Role of surface defect sites: from Pt model surfaces to shape-controlled nanoparticles
Qing-Song
Chen
ab,
Francisco J.
Vidal-Iglesias
a,
José
Solla-Gullón
a,
Shi-Gang
Sun
*b and
Juan M.
Feliu
*a
aInstitute of Electrochemistry, University of Alicante, E-03080, Alicante, Spain. E-mail: juan.feliu@ua.es; Fax: +34-965903537
bState Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: sgsun@xmu.edu.cn; Fax: +86 0592 2183047
First published on 26th September 2011
Abstract
In the present paper, preferentially oriented (111) Pt nanoparticles (mostly octahedral and tetrahedral, namely {111}Pt nanoparticles) have been characterized and compared with a Pt(554) single-crystal electrode as their voltammetric features are quite similar in 0.5 M H2SO4. The anion and Bi adsorption behaviours suggest that the {111}Pt nanoparticles contain relatively wide hexagonal domains and also isolated sites which could adsorb solely hydrogen. Bi step decoration has been successfully extended to modify the defects of {111}Pt nanoparticles without blocking terrace sites. CO charge displacement has been applied to determine the potential of zero total charge (pztc) of non-decorated and Bi decorated surfaces. It has found that the positive shift of pztc on defect-decorated {111}Pt nanoparticles is not so significant in comparison with that of Pt(554) due to the relative short mean length of (111) domains on the {111}Pt nanoparticles. CO stripping demonstrates that {111}Pt nanoparticles exhibit higher reactivity toward CO oxidation. This reflects the role of the defect sites in nanoparticles, evidenced by the disappearance of the “pre-wave” in the stripping voltammogram once the defects were blocked by Bi. The stripping peaks shift to higher potential on Bi decorated surfaces, indicating the active role of both steps and defects for CO oxidation. By comparing the CO stripping charge and the change in hydrogen adsorption charge of surfaces with and without Bi decoration, including reasonable deconvolution, the local CO coverage on defect and terrace sites were obtained for the first time for the {111}Pt nanoparticles, and the results are in good agreement with those obtained on Pt(554). Chronoamperometry studies show tailing in all current–time transients of CO oxidation on all surfaces studied. The kinetics of CO oxidation can be satisfactorily simulated by a modified Langmuir–Hinshelwood model, demonstrating that CO oxidation on all studied surfaces follows the same mechanism.
Introduction
Surface electrochemistry has been greatly advanced since the wide use of metal single crystals and the improvement of surface technology, especially after the convenient flame-annealing technique having been invented to clean Pt single-crystal surfaces.1–3Pt single-crystal planes can provide a variety of surface structures with well-defined atomic arrangements, depending on their orientation. Numerous studies based on Pt single crystals have been dedicated to reveal the properties of the solid/liquid interface such as double layer structure, electronic effects, potential of zero total charge (pztc), anion adsorption, electrocatalytic activities, etc. Valuable information related to these properties have been attained and evidenced their structure-sensitive characteristics.4–8 Many studies on electrocatalytic reactions, for example CO oxidation, small organic molecule oxidation and oxygen reduction, demonstrate that single-crystal planes with higher step/kink density lead to the enhancement of electrocatalytic reactivity. In terms of application, guided by the knowledge of model electrocatalysts of well defined single-crystal planes, great efforts have been devoted to shape-control synthesis that produces nanoparticles enclosed by high index facets which have high density of atomic steps/kinks with low coordination numbers.9–11As a typical molecular probe and usual poisonous intermediate present in fuel cells, the oxidation of CO on different surface structures and different materials has received special attention in electrochemistry. It has been revealed that step, kink or defects with low coordinated sites are able to enhance CO electrooxidation independently of the supporting electrolyte used,8 although its oxidation mechanism is very different in acidic and alkaline solutions.12–14 The enhanced catalytic activity has been ascribed to the preferential formation of oxygen-containing species at the step/defect sites on surfaces vicinal to Pt(111). It has been well established that CO oxidation is initiated on step sites, CO on (111) terraces possesses high mobility in acidic solution and diffuses rapidly to the step or defect sites, to be oxidized into CO2. The mobility decreases dramatically in basic solution which has been attributed to the adsorption of carbonate species arising from CO oxidation. Thus, the rapid diffusion model can not be as straight-forward for CO oxidation in alkaline solutions.12,15,16
In the case of nanoparticles, CO oxidation was found to be not only structure sensitive but also size dependent. It has reported that the multiplicity of CO oxidation peaks is closely correlated to the structure of nanoparticles, which are also characterized by the variation in cyclic voltammograms.17–19CO oxidation on smaller scale Pt nanoparticles has been carried out by several research groups, however, contradictory results have been published. Some reports showed that decreasing the size of Pt nanoparticles could enhance the activity of CO oxidation, which is consistent with that expected from the behaviour on stepped single-crystal surfaces.20,21 Other papers reported an opposite trend as the diameter of Pt nanoparticles are decreased below 4 nm.22,23 Lee et al. considered that this discrepancy may be due to the relatively ambiguous surface structure of small nanoparticles and showed that high-index surfaces of Pt nanoparticles (∼2 nm) supported on multiwalled carbon nanotubes, play a very important role in enhancing intrinsic activity for CO and methanol oxidation.24
Irreversibly adsorbed adatoms have been widely applied to modify the surface composition and the electronic structure, and thus to improve the reactivity of metal materials, or the selectivity of the desired reaction.25–29 For well defined surfaces and preferentially oriented nanoparticles, irreversible adsorbed adatoms (e.g., Bi, Ge) can be used to detect a particular surface structure and measure the contributions of different surface sites.30–32 It was reported that some adatoms are able to selectively decorate step sites while leaving terrace sites unblocked.33 Based on this result, decoration of step sites of vicinal Pt(111) surfaces without blocking the terraces was investigated, and a positive shift of the pztc, different CO coverage and dominant CO bonding on different step symmetries were revealed.34 These results give key information to better understand the intrinsic properties of steps and their electronic effects of well defined single crystals.
The aim of the present study is devoted to better understand defect reactivity in practical materials. To achieve this, Bi step decoration is attempted to modify the defects of preferentially oriented {111}Pt nanoparticles, whose cyclic voltammogram is very similar to that of the bulk metal, oriented as Pt(554), in 0.5 M H2SO4. The size of the nanoparticles is ∼8.6 nm, which can avoid the complication of size effects in the electrocatalytic behaviour. In order to compare their similarity, experiments such as cycling voltammetry, CO charge displacement, CO stripping and chronoamperometry, were carried out for both materials with and without Bi decoration. The local and overall CO coverage have been determined with the knowledge of pztc and the help of Bi defect decoration. The validity of the Langmuir–Hinshelwood mechanism for CO oxidation on all surfaces has been investigated. Another point considered in this work deals with in situ surface site characterization analysis. Taking advantage of the structure sensitivity of hydrogen and anion de/adsorption, which are widely used as surface probes, and the similarity of the voltammetry of {111}Pt nanoparticles with that of stepped surfaces vicinal to Pt(111), the relative amount of bidimensional domains have been refined. This analysis would complement the previous estimation based on irreversibly adsorbed Bi which is necessarily more limited because the calibration curve can not involve all terrace atoms and small surface ensembles.35 It should be stressed that the characterization procedure, albeit based in electrochemical structure sensitive reactions, can be considered general. This means that the surface site distribution of the nanoparticles is an intrinsic property of a particular synthesis and thus valid for any future application of them. Moreover, the electrochemical characterization is simple and can be carried out in a short time.
Experimental
A hemispherical platinum single-crystal electrode Pt(554) with 9 atom-wide (111) terraces separated by monoatomic (110) step sites, with a geometric area of 3.66 mm2, was prepared according to Clavilier's method.36 Prior to each experiment the electrode was flame annealed and cooled to room temperature in a H2 + Ar atmosphere. After flame annealing, the single-crystal electrodes were quenched with water in equilibrium with this mixture of gases and then transferred to the cell under the protection of a droplet of deoxygenated water. The preparation and structure characterization of {111}Pt nanoparticles have been described in detail elsewhere.37 The Pt nanoparticle electrode was prepared by depositing the dispersed nanoparticles on a hemispherical polycrystalline gold substrate (ca. 3 mm2) from a droplet of 1 μL solution and drying under Ar atmosphere.The experiments were carried out in two conventional three-electrode glass cells: The first one is used for deposition of Bi and the second one, with an additional inlet for dosing CO, was used for electrochemical characterization, CO displacement and CO oxidation. The adsorption of Bi on platinum single-crystal steps has been described elsewhere33 and will be further discussed in the next section as well as the Bi defect decoration on {111}Pt nanoparticles. CO displacement experiments were performed by using the following procedure:38 The working electrodes were arranged in a meniscus configuration and a flow of CO was introduced at 0.1 V over the meniscus in the cell under Ar atmosphere. The resulting current–time transient was recorded simultaneously until the current dropped to zero and the displacement charge was measured from the integration of current–time curve. Normally, in the case of nanoparticles the first CO displacement was used to clean the nanoparticle surfaces as described in ref. 39. Special care was taken to avoid the presence of atmospheric oxygen in the vicinity of the meniscus. After CO dosing, the electrode was immersed into the solution while keeping the potential constant at 0.1 V, and Ar was bubbled through the solution for at least 8 min to remove the dissolved CO in solution and the traces of CO that could remain in the cell atmosphere. Then the working electrode was returned to the meniscus configuration and the conventional CO stripping, or potential step experiment, was performed.
A large-area platinum wire was used as a counter electrode and a reversible hydrogen electrode (RHE) was used as a reference. The cell and all glassware were immersed in a potassium permanganate solution overnight, followed by rinsing with water and a solution of hydrogen peroxide with sulfuric acid. Finally, everything was boiled and rinsed with ultra-pure water several times. Working solutions were prepared from concentrated H2SO4 (Merck Suprapur), Bi2O3 (Merck) and Elga-Vivendi water (18 MΩ cm). Argon (N50, Air Liquide in all gas used) was used to deoxygenate all solutions and CO (N47) to dose CO. All electrochemical experiments were carried out at room temperature, using a waveform generator (EG&G PARC 175) together with a potentiostat (Amel 551) and a digital recorder (eDAQ, ED401).
Results and discussion
Structure characterization
It has been well established that the cyclic voltammograms can be used as fingerprints of Pt single-crystal electrodes because the hydrogen and anion de/adsorption are structure-sensitive on Pt surfaces.40–43Fig. 1a shows the atomic model and voltammetric profile of Pt(554) in 0.5 M sulfuric acid. From the model, it is clear that the Pt(554) surface is a combination of (111) terraces with 9 atom-width separated by monoatomic (110) step sites. In fact, the overall charge density may be considered as a linear combination of both contributions in this series of stepped surfaces.43 The coordination number (CN) of the atoms on (111) terraces is 9 while it is lower for the top atoms at the (110) step (CN = 7). The atoms with different CN have different electronic environment, which combined with their different arrangement, leads to the different de/adsorption behaviour of hydrogen and anion on different sites. As shown in Fig. 1a, the voltammogram of Pt(554) in 0.5 M H2SO4 is in good agreement with that previously reported.41,43 The symmetric sharp peaks around 0.12 V are associated with H de/adsorption on (110) step sites. The symmetric broad contributions located below 0.35 V are related to the H ad/desorption on (111) terraces. Moreover, the symmetric state around 0.5 V can be attributed to the sulfate de/adsorption on (111) terrace sites.44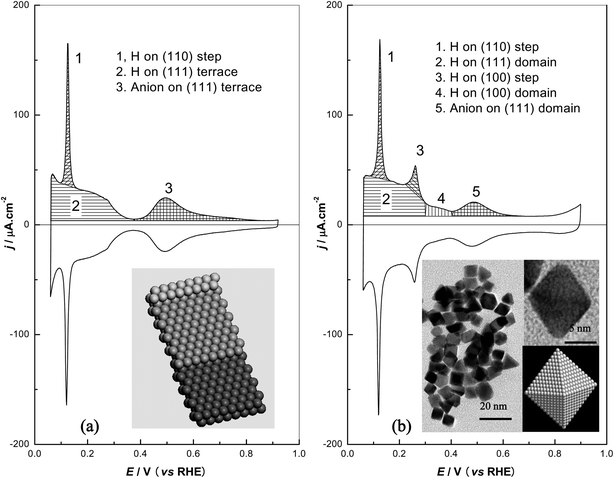 | ||
| Fig. 1 (a) Cyclic voltammogram and atomic model of Pt(554); (b) cyclic voltammogram, TEM image and a octahedral model of {111}Pt nanoparticles. Working solution: 0.5 M H2SO4, scan rate: 50 mV s−1. | ||
In the case of {111}Pt nanoparticles, the voltammogram shows the presence of additional contributions. The shape of the nanoparticles has been investigated by transmission electron microscopy (TEM), as also shown in Fig. 1b. The nanoparticles are dominated by octahedral shapes, including an important number of tetrahedra and a small number of irregular nanoparticles. It is known that the octahedra and tetrahedra are both enclosed by low-index facets with (111) symmetry. A representative high-magnification TEM image of one octahedral nanoparticle as well as the atomic model is also displayed in Fig. 1b. One atom in the (111) facet possesses 9 coordinated atoms, while the CN of atoms on the edge is only 7, the same to that of (110) sites, implying that they probably would have similar properties such as hydrogen adsorption potential. The CN of the vertex atom is 5 but, however, the contribution of these atoms can be considered negligible since there are only 6 vertices for each octahedron. Moreover, the vertices of the octahedral nanoparticles in some cases look more like a line than like a point, suggesting the possible presence of small (100) domains or other defects. The average size of the {111}Pt nanoparticles is about 8.6 nm, which points out that the mean length of the edge in the octahedron is ca. 20 atom-width. For one facet of ideal octahedral nanoparticles, the (111) sites form an equilateral triangle with 17 atoms in the side. By geometrical estimation the average length is 9 atom-width for 17 row terraces, similar to that of Pt(554). Nevertheless, it is inevitable that defects are formed during the synthesis of {111}Pt nanoparticles, so the effective terrace length may be even shorter than could be verified by measuring the amount of (111) domains. The edge atoms amount to about 15% of the total facet atoms, which is similar to that of step atoms in Pt(554) (ca. 11%). It should be noted that this quantitative analysis is possible on relatively large nanoparticles. In this case the particles have well-defined preferential shapes either cubic or octahedral-tetrahedral, depending on the synthetic procedure. In the same way, it can be assumed that the surface adsorption states of a sample will behave as those of the bulk metal, because the ratio of surface/bulk sites remains reasonably similar. However, when the nanoparticles have smaller dimension, large surface domains would disappear and the voltammogram would look as that of polycrystalline platinum.
The voltammograms of the {111}Pt nanoparticles in 0.5 M H2SO4 (Fig. 1b) is very similar to that of Pt(554), suggesting that similar sites to those of Pt(554) dominate on these particles. The sharpness and the symmetry of the adsorption states indicate the cleanliness of the surface. With the knowledge of the characteristic fingerprints of the single-crystal model electrodes,45 we can conclude that the symmetric contribution at 0.5 V can be attributed to the anion adsorption on two-dimensionally ordered (111) surface domains, the sharp peak at 0.12 V is related to the presence of (110) surface sites, perhaps both at the edges and corners between the (111) domains or the defects among them. The small shoulder that appears around 0.35 V is due to small amounts of two-dimensional (100) surface domains. Moreover, the small peak around 0.26 V can be ascribed to the (100) surface sites, likely steps between (111) domains and eventually at the edge of (100) domains. Thus, the voltammetric results further confirm what could be expected from the TEM images, but including a significantly larger number of nanoparticles.
Based on these voltammetric similarities the charge density involved in the different areas of Fig. 1 can be selectively measured. Table 1 summarizes the total, hydrogen and anion adsorption charges corresponding to different contributions as indicated in the voltammograms of Pt(554) and {111}Pt nanoparticles. The double layer correction was performed in a similar way to that performed previously,46 arbitrarily using a horizontal line through the minimum current in the positive-going voltammetric sweep as baseline. The active surface area of the {111}Pt nanoparticles was determined from the blank voltammogram by measuring the charge involved in the so-called hydrogen adsorption region assuming 230 μC cm−2 for the total charge after double layer correction recorded in 0.5 M H2SO4 solution.8 For Pt(554) electrode, the (110) step charge (qSH,(110)) was calculated by integrating the current under the peak around 0.12 V and gives 29 μC cm−2, close to the corresponding theoretical values of 26 μC cm−2 from the hard-sphere model.47 After double layer correction, integration of the voltammogram between 0.06 V and the potential where the minimum current appears gives a total charge (qtotal) of 233 μC cm−2, which is almost coincident with the value from the hard-sphere model (231 μC cm−2). The H and anion charge (qTH,(111) and qTA,(111)) on the (111) terrace are determined to be 141 and 63 μC cm−2, respectively. Their sum (203 μC cm−2) is close to the theoretical charge for a monoelectronic transfer per atomic terrace site (205 μC cm−2) on Pt(554). Each state corresponds to about 0.67 and 0.33 fraction, respectively, of the amount of terrace sites, in good agreement with previous studies.43
| q total | q sH,(110) | q TH,(111) | q SH,(100) | q TH,(100) | q TA,(111) | |
|---|---|---|---|---|---|---|
| Pt(554) | 233 | 29 | 141 | 0 | 0 | 63 |
| Percentage (%) | 13 | 60 | 0 | 0 | 27 | |
| {111}Pt | 230 | 35 | 136 | 17 | 11 | 31 |
| Percentage (%) | 15 | 60 | 7 | 5 | 13 |
If we accept that similar analysis can be applied to {111}Pt nanoparticles, the percentage of (110) step sites is 15%, a little bit higher than that of Pt(554) (13%). Besides, 7% of (100) step and 5% of (100) domains are present on {111}Pt nanoparticles, indicating that these nanoparticles possess a higher density and variety of surface sites than those present at the stepped surface. The amount of ordered (100) domains attained from the decomposing of the voltammogram is consistent with that (3%) obtained using irreversible Ge as an in situprobe.37 In general the error in charge measurements should be considered within 2–3%.
As a result of the charge balance, the sum of the charge of H and anion adsorption on sites with (111) symmetry would correspond to 73%. Nonetheless, the amount of (111) domains evaluated by Bi adsorption is only 42% of the whole sites,37 and this means a big deviation from that determined by the sum of H and anion adsorption charges. This may be due to the different adsorption behaviour of H, anion and Bi, or the superimposition of different H adsorption states, that would lead to difficulties to identify the H adsorption on the (111) terrace sites. An alternative way to determine the amount of (111) domains is to measure the anion adsorption charge solely, assuming that it involves 1/3 of the (111) terrace sites, according to previous results.43 In this framework, 40% for (111) domains was obtained through this simple assumption and it coincides well with that determined by Bi adsorption. The amount of (111) domains is apparently lower than that of a octahedron with 20 atom width edges (∼85%), which might be ascribed to imperfections and substrate dispersion problems of the {111}Pt nanoparticles. Furthermore, it should be pointed out that both peaks at 0.12 and 0.26 V can also be influenced by the presence of possible kink contributions, which are more difficult to be analyzed quantitatively, as no model analysis has been proposed yet.
Actual results suggest that the (111) symmetry sites can be split in two groups. In the first one, relatively large (111) domains are involved, able to adsorb anions and adatoms and following the generally fulfilled 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between hydrogen and anion adsorption. The second group would include isolated sites or small ensembles able to only adsorb hydrogen. A similar picture can be proposed from the analysis of Pt(111) surfaces disordered by oxygen adsorption.48
:1 stoichiometry between hydrogen and anion adsorption. The second group would include isolated sites or small ensembles able to only adsorb hydrogen. A similar picture can be proposed from the analysis of Pt(111) surfaces disordered by oxygen adsorption.48
It should be remarked that this analysis implies that the baseline for (110) sites is not the horizontal double layer considered in previous deconvolution studies,45,49 that agrees with that considered for Pt(110) and its vicinal surfaces.50 In the case of these preferential {111}Pt nanoparticles the (110) defects baseline would be that defined by the broad hydrogen adsorption on the overall sites with (111) symmetry.43 This would be the same situation used to measure the step density on stepped surfaces, such as Pt(554), and would then involve long and short (111) domains (in the nanoparticles or the n-atom wide terraces (in the stepped surfaces).
It should be remarked that this kind of analysis is only possible in samples having well defined (111)-like voltammetric features. The evaluation of (111) domains by irreversibly adsorbed Bi can be considered as a general method, applicable to all polycrystalline platinum samples, including different shaped nanoparticles. This evaluation enables to classify the different nanoparticles from the point of view of this particular type of sites. However, Bi is only able to account for (n-1) terrace sites in general.35 Deconvolution shows the existence of a broad state at 0.21 V that could be related to (111) contributions that have not been evaluated by irreversibly adatom adsorption.45 This state was even more important in previous deconvolutions that did not use Bi to eliminate (111) contributions on polycrystalline platinum.49 To precisely determine in more detail the presence of the different types of sites having the same symmetry but different neighborhood would require more experimental data, likely involving systematic studies with series of kinked surfaces. The results would be then exportable to selected sets of nanoparticles that could reveal the presence of different contributions. Studies on solutions of higher pH could help to define new criteria based on peak splitting that is usually observed in these conditions.13 More work is in progress to improve the analysis of the platinum blank voltammogram recorded under extremely well controlled conditions, in order to gain in situ understanding on the surface site distribution.
Decoration of Bi and measurement of pztc
Fig. 2 shows the typical Bi deposition CVs on Pt(554) and {111}Pt nanoparticles in 0.5 M H2SO4 containing 10−5∼10−4 Bi3+ solution, from which similar and different behaviours can be again observed. For the Pt(554) electrode, the variation in CVs during Bi deposition is in good agreement with those reported previously with a comparable electrode Pt(775) [6(111) × (110)].33 It can be seen that the sharp peaks around 0.12 V, due to H de/adsorption on (110) step sites, firstly decrease progressively with the voltammetric cycling, while the anion adsorption between 0.35 and 0.7 V, and the H adsorption state below 0.35 V on (111) terraces, both remain essentially unchanged. Parallel to this diminution of current, an increasing peak at 0.25 V appears, which has been ascribed to sulfate/bisulfate adsorbed33 on junction sites between the adsorbed Bi and the clean platinum atoms on the terraces. In order to control Bi decoration only at step sites, the deposition was stopped when the adsorption peak around 0.12 V almost disappeared, and special care was taken to avoid the appearance of redox peaks at 0.63 V as they evidence that Bi starts to deposit on the terraces.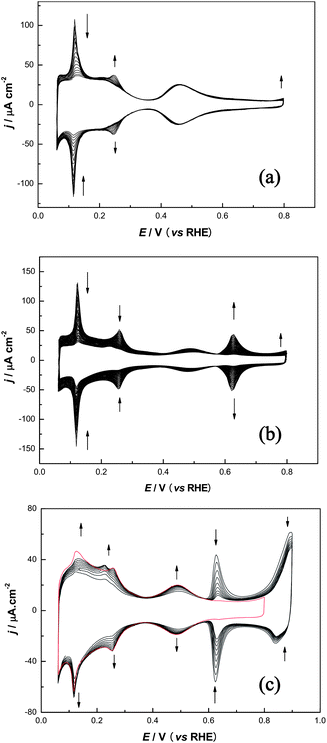 | ||
| Fig. 2 (a) Bi deposition on Pt(554) steps from 2.5 × 10−5 M Bi3+ + 0.5 M H2SO4 solution; (b) Bi deposition on {111}Pt nanoparticles steps/defects from 1 × 10−4 M Bi3+ + 0.5 M H2SO4 solution; (c) Bi stripping from the (111) terraces for {111}Pt nanoparticles by CV, upper potential limit 0.9 V; scan rate 50 mV s−1. | ||
When Bi is decorating the {111}Pt nanoparticle surfaces, as shown in Fig. 2b, it is obvious that besides the coincident decreasing trend of the 0.12 and 0.26 V peaks, reflecting the blockage of (110) and (100) surface sites, clear differences can also be observed. That is, the peak at 0.25 V is not so marked since it overlaps with the diminution of the (100) peak, and it is more evident that the contributions around 0.5 V begin to decrease, accompanied by the appearance of a pair of redox peaks at 0.63 V, far away before the complete blocking of defects, indicating that Bi starts to deposit on the ordered (111) domains before the defect sites are fully blocked. This is because of the heterogeneous structure of the {111}Pt nanoparticles and can be interpreted as following: (i) the Bi probably is able to preferentially adsorb on the edge sites of {111}Pt nanoparticles which have the same coordination number as that of the (110) step. However, their local neighborhoods are quite different, so the Bi maybe adsorbs on the two rows closest to the edge, which will disturb anion adsorption on the terrace, facilitating further Bi adsorption; (ii) it is worth noting that the facet of the {111}Pt nanoparticles is an equilateral triangle, the non-uniformity of the (111) terrace length may result in the different Bi decoration behaviour as compared to Pt(554). (iii) The amount of (111) domains is relatively small, even lower than the terrace percentage (50%) of single-crystal Pt(331), with 2 atom-width terrace rows separated by monoatomic (110) steps. This demonstrates that the (111) domains may be very short and are likely surrounded by many irregular defects. Therefore, as soon as Bi blocks one of the defects, it would start to deposit quickly on the (111) domains.
At first sight, it appeared to be impossible to fully decorate the defects without blocking the (111) domains by using the same direct protocol for Pt(554), although we have tried different methods, including the use of a more dilute solution or rotating the electrode. The most convenient method to solve this problem is to use a backtitration strategy: fully block the defects, disregarding the growth of Bi on the (111) domains and then take out the electrode, rinse it with ultrapure water and transfer it to another cell containing 0.5 M H2SO4 completely free of dissolved Bi(III) ions. In this second cell the electrode is subjected to voltammetric sweeps up to 0.9 V, a limit that does not destroy the surface structure of Pt nanoparticles, but high enough to dissolve the adatoms on terrace sites.51
As shown in Fig. 2c, with consecutively voltammetric cycling the contribution around 0.5 V recovers gradually while the peak at 0.63 V decreases at the same time, indicating that Bi on the (111) domains can be stripped off by voltammetric cycling to this relatively higher potential limit. The adsorption of hydrogen below 0.35 V, related to (111) sites, also increases. As can be seen, the peaks at 0.12 and 0.26 V also increase slightly, indicating that a very small fraction of Bi on defects (perhaps at the edges between (111) domains) was also removed during this treatment. These results illustrate that Bi adsorbed on (111) terraces or (111) small domains is more easily removed from the surface than Bi adsorbed on “true” step sites or defects, because the energy for Bi adsorption on (111) sites is lower than that of the step/defect sites with lower coordination number.
Fig. 3 compares the representative voltammograms of Pt(554) and {111}Pt nanoparticles before and after step/defect decoration with Bi in 0.5 M H2SO4. It is clear that the voltammogram of Pt(554) with decorated steps shows that the hydrogen de/adsorption peaks on (110) steps nearly disappears, while keeping the anion adsorption on terraces essentially unchanged, in agreement with previous studies.34 For {111}Pt nanoparticles, it can be seen that the defects present on the nanoparticle surfaces have been almost blocked, leaving the ordered (111) domains unblocked after the treatment mentioned above. The remaining unblocked defects appear not to be very symmetric, especially the peak at 0.12 V, which demonstrates that these defects are more difficult to be blocked and somewhat different to (110) step sites of the vicinal (111) single-crystal surface. These sites are probably located at the edges between (111) domains or other defects and hydrogen adsorption would likely affect the position of the adsorbed adatoms.
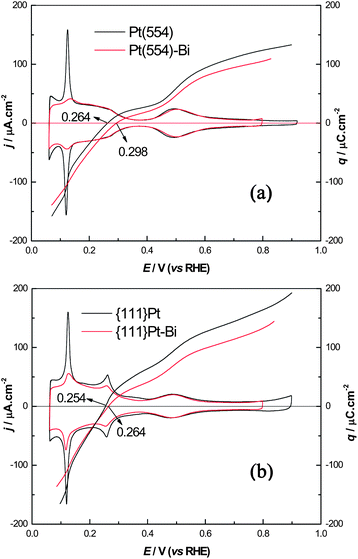 | ||
| Fig. 3 Comparisons of CVs and total charge curves before and after Bi steps/defects decoration on (a) Pt(554) and (b) {111}Pt nanoparticles in 0.5 M H2SO4, scan rate 50 mV s−1. | ||
The corresponding total charge density vs. potential curves are also displayed in Fig. 3 referred to right y-axis. These curves were obtained from the integration of the current density in voltammograms using eqn (1).52
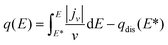 | (1) |
In contrast to this, it was shown that the local pztc is independent of step density for (100) vicinal planes in 0.1 M H2SO4.55 Based on the values of local pztc, the overall pztc can be inferred if reasonable assumptions are taken for the deconvolution of the different contributions to the voltammetric pseudocapacity. It was found that the overall pztc estimated by N2O reduction is consistent with the pztc determined by the CO charge displacement method and it always lies between the local pztc of the terrace and the step. Moreover, the Bi decoration of step sites is able to neutralize the charge associated to these step sites, resulting in a positive shift of the overall pztc towards the local value of the terrace. The invariant local pztc of the terrace also demonstrates that the Bi deposited on steps shows negligible effects towards terrace sites. In the present work, the positive shift of the pztc after the defects of {111}Pt nanoparticles were decorated with Bi is not so significant as that of Pt(554). As discussed previously, the (111) domains on {111}Pt nanoparticles may be inhomogeneous in length and randomly distributed. The average length of (111) domains should be very short, rather than large, since the percentage of (111) sites is formally even lower than that of Pt(331). Because of this, the local pztc of (111) domains, also reflects a weighed statistic response, maybe significantly lower than that of the Pt(554) terraces. This behaviour could also reflect the existence of isolated sites or small ensembles of the {111}Pt nanoparticles, that show different properties than those corresponding to the terraces present on vicinal Pt(111) stepped surfaces, as those related to Bi and anion adsorption and the local pztc values. In any case the interpretation of the pztc on nanoparticles is much more difficult than on stepped surfaces. In fact, all sites with different symmetry than (111) are considered as “defects”, independently of their origin, e.g. the (110) contributions could be due to defects in octahedral nanoparticles while those related to (100) symmetry could come from non-octahedral nanoparticles. In the same way, the pztc charges could be assigned to the different terrace width domains. All these distinctions could be considered arbitrary at this stage without the required structural information available. Attempts to analyze X-ray signals to acquire information on the surface contributions are necessary to understand overall pztc results.56
CO stripping
CO stripping was carried out as the final step of the CO displacement experiment, i.e. after a fully blocked adlayer of CO was completed on the electrode surface and the remaining CO in solution was removed by Ar bubbling. Fig. 4 shows the CO stripping voltammograms (first two cycles) of Pt(554), {111}Pt nanoparticles and their corresponding Bi decorated surfaces in 0.5 M H2SO4 at a scan rate of 20 mV s−1. The surface adsorption recovery and the absence of further CO oxidation in the second cycle illustrates that dissolved CO has been completely removed. The recuperation of the voltammogram after CO stripping also demonstrates that the Bi-decorated surface is stable in the whole experiment. When CO was stripped off from Pt(554), the voltammogram gives a symmetric oxidation peak centered around 0.712 V, this peak shifting positively to a higher potential after Bi step decoration. This result is in good agreement with previous studies of CO stripping on Pt(554) in 0.1 M HClO4.34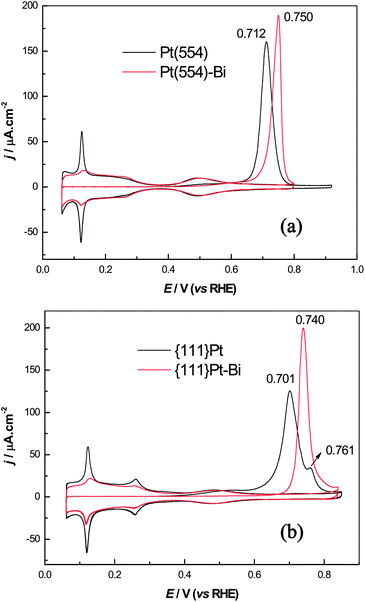 | ||
| Fig. 4 Comparisons of CO oxidation before and after Bi steps/defects decoration of (a) Pt(554) and (b) {111}Pt nanoparticles in 0.5 M H2SO4, scan rate: 20 mV s−1. | ||
Interestingly, the behaviour of CO stripping on {111}Pt nanoparticles is quite different to that of Pt(554) as evidenced by the appearance of peak multiplicity in the oxidation process, i.e., a “pre-wave” between 0.4 and 0.6 V, a main oxidation peak at 0.701 V followed by a small peak around 0.761 V. The lower onset potential and the negative shift of the main CO oxidation peak indicate that {111}Pt nanoparticles exhibit higher catalytic activity than that of Pt(554). The voltammogram of CO oxidation on the {111}Pt nanoparticles is quite different to that reported in ref. 17 on a similarly nominated electrode, especially because the peak around 0.761 V is much smaller. As it was pointed out, the peak around 0.761 V depends strongly on the amount of ordered (100) domains. In the present case, the amount of (100) terrace sites is only 3%, significantly lower than that reported for the sample in the previous reference (32%). This can also be confirmed by the apparent difference in the corresponding hydrogen adsorption regions of the blank voltammograms. This comparison suggests that the present nanoparticles are more representative of the {111}Pt character than the previously synthesized ones although all the same types of defect sites are present in both cases.
As previously stated, the multiplicity of CO oxidation peaks in acid media (H2SO4 and HClO4) is closely correlated to the structure of nanoparticles,.8,17–19 In addition, this multiplicity has been also observed in alkaline solution with well defined stepped surfaces13 as well as nanoparticles,57 and it has been again interpreted as CO oxidation on different surface sites. Up to now, it has been revealed that the activity of CO oxidation on different sites in alkaline media follows the sequence: kink > (110) step > (100) step > (111) terrace.11,16 However, results obtained in our laboratory with kinked surfaces suggest that kink sites do not confer special reactivity. In this framework, the “pre-wave” at the low potential region would be most likely associated with CO oxidation on isolated defects on the {111}Pt nanoparticles surface, likely the isolated (111) sites or small ensembles. This hypothesis can be supported by considering CO stripping on {111}Pt nanoparticles in acid media whose defects have been decorated with Bi. As it can clearly be seen, the “pre-wave” disappears completely after the defect of {111}Pt nanoparticles were blocked with Bi. Moreover, CO stripping on {111}Pt-Bi only gives a symmetric oxidation peak indicating that the (100) domains have also been blocked by Bi. This stripping peak also shifts positively to 0.740 V in comparison with the main stripping peak of CO on {111}Pt nanoparticles without decoration. As discussed previously, Bi decoration on step/defect sites shows negligible electronic effect towards adsorption on (111) terrace sites or (111) domains, only quenching the charge associated with these step/defect sites without modifying the behaviour on the terraces. In this respect, the positive shift of the CO oxidation peak points out that steps/defects act as active sites for CO electrooxidation.15,34 These results further confirm that CO oxidation is a structure-sensitive process and small modification on the surface cause significant changes on the reaction kinetics.
It is well known that CO coverage is an important parameter since it affects greatly the interaction between CO molecules and other co-adsorbates, thus further influencing their possibility of adsorption, binding energy and finally the oxidation kinetics.58–60 In our recent related study using adatom step decoration in 0.1 M HClO4, it was found that CO coverage on (110) and (100) step sites of CO saturated (111) vicinal surfaces was completely different, that is 0.7 for (110) steps while only 0.4 for (100) steps. The CO bond is also quite different, i.e., (110) was dominated by top CO but (100) was prevalent with bridge CO, indicating the different binding energy of CO with different step sites. Moreover the reaction kinetics was found to be different as a consequence of the different CO coverage and bond geometry on the different step sites34,61 Therefore, it is desirable to measure the CO coverage for the defects of preferentially oriented Pt nanoparticles. In order to determine the CO coverage, the first step deals with the determination of the true CO oxidation charge from the whole CO stripping charge, due to the non-negligible contribution of the double layer restoration, including anion re-adsorption.62 This problem can be easily solved, for both well defined single crystals and nanoparticles, with the knowledge of the pztc. The net charge of CO oxidation can be easily obtained by subtracting from the raw stripping charge the double layer correction charge from the integration of the voltammogram, in the first and second positive-going sweep respectively, between the pztc and the upper potential limit used for CO stripping.34 As discussed in our previous work, the hydrogen adsorption can be used as an alternative estimation of Pt step sites, following the previously known hydrogen coverage on steps (in both (110) and (100) steps it can be considered to be ∼1).34,41–43 The adsorbed H charge can be obtained from the total charge at the lower potential limit (0.06 V) in the absence of CO. The charge of CO oxidation and hydrogen adsorption before and after Bi decoration of the steps/defects can be obtained by subtracting the corresponding CO stripping and H adsorption charges before and after Bi decoration. Then CO coverage on step/defect sites (θSCO) can be calculated by eqn (2):
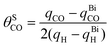 | (2) |
Therefore, θSCO of Pt(554) can be obtained by comparing the overall CO stripping charge with the change of H charge. The CO coverage on Pt(554) terraces can be evaluated with eqn (3):
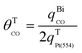 | (3) |
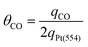 | (4) |
| θCO = xTθTCO + ySθSCO | (5) |
k as shown in Fig. 7
| Electrode | pztc/V | q CO /μC cm−2 | q H/μC cm−2 | q SCO/μC cm−2 | q SH/μC cm−2 | θ SCO | θ TCO | θ CO | θ (vs. qH) | Tafel slope, k1 | Tafel slope, k2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt(554) | 0.264 | 302.2 | 169.5 | 34.2 | 25.7 | 0.66 | 0.66 | 0.89 | 84 ± 6 | 84 ± 6 | |
| Pt(554)-Bi | 0.298 | 268.0 | 143.8 | 0.65 | 0.62 (× 0.67) | 70 ± 3 | 70 ± 3 | ||||
| {111}Pt | 0.254 | 351.8 | 197.7 | 65.2 | 49.6 | 0.66 | 0.66 | 0.89 | 95 ± 4 | 95 ± 6 | |
| {111}Pt-Bi | 0.264 | 286.6 | 148.1 | 0.65 | 0.65 (× 0.67) | 75 ± 6 | 67 ± 7 |
Kinetics study of CO electrooxidation
The kinetics of CO oxidation on the Pt(554) single-crystal electrode, {111}Pt nanoparticles as well as their Bi decorated ones were carried out by using chronoamperometry. Fig. 5 shows the corresponding current–time transients of CO oxidation on the different surfaces in 0.5 M H2SO4 as the potential was stepped from 0.1 V to a series of higher potentials. It is apparent that all the transients have the similar shape: after a fast double layer charge transient a main peak with tailing follows, indicating a similar CO oxidation transient on all the studied surfaces with and without Bi decoration. These transients are somewhat different to those observed on longer terrace electrodes vicinal to Pt(111) (symmetry of the main peak) but are similar to those obtained from higher step density vicinal Pt(111) electrode surfaces,12Pt(100) electrodes having surface defects63 and nanoparticles.20,21,63 The tailing in the main peak of CO monolayer oxidation on Pt stepped single crystals has been ascribed to a lesser reactivity of CO adsorbed on step edges, due to the limitations in the ability of CO to attain a reactive geometry.12,15 For Pt nanoparticles below 2 nm, it has been suggested that CO diffusion on the surface is slow, which limits the ability of CO to reach the sites active for OH formation.64 The possibility of blocking CO diffusion by the adsorption of anions on the free Pt sites that will appear as soon as CO molecules are removed from the surface has also been discussed.20 According to Inkaew et al., the tailing observed in the long time of the transients of Pt(100) with defects and Pt black catalyst particles, may be due to the different kinetics of CO adsorbed on terrace and step sites.63 It is obvious that the tailing transients can not be fitted by the typical mean field approximation.12 However, they can be well fitted with a modified mean-field Langmuir–Hinshelwood model (eqn (6)). The equation, which has also been successfully applied to fit multiple peaks for CO oxidation on Pt(510),65 is as following: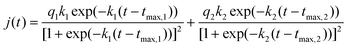 | (6) |
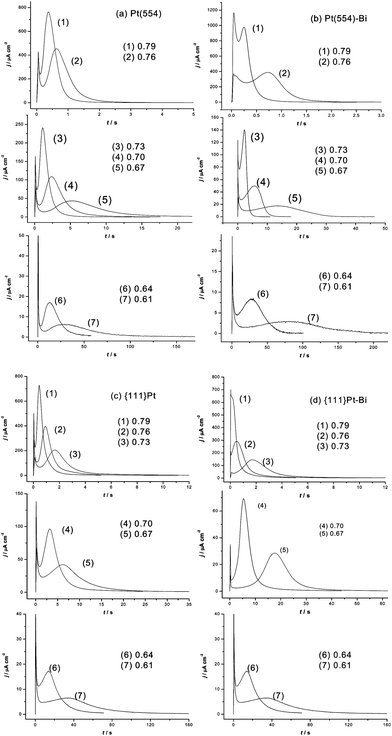 | ||
| Fig. 5 Current transients of CO oxidation at different potentials on Pt(554), Pt(554)-Bi, {111}Pt nanoparticles and {111}Pt-Bi in 0.5 M H2SO4. | ||
As shown in Fig. 6, the transients of CO oxidation when the potential is stepped at 0.73 V can all be fitted satisfactorily by eqn. (6), demonstrating that CO oxidation on Pt(554), preferentially oriented {111}Pt nanoparticles and their corresponding Bi decorated surfaces can be described by a similar mechanism composed of two components. A series of rate constants k1 and k2, measured at different step potentials, can be attained from the simulation process. The corresponding semilogarithmic plots are shown in Fig. 7. It can be seen that the CO oxidation rate decreases for Bi decorated Pt(554) and {111}Pt nanoparticles when compared to their corresponding blank surfaces, demonstrating that Pt step/defects sites play an enhancing role for CO oxidation. For Pt(554), the first contribution seems more related to the step effects since k1 remarkably decreases once the (110) step was blocked by Bi, while it shows negligible effects towards k2. The log(k1) and log(k2) both deviate from the linear relationship for Bi decorated Pt(554) and {111}Pt, at high potentials, may be due to anion adsorption on Bi decorated on step/defect sites at such high potentials (ca. 0.76 V), as can also be seen in the voltammograms (Fig. 3).
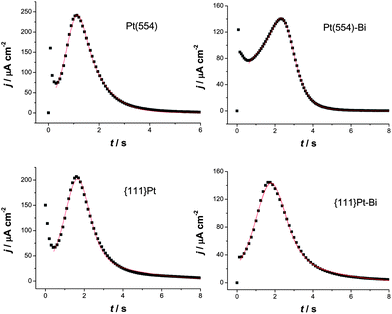 | ||
| Fig. 6 Current transients of CO oxidation at 0.73 V in 0.5 M H2SO4 on different surfaces as indicated in the figures. The symbols show the selective experimental data and the solid lines are the fit of the peaks by mean-field approximation. | ||
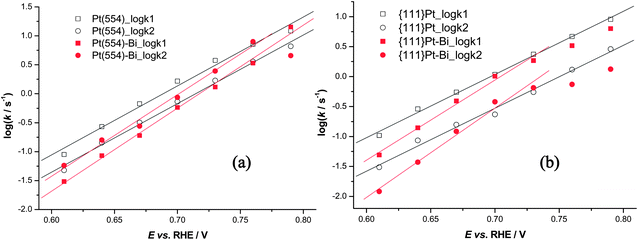 | ||
| Fig. 7 Dependence of the rate constants (k), determined by fitting the experimental data with eqn. (6), on step potential: (a) Pt(554) and Pt(554)-Bi; (b) {111}Pt nanoparticles and {111}Pt-Bi; in 0.5 M H2SO4. | ||
The values of Tafel slope obtained from the fitting lines are given in Table 2. Obviously, the Tafel slopes are the same for the two different contributions of both Pt(554) and {111}Pt nanoparticles, respectively. This corroborates that CO oxidation in the two contributions undertakes the same mechanism. After Bi decoration, the Tafel slope of Pt(554)-Bi for the two different contributions is still the same, although it decreases in comparison with those of Pt(554). The Tafel slopes show the same decreasing trend for Bi-decorated {111}Pt nanoparticles, except that there is a small difference on the values between the two contributions. It should be pointed out that the same Tafel slope in the two contributions of both Pt(554) and {111}Pt nanoparticles is different to that observed on Pt(510), which showed different values for different contributions, that were ascribed to CO oxidation on different types of sites.65 In the current case, the two contributions of CO oxidation may take place at the same reactive sites (steps/defects) and the differences would only be related to different kinetics of CO on terrace and step/defect sites, since their Tafel slopes are similar. It has been demonstrated that the Tafel slopes depend strongly on the step site symmetry (type of steps, density and decoration).61 The variation of the Tafel slopes in the present study resemble those in previous studies, i.e., they decrease with increasing terrace length as well as adatom decoration on step sites,61 and this has been interpreted by the change of OH (the oxygen containing species required for CO oxidation) adsorption isotherm on different surfaces.61,65 The Tafel slope of {111}Pt nanoparticles is somewhat larger than that of Pt(554) which can be attributed to its relatively shorter terraces, as aforementioned. The values of Tafel slopes are in the range 60–90 mV decade−1 pointing out that CO oxidation on Pt(554) and {111}Pt nanoparticles as well as their Bi decorated ones undergo the same mechanism, that is a Langmuir–Hinshelwood model involving a slow chemical step (COad + OHad → CO2 + H+ + e−).
Conclusions
In this paper, CO electrochemical oxidation on preferentially oriented {111}Pt nanoparticles was carried out and compared to that on single-crystal Pt(554). This is a particular situation because the characteristic fingerprint voltammograms are very similar, as nanoparticles only contain a small contribution from (100) symmetry sites, and thus Pt(554) can be used as model surface to understand the reactivity of {111}Pt nanoparticles. In this respect, Bi decoration on step and defect sites was performed to reveal the step/defect roles toward CO adsorption and oxidation. CO displacement experiments were performed to determine the pztc of Pt(554) and {111}Pt nanoparticles. The analysis of charges of individual site contributions, which can be attempted by using the criteria proposed for single-crystal stepped surfaces, suggests that for the {111}Pt nanoparticles, the (111) contributions are due to the presence of bidimensional domains. These (111) domains are able to adsorb Bi and anions, as done by stepped surfaces, but also contain an important fraction of isolated sites or small ensembles which can only adsorb hydrogen.The positive shift of pztc after Bi decoration is consistent with that observed on the single-crystal surface. The lower shift of the pztc of Bi decorated {111}Pt nanoparticles likely reflects a more complex nanoparticle structure and the presence of relatively short (111) terraces. The positive shift of CO stripping peak after Bi step/defect decoration demonstrates the catalytic effect of step/defect sites towards CO oxidation. With the knowledge of pztc and the help of Bi decoration, the local and total CO coverage of {111}Pt nanoparticles has been obtained. The different types of CO coverage determined for {111}Pt nanoparticles are quite coincident with those attained from Pt(554), further confirming their internal relationships as evidenced by their similar blank voltammograms. Chronoamperometry studies show similar current–time transients for Pt(554) and {111}Pt nanoparticles and their Bi decorated surfaces, and the corresponding transients can be simulated well with a modified Langmuir–Hinshelwood model. The Tafel slopes are located between 60–90 mV decade−1, confirming the similar mechanism of CO oxidation on the studied surfaces, i.e., Langmuir–Hinshelwood model with a slow chemical step. This study establishes a bridge from simple and basic single crystals to more complicated and practical nanoparticles. More experimental and theoretical efforts should be dedicated to unveil the intrinsic properties of CO oxidation on Pt nanoparticles, but the use of well defined, quantitative experiments, with model nanoparticles and stepped surfaces can contribute to this understanding.
Acknowledgements
This work has been financially supported by the MICINN of Spain through the project CTQ2010-16271 (Feder). Q. S. C. acknowledges the fellowship support of the China Scholarship Council and the support of NSFC (grant no. 20833005, 21021002). F. J. V. I. also thanks the support of the European Social Funding.Notes and references
- N. M. Markovic and P. N. Ross, Surf. Sci. Rep., 2002, 45, 121 CrossRef.
- J. Clavilier, R. Faure, G. Guinet and R. Durand, J. Electroanal. Chem., 1979, 107, 205 CrossRef.
- D. M. Kolb, Surf. Sci., 2002, 500, 722 CrossRef CAS.
- V. Climent, R. Gomez and J. M. Feliu, Electrochim. Acta, 1999, 45, 629 CrossRef CAS.
- R. Gomez, V. Climent, J. M. Feliu and M. J. Weaver, J. Phys. Chem. B, 2000, 104, 597 CrossRef CAS.
- L. Vattuone, L. Savio and M. Rocca, Surf. Sci. Rep., 2008, 63, 101 CrossRef CAS.
- G. A. Somorjai, Introduction to Surface Chemistry and Catalysis, John Wiley & Sons Inc., New York, 1994 Search PubMed.
- Q.-S. Chen, J. Solla-Gullón, S.-G. Sun and J. M. Feliu, Electrochim. Acta, 2010, 55, 7982 CrossRef CAS.
- Y. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732 CrossRef CAS.
- N. Tian, Z. Y. Zhou and S. G. Sun, J. Phys. Chem. C, 2008, 112, 19801 CAS.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Phys. Chem. B, 2002, 106, 12938 CrossRef CAS.
- G. Garcia and M. T. M. Koper, Phys. Chem. Chem. Phys., 2008, 10, 3802 RSC.
- G. Garcia and M. T. M. Koper, J. Am. Chem. Soc., 2009, 131, 5384 CrossRef CAS.
- N. P. Lebedeva, A. Rodes, J. M. Feliu, M. T. M. Koper and R. A. van Santen, J. Phys. Chem. B, 2002, 106, 9863 CrossRef CAS.
- G. Garcia and M. T. M. Koper, Phys. Chem. Chem. Phys., 2009, 11, 11437 RSC.
- J. Solla-Gullon, F. J. Vidal-Iglesias, E. Herrero, J. M. Feliu and A. Aldaz, Electrochem. Commun., 2006, 8, 189 CrossRef CAS.
- S. Brimaud, S. Pronier, C. Coutanceau and J. M. Leger, Electrochem. Commun., 2008, 10, 1703 CrossRef CAS.
- M. Nakamura, Y. Hanioka, W. Ouchida, M. Yamada and N. Hoshi, ChemPhysChem, 2009, 10, 2719 CrossRef CAS.
- M. Arenz, K. J. J. Mayrhofer, V. Stamenkovic, B. B. Blizanac, T. Tomoyuki, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2005, 127, 6819 CrossRef CAS.
- F. Maillard, M. Eikerling, O. V. Cherstiouk, S. Schreier, E. Savinova and U. Stimming, Faraday Discuss., 2004, 125, 357 RSC.
- K. Kinoshita, J. Electrochem. Soc., 1990, 137, 845 CrossRef CAS.
- A. Wieckowski, E. Savinowa and C. G. Vayenas, Catalysis and Electrocatalysis at Nanoparticle Surfaces, Marcel Dekker, Inc., New York, 2003 Search PubMed.
- S. W. Lee, S. O. Chen, W. C. Sheng, N. Yabuuchi, Y. T. Kim, T. Mitani, E. Vescovo and Y. Shao-Horn, J. Am. Chem. Soc., 2009, 131, 15669 CrossRef CAS.
- R. R. Adzic, in Modern Aspects of Electrochemistry, ed. J. O. Bockris, R. E. White and B. E. Conway, Plenum Press, New York, 1990, ch. 5, vol. 21 Search PubMed.
- E. Herrero, A. Fernández-Vega, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1993, 350, 73 CrossRef CAS.
- E. Herrero, J. M. Feliu and A. Aldaz, J. Catal., 1995, 152, 264 CrossRef CAS.
- E. Herrero, A. Rodes, J. M. Pérez, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1995, 393, 87 CrossRef.
- M. D. Macia, E. Herrero, J. M. Feliu and A. Aldaz, Electrochem. Commun., 1999, 1, 87 CrossRef CAS.
- J. M. Feliu, R. Gómez, M. J. Llorca and A. Aldaz, Surf. Sci., 1993, 289, 152 CrossRef CAS.
- J. M. Feliu, M. J. Llorca, R. Gómez and A. Aldaz, Surf. Sci., 1993, 297, 209 CrossRef CAS.
- J. Solla-Gullon, F. J. Vidal-Iglesias, P. Rodriguez, E. Herrero, J. M. Feliu, J. Clavilier and A. Aldaz, J. Phys. Chem. B, 2004, 108, 13573 CrossRef CAS.
- E. Herrero, V. Climent and J. M. Feliu, Electrochem. Commun., 2000, 2, 636 CrossRef CAS.
- Q.-S. Chen, A. Berna, V. Climent, S.-G. Sun and J. M. Feliu, Phys. Chem. Chem. Phys., 2010, 12, 11407 RSC.
- P. Rodriguez, J. Solla-Gullon, F. J. Vidal-Iglesias, E. Herrero, A. Aldaz and J. M. Feliu, Anal. Chem., 2005, 77, 5317 CrossRef CAS.
- J. Clavilier, D. Armand, S. G. Sun and M. Petit, J. Electroanal. Chem., 1986, 205, 267 CrossRef CAS.
- J. Solla-Gullon, F. J. Vidal-Iglesias, A. Lopez-Cudero, E. Garnier, J. M. Feliu and A. Aldaz, Phys. Chem. Chem. Phys., 2008, 10, 3689 RSC.
- J. Clavilier, R. Albalat, R. Gomez, J. M. Orts, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1992, 330, 489 CrossRef CAS.
- J. Solla-Gullon, V. Montiel, A. Aldaz and J. Clavilier, J. Electrochem. Soc., 2003, 150, E104 CrossRef CAS.
- N. Furuya and S. Koide, Surf. Sci., 1989, 220, 18 CrossRef CAS.
- J. Clavilier, K. El Achi and A. Rodes, J. Electroanal. Chem., 1989, 272, 253 CrossRef CAS.
- A. Rodes, K. El Achi, M. A. Zamakhchari and J. Clavilier, J. Electroanal. Chem., 1990, 284, 245 CrossRef CAS.
- J. Clavilier, K. El Achi and A. Rodes, Chem. Phys., 1990, 141, 1 CrossRef CAS.
- M. D. Macia, E. Herrero, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 2001, 500, 498 CrossRef CAS.
- J. Solla-Gullon, P. Rodriguez, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2008, 10, 1359 RSC.
- J. Clavilier, J. Electroanal. Chem., 1979, 107, 211 CrossRef.
- E. Herrero, V. Climent and J. M. Feliu, in Catalysis in Electrochemistry: From Fundamental Aspects to Strategies for Fuel Cell Development, ed. E. Santos and W. Schmickler, Wiley Series on Electrocatalysis and Electrochemistry, 2010 Search PubMed.
- A. Björling and J. M. Feliu, J. Electroanal. Chem. DOI:10.1016/j.jelechem.2011.01.045.
- D. Armand and J. Clavilier, J. Electroanal. Chem., 1987, 233, 251 CrossRef CAS.
- J. Souza-Garcia, V. Climent and J. M. Feliu, Electrochem. Commun., 2009, 11, 1515 CrossRef CAS.
- J. Clavilier, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1988, 243, 419 CrossRef CAS.
- V. Climent, R. Gomez, J. M. Orts, A. Aldaz and J. M. Feliu, Proc. Electrochem. Soc., 1997, 97-17, 222 CAS.
- V. Climent, E. Herrero and J. M. Feliu, Electrochem. Commun., 2001, 3, 590 CrossRef CAS.
- V. Climent, G. A. Attard and J. M. Feliu, J. Electroanal. Chem., 2002, 532, 67 CrossRef CAS.
- G. A. Attard, O. Hazzazi, P. B. Wells, V. Climent, E. Herrero and J. M. Feliu, J. Electroanal. Chem., 2004, 568, 329 CrossRef CAS.
- K. R. Beyerlin, J. Solla-Gullón, E. Herrero, E. Garnier, F. Pailloux, M. Leoni, P. Scardi, R. L. Snyder, A. Aldaz and J. M. Feliu, Mater. Sci. Eng., A, 2010, 528, 83 CrossRef.
- Q. S. Chen, S. G. Sun, Z. Y. Zhou, Y. X. Chen and S. B. Deng, Phys. Chem. Chem. Phys., 2008, 10, 3645 RSC.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Electroanal. Chem., 2002, 524–525, 242 CrossRef CAS.
- B. Shan, N. Kapur, J. Hyun, L. Wang, J. B. Nicholas and K. Cho, J. Phys. Chem. C, 2009, 113, 710 CAS.
- B. Shan, Y. J. Zhao, J. Hyun, N. Kapur, J. B. Nicholas and K. Cho, J. Phys. Chem. C, 2009, 113, 6088 CAS.
- Q.-S. Chen, J. M. Feliu, A. Berna, V. Climent and S.-G. Sun, Electrochim. Acta, 2011, 56, 5993 CrossRef CAS.
- R. Gomez, J. M. Feliu, A. Aldaz and M. J. Weaver, Surf. Sci., 1998, 410, 48 CrossRef CAS.
- P. Inkaew, W. Zhou and C. Korzeniewski, J. Electroanal. Chem., 2008, 614, 93 CrossRef CAS.
- B. Andreaus, F. Maillard, J. Kocylo, E. R. Savinova and M. Eikerling, J. Phys. Chem. B, 2006, 110, 21028 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullon, J. M. Campina, E. Herrero, A. Aldaz and J. M. Feliu, Electrochim. Acta, 2009, 54, 4459 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |