Ring-opening polymerization of an O-carboxyanhydride monomer derived from L-malic acid†
Ryan J.
Pounder
,
David J.
Fox
,
Ian A.
Barker
,
Michael J.
Bennison
and
Andrew P.
Dove
*
Department of Chemistry, University of Warwick, Coventry, UK. E-mail: a.p.dove@warwick.ac.uk; Tel: +44(0)24 7652 4107
First published on 19th July 2011
Abstract
The synthesis and ring-opening polymerization (ROP) of an O-carboxyanhydride (OCA) monomer derived from L-malic acid (L-malOCA) is reported. Application of 4-dimethylaminopyridine as catalyst led to the observation of a number of undesirable side products. Investigation of different para-substituted pyridines as catalysts identified 4-methoxypyridine to have the ideal balance of activity and selectivity to enable the controlled ROP of L-malOCA. Deprotection of the benzyl ester side groups of the resultant polymers was achieved by hydrogenolysis and the resulting hydrophilic poly(α-malic acid) was observed to fully degrade within 7 days in aqueous solution.
Introduction
The controlled synthesis of polymers derived from renewable resources presents a considerable challenge.1,2 The wealth of renewable resources containing stereochemical centers and latent functional groups however, make their utilization appealing for the synthesis of functional polymers. A particularly interesting potential source is malic acid, MA, the α-hydroxy acid equivalent of aspartic acid. Available commercially in both enantiomeric forms, MA contains a carboxylic acid β to an α-hydroxy acid unit that is a suitable site for the introduction of a range of functional groups thus making it an ideal renewable feedstock for the synthesis of functional poly(ester)s.3,4Poly(ester)s are commonly accessed by either polycondensation methods or by the ring-opening polymerization (ROP).5–8 Of these, the latter is generally preferred on account of the high levels of control that are possible over the molecular parameters of the resultant polymers including attainment of polymers with predictable molar mass, low molar mass distributions, high end-group fidelity and even control over polymer tacticity.7–11 Despite these favorable characteristics, poly(ester) synthesis by ROP is largely limited to monomers that do not contain high levels of functional groups. This largely results from the prevalence of transesterification side reactions that occur in the presence of even simple groups such as alcohols, amines and esters hence resulting in a loss of control over the molecular parameters.
A wide range of monomers have been synthesized and studied to incorporate functional groups along poly(ester) backbones.3,4,6,11–15 The derivation of monomers around a β-propriolactone structure has provided a particularly wide range of functional group incorporation.3,4,12,14,15 Notably, malic acid has been utilized to excellent effect to produce derivatives of poly(β-malic acid) with excellent effect.3,4,12,14–29 While several studies have investigated the ROP of 6-membered cyclic esters derived from a range of sources,6,13,15,30 relatively few have focused on the application of malic acid, either derived from aspartic acid or obtained directly.14 Kimura et al. reported the synthesis of 3-(S)-[(benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (BMD) with ROP mediated by stannous(II) octanoate (Sn(Oct)2) resulting in relatively poorly defined poly(ester)s (e.g. [M]/[I] = 100; Mn = 10 500 g mol−1; PDI = 2.0).31–33 Concurrently with Bourissou and coworkers who used a related monomer derived from glutamic acid,34 we have demonstrated that the application of highly specific catalysis has enabled the controlled ROP of this monomer.35 In common with many other hindered cyclic diester monomers, synthesis of poly(α-malic acid) derivatives by the controlled ROP of the symmetrical 3,6-(S)-[di(benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (malide) however remains challenging.35,36 Recent developments have detailed the synthesis and ROP of 1,3-dioxolane-2,5-diones or O-carboxyanhydrides (OCAs) that provide activated equivalents of cyclic diesters that are driven entropically by release of CO2 rather than enthalpically by ring-strain.37,38 Following their initial report that the ROP of 5-(S)-methyl-1,3-dioxolane-2,4-dione (L-lacOCA) using a mild 4-dimethylaminopyridine (DMAP) catalyst resulted in the isolation of a polymer equivalent to poly(L-lactide),37 Bourissou, Martin-Vaca and coworkers demonstrated that the ROP of the OCA derived from O-benzyl-L-glutamic acid (L-gluOCA) resulted in the synthesis of well controlled functional poly(ester)s.39 Most notably, the derivatized monomer was polymerized more rapidly than L-lacOCA, eliminating the retardation of ROP commonly observed with bulky substituents on 6-membered cyclic diesters and hence making this an extremely attractive route for the future synthesis of side-chain-functional poly(ester)s. A computational mechanistic investigation revealed the key role of multiple hydrogen bonding in the ROP with a general base activation of the initiating/propagating alcohol and monomer being more energetically favorable than the nucleophilic activation of the monomers.40,41 Herein we report the application of commercially available L-malic acid in the synthesis of functional poly(ester)s via an OCA monomer to provide a potentially versatile route to a range of stereopure side-chain functional poly(ester)s.
Experimental section
Materials
THF was refluxed over sodium then distilled, degassed and stored under a nitrogen atmosphere, CHCl3 was refluxed over CaH2 then distilled, degassed and stored under a nitrogen atmosphere. All alcohol initiators (including PEO macroinitiators) were obtained from Aldrich and dried over suitable dry agents before being distilled, degassed and/or sublimed as required. 2-hydroxy-succinic acid 4-benzyl ester35 was synthesized as previously reported and L-lacOCA37 was synthesized and purified/dried as previously reported. All other chemicals and solvents were obtained from Aldrich and used as received.General considerations
All manipulations were performed under moisture- and oxygen-free conditions either in a nitrogen-filled glovebox or by standard Schlenk techniques. Size-exclusion chromatography (SEC) was used to determine the molar masses and molar mass distributions (polydispersities, PDIs) of the synthesised polymers. SEC in THF was conducted on a system comprised of a Varian 390-LC-Multi detector suite fitted with differential refractive index (DRI), light scattering (LS) and ultra-violet (UV) detectors equipped with a guard column (Varian Polymer Laboratories PLGel 5 μM, 50 × 7.5 mm) and two mixed D columns (Varian Polymer Laboratories PLGel 5 μM, 300 × 7.5 mm). The mobile phase was tetrahydrofuran with 5% triethylamine eluent at a flow rate of 1.0 mL min−1, and samples were calibrated against Varian Polymer laboratories Easi-Vials linear poly(styrene) standards (162–2.4 × 105 g mol−1) using Cirrus v3.3. NMR spectra were recorded on a Bruker DPX-300, DPX-400, AC400, or DRX-500 spectrometer at 293 K unless stated otherwise. Chemical shifts are reported as δ in parts per million (ppm) and referenced to the chemical shift of the residual solvent resonances (CDCl31H: δ = 7.26 ppm; 13C δ = 77.16 ppm). Mass spectra were acquired by MALDI-TOF (matrix-assisted laser desorption and ionisation time-of-flight) mass spectrometry using a Bruker Daltonics Ultraflex II MALDI-TOF mass spectrometer, equipped with a nitrogen laser delivering 2 ns laser pulses at 337 nm with positive ion TOF detection performed using an accelerating voltage of 25 kV. Solutions of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propylidene]malonitrile (DCTB) as matrix (0.3 μL of a 10 g L−1acetone solution), sodium trifluoroacetate as cationisation agent (0.3 μL of a 10 g L−1acetone solution), and analyte (0.3 μL of a 1 g L−1DCM solution) were applied sequentially to the target followed by solvent evaporation to prepare a thin matrix/analyte film. The samples were measured in linear or reflectron ion mode and calibrated by comparison to 2 × 103 and 5 × 103 g mol−1poly(ethylene glycol) standards. Specific rotation measurements were recorded in CHCl3 on a Perkin-Elmer 241 polarimeter using a sodium source (λ = 589 nm) and a 1 cm rotation cell. Elemental analyses were performed by Warwick Analytical Services.Synthesis of 5-(S)-[(benzyloxycarbonyl)methyl]-1,3-dioxolane-2,4-dione (L-malOCA)
To a suspension of α-hydroxy acid, 2-hydroxy-succinic acid 4-benzyl ester, (4.78 g, 0.021 mol, 1 equiv) in dry THF (150 mL) was added diphosgene (3.1 mL, 0.026 mol, 1.2 equiv) under a nitrogen atmosphere. The resulting mixture was then treated with activated carbon and left to stir at room temperature for 18 h. The solution was then filtered off the activated carbon and concentrated in vacuo. The resulting residue was washed with pentanes (2 × 100 mL) and recrystallised from Et2O/petroleum ether (b.p. 40–60 °C) and dried over 4 Å molecular sieves to yield L-malOCA as a white solid. (3.62 g, 14.3 mmol 68%). 1H NMR (CDCl3, 400 MHz): δ = 7.42–7.33 (5H, m, –CHaromatic); 5.14 (2H, s, –CH2Ar); 5.09 (1H, dd, 2JHH = 5.57 Hz, 3JHH = 4.11 Hz, –CHCH2COOCH2Ar); 3.13 (2H, dd, 2JHH = 5.57 Hz, 3JHH = 4.11 Hz, –CH2COOCH2Ar). 13C NMR (CDCl3, 400 MHz): δ = 167.8 (–CH2COOCH2Ar); 166.7 (–OCOOCOCH–); 145.4 (–OCOOCOCH–); 134.4 (–Cipso aromatic); 128.9 (–Cmeta aromatic); 128.8 (–Cpara aromatic); 128.7 (–Cortho aromatic); 75.0 (–OCOOCOCH–); 68.2 (–CH2Ar); 34.4 (–CHCH2COOCH2Ar). Analysis: Calculated (Found) C: 57.6 (57.2); H: 4.0 (4.1). [α]33D values of −21.9° (in CHCl3, c = 5.96 g L−1).‡General procedure for polymerisation of L-malOCA ([M]/[I] = 20)
A solution of amine catalyst (1 equiv) and neo-pentanol (0.88 mg, 0.01 mmol, 1 equiv) was added to L-malOCA (50 mg, 0.2 mmol, 20 equiv) in CHCl3 (0.3 mL). The solution was left to stir at room temperature for the allotted time period before being diluted with DCM (4 mL), washed with cold 2 M HCl(aq) (2 × 5 mL) and brine (5 mL). The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The PBMA was precipitated into ice cold petroleum ether (b.p. 40–60 °C) to yield PBMA20 as a tacky solid (0.021 g, 0.005 mmol, 50%). 1H NMR (CDCl3, 400.0 MHz): δ = 7.42–7.20 (100H, m, –CHaromatic), 5.61–5.51 (20H, m, –CHCOO–), 5.17–5.04 (40H, m, –CH2Ar), 3.79 (2H, m, –CH2C(CH3)3), 3.05 – 2.78 (40H, m, –CH2COOCH2Ar), 0.89 (9H, s, –CH2C(CH3)3). SEC (THF, RI): Mn (PDI) = 3 860 g mol−1 (1.10) using 4-methoxypyridine as the ROP catalyst.General procedure for the deprotection of PBMA ([M]/[I] = 20)
A balloon of H2 was bubbled through a suspension of PBMA (0.05 g, 0.0095 mmol) and Pd/C (0.01 g, 10 wt. % loading) in THF (20 mL) for 15 min. The solution was then filtered to remove Pd/C and concentrated in vacuo. The PMA was extracted into MeOH and concentrated in vacuo to yield the desired product as a colorless oil (0.027 g, 0.012 mmol, 96%). 1H NMR (d8-THF, 400.0 MHz): δ = 5.60–5.53 (20H, m, –CHCOO–), 3.03–2.76 (40H, m, –CH2COOCH2Ar), 3.84 (2H, m, –CH2(CH3)3), 0.9 (9H, s, –CH2C(CH3)3). 13C{1H} NMR (d8-THF, 100.0 MHz): δ = 170.4 (–O(CO)CHO(CO)–), 168.4 (–CH2COOH), 70.6 (–O(CO)CHO(CO)–), 36.2 (–CH2COOH). SEC (THF (0.1 M citric acid), RI): Mn (PDI) = 1 100 g mol−1 (1.10).General procedure for the degradation of the PMA ([M]/[I] = 15)
PMA (56 mg, 0.0306 mmol) was dissolved in H2O (64 mL) and monitored via acid–base titration of a sample (0.2 mL) with an aqueous NaOH solution (0.45 mmol L−1) using phenolphthalein as a pH indicator, 1H NMR spectroscopy in D2O and ESI-MS analysis.Results and discussion
Monomer synthesis and polymerization
The synthesis of 5-(S)-[(benzyloxycarbonyl)methyl]-1,3-dioxolane-2,4-dione (L-malOCA) was achieved by the addition of diphosgene to β-benzyl α-(L)-malate which in turn was synthesized from L-malic acid, as we have previously reported.35
ROP of L-malOCA was initially attempted with neo-pentanol initator and DMAP as a catalyst at 25 °C in CH2Cl2 solution, comparable to the conditions successfully applied to the ROP of L-lacOCA and L-gluOCA. Initial studies focussed on a DMAP:initator ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with a monomer-to-initiator ratio of 20 ([M]/[I] = 20) with progress of the polymerization being monitored by the observation of the reduction of the methine resonance at δ = 5.09 ppm of the monomer and the appearance of the corresponding broadened multiplets at δ = 5.61–5.51 ppm in poly(β-benzyl α-(L)-malate), P(L-BMA), using 1H NMR spectroscopy. Upon completion of the allotted time, polymerizations were quenched via a 1.0 M HCl(aq) wash to remove DMAP before being precipitated into ice cold petroleum ether (b.p. 40–60 °C). Under these conditions, the ROP of L-malOCA achieved >90% monomer conversion in less than one minute with SEC analysis of the resultant P(L-BMA)20 indicating that the polymerization was well controlled, displaying a number-average molar mass (Mn) of 3 700 g mol−1 with a PDI of 1.19 (theoretical Mn = 4 208 g mol−1). Further investigation of the ‘living’ characteristics of the polymerization resulted in the observation of a linear correlation between Mn and initial monomer to initiator ratio and a second-feed experiment in which the chain extension of a P(L-BMA)20 macroinitiator (Mn = 3 700 g mol−1; PDI = 1.19) with 20 equiv. L-malOCA resulted in the isolation of a P(L-BMA)40 that exhibited a doubling in molar mass (Mn = 7 390 g mol−1) while maintaining a low PDI of 1.20. Furthermore, leaving the resultant P(L-BMA)40 for 2 h (24 times longer than required to reach >90% monomer conversion) in the presence of DMAP resulted in negligible changes in both the molar mass (Mn = 7 050 g mol−1) and molar mass distribution (PDI = 1.19) suggesting that transesterification side reactions were minimal despite full monomer consumption.
:1 with a monomer-to-initiator ratio of 20 ([M]/[I] = 20) with progress of the polymerization being monitored by the observation of the reduction of the methine resonance at δ = 5.09 ppm of the monomer and the appearance of the corresponding broadened multiplets at δ = 5.61–5.51 ppm in poly(β-benzyl α-(L)-malate), P(L-BMA), using 1H NMR spectroscopy. Upon completion of the allotted time, polymerizations were quenched via a 1.0 M HCl(aq) wash to remove DMAP before being precipitated into ice cold petroleum ether (b.p. 40–60 °C). Under these conditions, the ROP of L-malOCA achieved >90% monomer conversion in less than one minute with SEC analysis of the resultant P(L-BMA)20 indicating that the polymerization was well controlled, displaying a number-average molar mass (Mn) of 3 700 g mol−1 with a PDI of 1.19 (theoretical Mn = 4 208 g mol−1). Further investigation of the ‘living’ characteristics of the polymerization resulted in the observation of a linear correlation between Mn and initial monomer to initiator ratio and a second-feed experiment in which the chain extension of a P(L-BMA)20 macroinitiator (Mn = 3 700 g mol−1; PDI = 1.19) with 20 equiv. L-malOCA resulted in the isolation of a P(L-BMA)40 that exhibited a doubling in molar mass (Mn = 7 390 g mol−1) while maintaining a low PDI of 1.20. Furthermore, leaving the resultant P(L-BMA)40 for 2 h (24 times longer than required to reach >90% monomer conversion) in the presence of DMAP resulted in negligible changes in both the molar mass (Mn = 7 050 g mol−1) and molar mass distribution (PDI = 1.19) suggesting that transesterification side reactions were minimal despite full monomer consumption.
End group analysis by 1H NMR spectroscopy of a P(L-BMA) ([M]/[I] = 20; Mn = 4 210 g mol−1; PDI = 1.22) confirmed a DP = 23 polymer based on the integration of the tert-butylneo-pentyl ester resonances at δ = 0.87 ppm against those of the main chain methine protons at δ = 5.55 ppm (Fig. 1a) and further analysis by matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) revealed a single distribution centered around m/z = 4234.3 which corresponds to a sodium charged DP20 polymer chain with a neo-pentyl ester end group; a regular spacing equal to the molar mass of the repeat unit of benzyl α-(L)-malate (m/z = 206) demonstrates that no significant transesterification of the polymer chains occurs (Fig. 1b).
![(a) 1H NMR spectrum (400 MHz; CDCl3) and (b) MALDI-TOF MS of a P(l-BMA)20 (Mn = 4 210 g mol−1, PDI = 1.22) prepared by ROP of l-malOCA ([l-malOCA]0 = 0.32 M) catalyzed with 5 mol% DMAP using neo-pentanol as the initiator and the presence of impurities (*).](/image/article/2011/PY/c1py00254f/c1py00254f-f1.gif) | ||
| Fig. 1 (a) 1H NMR spectrum (400 MHz; CDCl3) and (b) MALDI-TOF MS of a P(L-BMA)20 (Mn = 4 210 g mol−1, PDI = 1.22) prepared by ROP of L-malOCA ([L-malOCA]0 = 0.32 M) catalyzed with 5 mol% DMAP using neo-pentanol as the initiator and the presence of impurities (*). | ||
Despite the evidence suggesting that the DMAP-catalyzed ROP of L-malOCA proceeds in a highly controlled manner, polymer impurities were clearly visible in both the 1H NMR and MALDI-TOF MS spectra of the polymer obtained (Fig. 1). By integration of the impurities at δ = 2.90 ppm against that of the equivalent desired polymer resonances at δ = 3.18 ppm in the 1H NMR spectrum the impurities were estimated to be up to 5% of the isolated product. The 13C NMR spectrum of the polymer also reveals several resonances in the carbonyl region (see ESI) which indicate the presence of polymer impurities resulting from side reactions. Initial attempts to remove these side products through sequential precipitation into ice cold acidified methanol was successful in the removal of the major impurities observed in the 1H NMR spectrum however analysis viaMALDI-TOF MS indicated that the larger main side product distribution remained. Furthermore, precipitation resulted in a significant detrimental effect on the yield, decreasing to 19%.
Catalyst optimization
We postulated that the impurities may be a consequence of deprotonation of the acidic methine proton in the monomer by DMAP. To this end, we studied the application of other less basic para-substituted pyridines to mediate the ROP of L-malOCA (Table 1).| R-Pyridine | pKa | Time (min) | Monomer Conversion (%) | M n c (g mol−1) | M w/Mnc | DP b |
|---|---|---|---|---|---|---|
| a [L-malOCA]0 = 0.32 M; [ROH] = 0.016 M; 5 mol% amine catalyst, 25 °C. b Determined by 1H NMR Spectroscopy. c Determined by SEC analysis. d Polymer impurity observed by MALDI-TOF MS analysis. e Polymer impurity observed by 1H NMR Spectroscopy. f Polymer impurity observed by MALDI-TOF MS analysis at low catalyst concentration only. | ||||||
| 4-dimethyl-amino-d,e | 9.7 | 5 | >95 | 3730 | 1.19 | 23 |
| 4-morpholino-d,e | 8.8 | 60 | 94 | 3870 | 1.14 | 19 |
| 4-methoxy-d | 6.6 | 90 | 95 | 3860 | 1.10 | 22 |
| 4-methyl-f | 6.0 | 160 | 95 | 2950 | 1.12 | 17 |
| H-f | 5.3 | 435 | 93 | 2100 | 1.13 | 16 |
ROP of L-malOCA ([M]/[I] = 20), initiated by neo-pentanol with 1 equivalent of substituted pyridine per alcohol was studied. Application of less basic pyridines led to a sequential decrease in polymerization activity. While SEC analysis of P(L-BMA)20 synthesized using 4-morpholinopyridine indicated that the polymerization was well controlled (Mn = 3 870 g mol−1; PDI = 1.14). Both 1H NMR spectroscopy and MALDI-TOF MS analysis indicated that side products were still present. Application of 4-methoxypyridine again resulted in a well controlled polymerization (Mn = 3 860 g mol−1; PDI = 1.10) however notably, 1H NMR spectroscopic analysis of the polymer did not reveal any side-product formation (Fig. 2a) and 13C NMR revealed only a minor amount of racemization of the monomer occurred during polymerization in comparison to the DMAP-catalyzed reaction. MALDI-TOF MS analysis indicated a main distribution centered around m/z = 4234.5 corresponding to a DP of 20 however, an impurity remained visible by MALDI-TOF MS analysis (Fig. 2b) but was able to be removed using column chromatography (Fig. 2c).
![(a) 1H NMR spectrum (400 MHz; CDCl3), (b) crude MALDI-TOF MS and (c) MALDI-TOF MS after column chromatography of a P(l-BMA) ([M]/[I] = 20) (Mn = 3 860 g mol−1, PDI = 1.10) prepared by ROP of l-malOCA ([l-malOCA]0 = 0.32 M) catalyzed with 5 mol% 4-methoxypyridine using neo-pentanol as the initiator.](/image/article/2011/PY/c1py00254f/c1py00254f-f2.gif) | ||
| Fig. 2 (a) 1H NMR spectrum (400 MHz; CDCl3), (b) crude MALDI-TOF MS and (c) MALDI-TOF MS after column chromatography of a P(L-BMA) ([M]/[I] = 20) (Mn = 3 860 g mol−1, PDI = 1.10) prepared by ROP of L-malOCA ([L-malOCA]0 = 0.32 M) catalyzed with 5 mol% 4-methoxypyridine using neo-pentanol as the initiator. | ||
Further decreasing the basicity of the pyridine catalyst led to no impurities being observed by 1H or 13C NMR spectroscopy or MALDI-TOF MS. Notably, the molecular weights and corresponding degrees of polymerization of the resultant polymers obtained using 4-methyl pyridine or pyridine as catalyst were markedly lower than that predicted by the monomer:initiator ratio (Table 1). Variation of the molar equivalence of pyridine catalyst resulted in the observation of molar masses closer to those predicted although concurrent increased levels of impurities were observed by MALDI-TOF MS (Fig. 3).
![MALDI-TOF MS analysis of P(l-BMA)s ([M]/[I] = 50) prepared by ROP of l-malOCA ([l-malOCA]0 = 0.32 M) catalyzed with pyridine at; 2.5 : 1 to neo-pentanol (A), 20 : 1 to neo-pentanol (B) and 50 : 1 to neo-pentanol (C).](/image/article/2011/PY/c1py00254f/c1py00254f-f3.gif) | ||
| Fig. 3
MALDI-TOF MS analysis of P(L-BMA)s ([M]/[I] = 50) prepared by ROP of L-malOCA ([L-malOCA]0 = 0.32 M) catalyzed with pyridine at; 2.5:1 to neo-pentanol (A), 20:1 to neo-pentanol (B) and 50:1 to neo-pentanol (C). | ||
Interestingly, inspection of the 1H NMR spectra measured during the polymerization of L-malOCA, catalyzed by pyridine, revealed notable differences in the chemical shift of the neo-pentyl group of the initiating/propagating species during the initial stages of the ROP of L-malOCA (See ESI). With high pyridine concentrations, the neo-pentyl group is observed as a singlet at δ = 0.86 ppm throughout the polymerization however, at lower pyridine concentrations this resonance is observed to change during the polymerization such that in the latter stages of the polymerization a singlet at δ = 0.86 ppm is observed while in the initial stages, multiple, resonances between δ = 0.94–0.84 ppm are observed. These observations indicate that at high pyridine concentrations, initiation is efficient whereas low pyridine concentrations lead to inefficient initiation and thus may lead to undesirable side reactions that consume monomer without yielding isolable polymer products.
Further investigation of 4-methoxypyridine catalyzed ROP
4-Methoxypyridine provided the best balance of control and activity of the para-substituted pyridine catalysts investigated in the ROP of L-malOCA. Further investigation into the ROP of L-malOCA with 4-methoxypyridine prepared and isolated as described above resulted in the observation of ‘living’ characteristics. By varying the monomer:initiator ratio, polymers up to ca. 25 kDa were able to be readily accessed (Table 2). At low [M]/[I], a linear relationship between Mn and initial monomer to initiator ratio (Fig. 4) was demonstrated although deviation at higher molar mass values was observed, attributed to impurities in the monomer.![Plot of [M]/[I] versus Mn and PDI for ROP of l-malOCA ([l-malOCA]0 = 0.32 M) using 5 mol% 4-methoxypyridine and neo-pentanol as the initiator at a ratio of 1 : 1.](/image/article/2011/PY/c1py00254f/c1py00254f-f4.gif) | ||
| Fig. 4 Plot of [M]/[I] versus Mn and PDI for ROP of L-malOCA ([L-malOCA]0 = 0.32 M) using 5 mol% 4-methoxypyridine and neo-pentanol as the initiator at a ratio of 1:1. | ||
Furthermore, as a consequence of the decreased rate of polymerization compared to DMAP-catalyzed ROP, it was also possible to observe a linear relationship between Mn and monomer conversion (Fig. 5). The ‘living’ characteristics of this polymerization were again confirmed through a second-feed experiment in which to a P(L-BMA)20 macroinitiator initiated from neo-pentanol ([M]/[I] = 20) (Mn = 3 860 g mol−1; PDI = 1.10), the further ROP of L-malOCA ([M]/[I] = 20) enabled a chain extended P(L-BMA)40 to be isolated that exhibited a double molar mass (Mn = 7 760 g mol−1) while maintaining a low PDI of 1.12. Leaving the resultant P(L-BMA)40 for 8 h (5 times longer than required to reach >90% monomer conversion) in the presence of 4-methoxypyridine resulted in negligible changes in both the molar mass (Mn = 8 030 g mol−1) and molar mass distribution (PDI = 1.11) again suggesting that transesterification side reactions were minimal despite full monomer consumption.
![Plot of monomer conversion versus Mn and PDI for ROP of l-malOCA ([l-malOCA]0 = 0.32 M) using 5 mol% 4-methoxypyridine and neo-pentanol as the initiator at a ratio of 1 : 1.](/image/article/2011/PY/c1py00254f/c1py00254f-f5.gif) | ||
| Fig. 5 Plot of monomer conversion versus Mn and PDI for ROP of L-malOCA ([L-malOCA]0 = 0.32 M) using 5 mol% 4-methoxypyridine and neo-pentanol as the initiator at a ratio of 1:1. | ||
MALDI-TOF MS side-product identification
The elimination of polymeric impurities from polymers prepared by ROP process catalyzed by 4-methoxypyridine using column chromatography enabled investigation into their identity. While analysis of the first fraction enabled isolation of the pure polymer, notably transesterification of the polymer chains did not occur during the purification process, analysis of the second product by 1H NMR spectroscopy revealed resonances resembling P(L-BMA) and confirmed the presence of a neo-pentyl ester end group implying that initiation had also proceeded through the expected mechanism. MALDI-TOF MS analysis however revealed a single distribution with the major peak at m/z = 4918.1 with a regular spacing equal to the molar mass of benzyl α-(L)-malate (m/z = 206) (See ESI). The observed mass increase from the first fraction by MALDI-TOF MS corresponded to a single molecule of L-malOCA (m/z = 250, i.e. the repeat unit + CO2) and an additional sodium atom (m/z = 23) to an already sodium charged P(L-BMA) initiated from neo-pentanol.We postulated that this impurity occurred as a consequence of a misinsertion of the propagating chain end into the disfavored 2-position of the OCA ring resulting in a carbonate linkage and a carboxylic acid ω-chain end group, capable of supporting a second sodium charge but incapable of further propagation (Scheme 1). To confirm the presence of the second sodium atom, the cationization agent used for the MALDI-TOF MS analysis was changed from sodium trifluoroacetate (NaTFA) to lithium chloride (LiCl). The resultant spectrum (See ESI) showed a major peak that was shifted from m/z = 4918.1 to m/z = 4886.5 respectively corresponding to a difference of m/z = 32, equal to the difference in mass between two sodium atoms and two lithium atoms thus confirming the proposed structure for this side product.
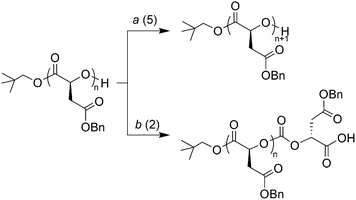 | ||
| Scheme 1 Products of ring-opening of L-malOCA at (a) 5-position of the OCA ring and (b) 2-position of the OCA ring. | ||
DFT calculations were performed in order to calculate the energy difference in the ring opening transition states for opening at the two different carbonyl groups.42 Two transition structures for the model DMAP-catalyzed ring opening of lactic acid OCA at the ester like carbonyl group (Fig. 6, pathway i) with methanol first reported by Cossio, Bourissou and coworkers were recalculated using the same basis set with insignificant differences from those reported.41 We then found two related transition structures for the ring opening at the carbonate-like carbonyl group (Fig. 7, pathway ii). At the B3LYP/6-31G(d) level (after ZPE correction) the relative energies for the two ester-forming (pathway i) transition structures were +2.8 kJ mol−1 (TS1) and 0 kJ mol−1 (TS2), and for the two carbonate-forming (pathway ii) transition structures +6.5 kJ mol−1 (TS3) and +16.4 kJ mol−1 (TS4). A combination of these four pathways can be used to estimate a relative reaction rate of roughly 17 to 1 in favor of ester-formation (pathway i) opening at a temperature of 300 K. Comparable calculations using 4-methoxypyridine suggest that this catalyst results in reduced selectivity compared to the DMP-catalyzed reaction (9 to 1 in favor of ester formation at 300 K - see ESI for more details). These results are consistent with the proposal that, while less frequent, opening of OCAs catalyzed by substituted pyridines at the carbonate-like carbonyl (pathway ii) is indeed energetically feasible.
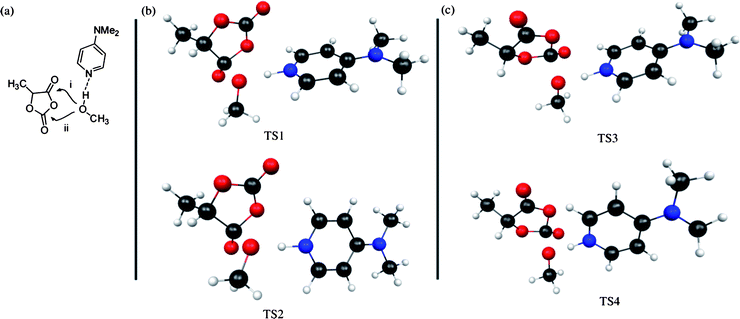 | ||
| Fig. 6 (a) The two pathways (i and ii) for DMAP-catalyzed ring opening of lacOCA with nucleophilic attack of methanol at either of the two carbonyl groups; (b) calculated structures for ester-forming transition states; (c) calculated structures for carbonate-forming transition states. Key: Grey = Carbon; Red = Oxygen; Blue = Nitrogen; White = Hydrogen. | ||
![Deconvoluted ESI MS of a PMA ([M]/[I] = 20) (Mn = 1 100 g mol−1; PDI = 1.10) prepared through the hydrogenation of P(l-BMA)20 prepared by ROP of l-malOCA ([l-malOCA]0 = 0.32 M) catalyzed with 5 mol% pyridine using neo-pentanol as the initiator. Right-hand side: expanded region between m/z = 2380 and 2480.](/image/article/2011/PY/c1py00254f/c1py00254f-f7.gif) | ||
| Fig. 7 Deconvoluted ESI MS of a PMA ([M]/[I] = 20) (Mn = 1 100 g mol−1; PDI = 1.10) prepared through the hydrogenation of P(L-BMA)20 prepared by ROP of L-malOCA ([L-malOCA]0 = 0.32 M) catalyzed with 5 mol% pyridine using neo-pentanol as the initiator. Right-hand side: expanded region between m/z = 2380 and 2480. | ||
Block copolymer synthesis
Manipulation of the initiating species was investigated to enable the synthesis of block copolymers. Initiation of the ROP of L-malOCA from commercially available MeO-PEO112-OH or MeO-PEO227-OH macroinitiators catalyzed by 4-methoxypyridine resulted in >99% monomer conversion after 225 min with subsequent SEC analysis of the block copolymers confirming chain growth (Table 3). 1H NMR spectroscopy confirmed the presence of PEO, methine and malate units at δ = 5.55, 3.62 and 3.05–2.78 ppm respectively at the expected ratio. Block copolymers were also demonstrated to be accessible by chain growth from a poly(L-lactide), PLLA, macroinitiator. PLLAs ([M]/[I] = 20 and 50) were synthesized by the ROP of L-lacOCA using identical conditions as described above with 4-methoxypyridine, initiated from neo-pentanol. Complete monomer conversion for [M]/[I] = 20 and 50 was achieved after 60 min and 150 min respectively. Chain growth of L-malOCA ([M]/[I] = 20) from the two PLLA-OH macroinitiators was confirmed by 1H NMR spectroscopy showing the presence of both the lactate methyl and malate resonances at δ = 1.58 and 3.08–2.85 ppm respectively. Additionally, SEC analysis revealed an increase in molar mass with a low molar mass distribution being maintained (Table 3).| Polymer | M n c (g mol−1) | M w/Mnc | DP b |
|---|---|---|---|
| a [L-malOCA]0 = 0.32 M; 5 mol% MeOPy catalyst, 25 °C. b Degree of Polymerization, determined by 1H NMR Spectroscopy. c Determined by SEC analysis. | |||
| MeO-PEO112-OH | 8 200 | 1.04 | — |
| MeO-PEO227-OH | 16 270 | 1.03 | — |
| PEO112-b-P(L-BMA)20 | 11 540 | 1.18 | 21 |
| PEO227-b-P(L-BMA)20 | 19 440 | 1.03 | 23 |
| PLLA20-OH | 2 640 | 1.11 | — |
| PLLA50-OH | 7 680 | 1.06 | — |
| PLLA20-b-P(L-BMA)20 | 5 890 | 1.12 | 22 |
Deprotection of P(L-BMA)
The pendant benzyl protected carboxylic acid groups of P(L-BMA)20 (Mn = 3 860 g mol−1, PDI = 1.10) were deprotected by hydrogenolysis using H2 over Pd/C in 15 min at 40 °C and resulted in hydrophilic poly(α-L-malic acid) (PMA) (Scheme 2). Clean and complete removal of the benzyl protecting groups was deduced from the disappearance of all of the aromatic and benzylic signals from both the 1H and 13C NMR spectra (See ESI). Further confirmation was obtained from the change in solubility of the resulting polymer from the P(L-BMA)20 (soluble in CHCl3, insoluble in MeOH) to the PMA20 (soluble in MeOH, insoluble in CHCl3). This process did not result in degradation of the poly(ester) backbone, as shown by lack of resonances associated with changes to the electronic environment of the methine protons associated with a neighboring hydroxyl proton in the 1H NMR spectrum that would be apparent upon cleavage of the backbone.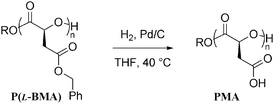 | ||
| Scheme 2 Deprotection of poly(benzyl α-(L)-malate), P(L-BMA), using hydrogenolysis to yield PMA. | ||
Electrospray MS analysis, obtained at high resolution provided additional confirmation of deprotection through observation of PMA20 as a doubly charged species with the major peak at m/z = 1203.5 that upon deconvolution (Fig. 7) resulted in a major peak at m/z = 2408.3 equal to the desired PMA20 along with additional peaks correlating to PMA20 species with a sodium charge (m/z = 2430.2) and with an additional sodium counterion resulting from exchange with a proton of one of the acid side-chain groups (m/z = 2452.1). A species displaying a lower molar mass was also present at m/z = 2390.2 matching ring closure of the PMA20 end group resulting in the loss of H2O. Additional analysis of both P(L-BMA)20 and PMA20 by SEC using a 0.1 M citric acid in THF solution as the eluent exhibited distributions with (Mn = 4 980 g mol−1; PDI = 1.06) and (Mn = 1 100 g mol−1; PDI = 1.10) respectively (compared to poly(styrene) standards). We postulated that the significant difference in molar mass that was observed between P(L-BMA)20 and PMA20viaSEC analysis is a result of interactions between the PMA20 and the eluent solvent system resulting in tightly coiled polymers thus leading to low observed molar mass values.
Degradation of PMA
Thermal degradation of both P(L-BMA)20 and PMA20 was studied using thermogravimetric analysis (TGA). While both poly(ester)s fully degraded within a reasonably short temperature range, P(L-BMA)20 was stable up to 280 °C whereas PMA20 began degrade at a significantly lower temperature, ∼180 °C, probably a result of the pendent carboxylic acid groups of PMA20 auto-catalyzing the degradation.Hydrolytic degradations were performed in H2O at room temperature on a PMA ([M]/[I] = 15) at a concentration of 0.48 mmol L−1. The degradations were monitored via acid–base titration using a 0.45 mmol L−1 aqueous NaOH solution with four drops of a phenolphthalein pH indicator in methanol solution. Degradation was complete after 10 days determined when two equivalents of the NaOH solution was required to neutralize the reaction. Examination of 1H NMR spectra during the degradation in D2O demonstrates a gradual reduction of resonances attributed to PMA15 at δ = 5.67 and 3.17–2.96 ppm with a corresponding increase of new resonances at δ = 4.53 and 2.93–2.76 ppm respectively resulting from the degradation products. Electrospray (ESI) MS (Fig. 8) provided a useful method to monitor the loss of molar mass during the PMA degradation. Significant reduction in molar mass was observed early in the degradation after 30 h that subsequently continued at a slower rate realising near complete degradation to L-malic acid (m/z = 133.0) after 7 days with the final oligomers requiring an additional 3 days to fully degrade.
![ESI MS analysis of the degradation of PMA ([M]/[I] = 15) ([PMA15]0 = 0.36 mmol L−1) in H2O at room temperature.](/image/article/2011/PY/c1py00254f/c1py00254f-f8.gif) | ||
| Fig. 8 ESI MS analysis of the degradation of PMA ([M]/[I] = 15) ([PMA15]0 = 0.36 mmol L−1) in H2O at room temperature. | ||
Conclusions
In conclusion, the synthesis of L-malOCA from L-malic acid has been demonstrated. Homopolymerization of L-malOCA catalyzed with a range of pyridine based catalysts enabled the synthesis of functional poly(ester)s with pendant benzyl protected carboxylic acid groups to high monomer conversions in the absence of transesterification side reactions. The choice of ROP catalyst was demonstrated to have a significant effect on the amount of side products produced with their removal shown to be successful viacolumn chromatography.The versatility of the polymerization system was shown through the successful initiation from alcohols including PEO and PLLA as macroinitiators in the preparation of block copolymers. Removal of the benzyl protecting groups was successful without any polymer backbone scission to yield hydrophilic poly(ester)s and degradation studies of the resultant PMA15 in H2O was demonstrated to occur fully within 10 days as determined by titration, 1H NMR and mass spectrometry.
Acknowledgements
The Research Councils UK (RCUK) are acknowledged for funding a fellowship to A.P.D. We gratefully acknowledge financial support from EPSRC (EP/C007999/1) for the purchase of the Bruker Ultraflex MALDI-TOF MS instrument. The SEC equipment used in this research was obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). Dr Lijiang Song is gratefully acknowledged for his assistance with measurement and analysis of ESI-MS data.Notes and references
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- R. Bizzarri, F. Chiellini, C. K. Ober, W. M. Saltzman, R. Solaro and E. Chiellini, Macromol. Symp., 2003, 197, 303–314 CrossRef CAS.
- M. Vert, Polym. Degrad. Stab., 1998, 59, 169–175 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Adv. Polym. Sci., 2002, 157, 1–40 CrossRef CAS.
- D. Bourissou, S. Moebs-Sanchez and B. Martin-Vaca, C. R. Chim., 2007, 10, 775–794 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- C. Jerome and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef CAS.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- G. L. Baker, E. B. Vogel and M. R. Smith, Polym. Rev., 2008, 48, 64–84 CrossRef CAS.
- O. Coulembier, P. Degee, J. L. Hedrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS.
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- C. Barbaud, F. Fay, F. Abdillah, S. Randriamahefa and P. Guerin, Macromol. Chem. Phys., 2004, 205, 199–207 CrossRef CAS.
- R. Bizzarri, F. Chiellini, R. Solaro, E. Chiellini, S. Cammas-Marion and P. Guerin, Macromolecules, 2002, 35, 1215–1223 CrossRef CAS.
- S. Cammas-Marion, M. M. Bear, A. Harada, P. Guerin and K. Kataoka, Macromol. Chem. Phys., 2000, 201, 355–364 CrossRef CAS.
- O. Coulembier, P. Degee, S. Cammas-Marion, P. Guerin and P. Dubois, Macromolecules, 2002, 35, 9896–9903 CrossRef CAS.
- O. Coulembier, P. Degee, P. Gerbaux, P. Wantier, C. Barbaud, R. Flammang, P. Guerin and P. Dubois, Macromolecules, 2005, 38, 3141–3150 CrossRef CAS.
- O. Coulembier, J. Ghisdal, P. Degee and P. Dubois, Arkivoc, 2007, 57–70 CAS.
- O. Coulembier, L. Mespouille, J. L. Hedrick, R. M. Waymouth and P. Dubois, Macromolecules, 2006, 39, 4001–4008 CrossRef CAS.
- P. Guerin, J. Francillette, C. Braud and M. Vert, Makromol. Chem., Macromol. Symp., 1986, 6, 305–314 CrossRef CAS.
- M. Helou, G. Moriceau, Z. W. Huang, S. Cammas-Marion and S. M. Guillaume, Polym. Chem., 2011, 2, 840–850 RSC.
- M. A. LeboucherDurand, V. Langlois and P. Guerin, React. Funct. Polym., 1996, 31, 57–65 CrossRef CAS.
- S. Matsumura, H. Beppu, K. Nakamura, S. Osanai and K. Toshima, Chem. Lett., 1996, 795–796 CrossRef CAS.
- L. Moine, S. Cammas, C. Amiel, P. Guerin and B. Sebille, Polymer, 1997, 38, 3121–3127 CrossRef CAS.
- B. Nottelet, C. Di Tommaso, K. Mondon, R. Gurny and M. Moller, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3244–3254 CrossRef CAS.
- J. Rieger, O. Coulembier, P. Dubois, K. V. Bernaerts, F. E. Du Prez, R. Jerome and C. Jerome, Macromolecules, 2005, 38, 10650–10657 CrossRef CAS.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- Y. Kimura, K. Shirotani, H. Yamane and T. Kitao, Macromolecules, 1988, 21, 3338–3340 CrossRef CAS.
- Y. Kimura, K. Shirotani, H. Yamane and T. Kitao, Polymer, 1993, 34, 1741–1748 CrossRef CAS.
- T. Yamaoka, Y. Hotta, K. Kobayashi and Y. Kimura, Int. J. Biol. Macromol., 1999, 25, 265–271 CrossRef CAS.
- O. T. du Boullay, N. Saffon, J. P. Diehl, B. Martin-Vaca and D. Bourissou, Biomacromolecules, 2010, 11, 1921–1929 CrossRef.
- R. J. Pounder and A. P. Dove, Biomacromolecules, 2010, 11, 1930–1939 CrossRef CAS.
- T. Ouchi and A. Fujino, Makromol. Chem., 1989, 190, 1523–1530 CrossRef CAS.
- O. T. du Boullay, E. Marchal, B. Martin-Vaca, F. P. Cossio and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 16442–16443 CrossRef.
- H. R. Kricheldorf and J. M. Jonte, Polym. Bull., 1983, 9, 276–283 CAS.
- O. T. du Boullay, C. Bonduelle, B. Martin-Vaca and D. Bourissou, Chem. Commun., 2008, 1786–1788 RSC.
- C. Bonduelle, B. Martin-Vaca and D. Bourissou, Biomacromolecules, 2009, 10, 3069–3073 CrossRef CAS.
- C. Bonduelle, B. Martin-Vaca, F. P. Cossio and D. Bourissou, Chem.–Eur. J., 2008, 14, 5304–5312 CrossRef CAS.
- Full computational details including software, references, and coordinates, energies and gradients for the structures discussed, are given in the ESI.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00254f |
| ‡ Retention of the stereochemistry was confirmed through measurement of their specific rotation viapolarimetry observing [α]33D values of −21.9° (in CHCl3, c = 5.96 g L−1); other methods to confirm this were not possible as a consequence of the reactivity of the monomer or lack of heavy atoms in the monomer. ROP using pyridine result in the observation of only 2 carbonyl resonances (using DMAP results in several such resonances) which indicates that the monomer is indeed stereopure. |
| This journal is © The Royal Society of Chemistry 2011 |