Engineering DNA aptamers for novel analytical and biomedical applications
Mingxu
You
a,
Yan
Chen
ab,
Lu
Peng
a,
Da
Han
a,
Bincheng
Yin
ac,
Bangce
Ye
*c and
Weihong
Tan
*ab
aCenter for Research at the Bio/Nano Interface, Department of Chemistry and Physiology and Shands Cancer Center, University of Florida, Gainesville, FL 32611-7200, USA. E-mail: tan@chem.ufl.edu
bState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Biology, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China
cLab of Biosystems and Microanalysis, State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Meilong Rd. 130, Shanghai, 200237, P. R. China. E-mail: bcye@ecust.edu.cn
First published on 17th February 2011
Abstract
As an alternative to antibodies, aptamers have shown promising applications in diagnostics and therapeutics. However, different from antibodies, the chemical nature of nucleic acids allows easy synthesis and modification of aptamers. As a result, there are various feasible ways to engineer aptamers with extended bioavailability (e.g., stability and binding affinity in complex environments), regulating ability, and multi-functional properties. In this review, recent advances in rational design and novel functionalization of aptamers, especially DNA aptamers, is described. The broad spectrum of ways for aptamer engineering and applications is paving the way for the future evolution of bioanalytical and biomedical developments.
Introduction
In the early 1990s, the products of systematic selection of nucleic acids that bind tightly to organic dyes and proteins were reported by three separate groups.1–3 The new antibody-like compounds, called aptamers, are single-stranded DNAs or RNAs that fold into unique three-dimensional binding pockets and clefts for molecular recognition. Aptamers are generated by a process known as Systematic Evolution of Ligands by Exponential Enrichment (SELEX) from a library pool of up to 1016DNA or RNA sequences. This large pool of nucleic acid molecules ensures high diversity in forming different three-dimensional structures to fit into the binding pockets of a wide variety of targets, from small molecules to proteins and whole cells.4–6 In 2004, research on aptamers gained further momentum by the discovery of riboswitches,7,8 which are comprised of a natural aptamer and an expression platform as regulatory elements for gene expression. At this time, aptamers have been applied to fulfil diverse functions, including sensing, purification, diagnostics and therapeutics.9–14 However, compared to antibodies, which are produced by living systems, laboratory-made aptamer molecules are still in their early stage of development. Several concerns need to be addressed before the aptamer-based technology is widely utilized: for example, biostability, availability of versatile aptamer choices for different targets, cost, and easy-to-manage platforms. This review describes the methodologies and recent developments in engineering aptamer molecules, as well as smart modification of these versatile molecules for advanced biomedical and bioanalytical functions.Current molecular engineering of DNA aptamers has focused on several aspects: improving aptamers' bioavailability through chemical modifications; engineering regulated aptamers by controlling their recognition and inhibition properties; and exploiting versatile functions.
(1) The bioavailability of aptamers can be improved through chemical modification to increase their in vivo stability, cellular uptake efficiency and target binding affinity. Aptamers find great potential in the applications of diagnostics and therapeutics. However, some critical problems hinder aptamer use for in vivo clinical applications: the natural nucleotides in aptamers are susceptible to nuclease-mediated degradation, and the negatively charged backbones result in inefficient cellular internalization. To overcome these inherent stability and internalization issues is an emerging goal for aptamer technology. As described further below, aptamers are commonly available with chemical modifications using non-natural nucleotides,15macromolecules16 or delivery vehicles17 to enhance their bioavailability for in vivo applications.
(2) A second area of interest is the design of regulable aptamers by exogenously controlling the recognition and inhibition properties to extend the applications of these molecules. The intercalation of chemical or physical triggers has assisted chemists and biologists to fine-tune aptamer functions or to regain control over molecules even in complex environments, such as the human body. The possibility of installing switches into aptamer molecules for better controlling functions is very favorable, given the ease of nucleic acid (especially DNA) synthesis and versatile modification methodologies. So far, nucleic acid aptamer probes have been modified with small competing molecules,18 complementary oligonucleotides19 and physical triggers (such as chromophores20) to achieve refined and extended functions.
(3) Aptamers are also being engineered to perform diverse functions, opening the way for design of advanced biosensors and clinical systems. In addition to their function as target recognition agents, more and more studies have demonstrated the therapeutic and allosteric effects of aptamers. The rational engineering of nucleic acid aptamers with multiple functions is interesting and meaningful, considering the efficient conversion of recognition events into physically detectable signals or therapeutic outputs (e.g., targeted drug delivery). From another point of view, if functional molecules are assembled using nucleic acids only, instead of combining different biological agents (such as proteins) or conjugations, it will be reasonable to expect a cost-efficient, simplified synthesis/modification process, and a homogenous, easily-manipulated and stable system. Advanced diagnostic and therapeutic applications will be accessible, thereby exploring the full range of potentials of aptamers to meet various requirements of biological systems.
Engineering DNA aptamers with enhanced bioavailability
Aptamers can be easily synthesized through solid-phase-based chemical synthesis or enzymatic procedures. To avoid problems with nuclease-mediated degradation and to improve bioavailability, numerous chemically modified non-natural nucleotides have been designed and incorporated into aptamers. These modified nucleobases or internucleotide linkages are used to enhance nuclease-resistance and binding affinity, and may even be able to generate aptamer structures for substrates not recognized by wild-type DNAs or RNAs.15,35In general, DNA aptamers are more stable towards nuclease digestion than RNA aptamers, since nature has introduced into RNA a 2′-hydroxyl group for chemical lability. Following nature's lead, the most useful modifications for aptamers have so far involved substitution at the 2′-ribose position by amino,21,22fluoro19,23 and O-methyl24,25pyrimidines or purines. It is especially noteworthy that 2′-fluoro pyrimidine and 2′-O-methyl purine modifications have paved the way for developing the first FDA approved aptamer-based drug, pegaptanib (Macugen),26,27 a vascular endothelial growth factor (VEGF)-specific aptamer drug for treating age-related macular degeneration (AMD). The Tan group and others are interested in generating more-stable aptamers by chemical modifications, especially using locked nucleic acids (LNAs), which contain a methylene bridge connecting the 2′-oxygen of ribose with the 4′-carbon.28,29LNA have advantages over natural nucleic acids (DNA) for diagnostic and therapeutic applications, as a result of the high nuclease resistance and target binding affinity afforded by the methylene bridge.30 Compared with other non-standard nucleotides, LNA's behave like DNA, with extremely high binding affinity to complementary DNA or RNA oligonucleotides, and can be used in assays requiring high specificity and reproducibility, such as the polymerase chain reaction (PCR). Recent studies by the Wengel group have demonstrated the accessibility of LNA for PCR amplification and in vitrotranscription,31,32 thereby laying the foundation for future aptamer SELEX using LNAs as building blocks. Performing SELEX on a library of modified oligonucleotides is preferred to direct chemical modification of existing aptamers, which bears the risk of weakening contact efficiency and target specificity.
The C5 position of pyrimidine derivatives is another promising modification site for DNA-based aptamers, due to the undisturbed helical structures with the acceptance by polymerase for efficient SELEX.33,34 The Sawai group reported a selection method for an aptamer that enantioselectively binds (R)-thalidomide based on DNA modified with a C5 cationic functional group.35 The stability against nucleases and the binding affinity to thalidomide were noticeably improved after modification. Nucleotide modification can also be made at the 4′-oxygen site of the ribose ring and at phosphate internucleotide linkages, and a summary of these modifications is presented in Fig. 1.
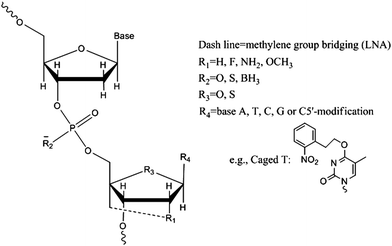 | ||
| Fig. 1 Commonly-used nucleotide modifications in aptamer structures. | ||
Other than nucleotide substitutions, the bioavailability of DNA aptamers can be improved by conjugation with high molecular-weight polymers (e.g., polyethylene glycol36) or cholesterols.37Aptamer conjugations in the 30–50 kDa size range meet the cutoff for renal glomerulus and significantly reduce renal elimination rates.36 As a result, an aptamer-polymer/cholesterol complex increased the aptamer's circulation half-life inside biological systems, thereby decreasing dosing densities and enhancing effectiveness towards vascular targets. Through rational understanding and choice of modification strategies based on specific biological requirements, the examples mentioned above demonstrate that enhanced bio-stability and diversity can be introduced into DNA aptamers to further increase their utility.
Besides nucleotide or polymer modification, multivalent aptamer binding is another way to engineer aptamers with enhanced binding affinities and target regulation activities, especially when dealing with complex biological systems. Through a cooperative interaction, often called synergism, the combined effects of aptamers acting together can surpass the sum of the effects of the individual aptamers.38–41 One example is the cooperative effect of two thrombin aptamers, involving a low binding affinity but functional 15-base aptamer (15Apt) and a high binding affinity but non-functional 27-base aptamer (27Apt)39(Fig. 2a). After the discovery of 15Apt in 1992,42 the anti-thrombin DNA aptamer was among the first therapeutic aptamers tested in animals.43,44 Together with ATP, thrombin is now the most widely employed proof-of-concept target molecule for various diagnostic and therapeutic designs.45 It has been proven that only 15Apt, which binds to exosite 1 of thrombin, can prevent coagulation by occupying the fibrinogen-binding site. However, the functional 15Apt has a much lower binding affinity (Kd≈450 nM) than the non-functional 27Apt (Kd≈0.7 nM). Therefore, by molecular assembly of 15Apt and 27Apt, the Tan group showed that the binding strength improved 62-fold, with 16.6-fold better anticoagulation efficiency.39 The high performance of the cooperative 15Apt and 27Apt aptamers provides a potent anti-thrombin agent for various diseases. The synergistic effect was further investigated using this 15Apt/27Apt molecular assembly as a model system. Using fluorescence resonance energy transfer (FRET) to signal the kinetics of thrombin-aptamer interactions, the relative koff value of bivalent 15Apt was observed to be 52 times smaller than that of monovalent 15Apt, while the kon values were similar. Thus, in this case the multivalent effect is believed to mainly involve koff, the dissociation rate constant.
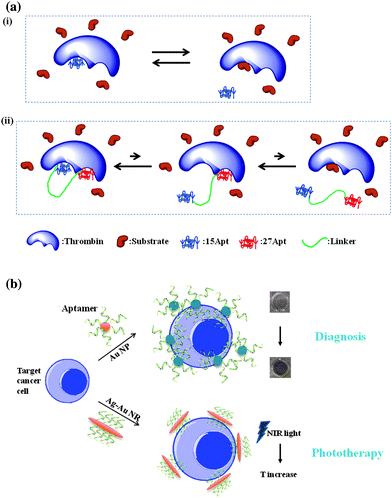 | ||
| Fig. 2 (a) Schematic of cooperative effect from (ii) bivalent thrombin aptamers. After linking to 27Apt, 15 Apt can rapidly diffuse back to the binding site after dissociation. As a result, the reaction equilibrium shifted to the left, compared with (i) monovalent aptamer. (b) Aptamer-nanomaterial assembly for diagnostic and therapeutic applications. | ||
Instead of direct linkage of aptamers, multiple aptamers can also be conjugated onto the surface of nanomaterials. Given their high surface area-to-volume ratios and variable sizes, nanomaterials are favored for multivalent binding of DNA aptamers to enhance the recognition signal and binding affinity.46 For example, an aptamer-gold nanoparticle assembly has been developed into a colorimetric assay for cancer cells.47 Given the relatively large size of a cancer cell, multiple aptamers conjugated on the 50nm-diameter gold nanoparticles could bind with the same cell simultaneously. The cooperative binding of these oligonucleotides enhanced the aptamer-cell affinity, and, at the same time, facilitated the assembly of gold nanoparticles, allowing overlap of their surface plasmon bands for sensitive detection of as few as 1000 cancer cells by the naked eye (Fig. 2b).
The therapeutic effect of aptamers can also been improved by multivalent conjugation. Using Au–Ag nanorods as a platform48 up to 80 fluorophore-labeled aptamers could be attached to one 12nm × 56nm nanorod surface, resulting in a 300-fold enhanced fluorescence signal and 26-fold higher binding affinity. The advanced recognition ability of these aptamer-nanorod conjugates was further demonstrated in targeted photothermal therapy. The aptamer-modified Au–Ag nanorods could efficiently absorb near-infrared (NIR) light and relax this energy into heat for specific cancer cell destruction.49Aptamer-based specific targeting has led to a phototherapy method with low side effects and high cancer therapy efficiency. After NIR light irradiation, only the aptamer-bound cancer cells were killed, leaving the control cell lines intact. These examples demonstrate the outstanding properties of aptamer-nanomaterial conjugates. Through multivalent capability, the integration of aptamers and nanomaterials can potentially address the recognition issue of some low affinity targets or binding sites. At the same time, the incompletely understood qualities of nano-sized materials have been reported to help avoid nuclease digestion of aptamers50 and to enhance passage of aptamers through the cell membranes.51 Linking of aptamer technology and nanotechnology is expected to enhance the practical accessibility of aptamers for diagnostic and therapeutic applications. The future is bright for engineering efforts in this direction.
Engineering DNA aptamers with regulatory switches
The ability to regulate the function of tools is an important criterion in the adaptability of a technology. Through engineering molecules responsive to physical stimuli (e.g., light, temperature, mechanical stress) or chemical stimuli, exogenous control over the bio-related therapeutic and diagnostic function of aptamers can be achieved, especially in a complicated biological system. This section will focus on regulatory switches based on complementary oligonucleotides, small molecules and light absorption.The unique regulatory agents usually employed in nucleic acid-based therapies are the complementary sequences, which can be used as either antidotes19 or agonists.52 Since the Sullenger group used the complementary oligonucleotide to an RNA aptamer that antagonized the blood clotting activity of coagulation factor IXa,19 such straightforward design of nucleic acid drug-antidote pairs has contributed greatly to aptamer-based therapeutic studies. Many DNA-based aptamer-antidote pairs have also been reported, with the thrombin aptamer serving as the most popular proof-of-concept model. For example, Simmel and colleagues have designed an aptamer-antidote based DNA machine to reversibly bind and release thrombin protein.53Via rational variation of the length of DNA hybridization, different competitive relationships between a DNA duplex and a DNA-protein complex could be fine-tuned (Fig. 3a). The highly specific DNA hybridization and biological nature of nucleic acids have rendered complementary oligonucleotide-based antidotes beneficial in preventing unnecessary adverse effects, which are commonly observed with other kinds of anticoagulants.54,55 Some small molecule-based chemical triggers can also regulate DNA aptamer functions. For example, quadruplex structure-binding cationic porphyrin TMPyP418 and hemin56 molecules have been demonstrated to manipulate the functions of the anti-thrombin aptamer, which contains a G-quadruplex structure. These novel small molecule antidotes have been proven capable of reversing the anticoagulant effect. Also, compared with cDNA based antidotes, a small molecule is more capable of transmembrane delivery. In the same regard, screening of small molecule drugs is an important area, sometimes using protein-bound aptamers as “parents”.57,58
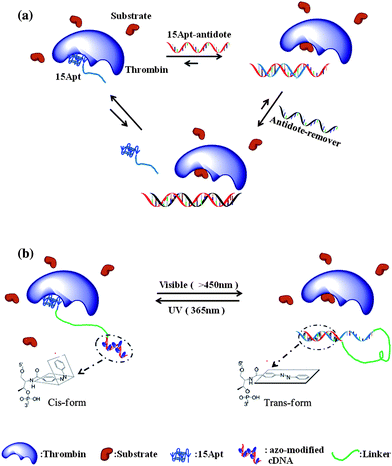 | ||
| Fig. 3 (a) Antidote function of complementary DNA could be used to fine-tune the therapeutic effects of aptamers. (b) Schematic of the photoregulatory inhibitor (PCI) design. Treating with visible light, the self-hybridization with the regulatory domain activates the thrombin function; under UV light, 15Apt becomes free to bind to the target, inducing low enzyme activity. | ||
Besides the regulatory effects of different concentrations of molecular stimuli, DNA aptamers responsive to external physical triggers comprise another promising direction for engineering. For example, functionalization of aptamers with photoresponsive groups can provide exact temporal and spatial control over the aptamer's function. Compared with a chemically triggered response, photoregulation enables remote control of timing, location and intensity of the light “dosage,” and minimizes the adverse effects from the addition of other chemical components.59 The Mayer group has realized this idea by developing “caged” anti-thrombin aptamers.60–62 These aptamers hold a photolabile protective group on the 5′-carbon of the nucleobases to temporally block the active conformation (Fig. 1). The rational design of positions for introducing photolabile groups is critical in these studies, and this information was provided by the crystal structure of thrombin-15Apt conjugation. This study is also a good example of the design of aptamer biological functions through an understanding of the chemical fundamentals. Opening of the “cage” group by 366 nm light enables the exogenous regulation of the blood-clotting cascade, either via an activation mode or a deactivation mode. The novel multifunctional anti-thrombin aptamers described above combine the highly specific recognition and anticoagulation properties with light-mediated effective and rapid regulation. The engineered spatio-temporal control is of potential benefit for remote regulation of aptamers inside cellular environments. Recently, the Mayer group further demonstrated that the caged bivalent thrombin aptamers enable the use of light to modulate individual domain activity in thrombin, achieving a more precise control over biomolecular functions.63
The Tan group further developed DNA engineering strategies to manipulate aptamer-target interactions using intercalating azobenzene moieties in DNA sequences. The azobenzene moiety is one of the most popular photoisomerization molecules.64 In contrast with single-use photolabile groups,65azobenzene-modified aptamers enable several rounds of reversible regulation using different wavelengths of light. A photocontrollable inhibitor (PCI) has been designed based on an azobenzene-modified aptamer for manipulating the thrombin inhibitory effect.66 The PCI design (Fig. 3b) involves the 15Apt anti-thrombin sequence (inhibitory domain) linked by polyethylene glycol (linking domain) to an azobenzene-modified DNA complementary to 15Apt (regulatory domain). Under visible light (longer than 450 nm), the hybridization of 15Apt with the regulatory domain disabled the thrombin inhibition effect; however, the azobenzenetrans-to-cis conversion after UV (365 nm) irradiation induced dehybridization of the DNA duplex, freeing 15Apt to bind and inhibit thrombin mediated coagulation. The temporal and spatial regulation of the clotting reactions by this photo-sensitive aptamer–antidote pair is expected to be useful for tissue-specific clotting and selective blocking of the blood vessels. The power of light regulation may also be extended to targeted, controllable drug delivery. The potential applications in this area have been demonstrated by recent studies in the Tan group of an azobenzene-based photo-responsive DNA hydrogel67 and a novel pyrene-disulfide assembly mediated photo-dissociable DNA micelle68 system. Besides photoinduced regulation, the design of other external triggers, even aptamers responsive to multiple triggers, are promising directions for future aptamer engineering.
DNA aptamers: Outstanding multifunctional agents
Although aptamers can be easily classified as recognition molecules, many studies are now demonstrating their utilization as multifunctional agents, involving therapeutics, delivery, signalling and assembly. One example is the direct therapeutic effect of aptamer molecules. Through the inhibition of protein functions, aptamers can be developed into drugs, such as the above-mentioned “Macugen” for age-related macular degeneration therapy. Aptamer-based drug screening in still in its infancy, but it has already demonstrated some promising applications.The therapeutic effect of aptamers can also be used for targeted intracellular delivery. An innovative application involves the use of aptamers for delivering other therapeutic genes (e.g., siRNAs, shRNAs or miRNAs). For example, one critical difficulty for RNA interference-based therapy is the in vivo delivery of siRNA. The Giangrande group has reported a novel aptamer–siRNA chimera to handle this problem (Fig. 4a).69,70 Prostate-specific membrane antigen (PSMA) is expressed on the surface of prostate cancer cells, and PSMA-specific aptamers can undergo cell internalization and mediate delivery viaendocytosis. By the hybridization of an elongated PSMA-specific RNA aptamer and siRNA, Giangrande et al. demonstrated the specific targeting of PSMA-expressing cancer cells and the silencing of two tumor overexpressed genes, polo-like kinase 1 (PLK1) and BCL-2. The conjugation of aptamers and other oligonucleotides has facilitated cell type-specific gene therapy. Compared with other gene delivery methods (e.g., proteins or cationic lipids), conjugation solely with nucleic acids simplified the synthesis procedure, decreased the immunogenicity, and allowed for site-specific linkage.
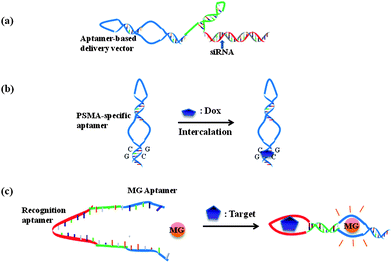 | ||
| Fig. 4 Multifunctional aptamer systems. Aptamers can be used as vectors for (a) siRNA and (b) Doxorubicin targeted delivery. Aptamers serve as both recognition agents and signaling agents in (c) allosteric aptamer sensors. | ||
The intracellular delivery function of aptamers has also been used for the delivery of other drugs, typically by covalent71 or noncovalent (e.g., biotin-steptaidin72) conjugation. One novel and unique type of aptamer-based drug delivery is by means of physical intercalation of some drugs within the DNA strands, such as the well-known anticancer drug Doxorubicin (Dox) (Fig. 4b). PSMA aptamer A10 has been used to deliver Dox specifically into PSMA-expressing LNCaP cells.73DNA aptamers can also be used in this manner. The Tan group has demonstrated one particular cancer cell-bound DNA aptamer, sgc8,74 which can be internalized by target cells. It is very likely that sgc8 will extend the toolbox of aptamers for intracellular gene or drug delivery, and recent studies have demonstrated that PTK7-specific sgc8c can conjugate doxorubicin for targeted drug delivery.75
The multifunctional ability of aptamer molecules has been most widely used in the design of sensing systems.76 A sensor system contains, at minimum, a target recognition element and a signal transduction element. Aptamers have found priority as target recognition components, given their properties of high specificity, target affinity and easy synthesis and modification. Following the binding events, aptamer-based sensors usually rely on conformational changes that alter the local environment of conjugated fluorophores (fluorescent sensors77), nanomaterials (e.g., Au nanoparticles for colorimetric sensors 47) or electrodes (electrochemical sensors78), as well as binding events resulting in detectable mass changes (e.g., quartz crystal microbalance (QCM) based assays79), competition effects (complementary DNA displacement-based method80) and direct binding strength difference (liquid chromatography,81capillary electrophoresis82 or mass spectrometry83-based separation and detection assay). Interested readers may consult the review of Liu et al. for more detailed information about these methods.76
An innovative sensor-engineering methodology will be the direct use of aptamers for both recognition and signal transduction, with the advantages of lower cost, simplified modification and increased response rate. For instance, the biological nature of aptamers has triggered the design of aptamer templated rolling circle amplification (RCA)84 or PCR.85 In these studies, the DNA aptamers serve first for target binding, then as the template for polymerase amplification, which can convert trace levels of target protein recognition events into detectable fluorescent signals. The potent SELEX procedures have also discovered some aptamers that directly influence the target signaling, leading to the design of allosteric aptamer sensors. Under some conditions, target binding by one aptamer can weaken or enhance the binding of a second aptamer. Such cooperative aptamer assemblies, called allosteric aptamers, have also been developed into sensors.86 Stojanovic et al. have designed a modular aptamer sensor, in which a recognition aptamer element was fused with a reporting aptamer element by a communication module.87 Target (ATP or FMN) binding by the DNA aptamer enhanced the second binding of the reporting aptamer with malachite green (MG) dye (Fig. 4c), resulting in an increase in the fluorescence quantum yield of MG for signal production. The rational “communication” design has rendered allosteric aptamer sensors with the multiple functions of recognition, signal transduction and detection.
From another viewpoint, the oligonucleotide nature of aptamers simplifies chemical modification processes by using routine solid-phase synthesis. Aptamers can be easily introduced as building blocks into different platforms. For example, hydrogels, which are 3-D networks of polymer chains, are promising for drug delivery and tissue engineering. A modified environmentally sensitive hydrogel is able to release its load when the changes are sensed. The Tan group has engineered a specific target-responsive hydrogel by making use of DNA aptamers for cross-linking linear polyacrylamide chains (Fig. 5b).88,89 The DNA aptamers served as both target recognition agents and building blocks to meet the requirements of hydrogel cross-linking densities. The dissolution of the hydrogel by competitive aptamer binding of a target (adenosine or thrombin) facilitated rapid release of the model drug (13 nm gold nanoparticles). It is expected that this system can be used as a targeted drug delivery platform. A subsequent study demonstrated that, by incorporating an amylose-I2-amylase system, the aptamer based hydrogel could be used for sensitive visual detection of targets.90
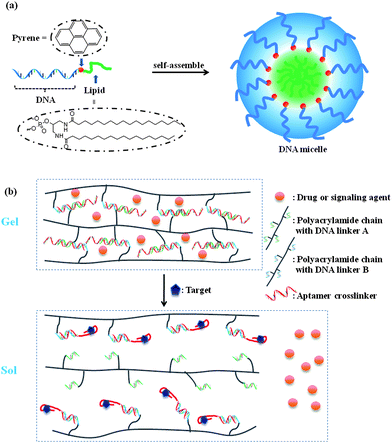 | ||
| Fig. 5 Aptamers as building blocks to generate (a) DNA micelle and (b) hydrogel structures for drug delivery and sensing. | ||
Based on the same thinking, the hydrophilicity of nucleic acids can also be employed to utilize aptamers for construction of nano-platforms. Micelle or liposome structures are good candidates for these nanostructures. Recently, functional DNA micelles composed of a hydrophilic single-stranded DNA shell and a hydrophobic polymer core have attracted attention for the delivery of antisense DNA and for cancer nanotechnology.91 The Tan group has shown that the aptamer-based micelle structure could bind specifically with the target cancer cell and deliver drugs (Fig. 5a).92 Interestingly, the multivalent effect described above also exists within aptamer-micelle structures. At the physiological temperature, normally unbound free aptamers can recognize and bind to target cancer cells. Moreover, using a flow channel system to mimic drug delivery in the human blood system, the dynamic specificity of an aptamer-micelle assembly was shown to be an effective detection/delivery vehicle. As these examples suggest, engineering DNA aptamers as building blocks for nanostructures can still preserve the binding affinity of aptamers, while the target recognition process can trigger larger scaffold structure alterations to realize novel functions, such as drug release and molecular detection.
Conclusion and future perspectives
The past 20 years have witnessed the rapid development of a maturing aptamer technology. The high stability, accessible synthesis and versatile modifications have rendered aptamers — aptly called “chemical antibodies,” but with more extensive functions than antibodies — as specific targeting components in diagnostic or therapeutic toolkits. This review has centered on the engineering features currently used for devising aptamers with novel and advanced functions to overcome theoretical and practical handicaps that inhibit real applications. So far, the number of commercially available aptamer-based sensors or drugs is still quite limited due to several issues.First is the limited number of aptamers which have been isolated for targets of interest; even though the extremely diverse libraries of nucleic acids could probably provide a molecule to bind almost any target.2 One hurdle may be the SELEX procedure, which is tedious and inefficient. The search for simplified, ready-to-use SELEX protocols is still underway. Engineering of chemically modified nucleic acids, beyond the four existing natural DNA bases, is a promising means for designing a modified SELEX procedure. Given rationally planned binding properties and extended library sizes, such predesigned SELEX may be useful for efficient aptamer isolation, sometimes even to targets that natural nucleic acids do not bind.
Second, most aptamers have been selected and studied in buffer systems, leading to short comings in terms of biostability and cell internalization efficiency. The performance of an aptamer in a complex environment, such as serum and plasma, decreases significantly compared to the behavior in aqueous buffer. As a result, various modifications aim to improve the bioavailability of aptamers. The Tan group has been performing cell-based selection of aptamers, replacing normal protein-based SELEX. The direct binding of targets in their bio-native conformations can greatly improve the biological functions of the isolated aptamers. Furthermore, implementation of fluorescence-activated cell sorting (FACS) technology93–95 into the selection procedure has facilitated cell-based SELEX in clinical applications. In addition, the recent advancement in bridging aptamer technology and nanotechnology shows great potential for improving both the recognition properties of aptamers and the power of nano-systems.
Third, even after the optimization of aptamers, mature technology for commercial use will require low cost, adaptability to diverse conditions, and the availability of easy-to-manage systems. Engineering regulable or multifunctional aptamer machines may be one way to achieve these goals.
In future years, the possibility of clinically using aptamers as drugs and diagnostic tools will be tested. Rational engineering pathways to boost the performance of aptamers, equip them with adjunctive functions, and reduce the cost of large-scale aptamer modification could assist aptamer-based systems to become a major technology with significant commercial potential in forefront research areas, such as drug screening, disease diagnosis and personalized medicine.
Acknowledgements
The authors would like to thank Dr Kathryn R. Williams for manuscript review. We acknowledge NIH and NSF for the funding this project. This work was supported by the National Key Scientific Program of China (2011CB911000) and China National Grand Program on Key Infectious Disease (2009ZX10004-312). This work was also supported by Grant 20627005 from the National Natural Science Foundation, the National Special Fund for SKLBE (2060204).References
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344, 467–468 CrossRef CAS.
- D. A. Daniels, H. Chen, B. J. Hicke, K. M. Swiderek and L. A. Gold, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15416–15421 CrossRef CAS.
- D. Shangguan, Y. Li, Z. Tang, Z. Cao, H. Chen, P. Mallikaratchy, K. Sefah, C. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS.
- M. Blank, T. Weinschenk, M. Priemer and H. Schluesener, J. Biol. Chem., 2001, 276, 16464–16468 CrossRef CAS.
- M. Mandal and R. R. Breaker, Nat. Rev. Mol. Cell Biol., 2004, 5, 451–463 CrossRef CAS.
- B. J. Tucker and R. R. Breaker, Curr. Opin. Struct. Biol., 2005, 15, 342–348 CrossRef CAS.
- D. H. Bunka and P. G. Stockey, Nat. Rev. Microbiol., 2006, 4, 588–596 Search PubMed.
- X. Fang and W. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS.
- M. Famulok, J. S. Hartig and G. Mayer, Chem. Rev., 2007, 107, 3715–3743 CrossRef CAS.
- G. Mayer, Angew. Chem., Int. Ed., 2009, 48, 2672–2689 CrossRef CAS.
- N. K. Navani and Y. Li, Curr. Opin. Chem. Biol., 2006, 10, 272–281 CrossRef CAS.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS.
- A. D. Keefe and S. T. Cload, Curr. Opin. Chem. Biol., 2008, 12, 448–456 CrossRef CAS.
- R. M. Boomer, S. D. Lewis, J. M. Healy, M. Kurz, C. Wilson and T. G. McCauley, Oligonucleotides, 2005, 15, 183–195 CrossRef CAS.
- Z. Cao, R. Tong, A. Mishra, W. Xu, G. C. Wong, J. Cheng and Y. Lu, Angew. Chem., Int. Ed., 2009, 48, 6494–6498 CrossRef CAS.
- A. Joachimi, G. Mayer and J. S. Hartig, J. Am. Chem. Soc., 2007, 129, 3036–3037 CrossRef CAS.
- C. P. Rusconi, E. Scardino, J. Layzer, G. A. Pitoc, T. L. Ortel, D. Monroe and B. A. Sullenger, Nature, 2002, 419, 90–94 CrossRef CAS.
- A. Heckel and G. Mayer, J. Am. Chem. Soc., 2005, 127, 822–823 CrossRef CAS.
- Y. Lin, Q. Qiu, S. C. Gill and S. D. Jayasena, Nucleic Acids Res., 1994, 22, 5229–5234.
- Y. Lin, D. Nieuwlandt, A. Magallanez, B. Feistner and S. D. Jayasena, Nucleic Acids Res., 1996, 24, 3407–3414 CrossRef CAS.
- U. Chakravarthy, A. P. Adamis, E. T. Jr Cunningham, M. Goldbaum, D. R. Guyer, B. Katz and M. Patel, Ophthalmology, 2006, 113, e1501–e1525.
- P. E. Burmeister, S. D. Lewis, R. F. Silva, J. R. Preiss, L. R. Horwitz, P. S. Pendergrast, T. G. McCauley, J. C. Kurz, D. M. Epstein and C. Wilson, Chem. Biol., 2005, 12, 25–33 CrossRef CAS.
- P. E. Burmeister, C. Wang, J. R. Killough, S. D. Lewis, L. R. Horwitz, A. Ferguson, K. M. Thompson, P. S. Pendergrast, T. G. McCauley and M. Kurz, Oligonucleotides, 2006, 16, 337–351 CrossRef CAS.
- E. S. Gragoudas, A. P. Adamis, E. T. Cunningham, M. Feinsod and D. R. Guyer, N. Engl. J. Med., 2004, 351, 2805–2816 CrossRef CAS.
- F. W. Fraunfelder, Drugs Today, 2005, 41, 703–709 CrossRef CAS.
- L. Wang, C. J. Yang, C. D. Medley, S. A. Benner and W. Tan, J. Am. Chem. Soc., 2005, 127, 15664–15665 CrossRef CAS.
- C. J. Yang, L. Wang, Y. Wu, Y. Kim, C. D. Medley, H. Lin and W. Tan, Nucleic Acids Res., 2007, 35, 4030–4041 CrossRef CAS.
- H. Kaur, B. R. Babu and S. Maiti, Chem. Rev., 2007, 107, 4672–4697 CrossRef.
- R. N. Veedu, B. Vester and J. Wengel, J. Am. Chem. Soc., 2008, 130, 8124–8125 CrossRef CAS.
- R. N. Veedu, B. Vester and J. Wengel, ChemBioChem, 2007, 8, 490–492 CrossRef CAS.
- S. Jager and M. Famulok, Angew. Chem., Int. Ed., 2004, 43, 3337–3340 CrossRef CAS.
- S. Jager, G. Rasched, H. Kornreich-Leshem, M. Engeser, O. Thum and M. A. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef.
- A. Shoji, M. Kuwahara, H. Ozaki and H. Sawai, J. Am. Chem. Soc., 2007, 129, 1456–1464 CrossRef CAS.
- J. M. Healy, S. D. Lewis, M. Kurz, R. M. Boomer, K. M. Thompson, C. Wilson and T. G. McCauley, Pharm. Res., 2004, 21, 2234–2246 CAS.
- C. P. Rusconi, J. D. Roberts, G. A. Pitoc, S. M. Nimjee, R. R. White, G. Q. Jr, E. Scardino, W. P. Fay and B. A. Sullenger, Nat. Biotechnol., 2004, 22, 1423–1428 CrossRef CAS.
- L. L. Kiessling, J. E. Getwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS.
- Y. Kim, Z. Cao and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5664–5669 CrossRef CAS.
- J. Mller, B. Wulffen, B. Potzsch and G. Mayer, ChemBioChem, 2007, 8, 2223–2226 CrossRef.
- Y. Kim, D. M. Dennis, T. Morey, L. Yang and W. Tan, Chem.–Asian J., 2010, 5, 56–59 CrossRef CAS.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS.
- L. C. Griffin, G. F. Tidmarsh, L. C. Bock, J. J. Toole and L. L. Leung, Blood, 1993, 81, 3271–3276 CAS.
- A. J. DeAnda, S. E. Coutre, M. R. Moon, C. M. Vial, L. C. Griffin, V. S. Law, M. Komeda, L. L. Leung and D. C. Miller, Ann. Thorac. Surg., 1994, 58, 344–350 CrossRef.
- B. Gatto, M. Palumbo and C. Sissi, Curr. Med. Chem., 2009, 16, 1248–1265 CrossRef CAS.
- H. Wang, R. Yang, L. Yang and W. Tan, ACS Nano, 2009, 3, 2451–2460 CrossRef CAS.
- C. Medley, J. Smith, Z. Tang, Y. Wu, S. Bamrungsap and W. Tan, Anal. Chem., 2008, 80, 1067–1072 CrossRef CAS.
- Y. Huang, H. Chang and W. Tan, Anal. Chem., 2008, 80, 567–572 CrossRef CAS.
- Y. Huang, K. Sefah, S. Bamrungsap, H. Chang and W. Tan, Langmuir, 2008, 24, 11860–11865 CrossRef CAS.
- Y. Wu, J. A. Phillips, H. Liu, R. Yang and W. Tan, ACS Nano, 2008, 2, 2023–2028 CrossRef CAS.
- N. W. Kam, M. O'Connell, J. A. Wisdom and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11600–11605 CrossRef CAS.
- C. M. Dollins, S. Nair, D. Boczkowski, J. Lee, J. M. Layzer, E. Gilboa and B. A. Sullenger, Chem. Biol., 2008, 15, 675–682 CrossRef CAS.
- W. U. Dittmer, A. Reuter and F. Simmel, Angew. Chem., Int. Ed., 2004, 43, 3550–3553 CrossRef CAS.
- J. Ebbesen, I. Buajordet, J. Erikssen, O. Brors, T. Hilberg, H. Svaar and L. Sandvik, Arch. Intern. Med., 2001, 161, 2317–2323 CrossRef CAS.
- M. C. Engoren, R. H. Habib, A. Zacharias, T. A. Schwann, C. J. Riordan and S. J. Durham, Ann. Thorac. Surg., 2002, 74, 1180–1186 CrossRef.
- J. Wang, Y. Cao, G. Chen and G. Li, ChemBioChem, 2009, 10, 2171–2176 CrossRef CAS.
- M. Hafner, A. Schmitz, I. Grune, S. G. Srivatsan, B. Paul, W. Kolanus, T. Quast, E. Kremmer and M. Bauer, Nature, 2006, 444, 941–944 CrossRef CAS.
- M. Hafner, E. Vianini, B. Albertoni, L. Marchetti, I. Grune, C. Gloeckner and M. Famulok, Nat. Protoc., 2008, 3, 579–587 Search PubMed.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900–4921 CrossRef CAS.
- G. Mayer, L. Krock, V. Mikat, M. Engeser and A. Heckel, ChemBioChem, 2005, 6, 1966–1970 CrossRef CAS.
- A. Heckel, M. C. Buff, M. S. Raddatz, J. Muller, B. Potzsch and G. Mayer, Angew. Chem., Int. Ed., 2006, 45, 6748–6750 CrossRef CAS.
- M. C. Buff, F. Schafer, B. Wulffen, J. Muller, B. Potzsch, A. Heckel and G. Mayer, Nucleic Acids Res., 2009, 38, 2111–2118.
- G. Mayer, J. Muller, T. Mack, D. F. Freitag, T. Hover, B. Potzsch and A. Heckel, ChemBioChem, 2009, 10, 654–657 CrossRef CAS.
- K. G. Yager and C. L. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250–261 CrossRef CAS.
- J. H. Kaplan, B. Forbush III and J. F. Hoffman, Biochemistry, 1978, 17, 1929–1935 CrossRef CAS.
- Y. Kim, J. A. Phillips, H. Liu, H. Kang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6489–6494 CrossRef CAS.
- H. Kang, H. Liu, X. Zhang, J. Yan, Z. Zhu, L. Peng, H. Yang, Y. Kim and W. Tan, Langmuir, 2011, 27, 399–408 CrossRef CAS.
- M. You, Z. Zhu, H. Liu, B. Gulbakan, D. Han, R. Wang, K. R. Williams and W. Tan, ACS Appl. Mater. Interfaces, 2010, 2, 3601–3605 CrossRef CAS.
- J. O. McNamara II, E. R. Andrechek, E. Y. Wang, K. D. Viles, R. E. Rempel, E. Gilboa, B. A. Sullenger and P. H. Giangrande, Nat. Biotechnol., 2006, 24, 1005–1015 CrossRef.
- J. D. Dassie, X. Liu, G. S. Thomas, R. M. Whitaker, K. W. Thiel, K. R. Stockdale, D. K. Meyerholz, A. P. McCaffrey, J. O. McNamara II and P. H. Giangrande, Nat. Biotechnol., 2009, 27, 839–846 CrossRef CAS.
- P. Mallikaratchy, H. Liu, Y. Huang, H. Wang, D. Lopez-Colon and W. Tan, Chem. Commun., 2009, 3056–3058 RSC.
- T. C. Chu, K. Y. Twu, A. D. Ellington and M. Levy, Nucleic Acids Res., 2006, 34, e73 CrossRef.
- V. Bagalkot, O. C. Farokhzad, R. Langer and S. Jon, Angew. Chem., Int. Ed., 2006, 45, 8149–8152 CrossRef CAS.
- Z. Xiao, D. Shangguan, Z. Cao, X. Fang and W. Tan, Chem.–Eur. J., 2008, 14, 1769–1775 CrossRef CAS.
- Y. Huang, D. Shangguan, H. Liu, J. A. Phillips, X. Zhang, Y. Chen and W. Tan, ChemBioChem, 2009, 10, 862–868 CrossRef CAS.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS.
- Z. Tang, P. Mallikaratchy, R. Yang, Y. Kim, Z. Zhu, H. Wang and W. Tan, J. Am. Chem. Soc., 2008, 130, 11268–11269 CrossRef CAS.
- I. Willner and M. Zayats, Aptamers in Bioanalysis, 2009, 63–86 Search PubMed.
- M. Liss, B. Petersen, H. Wolf and E. Prohaska, Anal. Chem., 2002, 74, 4488–4495 CrossRef CAS.
- J. Huang, Z. Zhu, S. Bamrungsap, G. Zhu, M. You, X. He, K. Wang and W. Tan, Anal. Chem., 2010, 82, 10158–10163 CrossRef CAS.
- M. Michaud, E. Jourdan, A. Villet, A. Ravel, C. Grosset and E. Peyrin, J. Am. Chem. Soc., 2003, 125, 8672–8679 CrossRef CAS.
- C. Huang, Z. Cao, H. Chang and W. Tan, Anal. Chem., 2004, 76, 6973–6981 CrossRef CAS.
- B. Gulbakan, E. Yasun, M. I. Shukoor, Z. Zhu, M. You, X. Tan, H. Sanchez, D. H. Powell, H. Dai and W. Tan, J. Am. Chem. Soc., 2010, 132, 17408–17410 CrossRef CAS.
- D. A. Di Giusto, W. A. Wlassoff, J. J. Gooding, B. A. Messerle and G. C. King, Nucleic Acids Res., 2005, 33, e64 CrossRef.
- H. Zhang, Z. Wang, X. Li and X. C. Le, Angew. Chem., Int. Ed., 2006, 45, 1576–1580 CrossRef CAS.
- W. Yoshida, K. Sode and K. Ikebukuro, Anal. Chem., 2006, 78, 3296–3305 CrossRef CAS.
- M. N. Stojanovic and D. M. Kolpashchikov, J. Am. Chem. Soc., 2004, 126, 9266–9300 CrossRef CAS.
- H. Yang, H. Liu, H. Kang and W. Tan, J. Am. Chem. Soc., 2008, 130, 6320–6321 CrossRef CAS.
- B. Wei, I. Cheng, K. Q. Luo and Y. Mi, Angew. Chem., Int. Ed., 2008, 47, 331–333 CrossRef CAS.
- Z. Zhu, C. Wu, H. Liu, Y. Zou, X. Zhang, H. Kang, C. J. Yang and W. Tan, Angew. Chem., Int. Ed., 2010, 49, 1052–1056 CrossRef CAS.
- H. Liu, Z. Zhu, H. Kang, Y. Wu, K. Sefah and W. Tan, Chem.–Eur. J., 2010, 16, 3791–3797 CrossRef CAS.
- Y. Wu, K. Sefah, H. Liu, R. Wang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2009, 107, 5–10.
- M. V. Berezovski, M. Lechmann, M. U. Musheev, T. W. Mak and S. N. Krylov, J. Am. Chem. Soc., 2008, 130, 9137–9143 CrossRef CAS.
- M.-S. L. Raddatz, A. Dolf, E. Endl, P. Knolle, M. Famulok and G. Mayer, Angew. Chem., Int. Ed., 2008, 47, 5190–5193 CrossRef CAS.
- G. Mayer, M.-S. L. Ahmed, A. Dolf, E. Endl, P. A. Knolle and M. Famulok, Nat. Protoc., 2010, 5, 1993–2004 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |