Versatile synthesis of temperature-sensitive polypeptides by click grafting of oligo(ethylene glycol)†
Yilong
Cheng
ab,
Chaoliang
He
a,
Chunsheng
Xiao
ab,
Jianxun
Ding
ab,
Xiuli
Zhuang
a and
Xuesi
Chen
*a
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: xschen@ciac.jl.cn; Fax: +86 431 8526 2112; Tel: +86 431 8526 2112
bGraduate University of Chinese Academy of Sciences, Beijing 100039, P. R. China
First published on 22nd August 2011
Abstract
A series of novel temperature-sensitive polypeptides were synthesized by ring opening polymerization (ROP) of γ-propargyl-L-glutamateN-carboxyanhydride (PLG-NCA) and subsequent click reaction between the pendant alkyne groups and 1-(2-methoxyethoxy)-2-azidoethane (MEO2-N3) or 1-(2-(2-methoxyethoxy)ethoxy)-2-azidoethane (MEO3-N3). The efficient click grafting and structure of the resultant copolymers were verified by 1H NMR, 13C NMR and GPC. All the copolymers hold α-helix conformation, and could self-assemble into amphiphilic nanoparticles in aqueous solution with hydrodynamic radii (Rh) of 32.3–62.8 nm. The graft copolymers exhibited sharp temperature-dependent phase transitions, and the LCST could be adjusted from 22.3 to 74.1 °C by varying the molecular weight, the length of the OEG side chain, the polymer concentration and salt concentration. MTT assays revealed that the graft copolymers exhibited no detectable cytotoxicity at all test concentrations up to 1 mg mL−1. In vitrodegradation tests demonstrated that the graft copolymers could be degraded by proteinase K. The drug release behaviors from the PPLG112-g-MEO2 nanoparticles were evaluated at 37 °C and 15 °C using doxorubicin (DOX) as a model drug. The drug release behavior displayed thermosensitivity, and a sustained release profile was observed at physiological temperature. These results suggested that the novel biodegradable and biocompatible polypeptide derivatives with adjustable temperature sensitivity could be a promising material for biomedical applications.
Introduction
Stimuli-responsive polymers have received significant attention for their potential applications in the biomedical field in the past few decades.1–3 The stimuli include temperature, light, electric fields, pH and so on, which are promising for many biomedical applications, including delivery systems of smart drugs and genes, tissue engineering scaffolds and biological separation.4–12 Among these intelligent polymers, temperature sensitive polymers have attracted considerable interest due to their unique advantages in practical applications.13,14Typical temperature sensitive polymers include poly(N,N′-diethyl acrylamide),15 poly(dimethylaminoethyl methacrylate),16 poly(N-acryloylpyrrolidine),17 poly(2-isopropyl-2-oxazoline),18poly(N-isopropylacrylamide) (PNIPAM),19,20 poly(L-glutamate)-g-oligo(2-(2-(2-methoxyethoxy)ethoxy)ethyl methacrylate) (PGA-g-MEO2MA)21,22 and poly(oligo(ethylene glycol) (meth)acrylate) (POEGMA).23,24 In aqueous solution, such temperature sensitive polymers undergo a soluble–insoluble phase transition at their lower critical solution temperatures (LCSTs). Recently, an oligo(ethylene glycol) (OEG)-grafted polymethacrylate derivative (POEGMA) was reported, and its LCST can be adjusted by changing the feed ratio of different MEOxMA monomers (MEOx indicates an OEG containing xethylene glycol repeat units).25 For instance, the random copolymers of MEO2MA and MEO9MA exhibited LCST values in between 26 and 90 °C. A LCST of 37 °C (body temperature) was observed for the copolymer possessing 8% MEO9MA units per chain.26 In our recent work, a type of temperature sensitive polypeptide derivative, PGA-g-MEO2MA, was synthesized through the combination of ring opening polymerization (ROP) of γ-2-chloroethyl-L-glutamate-N-carboxyanhydride (CELG-NCA) and atom transfer radical polymerization of MEO2MA. This type of polymer not only exhibited sharp temperature sensitivity, but also showed good biocompatibility due to the introduction of the polypeptide component.22
Most currently used temperature sensitive polymers are based on polyacrylate or polyacrylamide derivatives, which are nonbiodegradable and exhibit limited biocompatibility, resulting in limitations in some biomedical applications in vivo. Accordingly, in this work, a new class of temperature sensitive copolymers based on polypeptides and oligo(ethylene glycol)s was synthesized by the combination of ROP of an alkyne-substituted NCA and subsequent click reaction between the alkyne groups and azido-substituted OEG. It is well known that polypeptides exhibit good biocompatibility and can be degraded into nontoxic amino acid residues by enzymes in vivo through the cleavage of peptide bonds. In addition, oligo(ethylene glycol)s exhibit LCSTs tunable by the chain length and can be excreted by glomerular filtration without toxicity in vivo caused by long-term accumulation.27,28 Therefore, we prepared a series of biocompatible and biodegradable thermosensitive copolymers by the combination of polypeptides with OEGs via click chemistry. The self-assembly behavior, secondary conformation and temperature sensitive phase transitions of the copolymer aqueous solutions were investigated. The influence of the molecular weight of the polypeptide backbone, the length of the side chain, polymer concentration and salt concentration on the phase transition behavior of the copolymer are discussed in detail. In addition, the cytotoxicity, enzymatic degradation by proteinase K and the release behavior of a hydrophobic drug, doxorubicin, from the copolymer nanoparticles were evaluated in vitro.
Experimental section
Materials
γ-Propargyl-L-glutamate-N-carboxyanhydride (PLG-NCA) was synthesized according to our previously reported method.292-(2-Methoxyethoxy)ethanol (MEO2OH), 2-(2-(2-methoxyethoxy)ethoxy)ethanol (MEO3OH, 97%, Sigma) and n-hexylamine (HA, 99%, Aldrich) were used without further purification. THF was refluxed with sodium and distilled under nitrogen prior to use. N,N-Dimethylformamide (DMF) was stored over calcium hydride (CaH2) and purified by vacuum distillation with CaH2. All the other reagents and solvents were purchased from Sinopharm Chemical reagent Co. Ltd, China, and used as obtained.Characterization
1H NMR and 13C NMR spectra were recorded on a Bruker AV 400 NMR spectrometer in the mixed solvent of deuterated trifluoroacetic acid and deuterated chloroform (CF3COOD/CDCl3: 1/1 (v/v)) or deuterated trifluoroacetic acid. Fourier transform infrared (FT-IR) spectra were recorded on a Bio-Rad Win-IR instrument using a KBr method. Weight-average molecular weights (Mw) and polydispersity indices (PDI) were determined by gel permeation chromatography using a series of linear Tskgel Super columns (AW3000 and AW5000) and Waters 515 HPLC pump with an OPTILAB DSP Interferometric Refractometer (Wyatt Technology) as the detector. The eluent was DMF containing 0.01 M LiBr at a flow rate of 1.0 mL min−1 at 50 °C. Monodisperse polystyrene standards purchased from Waters Co. with a molecular weight range of 1790 to 2.0 × 105 were used to generate the calibration curve. CD spectra were recorded on a JASCO J-810 spectrometer in deionized water at a concentration of 0.01 g L−1 at 25 °C. TEM measurement was performed on a JEOL JEM-1011 transmission electron microscope with an accelerating voltage of 100 kV. A drop of the nanoparticles solution (0.50 g L−1) was deposited onto a 230 mesh copper grid coated with carbon and allowed to dry at room temperature before measurement. DLS measurements were performed on a WyattQELS instrument with a vertically polarized He–Ne laser (DAWN EOS, Wyatt Technology) and 90° collecting optics. All the samples were prepared in aqueous solution at a concentration of 0.5 g L−1. Before measurements, the solutions were filtered through a 0.45 μm Millipore filter. The LCSTs were determined on a Shimadzu UV-2401PC UV-Vis spectrometer equipped with a Shimadzu S-1700 temperature controller at a heating rate of 0.5 °C min−1. Fluorescence excitation spectra were recorded on a Perkin-Elmer LS50B luminescence spectrometer at the detection wavelength (λem) of 390 nm.Synthesis of 1-(2-methoxyethoxy)-2-azidoethane (MEO2-N3) and 1-(2-(2-methoxyethoxy)ethoxy)-2-azidoethane (MEO3-N3)
Azides of MEO2 and MEO3 were prepared similarly as described in the literature.30 Briefly, MEO2OH (18.3 mL, 0.15 mol), methanesulfonyl chloride (21.2 mL, 0.18 mol), and THF (200 mL) were placed in a 500 mL round-bottom flask in an ice bath. Then triethylamine (25 mL, 0.18 mol) was added dropwise over 20 min. The reaction mixture was gradually warmed to room temperature and stirred overnight. The mixture was diluted by the addition of 200 mL H2O and the pH was adjusted to neutral by adding NaHCO3. Sodium azide (11.8 g, 0.18 mol) was added, and the reaction mixture was heated to evaporate THF and then refluxed overnight. After cooling to room temperature, the mixture was extracted by ethyl acetate. The combined organic phase was dried over MgSO4, filtered, and evaporated to obtain a yellow oil. The crude product was chromatographed on silica gel with a CH2Cl2/MeOH mixed solution (CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (v/v) = 15:1) as the eluent. The reaction yielded 19.1 g (88%). MEO3-N3 was synthesized in a similar procedure with a yield of 84%.
:MeOH (v/v) = 15:1) as the eluent. The reaction yielded 19.1 g (88%). MEO3-N3 was synthesized in a similar procedure with a yield of 84%.
Synthesis of poly(γ-propargyl-L-glutamate) (PPLG)
Clickable PPLG was synthesized by ring opening polymerization of γ-propargyl-L-glutamateN-carboxyanhydride (PLG-NCA). Typically, PLG-NCA (2.04 g, 9.7 mmol) was dissolved in 20 mL dry DMF and introduced into a flame-dried flask, followed by addition of 1.1 mL of 0.076 M n-hexylamine in 1,4-dioxane solution (monomer/initiator molar ratio = 120:1). The polymerization was stopped after stirring for 72 h at room temperature, and then the solution was precipitated in an excess amount of a diethyl ether/methanol (2:1) mixture. The obtained product was further washed twice with diethyl ether and dried overnight under vacuum, yielding PPLG 0.9 g (61%).
Synthesis of PPLG-g-MEO2 and PPLG-g-MEO3
PPLG-g-MEO2 and PPLG-g-MEO3 were synthesized by “click” chemistry. A typical procedure for the preparation of PPLG-g-MEO2 is briefly described as follows: PPLG (200 mg containing about 1.2 mmol alkyne) and MEO2-N3 (174 mg, 1.2 mmol) were firstly dissolved in 20 mL DMSO, and then the mixture was bubbled with nitrogen for 30 min. Then CuSO4·5H2O (60 mg) was introduced into the flask and the mixture was further bubbled with nitrogen for 5 min until sodium ascorbate (NaAsc, 240 mg) was added. The flask was sealed under nitrogen and immersed in an oil bath at 40 °C for 3 days. The reaction was stopped by adding a small amount of deionized water. DOWEXHCRW2 resins (600 mg) were added and the mixture was stirred overnight to remove copper ions. After filtration, the yellowish solution was dialyzed against deionized water for 3 days using a dialysis bag (MWCO 3500 Da). The final product, PPLG-g-MEO2, was obtained by lyophilization, affording 84% yield (314 mg).Preparation of nanoparticles
The copolymer nanoparticles were prepared by the dialysis method. PPLG112-g-MEO2 (5.0 mg) was firstly dissolved in DMF (1 mL) and the solution was stirred for 2 h to make the copolymer dissolve completely. Then the solution was added dropwise to deionized water (2 mL) under gentle stirring. After stirring overnight, DMF was removed by dialysis using a dialysis bag (MWCO 3500 Da) against deionized water for 24 h to obtain the nanoparticles.Critical aggregation concentration (CAC) measurements
A predetermined amount of pyrene in acetone was added to a series of ampules, and the acetone was removed by gentle flowing N2 and then by vacuum. One millilitre of graft copolymer solutions was added into the ampules before 1 mL of deionized water was added. Different concentrations of graft copolymer ranging from 6.1 × 10−5 to 1.0 g L−1 were obtained, while the concentration of pyrene in each ampule was fixed at 6.0 × 10−7 mol L−1, slightly lower than the saturation solubility of pyrene in water.Cytotoxicity measurement
The relative cytotoxicity was assessed with a methyl thiazolyl tetrazolium (MTT) viability assay against HeLa cells. The cells were seeded in 96-well plates at 20000 cells per well in 100 μL of complete Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, supplemented with 50 U mL−1penicillin and 50 U mL−1streptomycin, and incubated at 37 °C in 5% CO2 atmosphere for 24 h, followed by removing the culture medium and adding copolymer solutions (100 μL in complete DMEM medium) at different concentrations (0.06–1 g L−1). The cells were subjected to MTT assay after being incubated for another 24 h. The absorbance of the solution was measured on a Bio-Rad 680 microplate reader at 492 nm. Cell viability (%) was calculated according to the following equation:
viability (%) = (Asample/Acontrol) × 100%, where Asample and Acontrol are the absorbances of the sample well and control well (without the copolymers), respectively. The measurements were performed in triplicate.
In vitro enzymatic degradation
The in vitro enzymatic degradation was performed at 37 °C in phosphate buffered saline (PBS, pH 7.4) solution containing proteinase K (0.2 mg mL−1). The concentration of the graft copolymer was 0.5 mg mL−1. At predetermined time intervals, samples were taken out and freeze-dried for GPC analysis.In vitro doxorubicin release experiment
Doxorubicin (Dox, 3.0 mg) was added to 15 mL of 1 mg mL−1copolymer solution in DMF, and the mixture was stirred for 2 h. Then deionized water (15 mL) was added dropwise to the solution under stirring. The mixture was stirred overnight before dialysis against deionized water for 24 h using a dialysis bag (MWCO 3500 Da). After freeze drying, Dox-loaded nanoparticles were obtained.The drug loading content (DLC%) and the drug loading efficiency (DLE%) were calculated by the following equations:
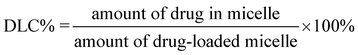
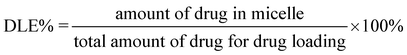
For the drug release study, the freeze-dried nanoparticles sample (5.0 mg) was resuspended in 5 mL phosphate buffered saline (PBS, pH 7.4) solution. The samples were sealed in dialysis bags (MWCO 3500 Da) and incubated in the release media (60 mL) at 37 °C and 15 °C, respectively, under oscillation at 60 r min−1. At given time intervals, the release medium (5 mL) was withdrawn for UV-Vis analysis and fresh buffer solution (5 mL) was supplemented. The released amount of Dox was determined from the absorbance at 481 nm based on a calibration curve of Dox in PBS. Then the accumulative weight and relative percentage of the released Dox were calculated as a function of incubation time.
Results and discussion
Synthesis and characterization of the graft copolymers
Because poly(γ-propargyl-L-glutamate) (PPLG) affords clickable alkyne pendants, various functional groups such as sugar azides and amino azides have been attached to the polypeptide backbone to impart functionality or intelligence to the polymer by click chemistry.29,31,32 In addition, it was found that oligo(ethylene glycol) (OEG)-grafted polymers could display a LCST in pure water or in physiological medium.33 Herein, a series of novel thermosensitive OEG-grafted polypeptides were synthesized with high efficiency by virtue of the versatile click method. First, PPLG was synthesized by ROP of γ-propargyl-L-glutamate-N-carboxyanhydride (PLG-NCA) with n-hexylamine as the initiator. A typical 1H NMR spectrum of PPLG is shown in Fig. 1A and all peaks are ascribed. The degree of polymerization (DP) of the polypeptide backbone was calculated by comparing the integration of the methyl peak of n-hexylamine at 0.91 ppm (–CH3) and a methylene peak of the glutamate unit at 2.58 ppm (–CH2CH2C(O)–), and the results are summarized in Table 1.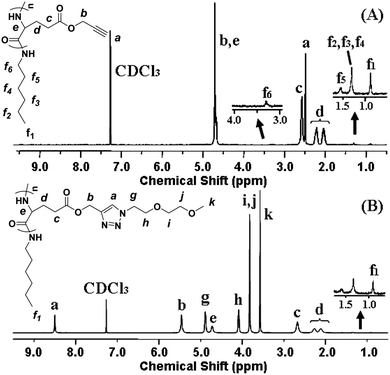 | ||
| Fig. 1 1H NMR spectra of (A) PPLG112 and (B) PPLG112-g-MEO2 (in CF3COOD + CDCl3 (1/1, v/v)). | ||
Two kinds of OEG side chains, 1-(2-methoxyethoxy)-2-azidoethane (MEO2-N3) and 1-(2-(2-methoxyethoxy)ethoxy)-2-azidoethane (MEO3-N3), were then grafted onto the PPLG backbone to form the thermosensitive graft polypeptides (Scheme 1). As shown in Fig. 1B, the propargyl peak at 2.49 ppm disappears completely and a new peak at 8.50 ppm (a) attributed to the proton in the triazole ring is observed, indicating the complete conversion of propargyl and azide groups into triazole rings. In addition, resonance peaks at 3.57 ppm (k), 3.80 ppm (i, j), 4.08 ppm (h) and 4.88 ppm (g) (Fig. 1B) are characteristic signals from the OEG side chain, which further confirm the successful grafting of OEG to the PPLG backbone. Similar results for PPLG112-g-MEO3 are shown in Fig. S1 of the ESI.† In addition, the chemical structures of the polymers were also confirmed by 13C NMR (Fig. S2, ESI†). GPC analysis was also used to characterize the synthesized PPLG and OEG-grafted polypeptide. Typical GPC chromatograms are shown in Fig. 2, and the PPLG homo-copolymers exhibited a relatively narrow PDI of around 1.03–1.16. Both PPLG112-g-MEO2 and PPLG112-g-MEO3 graft polypeptides showed reduced retention time compared to PPLG112, indicating higher molecular weights of the polypeptides after clicking with OEG side chains. A series of OEG-grafted polypeptides were synthesized and the results are showed in Table 2.
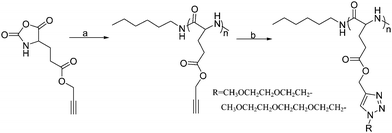 | ||
| Scheme 1 Synthesis route of the OEG-grafted poly(L-glutamate). Reagents and conditions: (a) n-hexylamine, DMF, 25 °C; (b) R-N3, CuSO4·5H2O, NaAsc, DMSO, 40 °C. | ||
| Code | Graft polypeptides | M n a | M w b | PDIb | LCST c/°C | CAC/10−2 g L−1 | R h/nm |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR. b Obtained by GPC. c The polymer concentration was 10 g L−1. | |||||||
| P1 | PPLG25-g-MEO2 | 7900 | 11800 | 1.05 | 36.9 | 4.57 | 40.6 ± 9.8 |
| P2 | PPLG25-g-MEO3 | 9000 | 14000 | 1.06 | 74.1 | 5.25 | 32.3 ± 7.5 |
| P3 | PPLG84-g-MEO2 | 26300 | 32900 | 1.06 | 28.9 | 3.47 | 44.8 ± 2.4 |
| P4 | PPLG84-g-MEO3 | 30000 | 36300 | 1.06 | 60.1 | 4.17 | 38.6 ± 6.1 |
| P5 | PPLG112-g-MEO2 | 35000 | 45600 | 1.14 | 24.7 | 2.88 | 46.1 ± 8.8 |
| P6 | PPLG112-g-MEO3 | 40000 | 55900 | 1.16 | 55.5 | 3.89 | 37.2 ± 7.9 |
| P7 | PPLG138-g-MEO2 | 43200 | 64600 | 1.13 | 22.3 | 2.63 | 62.8 ± 7.0 |
| P8 | PPLG138-g-MEO3 | 49200 | 73400 | 1.15 | 53.3 | 2.75 | 42.5 ± 8.3 |
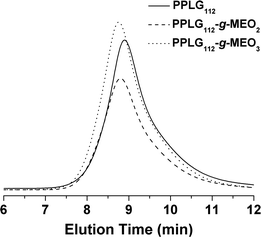 | ||
| Fig. 2 Typical GPC chromatograms of PPLG112, PPLG112-g-MEO2, and PPLG112-g-MEO3. | ||
Secondary structure characterization of the graft copolymers
To investigate their secondary structures, the graft polypeptides in solid state and aqueous solution were examined by FT-IR and CD analysis, respectively. As shown in Fig. 3A and Fig. S1A, ESI,†FT-IR spectra of the solid copolymer samples revealed the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O amine I absorption at 1654 cm−1 and N–H amine II band at 1549 cm−1, suggesting an α-helix structure.34 In agreement with this result, the CD spectra showed one maximum (λ = 192 nm) and two negative minima (λ = 208 and 222 nm) assigned to the perpendicularly polarized π–π* excitonic transition, the n–π* and the parallel polarized π–π* excitonic transitions, respectively (Fig. 3B).35–37 The molar ellipticities of PPLG84-g-MEO2, PPLG84-g-MEO3, PPLG112-g-MEO2 and PPLG112-g-MEO3 were determined to be 60500, 57700, 63300 and 58700, respectively, and the α-helix contents of the above mentioned graft polypeptides were almost 100%.21,29
O amine I absorption at 1654 cm−1 and N–H amine II band at 1549 cm−1, suggesting an α-helix structure.34 In agreement with this result, the CD spectra showed one maximum (λ = 192 nm) and two negative minima (λ = 208 and 222 nm) assigned to the perpendicularly polarized π–π* excitonic transition, the n–π* and the parallel polarized π–π* excitonic transitions, respectively (Fig. 3B).35–37 The molar ellipticities of PPLG84-g-MEO2, PPLG84-g-MEO3, PPLG112-g-MEO2 and PPLG112-g-MEO3 were determined to be 60500, 57700, 63300 and 58700, respectively, and the α-helix contents of the above mentioned graft polypeptides were almost 100%.21,29
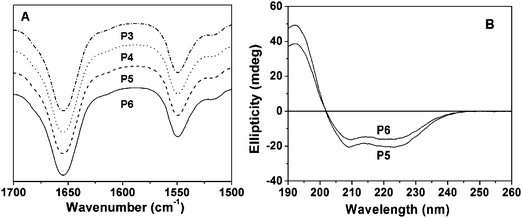 | ||
| Fig. 3 (A) Representative FT-IR spectra of P3 (PPLG84-g-MEO2), P4 (PPLG84-g-MEO3), P5 (PPLG112-g-MEO2) and P6 (PPLG112-g-MEO3), and (B) the CD spectra of P5 (PPLG112-g-MEO2) and P6 (PPLG112-g-MEO3), 0.01g L−1. | ||
Self-assembly behavior of the graft copolymers
It has been established that poly(L-glutamate) chains existing as α-helix structures exhibit hydrophobic interactions in aqueous solution.22,38,39 Therefore, the OEG-grafted poly(L-glutamate)s may form amphiphilic aggregates with a hydrophobic poly(L-glutamate) core and a hydrophilic OEG shell.22,39 In this study, both PPLG-g-MEO2 and PPLG-g-MEO3 formed stable amphiphilic nanoparticles with hydrodynamic radii (Rh) ranging from 32.3 to 62.8 nm at room temperature. The particle sizes were measured by DLS and the results are summarized in Table 2. The Rh increased from 40.6 to 62.8 nm with the increase of DP from 25 to 138 when the side chain was fixed as MEO2. A typical micrograph of PPLG112-g-MEO2nanoparticles observed by TEM is shown in Fig. 4A. The nanoparticles took a spherical shape with a diameter of about 50 nm, which was smaller than the hydrodynamic diameter determined by DLS, due to the shrinkage of the nanoparticles during the TEM sample drying process.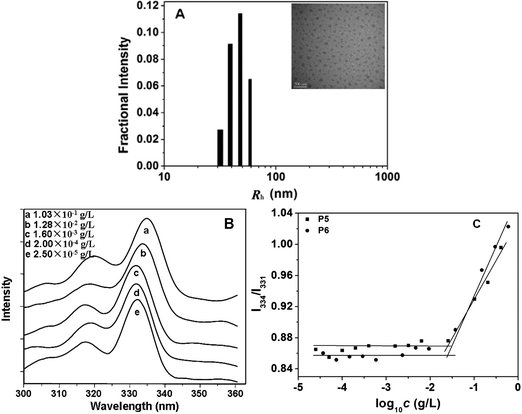 | ||
| Fig. 4 (A) Hydrodynamic radii (Rh) of PPLG112-g-MEO2 in aqueous solution at 25 °C determined by DLS. The inserted image is a representative TEM image of PPLG112-g-MEO2nanoparticles formed at room temperature, scale bar = 200 nm; (B) typical excitation spectra of pyrene in aqueous solution of PPLG112-g-MEO2 at different concentrations (λem = 390 nm); (C) the intensity ratio of I334/I331 as a function of concentration of P5 (PPLG112-g-MEO2) and P6 (PPLG112-g-MEO3). | ||
To investigate the critical aggregation concentrations (CAC) of the graft copolymers, a widely reported pyrene-probe-based fluorescence technique was used.40 The excitation spectra of pyrene as a function of the concentration of graft copolymers were measured. Fig. 4B shows the fluorescence excitation spectra of pyrene in the PPLG112-g-MEO2nanoparticles at different concentrations. A red shift of the (0, 0) absorption band from 331 to 334 nm was observed when the concentration of the copolymer increased from 2.5 × 10−5 to 1.03 × 10−1 g L−1. This red shift resulted from the transfer of pyrene molecules from a water environment to a hydrophobic core, indicating the formation of amphiphilic nanoparticles. In addition, from the plot of fluorescence intensity ratio of I334/I331versus log10c of the polymers (Fig. 4C), the CAC values were obtained. As listed in Table 2, the CACs of the graft copolymers ranged from 2.63 × 10−2 to 5.25 × 10−2 g L−1. When the side chain was fixed as MEO2 or MEO3, the CAC slightly decreased with the increasing DP of the polypeptide backbone, which indicated that the backbone had little influence on the stability of the nanoparticles. In addition, for the copolymers with fixed backbone lengths, the change of side chain from MEO2 to MEO3 resulted in higher CAC values (see Table 2 and Fig. 4C), due to the more hydrophilic MEO3 side chains compared to the MEO2 ones.
Lower critical solution temperatures (LCSTs) of the graft copolymers
The thermal-induced phase transition behaviors of the graft polypeptides were determined by monitoring the optical transmittance of the polymer aqueous solution at 550 nm. The LCST is defined as the temperature at which 50% decrease occurs in the optical transmittance. It is reported that the extent of dehydration or collapse of the shell of temperature responsive nanoparticles was dependent on the environmental temperature, which causes the change of the optical transmittance.41–43 As shown in Fig. 5, the graft copolymers exhibited sharp thermal-induced phase transitions with LCSTs ranging from 22.3 to 74.1 °C. The DP of the polypeptide backbone and the OEG side chain length exhibited marked influences on the LCST of the copolymers. For PPLG-g-MEO2, as the DP increased from 25 to 138, the LCST dropped sharply from 36.9 to 22.3 °C (Fig. 5B). This can be reasonably attributed to the fact that the increase of DP leads to the increase of the hydrophobic interactions of the polypeptide backbone. In contrast, graft copolymers with MEO3 side chains showed obviously higher LCSTs than those with MEO2 side chains, as shown in Table 2 (e.g. 55.5 °C for PPLG112-g-MEO3 and 24.7 °C for PPLG112-g-MEO2). This is owing to the higher hydrophilicity of the copolymers with longer OEG side chains.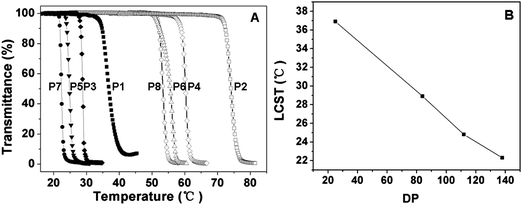 | ||
| Fig. 5 (A) Thermal phase transitions of P1 (PPLG25-g-MEO2), P2 (PPLG25-g-MEO3), P3 (PPLG84-g-MEO2), P4 (PPLG84-g-MEO3), P5 (PPLG112-g-MEO2), P6 (PPLG112-g-MEO3), P7 (PPLG138-g-MEO2) and P8 (PPLG138-g-MEO3), and the polymer concentration was 10 g L−1. (B) The lower critical solution temperatures (LCSTs) of PPLG-g-MEO2 as a function of the degree of polymerization. | ||
The influence of copolymer concentration on the LCST was investigated. As shown in Fig. 6 and Fig. S2, ESI,† the LCST showed a rapid decrease with increasing polymer concentration from 1 to 10 g L−1, as compared to only a few degrees decrease for PNIPAM and P(MEO2MA-co-OEGMA).42 In contrast, the rate of decrease of LCST reduced when the concentration was further increased from 10 to 20 g L−1. It can be explained that a higher concentration of polymer solution benefits the intermolecular aggregation which influences the LCST.44
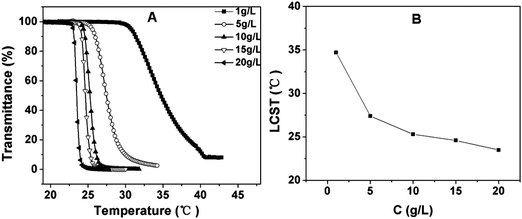 | ||
| Fig. 6 (A) Influence of the polymer concentration on the thermosensitive behavior of PPLG112-g-MEO2 in aqueous solution; (B) the LCST of PPLG112-g-MEO2 solution as a function of its concentration. | ||
It has been reported that some salts increase the LCST (“salting-in” effect), whereas some salts decrease the LCST (“salting-out” effect).33,45 In this study, the effect of salt concentration on the LCST was also evaluated. A typical “salting-out” effect was observed in our test procedures. As shown in Fig. 7 and Fig. S3, ESI,† the LCST decreased with the increase of salt concentration. For a 10 g L−1copolymer solution, the LCST of the copolymer in the presence of 9 g L−1sodium chloride (physiological salt concentration) was about 3 °C lower than that in pure water (Fig. 7A and Fig. S3A, ESI†). This can be reasonably explained, as the presence of sodium chloride results in a partial dehydration of the copolymers and therefore a decrease in the LCST.42 Although the change in LCST with varying salt concentration was not very sharp, it is interesting to note that the LCST had quite a good linear relationship with respect to the salt concentration in the experimental range (Fig. 7B and Fig. S3B, ESI†).
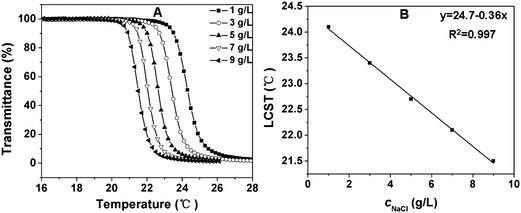 | ||
| Fig. 7 (A) Influence of sodium chloride concentration on the thermosensitive behavior of 10 g L−1 PPLG112-g-MEO2 solution; (B) the LCST of PPLG112-g-MEO2 solution as a function of sodium chloride concentration. | ||
In vitro cytotoxicity and enzymatic degradation of the graft copolymers
The biocompatibility and potential cytotoxicity of the polymeric materials is critical for the biomedical applications. It has been pointed out that PNIPAM showed relatively high cellular cytotoxicity at a concentration level of 5 mg ml−1.46 Furthermore, the N-isopropylacrylamide (NIPAM) monomer exhibited dramatically higher cytotoxicity than PNIPAM.43 Here the in vitro cytotoxicity and biocompatibility of the graft copolymers were evaluated by MTT assay.20 The cells were treated with the graft polypeptides (PPLG84-g-MEO2 and PPLG112-g-MEO2) at different concentrations for 24 h, and sodium dodecyl sulfate was used as the positive control. The relative cell viabilities are shown in Fig. 8. It was observed that the HeLa cells treated with our graft copolymers remained almost 100% viable at all test concentrations up to 1 mg mL−1, indicating noncytotoxicity and good biocompatibility of the graft copolymers.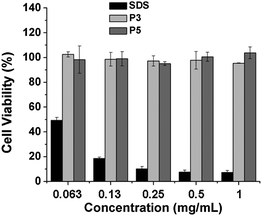 | ||
| Fig. 8 In vitro cytotoxicity of P3 (PPLG84-g-MEO2) and P5 (PPLG112-g-MEO2) to HeLa cells with SDS as a control. Data were presented as the average ± standard deviation (n = 3). | ||
In addition, the enzymatic degradation behavior of the grafted copolymers was evaluated by incubation of a copolymer with proteinase K (0.2 mg ml−1). As shown in Fig. S6, ESI,† a 13% decrease in the molecular weight of the copolymer was observed as the incubation time was increased to 12 h, indicating that the OEG-grafted polypeptide could be degraded by proteinase K.
In vitro doxorubicin (Dox) release experiment
To investigate the temperature-dependent drug release behavior of the drug-loaded copolymer nanoparticles, doxorubicin (Dox) was used as a model drug. The drug loading content (DLC%) was 2.4% with drug loading efficiency of 14.4% for PPLG112-g-MEO2. The release profiles of Dox from the PPLG112-g-MEO2nanoparticles in phosphate buffered saline solution (PBS, pH 7.4) were studied at 37 °C and 15 °C, which are above and below the LCST (24.7 °C), respectively. As shown in Fig. 9, over 80% of the Dox was released from the nanoparticles within 24 h at 37 °C. In contrast, at 15 °C below the LCST, only 40% of the Dox was released in the same period. This is consistent with the results of the PNIPAM-based thermosensitive systems reported by Okano and Liu,47,48 which suggested that the drug release from the temperature-sensitive amphiphilic nanoparticles could be accelerated by increasing the temperature above their LCSTs. In addition, the release of Dox displayed a constant rate in the first 24 h at 37 °C. This demonstrated that an ideal constant Dox release could be obtained in the nanoparticle system. The result suggested that the graft copolymers could be promising candidates as drug carriers for controlled drug delivery.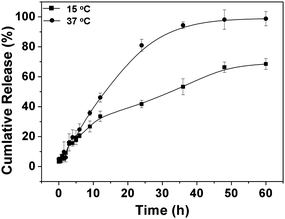 | ||
| Fig. 9 Release profile of doxorubicin from PPLG112-g-MEO2nanoparticles in PBS (pH 7.4) at 37 and 15 °C. | ||
Conclusions
A series of thermoresponsive OEG-grafted polypeptides were synthesized with high efficiency by the versatile click method. All the graft copolymers existed as α-helix structures, and they could self-assemble into amphiphilic nanoparticles in aqueous solution with Rh values of around 32.3–62.8 nm. The copolymers showed sharp thermal phase transitions with the LCSTs ranging from 22.3 to 74.1 °C, and the LCSTs were assessed to be dependent on the DP of the polypeptide backbone, the length of the side chains, polymer concentration and salt concentration. In addition, the graft polypeptides were confirmed to be biocompatible and non-toxic using the MTT method, and they were degradable in the presence of proteinase K. Drug loading and release were also conducted with these thermosensitive nanoparticles using doxorubicin as the model drug. A temperature-dependent sustained release profile was observed. Therefore, it is believed that these novel OEG-grafted polypeptides with tunable temperature responsiveness should be promising for smart biomedical applications.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (Key project 50733003, project 51003103 and 50973108), the Ministry of Science and technology of China (International cooperation and communication program 2010DFB50890), and the Scientific Development Program of Jilin Province (201101082).References and notes
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- C. L. He, S. W. Kim and D. S. Lee, J. Controlled Release, 2008, 127, 189–207 CrossRef CAS.
- P. Bawa, V. Pillay, Y. E. Choonara and L. C. du Toit, Biomed. Mater., 2009, 4(2), 022001 Search PubMed.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- M. Bikram, A. M. Gobin, R. E. Whitmire and J. L. West, J. Controlled Release, 2007, 123, 219–227 CrossRef CAS.
- W. S. Shim, J. H. Kim, K. Kim, Y. S. Kim, R. W. Park, I. S. Kim, I. C. Kwon and D. S. Lee, Int. J. Pharm., 2007, 331, 11–18 CrossRef CAS.
- X. Jiang, C. A. Lavender, J. W. Woodcock and B. Zhao, Macromolecules, 2008, 41, 2632–2643 CrossRef CAS.
- X. G. Jiang, S. Jin, Q. X. Zhong, M. D. Dadmun and B. Zhao, Macromolecules, 2009, 42, 8468–8476 CrossRef CAS.
- M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Biomacromolecules, 2008, 9, 1340–1347 CrossRef CAS.
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS.
- J. G. Ray, J. T. Ly and D. A. Savin, Polym. Chem., 2011, 2, 1536–1541 RSC.
- J. Wu, W. Wei, L. Y. Wang, Z. G. Su and G. H. Ma, Biomaterials, 2007, 28, 2220–2232 CrossRef CAS.
- Z. W. Cui, B. H. Lee and B. L. Vernon, Biomacromolecules, 2007, 8, 1280–1286 CrossRef CAS.
- W. S. Cai, L. H. Gan and K. C. Tam, Colloid Polym. Sci., 2001, 279, 793–799 CrossRef CAS.
- J. Li, W. D. He, S. C. Han, X. L. Sun, L. Y. Li and B. Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 786–796 CrossRef CAS.
- K. Skrabania, J. Kristen, A. Laschewsky, O. Akdemir, A. Hoth and J. F. Lutz, Langmuir, 2007, 23, 84–93 CrossRef CAS.
- J. S. Park and K. Kataoka, Macromolecules, 2006, 39, 6622–6630 CrossRef CAS.
- S. Ohya, H. Sonoda, Y. Nakayama and T. Matsuda, Biomaterials, 2005, 26, 655–659 CrossRef CAS.
- H. M. Li, M. Li, X. Yu, A. P. Bapat and B. S. Sumerlin, Polym. Chem., 2011, 2, 1531–1535 RSC.
- J. X. Ding, C. S. Xiao, Z. H. Tang, X. L. Zhuang and X. S. Chen, Macromol. Biosci., 2011, 11, 192–198 CrossRef CAS.
- J. X. Ding, C. S. Xiao, L. Zhao, Y. L. Cheng, L. L. Ma, Z. H. Tang, X. L. Zhuang and X. S. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2665–2676 CrossRef CAS.
- S. Sugihara, S. Kanaoka and S. Aoshima, Macromolecules, 2005, 38, 1919–1927 CrossRef CAS.
- J. F. Lutz, K. Weichenhan, O. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- K. Osada and K. Kataoka, in Peptide Hybrid Polymers, 2006, vol. 202, pp. 113–153 Search PubMed.
- S. N. S. Alconcel, A. S. Baas and H. D. Maynard, Polym. Chem., 2011, 2, 1442–1448 RSC.
- C. S. Xiao, C. W. Zhao, P. He, Z. H. Tang, X. S. Chen and X. B. Jing, Macromol. Rapid Commun., 2010, 31, 991–997 CrossRef CAS.
- B. C. Mei, K. Susumu, I. L. Medintz and H. Mattoussi, Nat. Protoc., 2009, 4, 412–423 Search PubMed.
- A. C. Engler, H. I. Lee and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 9334–9338 CrossRef CAS.
- A. C. Engler, A. Shukla, S. Puranam, H. G. Buss, N. Jreige and P. T. Hammond, Biomacromolecules, 2011, 12, 1666–1674 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. X. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS.
- E. R. Blout and A. Asadourian, J. Am. Chem. Soc., 1956, 78, 955–961 CrossRef CAS.
- G. Holzwart and P. Doty, J. Am. Chem. Soc., 1965, 87, 218–228 CrossRef CAS.
- C. T. Yang, Y. L. Wang and Y. C. Chang, Biomacromolecules, 2010, 11, 1308–1313 CrossRef CAS.
- M. Rinaudo and A. Domard, J. Am. Chem. Soc., 1976, 98, 6360–6364 CrossRef CAS.
- J. Y. Su, R. S. Hodges and C. M. Kay, Biochemistry, 1994, 33, 15501–15510 CrossRef CAS.
- C. L. He, C. W. Zhao, X. H. Guo, Z. J. Guo, X. S. Chen, X. L. Zhuang, S. Y. Liu and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4140–4150 CrossRef CAS.
- J. Cheng, J. X. Ding, Y. C. Wang and J. Wang, Polymer, 2008, 49, 4784–4790 CrossRef CAS.
- X. L. Zhao, W. G. Liu, D. Y. Chen, X. Z. Lin and W. W. Lu, Macromol. Chem. Phys., 2007, 208, 1773–1781 CrossRef CAS.
- J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- Y. Xia, X. C. Yin, N. A. D. Burke and H. D. H. Stover, Macromolecules, 2005, 38, 5937–5943 CrossRef CAS.
- K. Suwa, Y. Wada, Y. Kikunaga, K. Morishita, A. Kishida and M. Akashi, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1763–1768 CrossRef CAS.
- R. Freitag and F. Garret-Flaudy, Langmuir, 2002, 18, 3434–3440 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- J. E. Chung, M. Yokoyama, M. Yamato, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1999, 62, 115–127 CrossRef CAS.
- X. J. Wan, T. Liu and S. Y. Liu, Biomacromolecules, 2011, 12, 1146–1154 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00281c |
| This journal is © The Royal Society of Chemistry 2011 |