A redox-active reverse donor–acceptor bistable [2]rotaxane†
Sanjeev K.
Dey
a,
Ali
Coskun
a,
Albert C.
Fahrenbach
a,
Gokhan
Barin
a,
Ashish N.
Basuray
a,
Ali
Trabolsi
a,
Youssry Y.
Botros
bc and
J. Fraser
Stoddart
*a
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208-3113, USA. E-mail: stoddart@northwestern.edu
bIntel Labs, Building RNB-6-61, 2200 Mission College Blvd., Santa Clara, CA 95054-1549, USA
cNational Center for Nano Technology Research, King Abdulaziz City for Science and Technology (KACST), P.O. Box 6086, Riyadh, 11442, Kingdom of Saudi Arabia
First published on 30th March 2011
Abstract
The synthesis and the dynamic behavior of a bistable [2]rotaxane, based on a reverse donor–acceptor motif containing naphthalene diimide (NpI) and 4,4′-bipyridinium (BIPY2+) as two electron-deficient stations and bis-1,5-dioxynaphthalene[38]crown-10 (BDNP38C10) as the electron-rich ring, is described. A functionalized tetraarylmethane moiety has been incorporated between the two stations in order to control the free energy barrier for the shuttling of the BDNP38C10 on the dumbbell component. The bistable [2]rotaxane was synthesized using the so-called “threading-followed-by-stoppering” approach and characterized by NMR spectroscopy and mass spectrometry. Initially, the BDNP38C10 ring resides on the NpI station on account of the synthetic approach employed in the synthesis of the bistable [2]rotaxane. 1H NMR spectroscopy was used to follow the equilibration process between the two translational isomers of the bistable [2]rotaxane—namely, NpI ⊂ BDNP38C10 and BIPY2+ ⊂ BDNP38C10. After 72 h, equilibrium was reached with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of the two translational isomers in favor of the NpI ⊂ BDNP38C10 co-conformation in CD3CN. The rate of relaxation of the crown ether from NpI ⊂ BDNP38C10 back to BIPY2+ ⊂ BDNP38C10 was associated with a rate constant of 2.2 ± 0.3 × 10−5 s−1 (t1/2 = 3.4 h), corresponding to a free energy of activation of 23.8 ± 0.1 kcal mol−1. Cyclic voltammetry (CV) reveals that the BDNP38C10 ring can be enticed to pass over the speed bump onto the neutral BIPY0 unit upon the generation of the NpI2− dianion, even although the neutral BIPY0 has presumably little or no affinity for the BDNP38C10 ring.
:2 ratio of the two translational isomers in favor of the NpI ⊂ BDNP38C10 co-conformation in CD3CN. The rate of relaxation of the crown ether from NpI ⊂ BDNP38C10 back to BIPY2+ ⊂ BDNP38C10 was associated with a rate constant of 2.2 ± 0.3 × 10−5 s−1 (t1/2 = 3.4 h), corresponding to a free energy of activation of 23.8 ± 0.1 kcal mol−1. Cyclic voltammetry (CV) reveals that the BDNP38C10 ring can be enticed to pass over the speed bump onto the neutral BIPY0 unit upon the generation of the NpI2− dianion, even although the neutral BIPY0 has presumably little or no affinity for the BDNP38C10 ring.
Introduction
Mechanically interlocked molecules1 (MIMs), e.g., bistable [2]catenanes and [2]rotaxanes,2 have attracted a copious amount of interest in the area of molecular nanotechnology on account of their ability to serve as active components in molecular electronic devices (MEDs)3 and nanoelectromechanical systems (NEMs),4 in addition to mechanized nanoparticles (MNPs)5 for drug delivery. A bistable [2]rotaxane consists of two components—namely, a dumbbell containing two recognition sites and a macrocycle which binds preferentially to one of the recognition units in its ground state and then can be switched reversibly between the recognition sites by the application of external stimuli of a chemical,6 electrochemical7 or photochemical8 nature. In a bistable [2]rotaxane or catenane, consisting of 1,5-dioxynaphthalene (DNP) and tetrathiafulvalene (TTF) as electron-rich recognition units and cyclobis(paraquat-p-phenylene) (CBPQT4+) as the electron-deficient cyclophane, the CBPQT4+ ring prefers to reside on the TTF unit rather than on the DNP unit in its ground state co-conformation (GSCC) on account of a 1.4 kcal mol−1 difference in their binding affinities.9 Once the TTF unit is oxidized—chemically or electrochemically—to either its radical cation or to its TTF2+ dication, the CBPQT4+ ring moves on to the DNP unit as a consequence of Coulombic repulsion between the TTF2+ dication and the CBPQT4+ ring. Upon reduction of the TTF2+ dication, neutral TTF is regenerated, while the CBPQT4+ ring remains encircled around the DNP unit. This state, which slowly relaxes back to the GSCC, is defined as the metastable state co-conformation (MSCC). Increasing the life-time of the MSCC is crucial for the development of non-volatile flash memory devices. Inserting steric and/or electrostatic barriers10 between the two recognition sites has been shown11–13 to increase the life-time of the MSCC. Recently, it has been demonstrated that the rate of shuttling of the rings between two identical stations in a degenerate [2]rotaxane can be decreased dramatically by inserting functionalized azobenzenes,11 tosylimines12 or bulky alkyl groups.13A considerable amount of research effort has been expended with the focus of developing molecular switches based on the donor–acceptor [2]rotaxane system in which the electron-rich recognition motif serves as the dumbbell for the electron-deficient ring component. There are, however, few examples14 of switchable “reverse” donor–acceptor [2]rotaxanes in which electron-deficient recognition motifs serve as the dumbbell component for an electron-rich ring. The Sanders group6a,15 has recently demonstrated a template-directed synthesis16 of a neutral [2]catenane and a bistable [2]rotaxane, wherein pyromellitic diimide (PmI) and naphthalene diimide (NpI) serve as the two electron-deficient recognition motifs and BDNP38C10 serves as an electron-rich ring. The switching was stimulated both chemically, using Li+ ions, and electrochemically. Examples of electrochemical switching of reverse donor–acceptor [2]rotaxanes have been reported somewhat infrequently in the literature.17 We have recently demonstrated18 the synthesis of a reverse donor–acceptor bistable [2]catenane (Fig. 1a) containing NpI and 4,4′-bipyridinium (BIPY2+) units as two electron-deficient stations, BDNP38C10 as the ring component and di-tertbutyl functionalized tetraarylmethane, incorporated between the two stations, as a “speed bump” for circumrotation of the ring. Although this system was not electrochemically switchable on account of the size of the speed bump, it encouraged us to design and synthesize a bistable [2]rotaxane incorporating a speed bump somewhat smaller in size. Herein, we report the synthesis and full characterization of the reverse donor–acceptor bistable [2]rotaxaneR·2PF6 (Fig. 1b)—containing NpI and BIPY2+ as the two electron-deficient stations, BDNP38C10 as the ring component, and the centrally located diethyl-fuctionalized tetraarylmethane unit as a speed bump—which can be switched electrochemically. Reduction of the NpI unit to its NpI2− dianion resulted in the passage of the BDNP38C10 ring over the speed bump onto the neutral BIPY0 station as a consequence of the repulsive interaction between the dianion and the electron-rich ring.
![Structural formulas of (a) a [2]catenane, (b) the [2]rotaxaneR·2PF6 and (c) the dumbbell 12·2PF6 and their graphical representations.](/image/article/2011/SC/c0sc00586j/c0sc00586j-f1.gif) | ||
| Fig. 1 Structural formulas of (a) a [2]catenane, (b) the [2]rotaxaneR·2PF6 and (c) the dumbbell 12·2PF6 and their graphical representations. | ||
Experimental section
General methods and materials
Starting materials and reagents were purchased from Aldrich or Fisher and used as received. Compound 6,69·2PF618 and NpI were prepared following procedures reported in the literature. All reactions were performed under an atmosphere of nitrogen and in dry solvents, unless otherwise noted. Analytical thin-layer chromatography (TLC) was performed on aluminum sheets, precoated with silica gel 60-F254 (Merck 5554). Flash chromatography was carried out using silica gel 60 (Silicycle) as the stationary phase. HPLC purification was performed on a preparative RP-HPLC instrument, using a C18 column. 1H and 13C NMR spectra were recorded on either a Bruker Avance 500 MHz, or a Bruker Avance 600 MHz spectrometer. Chemical shifts are reported in ppm relative to the signals corresponding to the residual non-deuterated solvents (CDCl3: δ 7.26 ppm, CD3CN: δ 1.94 ppm). High resolution electrospray ionization (HR ESI) mass spectra were measured on Agilent 6210 LC-TOF with Agilent 1200 HPLC introduction. High-resolution matrix-assisted laser desorption/ionization (MALDI) mass spectra were measured on a Bruker Autoflex III mass spectrometer. Electrochemical experiments were carried out at room temperature in argon-purged DMF solutions, with a Gamry Reference 600 potentiostat interfaced to a PC. Cyclic voltammetry experiments were performed using a glassy carbon working electrode (0.071 cm2, Cypress Systems). Its surface was polished routinely with 0.05 μm alumina-water slurry on a felt surface immediately before use. The counter electrode was a Pt coil and the reference electrode was a standard Ag/AgCl electrode. The concentration of the sample and supporting electrolyte (TBAPF6) were 1 × 10−3 mol L−1 and 0.1 mol L−1, respectively.Compound R·2PF6
Compound 11 (97 mg, 0.068 mmol) and BDNP38C10 (48 mg, 0.074 mmol) were dissolved in CHCl3 (3 mL) in a 10 mL flask and stirred at 25 °C for 24 h. The solvent was removed under vacuum and the crude product was purified by column chromatography [SiO2:CHCl3] to obtain pseudorotaxaneR (69 mg). TBTA (18 mg) and Cu(MeCN)4PF6 (12 mg) were added to the solution of R in CHCl3 (3 mL), 10·2PF6 (80 mg, 0.034 mmol) dissolved in DMF (1 mL). The solution was stirred at 25 °C for 24 h. The solvent was evaporated off under vacuum and the solid product was purified by RP-HPLC (H2O–MeCN–0.1% TFA/80–100% in 40 min λ = 254 nm). The purple fraction was collected and concentrated. The resulting solid was dissolved in a minimal amount of MeCN and added to a saturated solution of NH4PF6 in H2O. The precipitate was collected and washed with H2O several times and dried under vacuum to afford the rotaxaneR·2PF6 as a reddish-white solid (27 mg, 23%). 1H NMR (CD3CN, 600 MHz, 298 K): δ = 8.75 (d, J = 7.2 Hz, 4H), 8.54 (s, 4H), 8.54 (s, 4H), 8.18 (s, J = 7.2 Hz, 4H), 8.12 (s, 4H), 7.78 (s, 1H), 7.77 (s, 1H), 7.53 (s, 1H), 7.52 (s, 1H), 7.29–6.58 (m, 116), 6.37 (d, J = 7.5 Hz, 2H), 6.07 (d, J = 7.5 Hz, 2H), 5.03 (t, J = 7.5 Hz, 2H), 4.91 (q, J = 7.5 Hz, 2H), 4.59–3.68 (m, 40H), 3.48 (t, J = 7.5 Hz, 2H), 3.37 (t, J = 7.5 Hz, 2H), 2.5 (q, J = 7.2 Hz, 4H), 2.40–1.25 (m, 48H), 1.1 (t, J = 7.5 Hz, 6H) ppm. 13C NMR (CD3CN, 125 MHz, 298 K): δ = 163.9, 163.8, 157.9, 157.7, 154.1, 153.9, 149.4, 149.3, 146.8, 145.6, 145.5, 145.5, 142.8, 142.6, 142.2, 140.6, 140.5, 140.4, 140.3, 132.6, 132.5, 131.5, 131.1, 127.8, 127.7, 126.6, 126.2, 126.1, 125.4, 125.3, 124.9, 124.7, 123.9, 114.6, 114.3, 114.2, 114.1, 114.0, 106.4, 104.5, 72.0, 71.9, 71.8, 70.7, 70.4, 69.0, 68.6, 68.3, 68.3, 63.9, 63.8, 63.7, 62.8, 62.0, 50.7, 50.6, 41.1, 41.0, 37.6, 36.0, 34.9, 32.6, 31.5, 30.5, 30.3, 30.1, 30.0, 29.9, 29.8, 29.5, 29.2, 28.7, 28.6, 28.4, 27.7, 27.6, 26.3, 25.6, 24.8, 23.7, 23.6, 23.3, 15.9, 15.8, 14.3. ESI-HRMS calcd for m/z = 1470.8275 [M − 2PF6]2+, found m/z = 1470.8265.Results and discussion
Synthesis
The synthesis of R·2PF6 is outlined in Scheme 1. The reaction of commercially available methyl 4-methoxybenzoate with 4-ethylbromobenzene in the presence of magnesium turnings in dry THF afforded (80%) compound 1, which was reacted subsequently with phenol in presence of conc. HCl to yield compound 2 in 65% yield. De-O-methylation was achieved using BBr3 in CH2Cl2 at RT, and subsequent alkylation employing 5-bromopentanol was carried out in dry MeCN in the presence of K2CO3 together with a catalytic amount of 18-crown-6 to yield compound 4, which was then reacted with tosyl chloride and Et3N in the presence of a catalytic amount of DMAP to afford the monotosylate derivative 5. Reaction of compound 5 and 66 with commercially available 1,4,5,8-naphthalenetetracarboxydiimide under Mitsunobu reaction conditions yielded the monotosylate derivative, which was subsequently converted into compound 11 by reacting the intermediate formed in the reaction in situ with NaN3 in dry DMF at 80 °C.![Synthesis of [2]rotaxaneR·2PF6 and its dumbbell component 12·2PF6.](/image/article/2011/SC/c0sc00586j/c0sc00586j-s1.gif) | ||
| Scheme 1 Synthesis of [2]rotaxaneR·2PF6 and its dumbbell component 12·2PF6. | ||
Similarly, the tosylation of compound 6, using tosyl chloride and Et3N in the presence of a catalytic amount of DMAP, afforded the tosyl derivative, which was converted into compound 8 by reacting it with NaN3 in dry DMF at 80 °C. Subsequently, compound 8 was reacted with the dialkyne-functionalized viologen 9·2PF618 under Cu(I)-catalyzed azide–alkynecycloaddition (CuAAc) reaction conditions19 to afford 10·2PF6. Reaction of 10·2PF6 with compound 11 in the presence of BDNP38C1020 using the so-called “threading-followed-by-stoppering” approach19c,21 yielded an inseparable mixture of both the [2]- and [3]rotaxanes, and so we decided to form the pseudorotaxane R prior to making the [2]rotaxaneR·2PF6. The pseudorotaxane R was obtained by mixing compound 11 with BDNP38C10 in CHCl3 at room temperature in 70% yield. Finally, the reaction of R with 10·2PF6 using CuAAc conditions afforded the [2]rotaxaneR·2PF6 in 23% yield. The dumbbell 12·2PF6 was obtained simply by reacting 11 with 10·2PF6 under the same CuAAc conditions.
Characterization by 1H NMR spectroscopy
All compounds have been fully characterized by 1H and 13C NMR spectroscopies and by mass spectrometry. The 1H NMR spectrum of the [2]rotaxaneR·2PF6 is recorded (Fig. 2) in CD3CN at 298 K.22 There are two sets of BDNP38C10 protons corresponding to the two translational isomers of R·2PF6—namely, NpI ⊂ BDNP38C10 and BIPY2+ ⊂ BDNP38C10. The set of signals observed at higher frequencies, that is, δ = 6.40 (d), 6.85 (t), 7.12 (d) ppm (Fig. 2) were assigned, in turn, to the H2/6, H3/7, H4/8protons of BDNP38C10 encircling the BIPY2+ station on account of the lower shielding effect between these two units. In contrast, the set of signals observed at lower frequencies, that is, δ = 6.10 (d), 6.60 (t) and 7.05 (d) ppm were assigned to the aromatic protons of BDNP38C10 encircling the NpI unit, i.e., NpI ⊂ BDNP38C10. Similarly, two sets of Hα and Hβprotons, corresponding to the encircled BIPY2+ and free BIPY2+ units, as well as another set of protons corresponding to the encircled and free form of the NpI station were observed. The relatively shielded signals at ∼ 8.50 (d) and 6.95 (d) ppm were assigned, respectively, to the Hα and Hβprotons of the encircled BIPY2+ and signals at δ = ∼ 8.78 (d) and 8.20 (d) ppm were assigned, respectively, to the Hα and Hβprotons of the free BIPY2+ unit. Likewise, the signals at δ = ∼ 8.10 (s) and 8.50 (s) ppm correspond to the encircled and free NpI stations, respectively. The assignment of these resonances was assisted by 1H–1H correlation spectroscopy (see ESI†).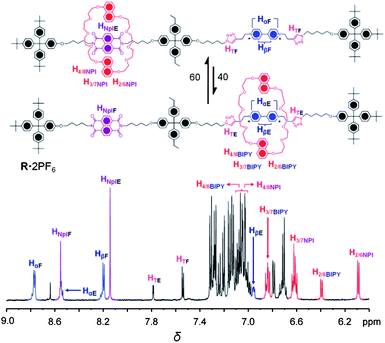 | ||
| Fig. 2 Partial 1H NMR of R·2PF6 and assignment of resonances recorded in CD3CN at 298 K. The subscripts “E” and “F” correspond to encircled and free, respectively. | ||
The synthetic strategy we employed allowed us to obtain the free energy of activation for the shuttling of the BDNP38C10 ring over the speed bump. Since the higher percentage of the BDNP38C10 ring encircling the NpI station decreases gradually over time, this equilibration process was followed (integration) by 1H NMR spectroscopy (Fig. 3) at 298 K in CD3CN. At the beginning, ∼85% of the BDNP38C10 ring encircles the NpI station based on the relative integrations of the peaks observed at δ = 6.40 and 6.10 ppm, resonances which correspond to the H2/6protons of the BDNP38C10 ring encircling BIPY2+ and NpI, respectively. The rate of equilibration between the two translational isomers—namely, NpI ⊂ BDNP38C10 and BIPY2+ ⊂ BDNP38C10—occurs in a relatively rapid manner at room temperature and then slows down with time in a manner consistent with a first-order decay process (see ESI†). The bistable [2]rotaxane was observed to have reached equilibrium after 72 h with a 3:2 ratio of the two translational isomers, where NpI ⊂ BDNP38C10 was found to be the major co-conformation. The slightly higher population of the NpI ⊂ BDNP38C10 co-conformation can be rationalized by a “side on” interaction18 between the BIPY2+ with the BDNP38C10 ring, while encircling the NpI unit as a consequence of the conformational flexibility inherent in the structure. The rate of relaxation of the BDNP38C10 ring from NpI ⊂ BDNP38C10 to BIPY2+ ⊂ BDNP38C10 is associated with a rate constant of 2.2 ± 0.3 × 10−5s−1, giving rise to a ΔG‡ value of 23.8 ± 0.1 kcal mol−1 (see ESI†). It is important to note that replacing the tert-butyl groups18 with ethyl groups on the central tetraarylmethane speed bump core decreases the activation barrier from 28.5 (measured at 343 K) to 23.8 kcal mol−1.
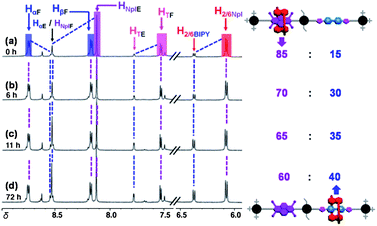 | ||
| Fig. 3 Partial 1H NMR of R·2PF6 in CD3CN at 298 K recorded at time (a) 0 h, (b) 6 h, (c) 11 h, and (d) 72 h. Annotations are shown in color. Blue line indicates the co-conformation in which the BIPY2+ is encircled by BDNP38C10. Purple line indicates the co-conformation in which the NpI is encircled by BDNP38C10. | ||
Switching behavior of the bistable [2]rotaxane
Electrochemical experiments—namely, cyclic voltammetry (CV), differential pulse voltammetry (DPV) and spectroelectrochemistry (SEC)—were carried out in argon-purged DMF solutions (0.1 M TBAPF6 as a supporting electrolyte) at 298 K. As a consequence of the fact that the barrier to shuttling of the BDNP38C10 is relatively large in MeCN as evidenced by 1H NMR spectroscopy, we chose to investigate the electrochemical switching behaviour of R·2PF6 in DMF,22 in which the 1H NMR spectrum reveals the shuttling behavior is much faster (see ESI†). All the potential values were referenced against the Ag/AgCl electrode (SSCE). Our focus in these experiments was centered on (i) corroborating the results from 1H NMR spectroscopy (in CD3CN) which shows that the BDNP38C10 ring exists approximately in a 1:1 ratio distributed between the two co-conformational isomers encircling either the NpI or the BIPY2+ station in DMF, a situation which corresponds to the ground state of the [2]rotaxaneR·2PF6 and (ii) whether it is possible to induce switching of the BDNP38C10 ring electrochemically.
Table 1 summarizes the results obtained from the CV experiments. Both the model compounds, NpI23 and 9·2PF618 exhibit (Fig. 4) two monoelectronic and reversible reduction processes. The dumbbell 12·2PF6, which incorporates both BIPY2+ and NpI redox-active recognition units, shows four reversible reduction processes at the same potentials as the model compounds, indicating that the redox behavior of each of the recognition units within the dumbbell component are independent of each other.
|
E(I)BIPY | E(I)NpI | E(II)BIPY | E(II)NpI |
|---|---|---|---|---|
| a Determined by cyclic voltammetry (argon-purged DMF, 298 K, referenced to SSCE). All values are in units of voltage. b Potential of the “free” recognition site. c Potential of the site after encirclement by the BDNP38C10 ring. | ||||
| 9·2PF6 | −0.46 (−0.42) | — | −0.86 (−0.83) | — |
| NpI | — | −0.58 (−0.54) | — | −1.07 (−1.04) |
| 12·2PF6 | −0.46 (−0.42) | −0.58 (−0.54) | −0.86 (−0.83) | −1.07(−1.04) |
| R·2PF6 | −0.46 (−0.42)b | −0.60 (−0.57)b | −0.86 (−0.83)b | −1.10 (−1.07)b |
| −0.61 (−0.58)c | −0.86 (−0.83)c | −0.89 (−0.86)c | −1.11 (−1.08)c |
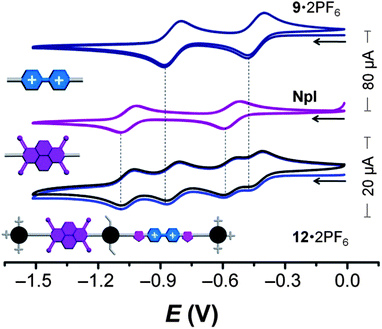 | ||
| Fig. 4 Cyclic voltammograms (argon-purged DMF; scan rate = 200 mV s−1) of the dumbbell 12·2PF6, as well as 9·2PF6 and NpI, which serve as model compounds for the recognition sites. Both the first and second scans for each compound are shown. | ||
The first scan of the CV of the rotaxaneR·2PF6 showed (Fig. 5) four reversible peaks in the reductive region. On the basis of assignments made with respect to the dumbbell 12·2PF6, the four reduction peaks have been partially assigned to the first and second monoelectronic reduction processes of the free BIPY2+ (−0.46 V and −0.86 V) and the free NpI (−0.60 V and −1.10 V) stations, respectively. As a consequence of the noncovalent bonding interactions of the BIPY2+ and NpI stations with the BDNP38C10 ring, the first reduction potentials of these units are known to become shifted and, as a consequence, overlap significantly with the first reduction potential of the free NpI (−0.60 V) and the second reduction potential of the BIPY2+ (−0.86 V) stations, respectively. The overlapping of these reduction processes is supported (see ESI†) by differential pulse voltammetry (DPV). The second reduction potentials of both the encircled and the free BIPY2+/NpI stations, respectively, were observed to be at approximately the same voltages, with only minor shifts towards more negative potentials. These minor shifts of potentials can be explained as a result of the loss of recognition of the BDNP38C10 ring after the first reduction processes relating to both the BIPY2+ and NpI stations. Furthermore, the return anodic scan displays four re-oxidation peaks, indicating that all these processes are completely reversible.
![Cyclic voltammograms (argon-purged DMF; scan rate = 200 mV s−1) of the dumbbell 12·2PF6 and the [2]rotaxaneR·2PF6. The first and second scans for both compounds are shown. Changes in the relative intensities between the first (red) and second (black) of the first two reduction processes for the rotaxaneR·2PF6 are observed, while no such changes are observed in the dumbbell component.](/image/article/2011/SC/c0sc00586j/c0sc00586j-f5.gif) | ||
| Fig. 5 Cyclic voltammograms (argon-purged DMF; scan rate = 200 mV s−1) of the dumbbell 12·2PF6 and the [2]rotaxaneR·2PF6. The first and second scans for both compounds are shown. Changes in the relative intensities between the first (red) and second (black) of the first two reduction processes for the rotaxaneR·2PF6 are observed, while no such changes are observed in the dumbbell component. | ||
Taking into consideration the overlapping potentials for the reduction of free NpI and encircled BIPY2+ stations, we estimate the relative amount of the translational isomers—namely, NpI ⊂ BDNP38C10 and BIPY2+ ⊂ BDNP38C10—based on the relative amounts of the free BIPY2+ to the encircled BIPY2+, to be approximately in a 1:1 ratio. Therefore, prior to the application of an electrochemical stimulus, the [2]rotaxaneR·2PF6 exists at equilibrium as an equimolar ratio of the two co-conformational isomers in DMF at 298 K. This equimolar ratio is consistent with binding constants obtained from host–guest titration studies of the BDNP38C10 ring with model BIPY2+ and NpI compounds (see ESI†).
The second scan of the rotaxaneR·2PF6 shows that the relative intensity of the peak corresponding to the reduction of free BIPY2+ (Fig. 5) decreased by a small amount relative to the intensity of the peak assigned to the simultaneous first reductions of the encircled BIPY2+ and the free NpI stations, in comparison to the first scan. This change in intensity between the first and second scans was not observed in the case of either the dumbbell or the model compounds. These observations suggest that the generation of the NpI2− dianion induces switching of the BDNP38C10 ring over the speed bump to encircle the neutral BIPY0 station on account of the repulsive interaction between the NpI2− dianion and the electron-rich ring. The BDNP38C10 ring becomes trapped upon full re-oxidation of the NpI and BIPY2+ stations on account of the central speed bump unit which prevents the relaxation of the BDNP38C10 ring back to the NpI station on the timescale of the CV experiment. In order to verify this hypothesis, a solution of the rotaxaneR·2PF6 was subjected to a potential of −1.5 V at the glassy carbon working electrode for ten seconds, before recording the CV scanning towards the limit of 0 V and returning back to −1.5 V. Indeed, on the return scan, the reduction peak (−0.46 V), corresponding to the reduction of free BIPY2+, was observed (Fig. 6) to decrease dramatically (almost undetectable) relative to the reduction peak (−0.60 V) corresponding to the encircled BIPY2+ and free NpI—a peak which was observed to increase. Moreover, consistent with the peak assignments, the reduction peak at −0.86 V, corresponding to the reduction of the encircled NpI, as well as the second reduction of both the free and encircled BIPY2+ stations, was observed to decrease. Furthermore, the peak assigned to the second reduction of the free and encircled BIPY2+ stations was observed to shift to a more negative potential by 30 mV, while the peak assigned to the reduction of the free and encircled NpI stations was observed to shift to a more positive potential by 30 mV, an observation which is consistent with a larger amount of the BDNP38C10 encircling the BIPY2+ prior to the second reductions of these stations. Spectroelectrochemistry was also performed on the rotaxaneR·2PF6. (For details see SI†).
![(a) CV of the [2]rotaxaneR·2PF6 recorded after applying a potential of −1.5 V for a 10 s equilibration time before recording the scan to 0 V and back (red), and overlay of the CV of the first scan of the rotaxane recorded starting at 0 V, scanning to −1.5 V and back (dotted line). Relative increases and decreases in the reduction peak intensities between the two different scans are shown (scan rate = 200 mV s−1). (b) Proposed switching mechanism of the [2]rotaxaneR·2PF6 deduced from the CV data.](/image/article/2011/SC/c0sc00586j/c0sc00586j-f6.gif) | ||
| Fig. 6 (a) CV of the [2]rotaxaneR·2PF6 recorded after applying a potential of −1.5 V for a 10 s equilibration time before recording the scan to 0 V and back (red), and overlay of the CV of the first scan of the rotaxane recorded starting at 0 V, scanning to −1.5 V and back (dotted line). Relative increases and decreases in the reduction peak intensities between the two different scans are shown (scan rate = 200 mV s−1). (b) Proposed switching mechanism of the [2]rotaxaneR·2PF6 deduced from the CV data. | ||
In order to further verify that the switching is a result of generation of the NpI2− dianion and not the NpI˙−radical anion, a CV of the rotaxane was recorded (see ESI†), initially from 0 V to the scan limit of −0.95 V. We did not observe any significant change in the relative intensity of the peak corresponding to the reduction of the free BIPY2+ on the second scan of the CV cycle. This observation indicates that the NpI˙−radical anion does not readily induce the switching of the BDNP38C10 ring over the speed bump to the neutral BIPY0 on the time scale of the CV experiment. These results strengthen the conclusion that the BDNP38C10 ring can be readily switched over the central speed bump onto the neutral BIPY0 unit through the generation of the NpI2− dianion, even although the neutral BIPY0 presumably has no affinity for the BDNP38C10 ring. Upon full re-oxidation of the [2]rotaxaneR·2PF6, the translational isomer BIPY2+ ⊂ BDNP38C10 predominates, and probably represents a metastable intermediate state of R·2PF6, since the ground state distribution corresponds to the 1:1 ratio of the two translational isomers.
Conclusions
In summary, a reverse donor–acceptor bistable [2]rotaxane, comprised of the electron-deficient recognition units NpI and BIPY2+, the electron-rich BDNP38C10 ring, and a speed bump between the two recognition units, has been designed and synthesized. The speed bump in this bistable [2]rotaxane is sterically of just the right size to allow the shuttling of BDNP38C10 between the two stations. Replacing the tert-butyl groups18 with ethyl groups on the central tetraarylmethane speed bump core decreases the free energy of activation for the shuttling quite dramatically from 28.5 to 23.8 kcal mol−1 in MeCN. These free energies of activation correspond to half-life times towards equilibration of 21 h (at 343 K) and 3.4 h, respectively. The [2]rotaxane was shown to be readily switchable electrochemically upon generation of the NpI2− dianion in DMF. The scope of electrochemical switching in this reverse donor–acceptor bistable [2]rotaxane validates its candidature for application in molecular (non-volatile) memory devices and provides valuable information for the design and synthesis of mechanically interlocked molecules based on these recognition motifs. Future work on these systems will reside in the development of principles that will make it possible to tune the distribution at equilibrium between the two different co-conformations, thus producing efficient molecular switches for memory applications.Acknowledgements
The research described in this full paper was sponsored in part by the National Center for Nano Technology Research at the King Abdulaziz City for Science and Technology (KACST) in Saudi Arabia. The authors thank Dr Turki M. Al-Saud and Dr Soliman H. Alkhowaiter at KACST for their generous support of this program of research at Northwestern University. The results are also based upon research sponsored by the Air Force Office of Scientific Research (AFOSR) under agreement number FA9550-07-1-0534. We also thank the National Science Foundation (NSF) for the award of a Graduate Research Fellowship to A. C. F. We wish to acknowledge that G. B. was supported as part of the Non-Equilibrium Research Center (NERC), which is an Energy Frontier Research Center (EFRC) funded by the U. S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Award No. DE-SC0000989. He thanks the International Center for Diffraction Data for the award of a 2011 Ludo Frevel Crystallography Scholarship.Notes and references
- (a) G. Schill; Catenanes, Rotaxanes, and Knots; Academic Press: New York, 1971 Search PubMed; (b) J.-P. Sauvage, C. Dietrich-BucheckerMolecular Catenanes, Rotaxanes, and Knots; Ed.; Wiley-VCH: Weinheim, 1999 Search PubMed; (c) J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS; (d) J. F. Stoddart and H. M. Colquhoun, Tetrahedron, 2008, 64, 8231–8263 CrossRef CAS; (e) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC; (f) J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC; (g) D. B. Amabilino and L. Pérez-García, Chem. Soc. Rev., 2009, 38, 1562–1571 RSC.
- (a) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS; (b) S. Nygaard, K. C.-F. Leung, I. Aprahamian, T. Ikeda, S. Saha, B. W. Laursen, S. Y. Kim, S. W. Hansen, P. C. Stein, A. H. Flood, J. F. Stoddart and J. O. Jeppesen, J. Am. Chem. Soc., 2007, 129, 960–970 CrossRef CAS; (c) I. Aprahamian, W. R. Dichtel, T. Ikeda, J. R. Heath and J. F. Stoddart, Org. Lett., 2007, 9, 1287–1290 CrossRef CAS; (d) C.-F. Lee, D. A. Leigh, R. G. Pritchard, D. Schultz, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Nature, 2009, 458, 314–318 CrossRef CAS; (e) J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422–442 RSC; (f) J.-P. Collin, F. Durola, J. Lux and J.-P. Sauvage, Angew. Chem., Int. Ed., 2009, 48, 8532–8535 CrossRef CAS; (g) H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2010, 49, 5331–5334 CrossRef CAS.
- (a) C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS; (b) A. H. Flood, J. F. Stoddart, D. W. Steuerman and J. R. Heath, Science, 2004, 306, 2055–2056 CrossRef CAS; (c) J. R. Heath, J. F. Stoddart and R. S. Williams, Science, 2004, 303, 1136–1137 CAS; (d) J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS; (e) A. Bandyopadhyay and S. Acharya, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3668–3672 CrossRef CAS; (f) J. R. Heath, Annu. Rev. Mater. Res., 2009, 39, 1–23 CrossRef CAS; (g) S. J. van der Molen and P. Liljeroth, J. Phys.: Condens. Matter, 2010, 22, 133001 CrossRef; (h) W. Zhang, E. DeIonno, W. R. Dichtel, L. Fang, A. Trabolsi, J.-C. Olsen, D. Benítez, J. R. Heath and J. F. Stoddart, J. Mater. Chem., 2011, 21, 1487–1495 RSC.
- (a) J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef CAS; (b) B. K. Juluri, A. S. Kumar, Y. Liu, T. Ye, Y.-W. Yang, A. H. Flood, L. Fang, J. F. Stoddart, P. S. Weiss and T. J. Huang, ACS Nano, 2009, 3, 291–300 CrossRef CAS; (c) Y. B. Zheng, Y.-W. Yang, L. Jensen, L. Fang, B. K. Juluri, A. H. Flood, P. S. Weiss, J. F. Stoddart and T. J. Huang, Nano Lett., 2009, 9, 819–825 CrossRef CAS; (d) T. Ye, A. S. Kumar, S. Saha, T. Takami, T. J. Huang, J. F. Stoddart and P. S. Weiss, ACS Nano, 2010, 4, 3697–3701 CrossRef CAS.
- (a) T. D. Nguyen, Y. Liu, S. Saha, K. C.-F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS; (b) S. Saha, K. C.-F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 685–693 CrossRef CAS; (c) S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222–2226 CrossRef CAS; (d) K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS; (e) K. K. Cotí, M. E. Belowich, M. Liong, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, N. M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16–39 RSC.
- (a) S. A. Vignon, T. Jarrosson, T. Iijima, H.-R. Tseng, J. K. M. Sanders and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 9884–9885 CrossRef CAS; (b) J. Li, Y. Li, Y. Guo, J. Xu, J. Lv, Y. Li, H. Liu, S. Wang and D. Zhu, Chem.–Asian J., 2008, 3, 2091–2096 CrossRef CAS; (c) K. C.-F. Leung, C.-P. Chak, C.-M. Lo, W.-Y. Wong, S. Xuan and C. H. K. Cheng, Chem.–Asian J., 2009, 4, 364–381 CrossRef CAS; (d) J. D. Crowley, K. D. Hänni, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem. Soc., 2010, 132, 5309–5314 CrossRef CAS.
- (a) H.-R. Tseng, S. A. Vignon and J. F. Stoddart, Angew. Chem., Int. Ed., 2003, 42, 1491–1495 CrossRef CAS; (b) H.-R. Tseng, S. A. Vignon, P. C. Celestre, J. Perkins, J. O. Jeppesen, A. Di Fabio, R. Ballardini, M. T. Gandolfi, M. Venturi, V. Balzani and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 155–172 CrossRef CAS; (c) A. Altieri, F. G. Gatti, E. R. Kay, D. A. Leigh, D. Martel, F. Paolucci, A. M. Z. Slawin and J. K. Y. Wong, J. Am. Chem. Soc., 2003, 125, 8644–8654 CrossRef CAS; (d) F. Durola and J.-P. Sauvage, Angew. Chem., Int. Ed., 2007, 46, 3537–3540 CrossRef CAS; (e) J. M. Spruell, A. Coskun, D. C. Friedman, R. S. Forgan, A. A. Sarjeant, A. Trabolsi, A. C. Fahrenbach, G. Barin, W. F. Paxton, S. K. Dey, M. A. Olson, D. Benítez, E. Tkatchouk, M. T. Colvin, R. Carmielli, S. T. Caldwell, G. M. Rosair, S. G. Hewage, F. Duclairoir, J. L. Seymour, A. M. Z. Slawin, W. A. Goddard III, M. R. Wasielewski, G. Cooke and J. F. Stoddart, Nature Chem., 2010, 2, 870–879 CrossRef CAS.
- (a) P. R. Ashton, V. Balzani, O. Kocian, L. Prodi, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1998, 120, 11190–11191 CrossRef CAS; (b) V. Balzani, A. Credi, F. Marchioni and J. F. Stoddart, Chem. Commun., 2001, 1860–1861 RSC; (c) A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124–2128 CrossRef CAS; (d) D.-H. Qu, Q.-C. Wang and H. Tian, Angew. Chem., Int. Ed., 2005, 44, 5296–5299 CrossRef CAS; (e) B. Ferrer, G. Rogez, A. Credi, R. Ballardini, M. T. Gandolfi, V. Balzani, Y. Liu, H.-R. Tseng and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18411–18416 CrossRef CAS; (f) S. Saha and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 77–92 RSC; (g) V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC.
- (a) S. Kang, S. A. Vignon, H.-R. Tseng and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 2555–2564 CrossRef CAS; (b) J. W. Choi, A. H. Flood, D. W. Steuerman, S. Nygaard, A. B. Braunschweig, N. N. P. Moonen, B. W. Laursen, Y. Luo, E. DeIonno, A. J. Peters, J. O. Jeppesen, K. Xu, J. F. Stoddart and J. R. Heath, Chem.–Eur. J., 2006, 12, 261–279 CrossRef CAS.
- A. Trabolsi, A. C. Fahrenbach, S. K. Dey, A. I. Share, D. C. Friedman, S. Basu, T. B. Gasa, N. M. Khashab, S. Saha, I. Aprahamian, H. A. Khatib, A. H. Flood and J. F. Stoddart, Chem. Commun., 2010, 46, 871–873 RSC.
- A. Coskun, D. C. Friedman, H. Li, K. Patel, H. A. Khatib and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 2493–2495 CrossRef CAS.
- A. S. Lane, D. A. Leigh and A. Murphy, J. Am. Chem. Soc., 1997, 119, 11092–11093 CrossRef CAS.
- M. Bělohradský, A. M. Elizarov and J. F. Stoddart, Collect. Czech. Chem. Commun., 2002, 67, 1719–1728 CrossRef CAS.
- (a) P. R. Ashton, R. Ballardini, V. Balzani, M. Bělohradský, M. T. Gandolfi, D. Philp, L. Prodi, F. M. Raymo, M. V. Reddington, N. Spencer, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1996, 118, 4931–4951 CrossRef CAS; (b) D. G. Hamilton, J. K. M. Sanders, J. E. Davies, W. Clegg and S. J. Teat, Chem. Commun., 1997, 897–898 RSC; (c) M. Asakawa, P. R. Ashton, R. Ballardini, V. Balzani, M. Bělohradský, M. T. Gandolfi, O. Kocian, L. Prodi, F. M. Raymo, J. F. Stoddart and M. Venturi, J. Am. Chem. Soc., 1997, 119, 302–310 CrossRef CAS; (d) D. G. Hamilton, J. E. Davies, L. Prodi and J. K. M. Sanders, Chem.–Eur. J., 1998, 4, 608–620 CrossRef CAS; (e) D. G. Hamilton, M. Montalti, L. Prodi, M. Fontani, P. Zanello and J. K. M. Sanders, Chem.–Eur. J., 2000, 6, 608–617 CrossRef CAS; (f) S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC; (g) G. Koshkakaryan, K. Parimal, J. M. He, X. Y. Zhang, Z. Abliz, A. H. Flood and Y. Liu, Chem.–Eur. J., 2008, 14, 10211–10218 CrossRef CAS; (h) G. Koshkakaryan, L. M. Klivansky, D. Cao, M. Snauko, S. J. Teat, J. O. Struppe and Y. Liu, J. Am. Chem. Soc., 2009, 131, 2078–2079 CrossRef CAS; (i) D. Cao, M. Amelia, L. M. Klivansky, G. Koshkakaryan, S. I. Khan, M. Semeraro, S. Silvi, M. Venturi, A. Credi and Y. Liu, J. Am. Chem. Soc., 2010, 132, 1110–1122 CrossRef CAS.
- (a) G. Kaiser, T. Jarrosson, S. Otto, Y.-F. Ng, A. D. Bond and J. K. M. Sanders, Angew. Chem., Int. Ed., 2004, 43, 1959–1962 CrossRef CAS; (b) T. Iijima, S. A. Vignon, H.-R. Tseng, T. Jarrosson, J. K. M. Sanders, F. Marchioni, M. Venturi, E. Apostoli, V. Balzani and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 6375–6392 CrossRef.
- (a) D. H. Busch and N. A. Stephenson, Coord. Chem. Rev., 1990, 100, 119–154 CrossRef CAS; (b) S. Anderson, H. L. Anderson and J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469–475 CrossRef CAS; (c) T. J. Hubin and D. H. Busch, Coord. Chem. Rev., 2000, 200, 5–52 CrossRef; (d) M.-J. Blanco, J.-C. Chambron, M.-C. Jimenez and J.-P. Sauvage, Top. Stereochem., 2003, 23, 125–173 Search PubMed; (e) C. A. Schalley, T. Weilandt, J. Bruggemann and F. Vögtle, Top. Curr. Chem., 2004, 248, 141–200; (f) F. Aricó, J. D. Badjić, S. J. Cantrill, A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 249, 203–259 CAS; (g) M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC.
- (a) B. Long, K. Nikitin and D. Fitzmaurice, J. Am. Chem. Soc., 2003, 125, 15490–15498 CrossRef CAS; (b) K. Nikitin, E. Lestini, J. K. Stolarczyk, H. Müller-Bunz and D. Fitzmaurice, Chem.–Eur. J., 2008, 14, 1117–1128 CrossRef CAS.
- A. Coskun, S. Saha, I. Aprahamian and J. F. Stoddart, Org. Lett., 2008, 10, 3187–3190 CrossRef CAS.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS; (c) W. R. Dichtel, O.Š. Miljanić, J. M. Spruell, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2006, 128, 10388–10390 CrossRef CAS; (d) O.Š. Miljanić, W. R. Dichtel, I. Aprahamian, R. D. Rohde, H. D. Agnew, J. R. Heath and J. F. Stoddart, QSAR Comb. Sci., 2007, 26, 1165–1174 CrossRef CAS.
- (a) P. R. Ashton, E. J. T. Chrystal, J. P. Mathias, K. P. Parry, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Tetrahedron Lett., 1987, 28, 6367–6370 CrossRef; (b) C. J. Bruns, S. Basu and J. F. Stoddart, Tetrahedron Lett., 2010, 51, 983–986 CrossRef CAS.
- (a) A. B. Braunschweig, W. R. Dichtel, O.Š. Miljanić, M. A. Olson, J. M. Spruell, S. I. Khan, J. R. Heath and J. F. Stoddart, Chem.–Asian J., 2007, 2, 634–647 CrossRef CAS; (b) W. R. Dichtel, O.Š. Miljanić, W. Zhang, J. M. Spruell, K. Patel, I. Aprahamian, J. R. Heath and J. F. Stoddart, Acc. Chem. Res., 2008, 41, 1750–1761 CrossRef CAS; (c) J. M. Spruell, W. R. Dichtel, J. R. Heath and J. F. Stoddart, Chem.–Eur. J., 2008, 14, 4168–4177 CrossRef CAS.
- Variable temperature 1H NMR spectrsocopy on the rotaxaneR·2PF6 in d7-DMF was also performed in order to demonstrate that the rate of shuttling of the BDNP38C10 ring is much faster in this solvent compared to that in CD3CN. This faster rate of shuttling can be explained by the decrease in affinity of the BDNP38C10 ring for both the BIPY2+ and NpI stations. UV/vis spectroscopic titration data of the BDNP38C10 ring with model compounds for BIPY2+ and NpI in DMF at 298 K reveal that the binding constants of these compounds with the ring are approximately 30 M−1. For details see ESI†.
- The model NpI compound employed for electrochemical studies was bis-N,N′-dipentylnaphthalene-1,4,5,8-tetracarboxylic diimide. G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011–1019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, 1D and 2D NMR spectra, cyclic voltammetry and the kinetic data based of 1H NMR experiments. See DOI: 10.1039/c0sc00586j |
| This journal is © The Royal Society of Chemistry 2011 |