DOI:
10.1039/C1SC00064K
(Edge Article)
Chem. Sci., 2011,
2, 1156-1165
A dendrimer-based platform for simultaneous dual fluorescence imaging of hydrogen peroxide and pH gradients produced in living cells†
Received
29th January 2011
, Accepted 23rd February 2011
First published on 17th March 2011
Abstract
We present a modular dendrimer-based platform for simultaneous dual fluorescence imaging of hydrogen peroxide (H2O2) and pH gradients produced in living cells. Acetyl-capped G5 PAMAM dendrimers functionalized with boronate-caged Peroxyfluor-1 (PF1) fluorophores for H2O2 detection and semi-naphthorhodafluor (SNARF2) dyes for pH sensing provide a single probe system that is capable of sensing multiple analytes at a time by multicolor fluorescence imaging. Spectroscopic measurements of the doubly-labeled dendrimer conjugates establish their ability to simultaneously monitor changes in both H2O2 and pH using different excitation/emission profiles. Moreover, this dual-probe platform allows for selective discrimination between H2O2 and pH changes in live RAW 264.7 macrophage cells when stimulated by an immune insult. Further imaging experiments show that pharmacological inhibition of NADPH oxidase (Nox) proteins triggers a decrease in both oxidative burst and in pH regulation within phagocytic compartments and leads to disruptions of endocytic activity, suggesting that Nox-derived H2O2 signaling is critical to the maintenance of multiple components of the immune response. This work establishes a general molecular platform for simultaneous, real-time imaging of multiple analytes associated with redox biology in living systems and should be applicable to a wide range of chemosensor constructs.
Introduction
Hydrogen peroxide (H2O2) and related reactive oxygen species (ROS) generated during aerobic metabolism can have diverse consequences for human health, aging, and disease. Oxidative stress stemming from imbalances in ROS production is connected to aging1–4 and terminal diseases ranging from cancer5 to neurodegeneration,6–9 as well as to metabolic disorders such as diabetes.10–14 Despite their potential toxicity, ROS are increasingly appreciated to play essential roles in the normal physiology of life.15–29 The classic example of a beneficial role for ROS is in immunology, where sentinel cells like macrophages and neutrophils employ ROS to kill pathogens or other harmful antigens during phagocytosis.18,30,31 Indeed, the absence or deficiency of ROS production in the immune system can be fatal as in the case of chronic granulomatous disease (CGD), in which patients suffer concurrent and severe bacterial and fungal infections due to an inability to produce sufficiently high levels of ROS in phagocytes due to genetic mutations in the NADPH oxidase (Nox) enzyme.32–34
The interplay between redox biology and cellular immune response is, however, more sophisticated than the simple killing of pathogens by ROS bursts.35–38 This complexity is well-illustrated by the dendritic cells, which are another class of phagocytes that participate in the initiation of cytotoxic immune responses through antigen cross-presentation. Foreign particles engulfed by dendritic cells are processed through milder degradation pathways, where the synergistic use of ROS and pH gradients modulates lysosomal protease activity in order to preserve an identifiable fragment for subsequent T-cell recognition.39–41 As such, many details of the phagocytosis regulatory pathway remain elusive.
Molecular imaging provides an attractive approach to address mechanistic questions surrounding phagocytic immune response. In this context, genetically encoded fluorescent biosensors are effective tools for understanding physical aspects of phagocytotic processes, such as mechanisms of protein trafficking,42–44membrane surface potential, and lipid dynamics.45,46 Small-molecule fluorophores offer a complementary set of tools for monitoring chemical changes within phagocytic vesicles, such as pH47 and ROS fluxes.48 With regard to the latter, we recently performed dual probe imaging of H2O2 and HOCl in PMA-stimulated macrophages, and discovered three distinct types of phagosome populations that produce either or both of these oxidants for pathogen killing.49 To further advance our ability to simultaneously track multiple analytes in living systems, with specific interest in studying phagosomes during immune response, we turned our attention to dendrimers as a platform for carrying multiple types of compounds, which can range from imaging agents to targeting molecules to therapeutic agents. Depending on a given dendrimer architecture, these active compounds can be encapsulated within the dendrimer scaffold or covalently attached to its surface,50–58 along with acylation or addition of PEG chains to increase the biocompability of these branched particles.59–61 Recent examples of acetylated PAMAMs in biological applications include dendrimer-based pH sensors62 and folate-functionalized PAMAMs for targeted anti-cancer drug delivery.63,64
In this report, we present the design, synthesis, spectral properties, and live-cell imaging studies of a new type of dendrimer-based multifunctional imaging probe that can simultaneously report on both pH and H2O2. We describe a simple protocol for decorating PAMAM-G5 dendrimers with both Peroxyfluor-1 (PF1),65 a H2O2-responsive fluorescent probe, and SNARF2, a pH-sensitive dye. PF1 is a member of a group of boronate-caged reporters for H2O2; this reaction-based approach offers selectivity for detecting H2O2 over other ROS produced in immune signaling such as superoxide (O2−), nitric oxide (NO), the hydroxyl radical (•OH), and hypochlorous acid (HOCl).49,66–71 This platform is capable of detecting both pH changes and H2O2 bursts in macrophages upon immune stimulation using multicolor fluorescence imaging, and can be applied to situations in which pharmacological inhibition of NOX leads to a decrease in ROS fluxes and pH regulation of the phagosome. In addition to providing a potentially powerful new tool for reporting two critical chemical species involved in immune response and in redox biology, we anticipate that the modularity of this dendrimer platform and related conjugates will afford a versatile approach to multifunctional imaging in living systems.
Results and discussion
Design and synthesis of a multifunctional PAMAM dendrimer platform for simultaneous dual imaging of hydrogen peroxide and pH
Our overall design strategy for developing new reporters for simultaneous dual imaging of hydrogen peroxide and pH is to conjugate multiple types of responsive small-molecule fluorescent probes to a modular chemical scaffold. Fig. 1 depicts our strategy for creating PAMAM-based imaging agents with conjugated H2O2 and pH probes. For H2O2 detection, we prepared a new carboxy-functionalized version of PF1, an established fluorescent indicator sensitive to oxidative stress levels of H2O2. H2O2-mediated deprotection of the boronate esters of PF1 and subsequent opening of the bottom ring lactone yields the highly fluorescent green fluorescein (Scheme 1). For pH detection we chose SNARF2, a semi-naphthorhodafluor that is sensitive to pH with a pKa of 7.5.72 Changes in proton concentration [H+] shift the equilibrium between acidic and basic forms of the dye, with corresponding changes in absorption and emission ratio values.
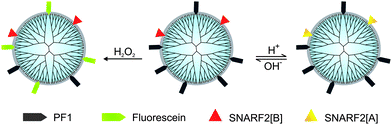 |
| | Fig. 1 PAMAM as a core structure for multifunctional nanoprobes for monitoring pH and H2O2. | |
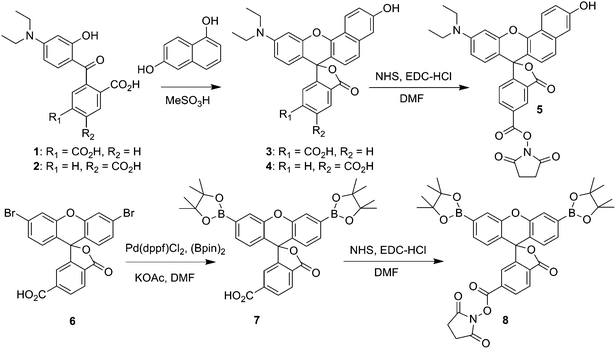 |
| | Scheme 1 Synthesis of SNARF2-NHS ester (5), and PF1-NHS ester (8). | |
PF1 and SNARF2 were covalently attached to PAMAM-G5 particles viaamide linkages, and the dual-conjugated PAMAMs were subsequently acetylated to produce low-toxicity, biocompatible nanoprobes (Scheme 2). Unreacted small-molecule fluorophores were separated from the dendrimers by Amicon 10 K centrifugal units (regenerated cellulose membrane MWCO 10,000) and by size-exclusive chromatography using a Sephadex LH-20 column. The average number of functional groups on the surface of the dendrimers was determined by MALDI-TOF analysis (Fig. S1, ESI†) and UV-Vis spectroscopy (Fig. S2, ESI†); the results are summarized in Table 1. G5-SNARF2-Ac is decorated, on average, with two SNARF2 dyes per dendrimer, whereas G5-SNARF2-PF1-Ac displays an average number of two SNARF2 and six PF1 dyes per dendrimer. Dynamic light scattering (DLS) experiments show that G5-SNARF2-Ac and G5-SNARF2-PF1-Ac reporters are narrowly distributed spherical nanoparticles with average hydrodynamic diameters of 3.63 and 3.56 nm, respectively (Fig. S4, ESI†). The modified dendrimers have a more compact structure than PAMAM-G5, which has an average hydrodynamic diameter of 5 nm, owing to the decrease in charge repulsion from fewer numbers of protonated primary amines in the parent unmodified dendrimer.59
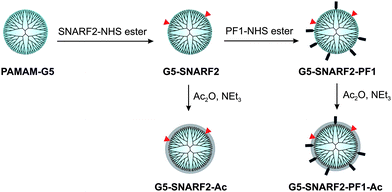 |
| | Scheme 2 Synthesis of functionalized PAMAMs. | |
Table 1 Molecular weight of functionalized PAMAM-G5 dendrimers
|
Dendrimer
|
m/z |
Fuctionality |
|
PAMAM-G5
|
26300 |
|
|
G5-SNARF2
|
27200 |
SNARF2(2)
|
|
G5-SNARF2-Ac
|
31000 |
SNARF2(2), Ac(88) |
|
G5-SNARF2-PF1
|
30660 |
SNARF2(2), PF1(6) |
|
G5-SNARF2-PF1-Ac
|
32500 |
SNARF2(2), PF1(6), Ac(44) |
Spectroscopic properties and responses to pH and hydrogen peroxide
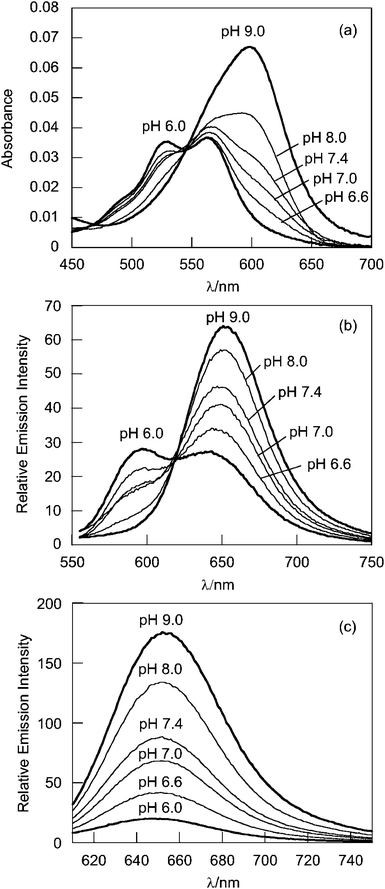
G5-SNARF2-Ac shows red shifts in absorption and emission maxima compared to SNARF2 alone (Table 2). The dendrimer-conjugated basic form of SNARF2, (SNARF2[B]), displays absorption and emission maxima at 600 and 650 nm, respectively, whereas the dendrimer-conjugated acidic form of SNARF2, SNARF2[A], exhibit absorption maxima at 525 and 563 nm and an emission maximum at 590 nm (Fig. 2). The pKa value of G5-SNARF2-Ac is 7.76 as determined by absorbance measurements in phosphate buffered solution.
Table 2 Spectroscopic properties of G5-SNARF2-Aca
| Compound |
A
λ
Absmax(nm) |
B
λ
Absmax(nm) |
B
λ
Emmax(nm) |
BλEmmax(nm) |
A
Φ
|
B
Φ
|
pKa |
|
The superscript A and B designate acidic (PH 5–6) and basic (PH 10–12) aqueous solutions, respectively. Quantum yields are relative to Rhodamine 101 in ethanol (ϕ = 0.92).
|
|
SNARF2
|
518,550 |
576 |
584 |
633 |
0.022 |
0.110 |
7.50 |
|
SNARF2-Ac
|
527,563 |
596 |
597 |
650 |
0.013 |
0.028 |
7.76 |
|
G5-SNARF2-PF1-Ac
|
527,564 |
599 |
598 |
650 |
0.051 |
0.043 |
7.78 |
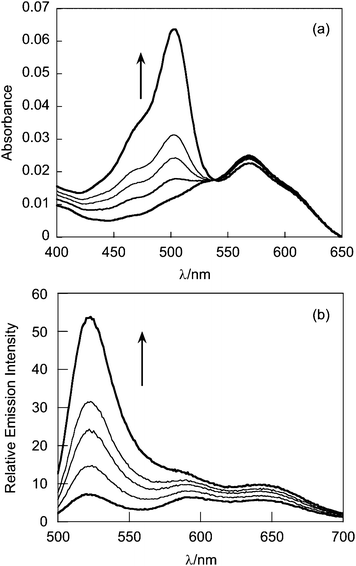
The absorption spectrum of G5-SNARF2-PF1-Ac in phosphate buffer at pH 7.4 is virtually identical to that of the G5-SNARF2-Ac absorption in the visible region for the colorless PF1 dye in its closed lactone form. Reaction of the G5-SNARF2-PF1-Ac probe with H2O2 triggers the appearance of a fluorescein-derived absorption peak near 500 nm (Fig. 3a). We observe a 4-fold turn-on in fluorescence at 520 nm with 488 nm excitation after 30 min incubation of the probe with 100 μM H2O2 (Fig. 3b). The observed rate constant of the H2O2-mediated deprotection of G5-SNARF1-PF1-Ac under pseudo-first order conditions (10 μg mL−1G5-SNARF2-PF1-Ac, ∼1.5 μM PF1, 1 mM H2O2) is kobs = 2.8 × 10−4s−1 (Fig. S5, ESI†). The quantum yield of G5-SNARF2-PF1-Ac when PF1 is fully deprotected to fluorescein is 0.16, which is lower than the quantum yield of fluorescein in solution (Φ = 0.93).73 The observed decrease in quantum efficiency is likely due to interactions of the fluorophore with the densely packed amide and amine groups on the dendrimer surface. Further evidence that the G5-SNARF2-PF1-Ac probe is responsive to H2O2 is provided by fluorescence images of SDS-PAGE samples of G5-SNARF2-PF1-Ac treated with various concentrations of H2O2 (Fig. 4). Finally, the reaction of G5-SNARF2-PF1-Ac with H2O2 has no effect on the fluorescence emission profile of the SNARF2 pH reporter and this probe maintains the ability to sense pH changes in aqueous solution, confirming that the dual-conjugated probe can independently and simultaneously detect changes in both H2O2 levels and pH using different excitation wavelengths (Fig. S6, ESI†).
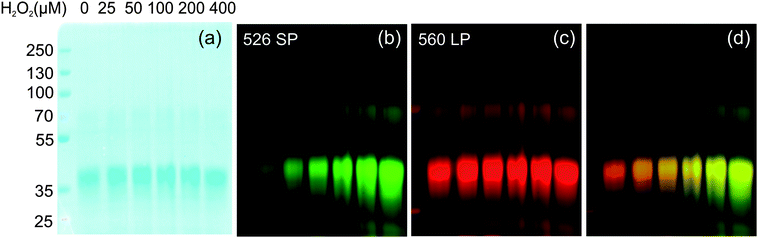 |
| | Fig. 4
SDS-PAGE of G5-SNARF2-PF1-Ac after reaction with various concentrations of added H2O2. (a) Coomassie stain. (b) Fluorescence emission from PF1 on the dendrimer. (c) Fluorescence emission from SNARF2. (d) Merged image of PF1 and SNARF2 channel. (λexc = 532 nm). | |
Fluorescence imaging of phagosomal acidification in RAW 264.7 macrophages with G5-SNARF2-Ac
We proceeded to test the dendrimer-based fluorescent reporters in live-cell imaging experiments. First, WST-1 assays showed no significant cytotoxicity after 3 h incubations of live RAW 264.7 macrophages with 500–1000 μg mL−1 levels of G5-SNARF2-Ac or G5-SNARF2-PF1-Ac (Fig. S7, ESI†). We then established that G5-SNARF2-Ac is able to report the progressive acidification of the phagosomal lumen in RAW 264.7 cells stimulated to an immune response with phorbol 12-myristate 13-acetate (PMA) (Fig. 5). For these experiments we employed the fluorescent filter set; Ch1-SNARF2[B] (λexc = 600 nm, λem = 660–710) and Ch2-SNARF2[A] (λexc = 550 nm, λem = 570–640) to allow spectral separation of the acidic and basic forms of SNARF2, respectively, without spectral overlap with the optical window for subsequent PF1-derived H2O2 detection. Images of a 50 μg mL−1 solution of G5-SNARF2-Ac in 50 mM phosphate buffer enabled a color-coded pH calibration as shown in Fig. 5. Fifteen minutes after PMA stimulation, the RAW 264.7 macrophage cells are largely populated with early phagosomes, which maintain slightly basic to neutral pH, and hence give a strong signal from the Ch1-SNARF2[B] channel. Apart from these neutral phagosomes, the live-cell images also show a population of smaller but more acidic vesicles, which we assign to cytosolic endosomes that have obtained G5-SNARF2-Ac either by pinocytosis or via a kiss-and-run interaction with phagosomes.74,75 During endocytic trafficking, phagosomes also acquire V-ATPase, a transmembrane proton pump through fusion with endomembrane compartments. V-ATPase is responsible for the acidification of phagosomes and provides an optimal pH for the activation and function of other microbicide agents, including hydrolytic enzymes and cationic peptides.76 Forty-five minutes after PMA stimulation, we observed the acidification of mature phagosomes, as indicated by a prominent emission from the Ch2-SNARF2[A] channel. These experiments demonstrate that the dendrimer-based probes are capable of imaging changes in pH in living cells and show a progression of pH-dependent events during a stimulated phagocytic immune response.
![Fluorescence imaging of pH changes in phagosomal lumen with G5-SNARF2-Ac. RAW 264.7 cells were at rest in DMEM containing 300 μg mL−1G5-SNARF2-Ac before PMA (4 μg mL−1) was added to stimulate phagocytosis. Fluorescence emission from basic and acid forms of SNARF2 are displayed in Ch1-SNARF2[B] and Ch2-SNARF2[A], respectively. A pH profile is generated from merging the signal from Ch1 and Ch2. Row (a) shows an image taken 15 min after the onset of phagocytosis. Cells are populated by early phagosomes which have a basic to neutral pH. Row (b) shows progressive acidification in phagosomal lumen after cells in row (a) were incubated in fresh media for another 30 min. Column (1) Ch1 filter set (600: 670–710), (2) Ch2 filter set (550: 570–640), (3) DIC image, scale bar = 10 μm, and (4) overlay image of Ch1 and Ch2, nuclear staining with Hoechst 33342.](/image/article/2011/SC/c1sc00064k/c1sc00064k-f5.gif) |
| | Fig. 5
Fluorescence imaging of pH changes in phagosomal lumen with G5-SNARF2-Ac. RAW 264.7 cells were at rest in DMEM containing 300 μg mL−1G5-SNARF2-Ac before PMA (4 μg mL−1) was added to stimulate phagocytosis. Fluorescence emission from basic and acid forms of SNARF2 are displayed in Ch1-SNARF2[B] and Ch2-SNARF2[A], respectively. A pH profile is generated from merging the signal from Ch1 and Ch2. Row (a) shows an image taken 15 min after the onset of phagocytosis. Cells are populated by early phagosomes which have a basic to neutral pH. Row (b) shows progressive acidification in phagosomal lumen after cells in row (a) were incubated in fresh media for another 30 min. Column (1) Ch1 filter set (600: 670–710), (2) Ch2 filter set (550: 570–640), (3) DIC image, scale bar = 10 μm, and (4) overlay image of Ch1 and Ch2, nuclear staining with Hoechst 33342. | |
Simultaneous multicolor imaging of H2O2 and pH fluxes during phagocytosis with G5-SNARF2-PF1-Ac
Phagocytosis triggers the assembly of NADPH oxidase (Nox), a family of multi-subunit enzymes that possess both membrane-bound and cytosolic components. By facilitating electron transfer to molecular oxygen, Nox generates fluxes of superoxide (O2−) within the phagosome that can reach millimolar levels and then undergo rapid dismutation to give H2O2 as the primary microbicide.77 Using G5-SNARF2-PF1-Ac, we were able to directly observe H2O2 generation in early phagosomes upon PMA-stimulation of RAW 264.7 macrophages based on the colocalization of emission signals derived from PF1 and basic SNARF2 signatures (Fig. 6a). Similar results were obtained when the cells were stimulated in the presence of superoxide dismutase (5,000 U mL−1), which catalyzes the dismutation of O2− to H2O2. This result confirms the selectivity of G5-SNARF2-PF1-Ac for H2O2 over O2− in living cells (Fig. 6b). A further control experiment shows that addition of both superoxide dismutase (5,000 U mL−1) and catalase (5,000 U mL−1), which catalyzes the decomposition of H2O2, led to a marked decrease in the PF1 signal corresponding to diminished phagosomal H2O2 production in these stimulated cells (Fig. 6c).
![Fluorescence imaging of oxidative burst and pH change in early phagosome with G5-SNARF2-PF1-Ac (300 μg mL−1 in DMEM). Row (a) shows RAW 264.7 cells stimulated with PMA for 15 min. Generation of H2O2 in early phagosomes is displayed by the colocalization of SNARF2[B] with fluorescein, a product of the reaction of PF1 with H2O2. Row (b) displays PMA stimulated RAW 264.7 in the presence of superoxide dismutase (SOD 5,000 U mL−1). Row (c) shows PMA stimulated cells in the presence of SOD (5,000 U mL−1) and catalase (5,000 U mL−1). Column (1) Ch1 filter set (600: 670–710), (2) Ch2 filter set (550: 570–640), (3) Ch3 filter set (470: 500–550), (4) DIC image, scale bar = 10 μm, and (5) overlay image of Ch1 and Ch2, nuclear staining with Hoechst 33342.](/image/article/2011/SC/c1sc00064k/c1sc00064k-f6.gif) |
| | Fig. 6
Fluorescence imaging of oxidative burst and pH change in early phagosome with G5-SNARF2-PF1-Ac (300 μg mL−1 in DMEM). Row (a) shows RAW 264.7 cells stimulated with PMA for 15 min. Generation of H2O2 in early phagosomes is displayed by the colocalization of SNARF2[B] with fluorescein, a product of the reaction of PF1 with H2O2. Row (b) displays PMA stimulated RAW 264.7 in the presence of superoxide dismutase (SOD 5,000 U mL−1). Row (c) shows PMA stimulated cells in the presence of SOD (5,000 U mL−1) and catalase (5,000 U mL−1). Column (1) Ch1 filter set (600: 670–710), (2) Ch2 filter set (550: 570–640), (3) Ch3 filter set (470: 500–550), (4) DIC image, scale bar = 10 μm, and (5) overlay image of Ch1 and Ch2, nuclear staining with Hoechst 33342. | |
Dendrimer-based multiplex imaging reveals the effects of Nox activity on phagosomal ROS and pH fluxes
We next applied the G5-SNARF2-PF1-Ac probe to fluorescence imaging of PMA-stimulated RAW 264.7 macrophages treated with various doses of diphenylene iodonium (DPI), a broad-spectrum small-molecule Nox inhibitor. Unlike superoxide dismutase and catalase enzymes that scavenge ROS without largely perturbing the physiological environment of phagosome, inhibition of Nox activity disrupts various equilibria within and surrounding the phagosomal lumen and simultaneously influences ROS levels, local pH, and membrane potential. By mediating electron transfer to molecular oxygen from the cytosol to the phagosome, O2− production by Nox is accompanied by a transient depolarization of the plasma membrane.78,79 In addition, dismutation and/or reduction of O2− also consumes protons leading to temporary alkalinization of the phagosomal lumen; this process is most prominent in neutrophils, which produce phagocytes with the highest ROS fluxes.80 Furthermore, phagosomal pH is regulated by a complex system involving the interplay of many membrane-bound proteins, including Nox, V-ATPase, voltage-gated channels, and other unidentified H+ channels.81
Live-cell imaging with G5-SNARF2-PF1-Ac in PMA-stimulated RAW 264.7 cells shows a marked decrease in the H2O2-responsive, PF1-derived fluorescein signal, indicating lower H2O2 production, upon addition of DPI in a dose-dependent manner (Fig. 7(3)). In addition to reducing levels of H2O2 production during oxidative bursts, inhibition of Nox activity also results in more rapid acidification of phagosomes due to a loss of pH regulation (Fig. 7(5)).82 Fifteen minutes after the onset of phagocytosis, phagosomes of stimulated macrophages with normal Nox activity are mostly neutral in pH with a concomitant robust H2O2 flux. However, cells treated with the Nox inhibitor DPI produce fewer phagosomes; these early phagosomes possess more acidic lumen than cells without DPI treatment. The effects of DPI on the frequency of phagosome formation becomes particularly evident in macrophages treated with 5 μM DPI, where there is an apparent elimination of phagocytosis. Our findings are in line with previous reports that DPI-treated macrophages show suppression in phagocytosis of myelin proteins83 and apoptotic neutrophils.84 Evidently, Nox plays an important role in the process of programmed cell clearance in macrophages. We speculate that high doses of DPI might disrupt the PI3K signaling pathway, which is involved in membrane remodeling and trafficking required for pseudopod extension, during both phagocytosis and pinocytosis,85–87 as H2O2 is a regulator of PI3K signaling through redox-mediated inhibition of the opposing phosphatase PTEN.88,89 In complementary experiments, flow cytometry analysis of PMA-stimulated macrophages treated with DPI also shows a dose-dependent decrease in the H2O2-responsive, PF1-derived fluorescein signal. Finally, in accordance with the imaging data, 5 μM DPI inhibition of endocytosis results in a lower average fluorescein intensity in comparison to control cells in which G5-SNARF2-PF1-Ac uptake is facilitated by pinocytosis (Fig. 8).
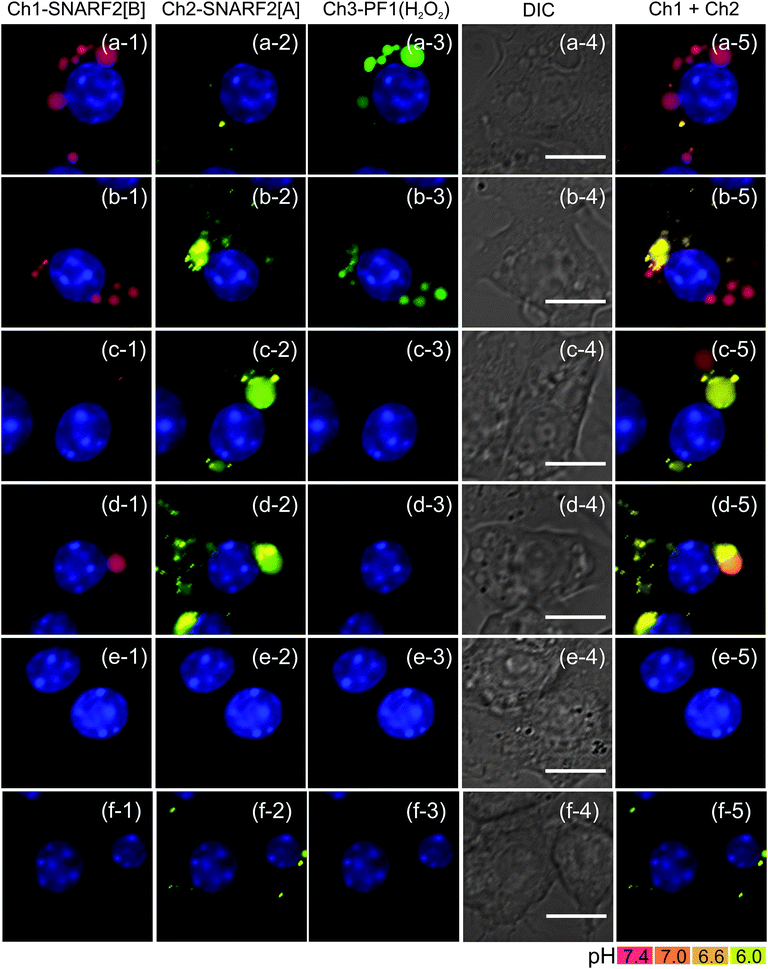 |
| | Fig. 7 Inhibition of Nox activity in PMA-stimulated RAW 264.7 cells with various doses of DPI. (a) no DPI, (b) 200 nM, (c) 500 nM, (d) 1 μM, (e) 5 μM, and (f) unstimulated cells with no PMA and no DPI. Phagosomal H2O2 decreases in a dose-dependent manner with DPI. DPI treated cells also show faster acidification of phagosome. At 5 μM DPI, endocytic activity is disrupted, showing the absence of pinocytosis compared to the unstimulated cells. Column (1) Ch1 filter set (600: 670–710), (2) Ch2 filter set (550: 570–640), (3) Ch3 filter set (470: 500–550), (4) DIC image, scale bar = 10 μm, and (5) overlay image of Ch1 and Ch2, nuclear staining with Hoechst 33342. | |
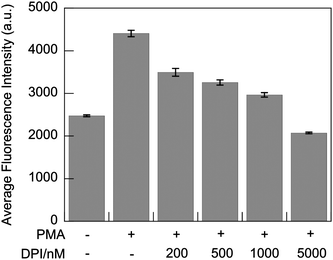 |
| | Fig. 8
Flow cytometry
detection of fluorescent emission intensity of PF1-derived fluorescein in PMA-stimulated RAW 264.7 macrophages. Cells were harvested after 30 min incubation in media containing G5-SNARF2-PF1-Ac and the indicated concentration of PMA and DPI. (λexc = 532 nm, λem = 500–550 nm). Error bars represent s.e.m. | |
Dendrimer-based multiplex imaging shows pH regulation of the phagosomal lumen by Nox and V-ATPase
We then applied the G5-SNARF2-PF1-Ac probe to study the roles of components that maintain pH regulation in the phagosomal lumen, using PMA-stimulated RAW 264.7 macrophages as a model. Forty-five minutes after PMA stimulation, acidification of the mature phagosome results in the absence of signal from the SNARF2[B] channel, and the appearance of a SNARF2[A] emission that colocalizes well with the H2O2-responsive PF1 fluorescence (Fig. 9a). In contrast, addition of concanamycin A, a V-ATPase inhibitor,90 halts the acidification of phagosomes without disrupting ROS generation, as neutral phagosomes persist 45 min after PMA stimulation with 400 nM concanamycin treatment (Fig. 9b). In addition, concanamycin A alters endosomal trafficking, as evidenced by the drastic changes in vesicle size distributions, with concanamycin A-treated macrophages producing larger vesicles. To compensate for the loss of V-ATPase activity and to enable maintainance of a neutral phagosome pH, alkalinization of the phagosomal lumen by Nox activity is accompanied by passive proton flux and activation of other transporters, including voltage-gated H+ channel. As a consequence, when both V-ATPase and Nox are inhibited (Fig. 9c), most vesicles exhibit neutral to slightly acidic pH, sensed by the SNARF2 channel, accompanied by low H2O2 production as detected by the PF1 channel.
![Fluorescence images of concanamycin A-treated macrophages that lead to the inhibition of the phagosomal acidification. RAW 264.7 cells were incubated with G5-SNARF2-PF1-Ac (300 μg mL−1 in DMEM), PMA (4 μg mL−1), with or without concanamycin A (400 nM) and DPI (500 nM). After 15 min, the labeling solution was removed. Cells were further incubated in label-free media for another 30 min before imaging. Row (a) shows PMA-stimulated RAW 264.7 cells 45 min after PMA stimulation. Overlay image of SNARF[A] and SNARF[B] shows marked acidic environment in phagosomal lumen. Row (b) displays concanamycin A-treated cells after 45 min PMA stimulation showing mostly neutral phagosome. Row (c) displays PMA stimulated cell treated with both PMA and DPI, in which mixed population of acidic and neutral pH phagosomes can be observed. Column (1) Ch1 filter set (600: 670–710), (2) Ch2 filter set (550: 570–640), (3) Ch3 filter set (470: 500–550), (4) DIC image, scale bar = 10 μm, and (5) overlay image of Ch1 and Ch2, nuclear staining with Hoechst 33342.](/image/article/2011/SC/c1sc00064k/c1sc00064k-f9.gif) |
| | Fig. 9
Fluorescence images of concanamycin A-treated macrophages that lead to the inhibition of the phagosomal acidification. RAW 264.7 cells were incubated with G5-SNARF2-PF1-Ac (300 μg mL−1 in DMEM), PMA (4 μg mL−1), with or without concanamycin A (400 nM) and DPI (500 nM). After 15 min, the labeling solution was removed. Cells were further incubated in label-free media for another 30 min before imaging. Row (a) shows PMA-stimulated RAW 264.7 cells 45 min after PMA stimulation. Overlay image of SNARF[A] and SNARF[B] shows marked acidic environment in phagosomal lumen. Row (b) displays concanamycin A-treated cells after 45 min PMA stimulation showing mostly neutral phagosome. Row (c) displays PMA stimulated cell treated with both PMA and DPI, in which mixed population of acidic and neutral pH phagosomes can be observed. Column (1) Ch1 filter set (600: 670–710), (2) Ch2 filter set (550: 570–640), (3) Ch3 filter set (470: 500–550), (4) DIC image, scale bar = 10 μm, and (5) overlay image of Ch1 and Ch2, nuclear staining with Hoechst 33342. | |
Concluding remarks
In summary, we have described the synthesis, properties, and biological applications of G5-SNARF2-PF1-Ac, a new dendrimer-based, dual-responsive fluorescent reporter for the simultaneous imaging of both H2O2 and pH gradients in living cells. This work demonstrates the utility of PAMAM dendrimers as modular and versatile scaffolds for the loading of multiple chemically-responsive probes for multiplex imaging. Using this new G5-PAMAM imaging platform, we studied the contributions of ROS and pH fluxes to phagocytosis during immune insult using PMA-stimulated RAW 264.7 macrophages as a model system. G5-SNARF2-PF1-Ac affords a method for the direct imaging of confined H2O2 bursts in early endosomes, as well as a way to monitor the progression of acidification during endosome trafficking and maturation. Additional imaging experiments show that the regulation of ROS through discrete molecular sources plays a critical role in the physiological immune response, as DPI-induced inhibition of Nox, the major source of phagocytic H2O2 bursts, not only affects transient ROS levels but also alters the course of phagosomal trafficking. Moreover, decreased Nox activity also results in a loss of pH regulation and more rapid rate of phagosomal acidification, with V-ATPase acting as another primary regulator for proper phagosome maturation.
This work contributes to the mounting evidence that the roles of H2O2 in immunology are more sophisticated and extend beyond the simplistic, primary view of ROS as microbicides. Our data also highlights the value of multifunctional fluorescent probes as powerful tools that can help to elucidate complex and transient changes in multiple molecular species with spatial and temporal resolution. Ongoing efforts include the application of this dual-imaging probe to studies of Nox chemistry in a variety of biological models, the development of second generation probes with improved optical brightness and photostability properties, and an expansion of the range of dendrimers and other molecular scaffolds for attachment of multiple responsive sensors and dosimeters.
Acknowledgements
We thank the Packard Foundation, Amgen, AstraZeneca, Novartis, and the National Institute of General Medical Sciences (NIH GM 79465) for funding this work. C.J.C. is an Investigator with the Howard Hughes Medical Institute. D.S. was supported by a scholarship from the Ministry of Science, Thailand. A.E.A. thanks the NIH Chemical Biology Graduate Program (T32 GM066698) for support, as well as the ACS Organic Division for an Emmanuil Troyansky Graduate Fellowship and UC Berkeley for a Chancellor's Opportunity Fellowship. We thank Holly Aaron (UCB Molecular Imaging Center) and Ann Fischer (UCB Tissue Culture Facility) for expert technical assistance, and Prof. Jean Frechet for use of his lab's MALDI instrument.
References
- D. Harman, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 7124–7128 CrossRef CAS.
- R. A. Floyd, Science, 1991, 254, 1597–1597 CrossRef CAS.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS.
- E. R. Stadtman, Free Radical Res., 2006, 40, 1250–1258 CrossRef CAS.
- T. Finkel, M. Serrano and M. A. Blasco, Nature, 2007, 448, 767–774 CrossRef CAS.
- R. A. Floyd and K. Hensley, Neurobiol. Aging, 2002, 23, 795–807 CrossRef CAS.
- J. K. Andersen, Nat. Rev. Neurosci., 2004, 10, S18–S25 CrossRef.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS.
- K. Bedard and K. H. Krause, Physiol. Rev., 2007, 87, 245–313 CrossRef CAS.
- J. M. May and C. D. Haen, J. Biol. Chem., 1979, 254, 2214–2220 CAS.
- D. B. Muchmore, S. A. Little and C. Dehaen, Biochemistry, 1982, 21, 3886–3892 CrossRef CAS.
- S. P. Mukherjee, R. H. Lane and W. S. Lynn, Biochem. Pharmacol., 1978, 27, 2589–2594 CrossRef CAS.
- B. J. Goldstein, M. Kalyankar and X. D. Wu, Diabetes, 2004, 54, 311–321 CrossRef.
- Y. Song, N. Driessens, M. Costa, X. De Deken, V. Detours, B. Corvilain, C. Maenhaut, F. Miot, J. Van Sande, M. C. Many and J. E. Dumont, J. Clin. Endocrinol. Metab., 2007, 92, 3764–3773 CrossRef CAS.
- M. Sundaresan, Z. X. Yu, V. J. Ferrans, K. Irani and T. Finkel, Science, 1995, 270, 296–299 CAS.
- Y. A. Suh, R. S. Arnold, B. Lassegue, J. Shi, X. X. Xu, D. Sorescu, A. B. Chung, K. K. Griendling and J. D. Lambeth, Nature, 1999, 401, 79–82 CrossRef CAS.
- M. Geiszt, J. B. Kopp, P. Varnai and T. L. Leto, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8010–8014 CrossRef CAS.
- J. D. Lambeth, Nat. Rev. Immunol., 2004, 4, 181–189 CrossRef CAS.
- S. G. Rhee, Science, 2006, 312, 1882–1883 CrossRef.
- J. R. Stone and S. Yang, Antioxid. Redox Signaling, 2006, 8, 243–270 Search PubMed.
- B. D'AutrEaux and M. B. Toledano, Nat. Rev. Mol. Cell Biol., 2007, 8, 813–824 CrossRef CAS.
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS.
- E. A. Veal, A. M. Day and B. A. Morgan, Mol. Cell, 2007, 26, 1–14 CrossRef CAS.
- L. B. Poole and K. J. Nelson, Curr. Opin. Chem. Biol., 2008, 12, 18–24 CrossRef CAS.
- P. Niethammer, C. Grabher, A. T. Look and T. J. Mitchison, Nature, 2009, 459, 996–U123 CrossRef CAS.
- C. E. Paulsen and K. S. Carroll, ACS Chem. Biol., 2010, 5, 47–62 CrossRef CAS.
- E. W. Miller, B. C. Dickinson and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15681–15686 CrossRef CAS.
- H. A. Woo, S. H. Yim, D. H. Shin, D. Kang, D. Y. Yu and S. G. Rhee, Cell, 2010, 140, 517–528 CrossRef CAS.
- B. C. Dickinson, J. Peltier, D. Stone, D. V. Schaffer and C. J. Chang, Nat. Chem. Biol., 2010, 7, 106–112.
- M. Djaldetti, H. Salman, M. Bergman, R. Djaldetti and H. Bessler, Microsc. Res. Tech., 2002, 57, 421–431 CrossRef.
- A. W. Segal, Annu. Rev. Immunol., 2005, 23, 197–223 CrossRef CAS.
- M. Geiszt, A. Kapus and E. Ligeti, J. Leukoc. Biol., 2001, 69, 191–196 CAS.
- P. G. Heyworth, A. R. Cross and J. T. Curnutte, Curr. Opin. Immunol., 2003, 15, 578–584 CrossRef CAS.
- S. M. Holland, Clin. Rev. Allergy Immunol., 2009, 38, 3–10 Search PubMed.
- H. J. Forman and M. Torres, Am. J. Respir. Crit. Care Med., 2002, 166, 4S–S8 CrossRef.
- M. T. Quinn and K. A. Gauss, J. Leukocyte Biol., 2004, 76, 760–781 Search PubMed.
- W. M. Nauseef, J. Biol. Chem., 2008, 283, 16961–16965 CrossRef CAS.
- D. I. Brown and K. K. Griendling, Free Radical Biol. Med., 2009, 47, 1239–1253 CrossRef CAS.
- A. Savina and S. Amigorena, Immunol. Rev., 2007, 219, 143–156 CrossRef CAS.
- R. D. Michalek, K. J. Nelson, B. C. Holbrook, J. S. Yi, D. Stridiron, L. W. Daniel, J. S. Fetrow, S. B. King, L. B. Poole and J. M. Grayson, J. Immunol., 2007, 179, 6456–6467 CAS.
- Z. H. Yan, S. K. Garg, J. Kipnis and R. Banerjee, Nat. Chem. Biol., 2009, 5, 721–723 CrossRef CAS.
- M. Yan, R. F. Collins, S. Grinstein and W. S. Trimble, Mol. Biol. Cell, 2005, 16, 3077–3087 CrossRef CAS.
- W. Tian, X. J. Li, N. D. Stull, W. Ming, C. I. Suh, S. A. Bissonnette, M. B. Yaffe, S. Grinstein, S. J. Atkinson and M. C. Dinauer, Blood, 2008, 112, 3867–3877 CrossRef CAS.
- K. K. Huynh and S. Grinstein, Curr. Biol., 2008, 18, R563–R565 CrossRef CAS.
- T. Yeung and S. Grinstein, Immunol. Rev., 2007, 219, 17–36 CrossRef CAS.
- G. D. Fairn, K. Ogata, R. J. Botelho, P. D. Stahl, R. A. Anderson, P. De Camilli, T. Meyer, S. Wodak and S. Grinstein, J. Cell Biol., 2009, 187, 701–714 CrossRef CAS.
- J. Han and K. Burgess, Chem. Rev., 2009, 110, 2709–2728.
- K. Setsukinai, Y. Urano, K. Kakinuma, H. J. Majima and T. Nagano, J. Biol. Chem., 2003, 278, 3170–3175 CrossRef CAS.
- B. C. Dickinson, C. Huynh and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 5906–5915 CrossRef CAS.
- J. F. KukowskaLatallo, A. U. Bielinska, J. Johnson, R. Spindler, D. A. Tomalia and J. R. Baker, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4897–4902 CrossRef CAS.
- S. Hecht and J. M. J. Frechet, Angew. Chem., Int. Ed., 2001, 40, 74–91 CrossRef CAS.
- R. Esfand and D. A. Tomalia, Drug Discovery Today, 2001, 6, 427–436 CrossRef CAS.
- C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS.
- C. Dufès, I. F. Uchegbu and A. G. Schätzlein, Adv. Drug Delivery Rev., 2005, 57, 2177–2202 CrossRef CAS.
- S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106–2129 CrossRef CAS.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS.
- R. K. Tekade, P. V. Kumar and N. K. Jain, Chem. Rev., 2009, 109, 49–87 CrossRef CAS.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS.
- I. J. Majoros, B. Keszler, S. Woehler, T. Bull and J. R. Baker, Macromolecules, 2003, 36, 5526–5529 CrossRef CAS.
- R. B. Kolhatkar, K. M. Kitchens, P. W. Swaan and H. Ghandehari, Bioconjugate Chem., 2007, 18, 2054–2060 CrossRef CAS.
- M. Labieniec and C. Watala, Cent. Eur. J. Biol., 2009, 4, 434–451 Search PubMed.
- L. Albertazzi, B. Storti, L. Marchetti and F. Beltram, J. Am. Chem. Soc., 2010, 132, 18158–18167 CrossRef CAS.
- I. J. Majoros, C. R. Williams, A. Becker and J. R. Baker, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2009, 1, 502–510 Search PubMed.
- S. K. Choi, T. Thomas, M. H. Li, A. Kotlyar, A. Desai and J. R. Baker, Chem. Commun., 2010, 46, 2632–2634 RSC.
- M. C. Y. Chang, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2004, 126, 15392–15393 CrossRef CAS.
- E. W. Miller, A. E. Albers, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16652–16659 CrossRef CAS.
- A. E. Albers, V. S. Okreglak and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9640–9641 CrossRef CAS.
- A. E. Albers, B. C. Dickinson, E. W. Miller and C. J. Chang, Bioorg. Med. Chem. Lett., 2008, 18, 5948–5950 CrossRef CAS.
- E. W. Miller, O. Tulyanthan, E. Y. Isacoff and C. J. Chang, Nat. Chem. Biol., 2007, 3, 263–267 CrossRef CAS.
- B. C. Dickinson and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 11561–11562 CrossRef CAS.
- D. Srikun, E. W. Miller, D. W. Domaille and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 4596–4597 CrossRef CAS.
- J. E. Whitaker, R. P. Haugland and F. G. Prendergast, Anal. Biochem., 1991, 194, 330–344 CrossRef CAS.
- R. Sjoback, J. Nygren and M. Kubista, Spectrochim. Acta, Part A, 1995, 51, L7–L21 CrossRef.
- Y. L. Wang and M. B. Goren, J. Cell Biol., 1987, 104, 1749–1754 CrossRef CAS.
- M. Desjardins, Trends Cell Biol., 1995, 5, 183–186 CrossRef CAS.
- O. V. Vieira, R. J. Botelho and S. Grinstein, Biochem. J., 2002, 366, 689–704 CAS.
- A. W. Segal and A. Abo, Trends Biochem. Sci., 1993, 18, 43–47 CrossRef CAS.
- B. K. Rada, M. Geiszt, K. Kaldi, C. Timar and E. Ligeti, Blood, 2004, 104, 2947–2953 CrossRef CAS.
- N. Demaurex and G. L. Petheo, Philos. Trans. R. Soc. London, Ser. B, 2005, 360, 2315–2325 CrossRef CAS.
- P. Cech and R. I. Lehrer, Blood, 1984, 63, 88–95 CAS.
- D. Morgan, M. Capasso, B. Musset, V. V. Cherny, E. Rios, M. J. S. Dyer and T. E. DeCoursey, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18022–18027 CrossRef CAS.
- A. Jankowski, C. C. Scott and S. Grinstein, J. Biol. Chem., 2001, 277, 6059–6066.
- A. van der Goes, W. Boorsma, K. Hoekstra, L. Montagne, C. J. A. de Groot and C. D. Dijkstra, J. Neuroimmunol., 2005, 161, 12–20 CrossRef CAS.
- D. Sanmun, E. Witasp, S. Jitkaew, Y. Y. Tyurina, V. E. Kagan, A. Ahlin, J. Palmblad and B. Fadeel, Am. J. Physiol.: Cell Physiol., 2009, 297, C621–C631 CrossRef CAS.
- L. Stephens, C. Ellson and P. Hawkins, Curr. Opin. Cell Biol., 2002, 14, 203–213 CrossRef CAS.
- D. Cox, J. S. Berg, M. Cammer, J. O. Chinegwundoh, B. M. Dale, R. E. Cheney and S. Greenberg, Nat. Cell Biol., 2002, 4, 469–477 CAS.
- J. A. Deane and D. A. Fruman, Annu. Rev. Immunol., 2004, 22, 563–598 CrossRef CAS.
- S. R. Lee, K. S. Yang, J. Kwon, C. Lee, W. Jeong and S. G. Rhee, J. Biol. Chem., 2002, 277, 20336–20342 CrossRef CAS.
- J. Kwon, S. R. Lee, K. S. Yang, Y. Ahn, Y. J. Kim, E. R. Stadtman and S. G. Rhee, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16419–16424 CrossRef CAS.
- M. Huss and H. Wieczorek, J. Exp. Biol., 2009, 212, 341–346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00064k |
|
| This journal is © The Royal Society of Chemistry 2011 |