Facile, modular transformations of RAFT block copolymersvia sequential isocyanate and thiol-ene reactions†‡
Joel D.
Flores
a,
Nicolas J.
Treat
a,
Adam W.
York
a and
Charles L.
McCormick
*ab
aDepartment of Polymer Science, The University of Southern Mississippi, Hattiesburg, Mississippi 39406, USA. E-mail: charles.mccormick@usm.edu.
bDepartment of Chemistry and Biochemistry, The University of Southern Mississippi, Hattiesburg, Mississippi 39406, USA
First published on 15th June 2011
Abstract
We describe a robust strategy utilizing reversible addition-fragmentation chain transfer (RAFT) polymerization and sequential transformations involving carbamate formation and thiol-ene click addition to synthesize well-defined functional block copolymers. The hydroxy-functional block copolymer scaffold, poly[(N,N-dimethylacrylamide)-b-(N-(2-hydroxyethyl)acrylamide)] (PDMAn-b-PHEAm) was first prepared via RAFT, requiring no protecting group chemistry. The hydroxyl groups of the HEA block were then reacted with 2-(acryloyloxy)ethylisocyanate (AOI) or allylisocyanate (AI) resulting in acrylate- and allyl-functionalized copolymer precursors, respectively. The efficiencies of both Michael and free radical-mediated thiol-ene addition reactions were investigated using model thiol compounds having alkyl, aryl, hydroxyl, carboxylic acid, amine and amino acid functionalities. The steps of RAFT polymerization, isocyanate-hydroxyl coupling and thiol-ene addition can be accomplished under mild conditions, thus offering a facile, modular route to the synthesis of functional copolymers from a single polymeric precursor.
Introduction
Over the past decade, controlled polymerization (CP) methods and click-type chemical reactions have provided unprecedented opportunities for rational design and synthesis of materials particularly useful in the fields of microelectronics, personal care, water purification and nanomedicine, among others.1–5 Recent advances in CP have allowed for the direct synthesis of polymers containing a wide array of functional groups with remarkable molecular weight control. However, in order to attain targeted properties, efficient post-polymerization transformation strategies are often required. The click chemistry concept, as described by Sharpless and coworkers,6 and related strategies have provided attractive routes for the modification of structoterminal and structopendent groups of polymeric precursors.7–11There are a number of chemical reactions that meet the requisite features of click chemistry, with the copper(I)-mediated azide-alkyne 1,3-dipolar cycloaddition being the most recognized.5 Another resurgent technique, the well-established addition of thiols to unsaturated carbon-carbon bonds, is now being referred to as thiol-ene click chemistry.12–18Thiol-ene addition reactions are especially attractive since they occur in a facile manner under mild conditions and do not require metal catalysts.8 In addition, thiol-containing proteins, glycoproteins and other bio-relevant species can be conjugated easily to synthetic scaffolds, often without requiring protecting group chemistry.9Thiol-ene click chemistry enables a modular approach for attaching diverse functionalities onto the polymer chain and thus tuning of chemical and physical properties. Several examples are reported in the literature that involve conversion of the terminal thiocarbonylthio functionality of polymers prepared via reversible addition-fragmentation chain transfer (RAFT) polymerization to the thiol groups for subsequent thiol-ene click reactions.19–27 Likewise, pendent group transformations viathiol-ene click chemistry are also powerful synthetic tools for altering (co)polymer structure.9,28–36
A review of the literature reveals a number of attempts to prepare linear polymers with pendent alkenes. For example, the selective polymerization of asymmetric bifunctional vinyl monomers using controlled radical polymerization techniques has been reported.36–39 These techniques are successful only when the reactivity of the pendent alkene is sufficiently different from that of the alkene incorporated into the backbone. Many attempts to directly polymerize asymmetric bifunctional vinyl monomers have resulted in broad molecular weight distributions and formation of branched or cross-linked structures.40–45 Alternatively, well-defined polymer precursors with pendent alkenes may be prepared by ring-opening and/or ionic polymerizations of appropriate monomers.28–30,33,34,46–53 However, these polymerization methods typically employ metal catalysts, require stringent reaction conditions and are less tolerant of functional groups and impurities. Thus, more versatile synthetic routes for preparing structopendent alkene-containing polymeric precursors are needed.
Herein, we detail a synthetic protocol, graphically depicted in Scheme 1, that utilizes RAFT polymerization and sequential reactions involving carbamate (urethane) linkage formation and thiol-ene click addition for the syntheses of well-defined, functional block copolymers. First, a hydroxyl-containing diblock copolymer precursor was prepared viaRAFT polymerization. Pendent alkene functional groups were then obtained by reacting the precursor diblock copolymer with isocyanates having either acrylate or allyl groups. Since these in situ reactions do not generate by-products, there are fewer impurities to remove during workup. Thiol-ene addition reactions were then carried out using selected small molecule thiols having alkyl, aryl, hydroxyl, amine, carboxylic acid and amino acid functionalities to demonstrate the utility of the procedure (see Scheme 2). The combination of RAFT polymerization, efficient carbamate formation and subsequent thiol-ene click addition thus provides a facile route for preparing functional copolymers for applications that require precise control over polymer architectures.
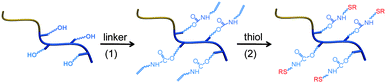 | ||
| Scheme 1 Sequential modification of a hydroxyl-containing diblock copolymer precursor: (1) reaction between pendent hydroxyl groups and isocyanate-alkene linker; (2) thiol-ene click addition with selected thiols. | ||
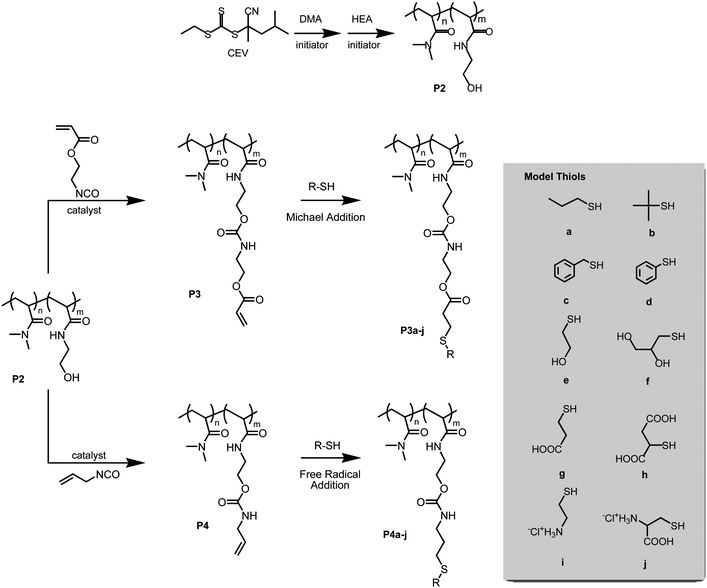 | ||
| Scheme 2 Block copolymerization of N,N-dimethylacrylamide (DMA) and N-(2-hydroxyethyl)acrylamide (HEA) to form precursor copolymer P2 and modification through reaction with 2-(acryloyloxy)ethylisocyanate (AOI) and allylisocyanate (AI). Structopendent alkene copolymers P3 and P4 were then utilized in Michael and free radical thiol-ene addition reactions, respectively, with selected thiols to obtain a series of functionalized copolymers. | ||
Experimental
Materials
N,N-Dimethylacrylamide (DMA, 99%, Sigma-Aldrich, St. Louis, MO, USA) and 2-(acryloyloxy)ethylisocyanate (AOI, Showa Denko K.K., Tokyo, Japan) were distilled under reduced pressure and stored below 0 °C until use. N-(2-Hydroxyethyl)acrylamide (HEA, 97%, Sigma-Aldrich, St. Louis, MO, USA) was purified using inhibitor remover (Sigma-Aldrich, St. Louis, MO, USA) before polymerization. Allylisocyanate (AI, 98%), ethanethiol (97%), 3-propanethiol (99%), carbon disulfide (99.9%), sodium hydride (60% dispersion in mineral oil), sodium thiosulfate (99%), iodine (>99%), 3-mercaptopropionic acid (99 + %), cysteamine hydrochloride, L-cysteine hydrochloride monohydrate (98 + %), benzylmercaptan (99%), thiophenol (99 + %), thioglycerol (99%), 2-methyl-2-propanethiol (98%), mercaptosuccinic acid (99%), sodium 1-thio-β-d-glucose, deuterium oxide (D2O, 99.9 atom % D), chloroform-d1 (CDCl3, 99.8 atom % D + 0.1 (v/v) % TMS), 4,4′-methylenebis(2,6-di-tert-butylphenol) (98%), dibutyltin dilaurate (DBTDL, 95%), lectin-fluorescein isothiocyanate conjugate (FITC-Con A, from Canavalia ensiformis, lyophilized powder) and transfer RNA (tRNA, from Saccharomyces cerevisiae, type X, lyophilized powder) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received. Triethylamine (TEA, >99.5%), 2-mercaptoethanol (>99%), 4,4′-azobis(4-cyanopentanoic acid) (ACPA, 99%) were obtained from Fluka (Buchs, Switzerland). 2,2′-Azobis(2,4-dimethylvaleronitrile) (ADVN, 97%) was purchased from Wako Pure Chemicals Industries, Ltd. (Osaka, Japan) and used as received. Deuterated dimethylsulfoxide (DMSO-d6, 99.9 atom % D + 1% v/v TMS) was purchased from Cambridge Isotope Laboratories, Inc (Andover, MA, USA). The solvents and other reagents were purchased from Sigma-Aldrich or Fisher Scientific in A.C.S. grade or its equivalent.Instrumentation
Spectra of the CTA, monomers and (co)polymers were generated using a Varian INOVA 300 MHz NMR spectrometer. Fourier transform infrared (FT-IR) spectroscopic analysis was carried out using a Nicolet 8700 spectrophotometer in transmission mode. The samples were prepared by casting solutions (in chloroform, methanol or trifluoroethanol) of the copolymers on NaCl plates. The solvent was allowed to evaporate and the plates were placed in vacuum overnight prior to analysis. Molecular weights and polydispersities were determined either by organic solvent or aqueous size exclusion chromatography (SEC). The former utilized 0.02 M lithium bromide in DMF as eluent at a flow rate of 1.0 mL/min. The instrument was equipped with Viscotek I-Series Mixed Bed low-MW (exclusion limit > 20 K PS) and mid-MW (exclusion limit > 200 K PS) columns, Viscotek triple detector array (302 nm RI, viscosity, 7 mW 90° and 7° true low angle light scattering detectors (670 nm)) equilibrated at 35 °C. The cationic aqueous SEC system utilized an eluent of 1 wt % acetic acid/0.10 M Na2SO4 (aq) at a flow rate of 0.25 mL min−1 at 25 °C, Eprogen, Inc. CATSEC columns (100, 300, and 1000 Å), a Polymer Laboratories LC1200 UV-visible detector, a Wyatt Optilab DSP interferometric refractometer (λ = 690 nm), and a Wyatt DAWN-DSP multiangle laser light scattering (MALLS) detector (λ = 633 nm). Anionic copolymers were analyzed in an aqueous SEC system utilizing 20%/80% acetonitrile/0.05M Na2SO4(aq) as eluent (flow rate of 0.3 mL min−1), TOSOH Biosciences TSK-GEL columns (G3000 PWXL, <50,000 g mol−1, 200 Å) and (G4000PWXL, 2,000–300,000 g mol−1, 500 Å), Wyatt OptilabDSP interferometric refractometer and a Wyatt DAWN EOS multiangle laser light scattering detector (λ = 690 nm). Absolute molecular weights and polydispersities were calculated using Wyatt OmniSEC software. Dynamic light scattering (DLS) studies were conducted at 25 °C using a Malvern Instruments Zetasizer Nano series instrument equipped with a 22 mW He-Ne laser (λ = 632.8 nm), an avalanche photodiode detector and an ALV/LSE-5003 multiple tau digital correlator. Dispersion Technology Software v5.03 (Malvern Instruments Ltd) was used to record and analyze the data to determine particle size distributions. The permanently water-soluble copolymers (1.0 mg mL−1) were directly dissolved in HPLC water, while aliquots of hydrophobically-modified copolymer solutions in DMSO were added into HPLC water to obtain 1.0 mg mL−1copolymer concentrations. The solution pH was adjusted using 0.1 M HCl or 0.1 M NaOH. The solutions were shaken in a vortex mixer, sonicated for 1 min and filtered (0.2 μm, Millipore) directly into disposable cuvettes for DLS measurements. Fluorescence studies were performed using Photon Technology International QuantaMaster™ fluorimeter and FeliX32™ software.Synthesis of 2-(ethylsulfanylthiocarbonylsulfanyl)-2,4-dime- thylvaleronitrile (CEV)
CEV was synthesized following a modified literature procedure.54,55 Briefly, a slurry of sodium hydride (1.9 g, 0.08 mol) in diethyl ether (200 mL) was cooled in an ice bath for 15 min. To this mixture, ethanethiol (5 g, 0.080 mol) was added dropwise. The mixture was stirred for 15 min and carbon disulfide (6.2 g, 0.080 mol) was then added slowly forming a yellow suspension. After stirring for 1 h, the mixture was allowed to warm to room temperature before being concentrated under reduced pressure on a rotatory evaporator. The resulting residue was suspended in diethyl ether (200 mL). Iodine (10.3 g, 0.041 mmol) was then added to convert the trithiocarbonate salt into the disulfide. The solution was then washed with a sodium thiosulfate solution (2 × 200 mL, 5 wt %). The yellow organic layer was collected and dried over magnesium sulfate. After removing the drying agent, the solvent was removed under reduced pressure yielding a yellow oil. This crude product and ADVN (15.0 g, 0.060 mol) were dissolved in ethyl acetate (100 mL) and the mixture was allowed to react with stirring overnight under reflux at 70 °C. The solvent was removed under reduced pressure and the oil was purified by column chromatography. Yield = 7.4 g (38%). Rf 0.40 (SiO2, ethyl acetate/hexanes, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95 (v/v)). δH (300 MHz, CDCl3): 1.02–1.12 (dd, 6H, -CH(-CH3)2), 1.32–1.40 (t, 3H, -S-CH2-CH3), 1.78–1.86 (dd, 1H, -CH2-CH(-CH3)2), 1.90–1.92 (s, 3H, -C(-CH3)(-CN)-), 1.94–2.10 (m, 1H, -CH2-CH(-CH3)2), 2.10–2.18 (dd, 1H, -CH2-CH(-CH3)2), 3.30–3.40 (q, 2H, -S-CH2-CH3). 13C NMR (300 MHz, CDCl3) δ: 12.90 (-S-CH2-CH3), 23.60 (-CH2-CH(-CH3)2), 24.00(-C(-CH3)(-CN)-), 25.50 (-CH(-CH3)2), 25.80 (-CH(-CH3)2), 31.20 (-S-CH2-CH3), 46.50 (-S-C(-CN)(-CH3)-), 47.30 (-CH2-CH(-CH3)2), 120.00 (-CN), 217.60 (-C
:95 (v/v)). δH (300 MHz, CDCl3): 1.02–1.12 (dd, 6H, -CH(-CH3)2), 1.32–1.40 (t, 3H, -S-CH2-CH3), 1.78–1.86 (dd, 1H, -CH2-CH(-CH3)2), 1.90–1.92 (s, 3H, -C(-CH3)(-CN)-), 1.94–2.10 (m, 1H, -CH2-CH(-CH3)2), 2.10–2.18 (dd, 1H, -CH2-CH(-CH3)2), 3.30–3.40 (q, 2H, -S-CH2-CH3). 13C NMR (300 MHz, CDCl3) δ: 12.90 (-S-CH2-CH3), 23.60 (-CH2-CH(-CH3)2), 24.00(-C(-CH3)(-CN)-), 25.50 (-CH(-CH3)2), 25.80 (-CH(-CH3)2), 31.20 (-S-CH2-CH3), 46.50 (-S-C(-CN)(-CH3)-), 47.30 (-CH2-CH(-CH3)2), 120.00 (-CN), 217.60 (-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S).
S).
RAFT polymerization (P1, P2)
DMA (10.0 g, 101 mmol), CEV (247 mg, 1.0 mmol) and ADVN (50.0 mg, 0.20 mmol) were dissolved in dioxane (50 mL) in a round bottom flask, equipped with a magnetic stir bar and sealed with a rubber septum. The mixture was purged with nitrogen for 45 min. The polymerization was allowed to proceed at 50 °C for 18 h (conversion by 1H NMR = 95%). The PDMAn-CEV macroCTA (P1, Mn(SEC) = 10,300, PDI = 1.04, dn/dc = 0.091) was purified by precipitation (twice) in diethyl ether (polymer yield = 9.3 g, 98%). A mixture of HEA (11.0 g, 95.0 mmol), MEHQ inhibitor remover and dioxane (25 mL) in a conical flask was stirred on a magnetic stir plate for 4 h at room temperature. After filtration, ACPA (62.0 mg, 0.22 mmol) was added to the clear filtrate in a round bottom flask (250 mL) and allowed to dissolve with stirring. P1 (8.0 g, 0.78 mmol) was dissolved in DI water (100 mL) and the resulting solution was slowly added to the HEA/ACPA mixture. The pH of the solution was adjusted to 5.0 using 0.1 M HCl. The mixture was then purged with nitrogen for 1.5 h. The polymerization was carried out at 50 °C for 10 h (conversion by 1H NMR = 75%). The resulting solution was diluted with DI water and dialyzed against water at pH 3–5 for 3 days using a dialysis membrane (Spectra/Por Regenerated Cellulose, Spectrum Laboratories Inc, CA, USA) with MWCO of 3,500. Lyophilization yielded PDMAn-b-PHEAm diblock copolymer (P2). Polymer yield = 14.5 g (89%). Mn (SEC) = 23,500, PDI = 1.11, dn/dc = 0.062. δH (300 MHz, DMSO-d6): 0.90–2.40 (b, n × 3H, m × 3H; backbone, -CH2-CH-), 2.58–3.24 (b, n × 6H, N-CH3; b, m × 2H, NH-CH2-), 3.39–3.51 (b, m × 2H, -CH2-O-), 4.64–5.18 (b, m × 1H, -OH), 7.19–7.96 (b, m × 1H, -NH). Characteristic bands in FT-IR, ν (NaCl, cm−1): 3423 (b, O–H), 3309 (b, N–H), 1643 (s, CO, amide), 1060 (m, C–O, hydroxyl). The full FT-IR spectrum of P2 is available in the ESI.†
Alkene functionalization (P3, P4)
Alkene-functionalized copolymers P3 and P4 were obtained through reaction of P2 with AOI and AI, respectively (see Scheme 2). Copolymer P2 (4.0 g, 14.0 mmol hydroxyl groups) and 4,4′-methylenebis(2,6-di-tert-butylphenol) (0.35 g, 0.9 wt %) were dissolved in anhydrous DMF (35 mL). An excess of isocyanate reactant (30 mmol) and DBTDL catalyst (50 mg, 0.1 wt %) were added to the polymer solution in a septum-sealed flask equipped with a magnetic stir bar. The mixture was allowed to react at 40 °C for 24–36 h. The polymers were precipitated twice in cold diethyl ether, filtered and dried under reduced pressure for 2 h. P3: yield = 4.9 g (80%); Mn(SEC) = 39,600 PDI = 1.18; δH (300 MHz, DMSO-d6): 0.90–2.40 (b, n × 3H, m × 3H; backbone, -CH2-CH-), 2.60–3.29 (b, n × 6H, N-CH3; b, m × 2H, -CH-C(O)-NH-CH2-; b, m × 2H, -O-C(O)-NH-CH2-), 3.66–4.27 (b, m × 2H, -C(O)-O-CH2-; b, m × 2H, -NH-C(O)-O-CH2-), 5.79–6.41 (multiple resonances; m × 3H; CH2 = CH-), 6.80–7.87 (b, m × 2H, -NH-); Characteristic bands in FT-IR, ν (NaCl, cm−1): 3305 (b, N–H), 1724 (s, CO, carbamate, ester), 1643 (s, CO, amide), 1253 and 1189 (s, C–O, carbamate, ester), 981 (s, vinyl). P4: yield = 5.3 g (94%); Mn(SEC) = 35,300 PDI = 1.14; δH (300 MHz, DMSO-d6): 0.90–2.40 (b, n × 3H, m × 3H; backbone, -CH2-CH-), 2.60–3.3 (b, n × 6H, N-CH3; b, m × 2H, C(O)-NH-CH2-), 3.48–3.74 (b, m × 2H, -O-C(O)-NH-CH2-), 3.76–4.14 (b, m × 2H, -NH-C(O)-O-CH2-), 4.93–5.23 (dd, m × 2H, CH2 = CH-), 5.65–5.93 (m, m × 1H; CH2 = CH-), 6.86–7.92 (b, m × 2H, -NH-); Characteristic bands in FT-IR, ν (NaCl, cm−1): 3315 (b, N–H), 1710 (s, CO, carbamate), 1643 (s, CO, amide), 1249 and 1137 (s, C–O, carbamate), 989 and 916 (m, vinyl). See the ESI for the FT-IR spectra of copolymers P3 and P4.†
General procedure for thiol michael addition (P3a-j)
Precursor copolymer P3 (300 mg, 0.88 mmol ene) was dissolved in DMSO (3 mL). Into the copolymer solutions, the thiol reactant (1.1 mmol; 3-propanethiol (a, 84 mg), 2-methyl-2-propanethiol (b, 99 mg), benzylmercaptan (c, 136 mg), thiophenol (d, 121 mg), 2-mercaptoethanol (e, 86 mg), thioglycerol (f, 119 mg), 3-mercaptopropionic acid (g, 117 mg), mercaptosuccinic acid (h, 165 mg), cysteamine hydrochloride (i, 138 mg), L-cysteine hydrochloride monohydrate (j, 190 mg)) and triethylamine (9 mg, 0.09 mmol) were added. The reaction mixtures were stirred using a magnetic stir plate at room temperature for 12 h. The disappearance of the vinyl resonances was confirmed using 1H NMR spectroscopy. After the reactions, the copolymer solutions were transferred into dialysis tubing (MWCO 6–8 kDa, Spectra/Por Regenerated Cellulose, Spectrum Laboratories Inc, CA, USA) and dialyzed against DI water with pH adjusted to 3–5 using 1.0 M HCl, changing the dialysate every 2 h for 1 day. For hydrophobically modified copolymers, the solutions were first dialyzed against 90% (v/v) ethanol aqueous solution for 24 h, followed by dialysis against DI water for another 24 h. Dry copolymers obtained after lyophilization were characterized by SEC, NMR and FT-IR spectroscopy. The 1H NMR and FT-IR spectra of all the copolymers are available in the ESI.† Molecular weights and polydispersities are shown in Table 2. Copolymer yields after dialysis and lyophilization: P3a (300 mg, 85%), P3b (366 mg, >99%), P3c (380 mg, 99%), P3d (390 mg, >99%), P3e (340 mg, 96%), P3f (420 mg, >99%), P3g (330 mg, 88%), P3h (399 mg, 98%), P3i (350 mg, 92%), P3j (310 mg, 75%).General procedure for free radical thiol-ene addition (P4a-j)
Precursor copolymer P4 (200 mg, 0.71 mmol ene), thiol (7.1 mmol; 3-propanethiol (a, 540 mg), 2-methyl-2-propanethiol (b, 639 mg), benzylmercaptan (c, 880 mg), thiophenol (d, 781 mg), 2-mercaptoethanol (e, 554 mg), thioglycerol (f, 767 mg), 3-mercaptopropionic acid (g, 753 mg), mercaptosuccinic acid (h, 1065 mg), cysteamine hydrochloride (i, 887 mg), L-cysteine hydrochloride monohydrate (j, 1242 mg)) and ADVN free radical initiator (53 mg, 0.21 mmol) were dissolved in DMSO (2 mL). The solutions were deoxygenated by purging with nitrogen for 45 min. The solutions were then heated at 50 °C for 12 h, followed by exposure to air to quench the reactions. The solutions were transferred to dialysis tubing (MWCO 6–8 kDa, Spectra/Por Regenerated Cellulose, Spectrum Laboratories Inc, CA, USA) and dialyzed against 90% (v/v) ethanol aqueous solution for 24 h and then against DI water for another 24 h, changing the dialysate every 2 h. The solutions were then lyophilized and the purified copolymers were analyzed by SEC, NMR and FT-IR spectroscopy. The spectra of all the copolymers are available in the ESI.† Molecular weights and polydispersities are shown in Table 3. Yields after dialysis and lyophilization: P4a (207 mg, 84%), P4b (182 mg, 71%), P4c (160 mg, 49%), P4e (180 mg, 73%), P4f (230 mg, 85%), P4g (209 mg, 79%), P4h (189 mg, 54%), P4i (256 mg, 93%).Results and discussion
RAFT polymerization
The precursor diblock copolymer P2 poly[(N,N-dimethylacryl amide)-b-(N-(2-hydroxyethyl)acrylamide)] (PDMAn-b- PHEAm, Mn(SEC) = 23,500 PDI = 1.11) was synthesized directly (in the absence of protecting groups) by sequential RAFT polymerization of DMA and HEA monomers using CEV as the chain transfer agent (CTA). CEV was utilized in the RAFT polymerization as it affords excellent control and produces polymers with termini that do not interfere in the reaction with isocyanates. The SEC traces and molecular weights of PDMAn-CEV macroCTA (P1) and PDMAn-b-PHEAm diblock copolymer (P2) are shown in Fig. 1 and Table 1, respectively.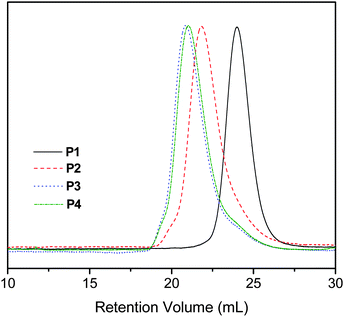 | ||
| Fig. 1 SEC traces (RI) of PDMAn macroCTA (P1), PDMAn-b-PHEAm diblock copolymer (P2) and the resulting alkene-functionalized copolymers P3 and P4. Polymer molecular weights and polydispersities are shown in Table 1. | ||
| (Co)polymer a | Mn (theo)b | Mn (NMR) | Mn (SEC)c | PDI | |
|---|---|---|---|---|---|
| a Degrees of polymerization (DP) were calculated using theoretical molecular weights. b Mn(theo) = ([monomer]0/[CTA]0) × MWmonomer × Conversion + MWCTA. c Determined using SEC (DMF). | |||||
| P1 | PDMA96 | 9,800 | 9,500 | 10,300 | 1.04 |
| P2 | PDMA96-b-PHEA115 | 23,100 | 19,000 | 23,500 | 1.11 |
| P3 | PDMA96-b- PHEA(acrylate)115 | 39,300 | 30,600 | 39,600 | 1.18 |
| P4 | PDMA96-b-PHEA(allyl)115 | 32,600 | 25,800 | 35,300 | 1.14 |
Alkene functionalization
The hydroxyl groups of the HEA block were reacted in separate reactions with two alkene-containing isocyanates 2-(acryloyloxy)ethylisocyanate (AOI) and allylisocyanate (AI) in the presence of dibutyltin dilaurate (DBTDL) catalyst, resulting in acrylate- (P3) and allyl-functionalized (P4) precursor copolymers, respectively.56 These isocyanate reactions were conducted at 40 °C in the presence of 4,4′-methylenebis(2,6-di-tert-butylphenol) inhibitor to prevent the alkene groups from polymerizing. 1H NMR spectra of P2, P3 and P4 are shown in Fig. 2. The quantitative conversions were confirmed by the following: (a) complete disappearance of the hydroxyl group resonances at 4.64–5.18 ppm, (b) shift of neighboring methylene proton resonances from 3.39–3.51 to 3.7–4.2 ppm, (c) appearance of resonances due to vinylic protons at 5.7–6.5 for acrylate and 5–6 ppm for allyl groups, and (d) the appearance of the carbamate N–H signal at 7.20–7.40 ppm. The consumption of the hydroxyl groups was also qualitatively observed by the changes in FT-IR spectra of P2, P3 and P4 (spectra are available in the ESI†). In addition to the change in shape of the broad band at 3600–3200 cm−1 attributed to the reaction of hydroxyl groups, copolymers P3 and P4 also showed vibrations at 1724 cm−1 from carbamate and ester carbonyl groups, C–O stretching bands at 1250–1150 cm−1 and characteristic vibrations of the alkene moieties at 1000–900 cm−1. Lastly, the success of the coupling reaction was indicated by the increase in molecular weight of copolymers P3 and P4 as compared to its precursor copolymer P2 (see Fig. 1, Table 1). The experimental Mn values agree with the theoretically predicted molecular weights.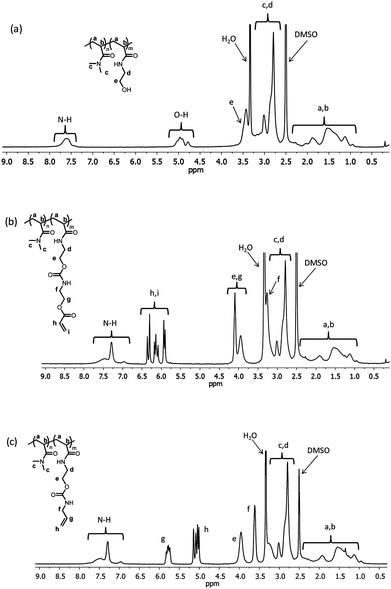 | ||
| Fig. 2 1H NMR spectra (in DMSO-d6) of (a) PDMAn-b-PHEAm (P2) diblock copolymer and alkene-functionalized precursor copolymers (b) P3 and (c) P4. Efficiencies of functionalization were determined from the disappearance and appearance of characteristic resonances due to the reactions of alkene isocyanate linkers (i.e.2-(acryloyloxy)ethylisocyanate (AOI) and allylisocyanate (AI)) with the pendent hydroxyl groups. | ||
Thiol-ene click addition
Hydrothiolation of alkenes can be generally categorized as anionic Michael-type additions or as free radically-mediated reactions. Driven by the high nucleophilicity of the thiolate anion, the former route is effective with electron-deficient alkenes (e.g. (meth)acrylates, (meth)acrylamides, maleimides, etc). However, free radical type addition of thiols to electron deficient alkenes is not as effective due to inherently low reaction rates and competing side reactions such as homopolymerization. With these considerations in mind, precursor copolymers P3 and P4 were modified via Michael and free radical thiol-ene addition reactions, respectively, using selected thiols (see Scheme 2).Amines are effective catalysts for thiol Michael-type addition.24,57,58 In the base-catalyzed reaction, a proton is abstracted from the thiol forming a thiolate anion and the conjugate acid. The thiolate anion then adds to the less hindered beta carbon of the alkene and generates the carbon-centered anion (enolate) intermediate that immediately abstracts a proton from a donor (i.e. the conjugate acid or a thiol), yielding the thiol Michael addition product and regenerating the base or thiolate anion. In the free radical-mediated thiol-ene addition, the free radical may be generated photochemically or thermally using appropriate initiator. The generated free radical first abstracts a hydrogen atom from the thiol. The formed thiyl free radical then adds to the carbon-carbon double bond in anti-Markownikov fashion. The carbon-centered free radical subsequently abstracts a hydrogen atom from another thiol, regenerating the thiyl free radical. This chain reaction continues until all the reactants are consumed.13
The thiol conjugations to copolymer P3via Michael-type addition were carried out in DMSO using triethylamine (TEA) as the base catalyst. In the reactions of neutral thiols (copolymers P3a-f), successful conjugations were observed using 0.1 equivalent of the catalyst. However, this amount of catalyst failed to achieve the desired conversions in the reactions of 3-mercaptopropionic acid, mercaptosuccinic acid, cysteamine HCl and L-cysteine HCl. These thiol compounds contain protons that are more labile (lower pKa values) than those of the other thiols. The base catalyst and/or the enolate anion intermediate, a strong base, may preferentially abstract a proton from these highly labile donors, hindering the (re)generation of thiolate anion and quenching the reaction cycle. To circumvent this issue, an excess TEA was added to neutralize these acidic groups. For example, the use of 1.1 equivalents of TEA in the reaction of 3-mercaptopropionic acid yielded >99% conjugation.
As indicated in Table 2, the use of slight excess (1.3 equivalents) of the thiol reactant yielded quantitative conjugations to copolymer P3. However, the reaction of 2-methyl-2-propanethiol (copolymer P3b) resulted only in ∼83% conjugation. Increasing the amount of thiol to 5.0 equivalents resulted in >99% alkene conversion as observed in 1H NMR spectroscopy.
| Copolymer | Thiol | Ene: Thiol: Catalyst | Conversion (%) | Mn (theo) | Mn (SEC) | PDI |
|---|---|---|---|---|---|---|
| a Use of 1.3 equivalents of thiol yielded 83% conjugation. b Determined using SEC (DMF). c Determined using aqueous SEC. The dn/dc values of the precursor copolymers were used in the SEC analyses. | ||||||
| P3a |

|
1.0: 1.3: 0.1 | >99 | 48,000 | 50,600 b | 1.18 |
| P3b |

|
1.0: 5.0: 0.1 | >99 (83) a | 49,700 | 52,400 b | 1.17 |
| P3c |

|
1.0: 1.3: 0.1 | >99 | 53,600 | 74,200 b | 1.14 |
| P3d |

|
1.0: 1.3: 0.1 | >99 | 52,000 | 71,500 b | 1.17 |
| P3e |

|
1.0: 1.3: 0.1 | >99 | 48,300 | 59,400 b | 1.18 |
| P3f |

|
1.0: 1.3: 0.1 | >99 | 51,700 | 48,600 c | 1.02 |
| P3g |

|
1.0: 1.3: 1.1 | >99 | 51,500 | — | — |
| P3h |

|
1.0: 1.3: 2.1 | >99 | 56,600 | 44,300 c | 1.18 |
| P3i |

|
1.0: 1.3: 1.1 | >99 | 52,400 | 42,900 c | 1.02 |
| P3j |

|
1.0: 1.3: 2.1 | >99 | 57,400 | 52,400 c | 1.01 |
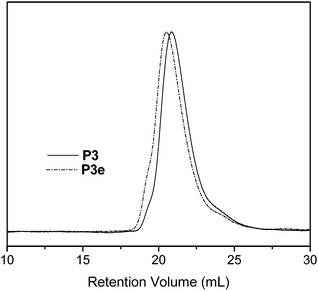
The efficiencies of the Michael-type thiol addition reactions were indicated by the complete disappearance of the vinyl resonances (5.7–6.5 ppm) as well as the appearance of new resonances due to the conjugated thiols in the 1H NMR spectra. For example, copolymer P3e shows resonances at 3.5 and 4.75 ppm from the methylene and hydroxyl protons, respectively, of the conjugated 2-mercaptoethanol (Fig. 3). In addition, new resonances between 2.5–2.75 are attributed to the methylene protons located next to the thioether linkage. 1H NMR and FT-IR spectra of all functionalized copolymers are found in the accompanying ESI.† The corresponding molecular weights and polydispersities are shown in Table 2. All functionalized copolymers maintained low polydispersities that are comparable to those of the precursor copolymer P3. For example, the SEC traces for copolymers P3 and P3e are shown in Fig. 4.
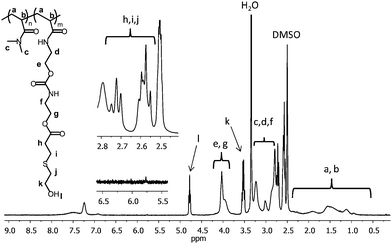 | ||
| Fig. 3 1H NMR spectrum (in DMSO-d6) of copolymer P3e. Quantitative conjugation of the thiol is indicated by the complete disappearance of the vinyl resonances at 5.7–6.5 ppm as well as the appearance of new resonances associated to the conjugated thiol. | ||
The functionalization of P4 was conducted in solution via thermal initiation following literature procedure.14 The thiol-ene addition reactions (alkene/thiol/initiator = 1/10/0.3) were performed in DMSO solution. The hydrophobic thiols with the exception of thiophenol were successfully conjugated to the precursor copolymer P4 (see Table 3). Thiophenol is an aromatic thiol and its thiyl radical is more stable than that of an alkyl thiol. In contrast to thiol Michael-type addition, the free radical additions of polar thiols to P4 are sensitive to the chemical structure of the thiols. The primary thiols (2-mercaptoethanol, 3-mercaptoproprionic acid and cysteamine HCl) yielded quantitative conversions of the allyl groups. On the other hand, the reaction of the bulkier thiols (1-thioglycerol and mercaptosuccinic acid) resulted in less than 100% conjugation. Lastly, attempts to conjugate L-cysteine HCl to P4 using various solvents such as DMSO, DMSO/buffer and DMSO/DI water mixtures failed. It should be noted that the pH of the reaction mixtures was carefully adjusted to the acidic range, and monitored before and after reactions to make certain that the thiol groups were protonated.
| Copolymer | Thiol | Conversion (%) | Mn (theo) | Mn (SEC) | PDI |
|---|---|---|---|---|---|
| a Determined using SEC (DMF). b Determined using aqueous SEC. The dn/dc values of the precursor copolymers were used in the SEC analyses. | |||||
| P4a |

|
>99 | 41,300 | 47,900 a | 1.16 |
| P4b |

|
>99 | 43,000 | 50,200 a | 1.15 |
| P4c |

|
>99 | 46,900 | 73,000 a | 1.14 |
| P4d |

|
0 | 45,300 | — | — |
| P4e |

|
>99 | 41,600 | 52, 600 a | 1.13 |
| P4f |

|
98 | 44,100 | 41,700 b | 1.01 |
| P4g |

|
>99 | 44,800 | — | — |
| P4h |

|
83 | 41,400 | 42,300 b | 1.17 |
| P4i |

|
>99 | 47,000 | 41,900 b | 1.03 |
| P4j |

|
0 | 46,500 | — | — |
Potential applications
In the previous sections and ESI,† we have demonstrated the efficiency of sequential RAFT polymerization, structopendent isocyanate coupling, and thiol click addition in preparing well-controlled structures. The reaction sequence described here offers yet another synthetic route for preparation of stimuli-responsive amphiphilic block copolymers for utility in controlled/targeted release, diagnostics, formulation, water remediation, enhanced oil recovery, etc. Such additional ways of introducing selected hydrophobic, pH-, salt- or temperature-responsive segments will add to the growing “toolbox” available to chemists for constructing reversible micelles, vesicles, rods, and other nanostructures.59–63 Some obvious extensions of these “proof of concept” studies are generalized in Scheme 3. | ||
| Scheme 3 Conceptual examples demonstrating the utility of the synthetic pathway involving sequential isocyanate and thiol-ene reactions from a RAFT-synthesized polymeric scaffold. | ||
Analyses by dynamic light scattering (DLS) were performed to show how changes in amphiphilicity of the precursor (scaffold) block copolymer affect hydrodynamic dimensions in water. The RAFT precursor copolymer P2 is water soluble (<10 nm) but is transformed into amphiphilic, micelle-forming block copolymers P3 and P4 with hydrodynamic diameters of 30 nm and 38 nm by reactions with 2-(acryloyloxy)ethylisocyanate and allylisocyanate, respectively (Scheme 2). The click additions of carboxylic acid- and amino-containing thiols yield responsive block copolymers with pH-dependent transitions. Fig. 5 shows comparisons of the pH-dependent behavior of carboxylic acid (P3g), amine (P3i) and zwitterionic (P3j) conjugates of P3 prepared by thiol click additions of 3-mercaptopropionic acid, cysteamine and L-cysteine, respectively.
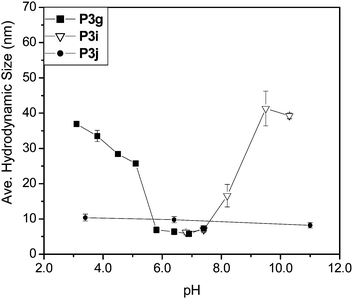 | ||
| Fig. 5 pH response of functional copolymers P3g (carboxylic acid), P3i (amine) and P3j (zwitterion) as measured by DLS in water at 1.0 mg mL−1copolymer concentrations. | ||
Copolymer P4i was prepared from the thiol click addition of cysteamine to the allyl-substituted precursor P4. The formation of interpolyelectrolyte complexes of this protonated cationic block copolymer with transfer ribonucleic acid (tRNA) was studied (see Fig. 6). Gel electrophoresis experiments indicate behavior dependent on cationic block length (N/P ratio) and suggest application as alternative scaffolds to those recently reported for gene delivery of RNA or DNA.64–67
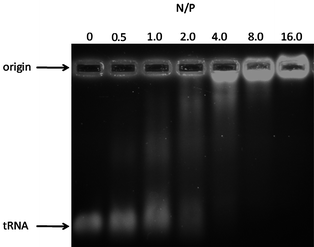 | ||
| Fig. 6 Agarose gel image of tRNA interpolyelectrolyte complexes (IPECs) with the amine-functionalized copolymer P4i at various nitrogen to phosphate (N/P) ratios. | ||
In a final example, glycopolymer derivatives were prepared by conjugating 1-thio-β-D-glucosevia the free radical pathway. Synthetic glycopolymers are promising materials for designing therapeutic carriers since sugar-binding proteins such as lectins found on cell surfaces are responsible for various intercellular recognition processes.68–71 Synthetic polymeric glycoconjugates exhibit enhanced signal recognition by lectins due to the multivalency of the saccharide moieties, a behavior termed as cluster glycoside effect.71 Data from lectin-binding assay of the glycocopolymers are shown in the ESI.†
Conclusions
The combination of RAFT polymerization and sequential reactions involving carbamate formation and thiol-ene click addition to modify the pendent groups of well-defined copolymer proves to be a facile, modular approach for synthesis of a library of functional copolymers from a single copolymer scaffold. The RAFT technique provides extensive options for monomer selection while the isocyanate-hydroxyl group reactions and thiol additions to alkene offer highly versatile routes for structopendent group transformations. The model thiols of this study reacted efficiently via Michael-type and free radical-mediated thiol-ene addition reactions. The modular capability of the method allows the attachment of various groups to the polymer chains and hence preparation of multifunctional scaffolds. This approach may be envisioned for the conjugation of thiol-containing molecules such as proteins and other bio-relevant species in combination with other moieties for targeting, imaging and therapeutics. Efforts to prepare multifunctional polymeric architectures using the strategy outlined here are being explored in our laboratories.Acknowledgements
We gratefully acknowledge the financial support provided by the MRSEC (DMR-0213883) and the EPSCOR (EPS-0903787) programs of the National Science Foundation.References
- J. F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- M. van Dijk, D. T. S. Rijkers, R. M. J. Liskamp, C. F. van Nostrum and W. E. Hennink, Bioconjugate Chem., 2009, 20, 2001–2016 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS.
- J. Justynska and H. Schlaad, Macromol. Rapid Commun., 2004, 25, 1478–1481 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- J. Justynska, Z. Hordyjewicz and H. Schlaad, Polymer, 2005, 46, 12057–12064 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- V. Lima, X. L. Jiang, J. Brokken-Zijp, P. J. Schoenmakers, B. Klumperman and R. Van Der Linde, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 959–973 CrossRef CAS.
- X. P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648–1653 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- X. P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069–7071 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854–859 RSC.
- Y. Geng, D. E. Discher, J. Justynska and H. Schlaad, Angew. Chem., Int. Ed., 2006, 45, 7578–7581 CrossRef CAS.
- A. Gress, A. Volkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- Z. Hordyjewicz-Baran, L. C. You, B. Smarsly, R. Sigel and H. Schlaad, Macromolecules, 2007, 40, 3901–3903 CrossRef CAS.
- C. Konak, V. Subr, L. Kostka, P. Stepanek, K. Ulbrich and H. Schlaad, Langmuir, 2008, 24, 7092–7098 Search PubMed.
- R. L. A. David and J. A. Kornfield, Macromolecules, 2008, 41, 1151–1161 CrossRef CAS.
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445–4448 CrossRef CAS.
- L. C. You and H. Schlaad, J. Am. Chem. Soc., 2006, 128, 13336–13337 CrossRef.
- G. J. Chen, S. Amajjahe and M. H. Stenzel, Chem. Commun., 2009, 1198–1200 RSC.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344–1350 CrossRef CAS.
- J. Ma, C. Cheng, G. R. Sun and K. L. Wooley, Macromolecules, 2008, 41, 9080–9089 CrossRef CAS.
- J. Ma, C. Cheng and K. L. Wooley, Macromolecules, 2009, 42, 1565–1573 CrossRef CAS.
- A. Li, J. Ma and K. L. Wooley, Macromolecules, 2009, 42, 5433–5436 Search PubMed.
- R. Venkatesh, F. Vergouwen and B. Klumperman, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3271–3284 Search PubMed.
- R. Paris and J. L. de la Fuente, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6247–6261 CrossRef CAS.
- R. Paris and J. L. de la Fuente, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2395–2406 CrossRef CAS.
- Y. Lin, X. H. Liu, X. R. Li, J. Zhan and Y. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 26–40 Search PubMed.
- Z. M. Dong, X. H. Liu, Y. Lin and Y. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6023–6034 CrossRef CAS.
- Z. Jia, J. Liu, T. P. Davis and V. Bulmus, Polymer, 2009, 50, 5928–5832 CrossRef CAS.
- B. Parrish and T. Emrick, Macromolecules, 2004, 37, 5863–5865 CrossRef CAS.
- X. L. Hu, X. S. Chen, S. Liu, Q. Shi and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1852–1861 CrossRef CAS.
- F. Jing and M. A. Hillmyer, J. Am. Chem. Soc., 2008, 130, 13826–13827 CrossRef CAS.
- A. E. Cherian, F. C. Sun, S. S. Sheiko and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11350–11351 CrossRef CAS.
- B. R. Maughon and R. H. Grubbs, Macromolecules, 1996, 29, 5765–5769 CrossRef CAS.
- D. J. Liaw, C. C. Huang and S. M. Hong, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6287–6298 Search PubMed.
- H. M. Zhang and E. Ruckenstein, Macromolecules, 1999, 32, 5495–5500 Search PubMed.
- H. M. Zhang and E. Ruckenstein, Macromolecules, 2001, 34, 3587–3593 Search PubMed.
- A. J. Convertine, D. S. W. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2009, 133, 221–229 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- J. D. Flores, J. Shin, C. E. Hoyle and C. L. McCormick, Polym. Chem., 2010, 1, 213–220 RSC.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Polymer, 2009, 50, 3158–3168 CrossRef CAS.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS.
- C. L. McCormick, S. E. Kirkland and A. W. York, Polym. Rev., 2006, 46, 421–443 Search PubMed.
- C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC.
- A. E. Smith, X. W. Xu and C. L. Mccormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- C. W. Scales, F. Q. Huang, N. Li, Y. A. Vasilieva, J. Ray, A. J. Convertine and C. L. McCormick, Macromolecules, 2006, 39, 6871–6881 CrossRef CAS.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS.
- A. W. York, Y. L. Zhang, A. C. Holley, Y. L. Guo, F. Q. Huang and C. L. McCormick, Biomacromolecules, 2009, 10, 936–943 CrossRef CAS.
- D. Smith, A. C. Holley and C. L. McCormick, Polym. Chem., 2011, 2, 1428–1441 RSC.
- I. Otsuka, T. Hongo, H. Nakade, A. Narumi, R. Sakai, T. Satoh, H. Kaga and T. Kakuchi, Macromolecules, 2007, 40, 8930–8937 CrossRef CAS.
- Y. Miura, T. Ikeda and K. Kobayashi, Biomacromolecules, 2003, 4, 410–415 CrossRef CAS.
- Y. Miura, N. Wada, Y. Nishida, H. Mori and K. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4598–4606 CrossRef CAS.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
Footnotes |
| † Paper number 152 in a series on Water Soluble Polymers. |
| ‡ Electronic supplementary information (ESI) available: 1H NMR and FT-IR spectra of precursor and functionalized copolymers; DLS data; and lectin-binding assay data. See DOI: 10.1039/c1py00182e |
| This journal is © The Royal Society of Chemistry 2011 |