kolb and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 2120–2123 CrossRef CAS.
- Z. Zhu, D. Su, G. Weinberg and R. Schlögl, Nano Lett., 2004, 4, 2255–2259 CrossRef CAS.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano. Lett., 2011, 11, 2644–2647 CrossRef CAS.
- A. Chuvilin, E. Bichoutskaia, M. C. Gimenez-Lopez, T. W. Chamberlain, G. A. Rance, N. Kuganathan, J. Biskupek, U. Kaiser and A. N. Khlobystov, Nat. Mater., 2011, 10, 687–692 CrossRef CAS.
- C. Nethravathi, B. Viswanath, C. Shivakumara, N. Mahadevaiah and M. Rajamathi, Carbon, 2008, 46, 1773–1781 CrossRef CAS.
- L. J. Cote, F. Kim and J. Huang, J. Am. Chem. Soc., 2008, 131, 1043–1049 CrossRef.
- S. Biswas and L. T. Drzal, Nano Lett., 2008, 9, 167–172 CrossRef.
- J. Zhao, S. Pei, W. Ren, L. Gao and H.-M. Cheng, ACS Nano, 2010, 4, 5245–5252 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, R. Voggu and K. S. Subrahmanyam, J. Phys. Chem. Lett., 2010, 1, 572–580 CrossRef CAS.
- O. Prezhdo, P. V. Kamat and G. C. Schatz, J. Phys. Chem. C, 2011, 115, 3195–3197 CAS.
- Q. H. Wang and M. C. Hersam, Nano. Lett., 2011, 11, 589–593 CrossRef CAS.
- Q. H. Wang and M. C. Hersam, Nat. Chem., 2009, 1, 206–211 CrossRef CAS.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Lia, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. USA DOI:10.1073/pnas.1105270108.
- A. A. Green and M. C. Hersam, J. Phys. Chem. Lett., 2010, 1, 544–549 CrossRef CAS.
- A. A. Green and M. C. Hersam, Nano Lett., 2009, 9, 4031–4036 CrossRef CAS.
- J. A. Kellar, J. M. P. Alaboson, Q. H. Wang and M. C. Hersam, Appl. Phys. Lett., 2010, 96, 143103 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |