In situ growth of a thermoresponsive polymer from a genetically engineered elastin-like polypeptide†
Weiping
Gao
a,
Donghua
Xu
b,
Dong Woo
Lim
c,
Stephen L.
Craig
b and
Ashutosh
Chilkoti
*a
aDepartment of Biomedical Engineering and Center for Biologically Inspired Materials and Material Systems, Duke University, Durham, NC 27708, USA. E-mail: chilkoti@duke.edu; Fax: +1-919-660-5409; Tel: +1-919-660-5373
bDepartment of Chemistry, Duke University, Durham, NC 27708, USA
cDepartment of BioNano Engineering, Hanyang University, Gyeonggi-do, Republic of Korea
First published on 17th May 2011
Abstract
We report the in situgrowth of a thermoresponsive dumbbell-like polymer conjugate of a genetically engineered triblock elastin-like polypeptide (tELP). Atom transfer radical polymerization (ATRP) was used to directly grow a PEG-like polymer selectively from the first and third blocks of the tELP to form a poly(oligo(ethylene glycol) methyl ether methacrylate) (poly(OEGMA)) brush conjugate with quantitative yield. We found that in situgrowth of poly(OEGMA) from tELP significantly changed the inverse phase transition and rheological behaviors of tELP. Dynamic light scattering (DLS) and turbidity measurements as a function of temperature showed that the inverse phase transition behavior of the conjugate was not determined by the tELP but by poly(OEGMA). Oscillatory rheological measurements indicated that the conjugate started to form a physical hydrogel at a temperature of 55 °C. In vitro enzymatic degradation studies showed that the conjugate could be degraded by collagenase. These results suggest that this class of conjugates may be potentially useful as an injectable, thermoresponsive drug carrier for local drug delivery and as a scaffold for tissue engineering.
Introduction
Polymer–protein/peptide hybrids are a novel class of biomaterials that are receiving increasing attention for a number of applications in biomedicine, biotechnology, and nanotechnology.1–6 These biohybrids combine the structural and functional control of proteins/peptides with the versatility of synthetic polymers, and have the potential to exhibit technologically useful properties. For example, conjugating synthetic polymers to proteins has been shown to significantly improve their properties such as thermal and chemical stability, solubility, biocompatibility, and pharmacokinetics.1–4,7,8Attaching stimulus-responsive polymers to proteins generates “smart” polymer bioconjugates that can be employed to modulate protein activity, with applications in affinity separation, microfluidic protein analysis and capture, and biomolecular sensors.9–15 Conjugating proteins with hydrophobic polymers forms polymer–protein hybrid amphiphiles that can self-assemble in aqueous solutions into nanostructures,16 while attaching poly(ethylene glycol) (PEG) to self-assembling peptides can drive the formation of nanofibrils.17–19 A coiled coil peptide was conjugated to the two ends of PEG to form a triblock peptide–PEG–peptide conjugate that self-assembled into viscoelastic hydrogels.20 A protein was used to chemoselectively cross-link a functional polymer to form a protein–polymer hybrid gel.21
Despite these advances, to our knowledge, there are few examples of conjugates of peptide polymers and synthetic polymers and none that use an in situ methodology to directly grow the synthetic polymer from one or more spatially defined sites on the peptide polymer. This lacuna motivated us to explore the in situ synthesis of a synthetic polymer from a genetically encoded peptide polymer. Herein, we report on the synthesis and characterization of a new functional biohybrid architecture—a thermally responsive and dumbbell-like polymer conjugate of a genetically engineered elastin-like polypeptide (ELP) that forms a physically crosslinked hydrogel in response to temperature change.
We chose an ELP as the peptide polymer for this study, because ELPs exhibit lower critical solution temperature (LCST) transition behavior, wherein they reversibly aggregate in aqueous solution above their LCST (which is often also called their inverse transition temperature (Tt)). The stimulus responsiveness of ELPs provides a useful property to modulate the behavior of the conjugate that can also be easily assayed by a number of methods. Although a number of ELP architectures were available to us from previous studies, we specifically chose an ABA triblock ELP (tELP) for these studies, in which the A block has 8 lysine residues and the B block is neutral, hydrophilic, and does not contain any Lys residues. We have previously demonstrated that the lysine residues in the A blocks of this ABA triblock copolymer can be chemically cross-linked to form a hydrogel.22 We hypothesized that the lysine residues could be similarly modified to present ATRP initiation sites for the in situgrowth of a polymer brush from the A blocks to form an ELP–polymer conjugate with a dumbbell-like architecture.
We chose to grow poly(OEGMA) brush from the tELP because we previously found that poly(OEGMA) brushes grown by in situATRP from macroscopic surfaces are “non-fouling”—protein and cell resistant.23,24 Very recently, we demonstrated that site-specific and stoichiometric protein–poly(OEGMA) conjugates by in situATRP show significantly improved in vivo pharmacokinetics.7,8 Therefore, we hypothesized that the in situgrowth of poly(OEGMA) brush from ELP may lead to useful biomaterials for biomedical applications, wherein the poly(OEGMA) brush would provide a biologically inert component, while the ELP segments would provide stimulus responsiveness and protease degradation, properties that are relevant for biomedical applications of this class of hybrid biomacromolecules.
Experimental
Construction of the ABA triblock ELP (tELP)
The ABA triblock ELP was synthesized from a plasmid-borne gene in E. coli. Blocks A and B are named ELP(KV7F-72) and ELP(VG7A8-64), respectively. ELP(KV7F-72) is an ELP that contains 72 repeat units of VPG![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) G, VPGVG and VPG
G, VPGVG and VPG![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) G, where the ratio of the guest residues at the fourth position in the monomers is 1
G, where the ratio of the guest residues at the fourth position in the monomers is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:1. ELP(VG7A8-64) has 64 repeat units of VPGVG, VPG
:7:1. ELP(VG7A8-64) has 64 repeat units of VPGVG, VPG![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) G and VPG
G and VPG![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) G with a V:G:A ratio of 1:7:8. The ABA triblock ELP, ELP(KV7F-72)–(VG7A8-64)–(KV7F-72), was purified by inverse transition cycling (ITC) after over-expression in E. coli. Each A block has 8 Lys residues, which are potential initiation sites for growth of poly(OEGMA), and there is an additional Lys residue in a short N-terminal peptide sequence (Ser-Lys-Pro-Gly) that precedes the N-terminal A block of the ABA triblock ELP.
G with a V:G:A ratio of 1:7:8. The ABA triblock ELP, ELP(KV7F-72)–(VG7A8-64)–(KV7F-72), was purified by inverse transition cycling (ITC) after over-expression in E. coli. Each A block has 8 Lys residues, which are potential initiation sites for growth of poly(OEGMA), and there is an additional Lys residue in a short N-terminal peptide sequence (Ser-Lys-Pro-Gly) that precedes the N-terminal A block of the ABA triblock ELP.
Synthesis of an ATRP initiator N-succinimidyl 2-bromo-2-methylpropionate (SBM)
Hydroxysuccinimide (12.4 g, 107.7 mmol) and triethylamine (10 g, 100 mmol) in 50 mL dichloromethane were dropwise added to a solution of 2-bromoisobutyryl bromide (20.7 g, 90.0 mmol) in dry dichloromethane (50 mL), at 0 °C. The resulting mixture was allowed to warm up to room temperature and stirred overnight. After the solid precipitate was filtered off, chromatographic separation (silica gel, hexane/ethyl acetate 3:1 v/v) yielded the target product N-succinimidyl 2-bromo-2-methylpropionate (SBM). 1H NMR (CDCl3): 2.0 (s, 6H, CH3), 2.8 (s, 4H, CH2). 13C NMR (CDCl3): 25.5, 30.6, 51.2, 167,4, 168.7. EIMS m/z: 264 [M + H]+.
Attaching the ATRP initiator to tELP
To the solution of tELP (100 μM, 20 mL) in PBS (100 mM, pH 8.5), SBM (1.0 g, 38 mmol) was added dropwise in 20 mL dimethylformamide at 0 °C. The reaction solution was allowed to warm up to room temperature and was stirred overnight. The crude product was purified by adding sodium chloride to the reaction solution to precipitate out the product (tELP-Br). Centrifugation at 35000g for 15 min resulted in the formation of pellet that was enriched in tELP-Br. The pellet was redissolved in PBS, and this process was repeated three times to obtained pure tELP-Br.
In situ ATRP from tELP-Br
A deoxygenated solution of 1 mL PBS, 2.7 mmol oligo(ethylene glycol) methyl ether methacrylate (OEGMA) (MW = 300, Sigma-Aldrich), 0.02 mmol CuCl, 0.088 mmol CuCl2, and 0.13 mmol 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA) was transferred into a deoxygenated tELP-Br solution (50 μM, 10 mL) in PBS containing 15% dimethylformamide. The polymerization was allowed to proceed for 1 h under argon and then exposed to air. The polymerization mixture was further purified by dialysis against water.Size exclusion chromatography (SEC)
SEC was performed on a Shimadzu HPLC system equipped with an Asahipak GS-620 HQ (with a guard column), a UV-vis detector (SPD-10A VP), a light scattering detector (DAWN, HELEOS-II, Wyatt) and a refractive index detector (Optilab rex, Wyatt). Tris–HCl buffer (50 mM Tris, pH7.4) was used as the eluent at 10 °C (flow rate: 0.6 mL min−1). The dn/dc was determined by the refractive index (RI) detector and was used to calculate the molecular weight and polydispersity by the light scattering detector. Briefly, a known sample volume with an analyte concentration of 0.1 wt% was injected into the HPLC column, and the RI profile of the elution peak of the sample was detected by the RI detector. The software from Wyatt Instruments was used to calculate the dn/dc (0.086 mL g−1), according to the RI profile.Turbidity measurement
The inverse phase transition behaviors of tELP, tELP-Br, tELP-g-poly(OEGMA) were measured at a concentration of 10 μM between 10 and 90 °C at a heating rate of 1 °C min−1. The OD350 was measured by a UV-visible spectrophotometer (Cary 300 Varian Instruments). The inverse phase transition temperature (Tt) is defined as the temperature at which the first derivative of the turbidity as a function of temperature (d(OD)/dT) is maximum.Dynamic light scattering (DLS)
DLS of tELP, tELP-Br and tELP-g-poly(OEGMA) at 5 μM in PBS were performed using a DynaPro LSR instrument (Protein Solutions, Charlottesville, VA). The intensity versus intensity time correlation functions were measured at a scattering angle of 90° at temperatures ranging from 10 to 70 °C.Oscillatory rheology
Rheological data were obtained by using an AR G2 rheometer (TA Instruments) with a cone-plate geometry (diameter of 20 mm, cone angle of 2°, truncation height of 49 μm). After loading samples, the edge of sample was sealed with mineral oil to prevent the evaporation of water from the sample during temperature sweep experiments. Temperature sweep experiments were performed at a frequency of 10 rad s−1 and at 5% strain amplitude over a range of temperatures from 25 °C to 75 °C (or 90 °C for the conjugate).In vitro degradation
Collagenase from Clostridium histolyticum (Sigma) was dissolved in PBS to a concentration of 80 units mL−1. 5 μL of the collagenase solution was added to 200 μL of a 2 μM tELP and tELP-g-poly(OEGMA) solution in PBS. The mixtures were incubated for 1 h at 37 °C and then were analyzed by SDS-PAGE.Results and discussion
Attachment of an ATRP initiator to the tELP, ELP(KV7F-72)–(VG7A8-64)–(KV7F-72)
In order to grow a poly(OEGMA) brush from tELP by in situATRP, a hydroxysuccinimide derivatized ATRP initiator, N-succinimidyl 2-bromo-2-methylpropionate (SBM), was synthesized and was attached to the amine groups of the lysine residues and the N-terminus in the tELP to form a macroinitiator (tELP-Br, Scheme 1). The product was purified by inverse transition cycling (ITC), as described in the Experimental Section. The yield of attachment of the ATRP initiator was determined by NMR (Fig. S1†). A characteristic NMR signal from the ATRP initiator was clearly observed at 1.8 ppm, corresponding to CHBr–CH3, and the peak for the α-methylene (N–CH2–CH2) of the lysine side-chain shifted from 2.7 ppm to 3.0 ppm after the amidation reaction. Integration of the peak intensities at δ 2.7 ppm and 3.0 ppm suggests that the efficiency of the attachment of the ATRP initiator to the lysine residues was ∼90%, or ∼8 initiator moieties attached to each A block of the ELP.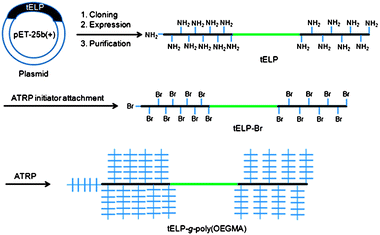 | ||
| Scheme 1 Schematic illustration of synthesis of a conjugate of poly(OEGMA) from a genetically encoded triblock elastin-like polypeptide (tELP-g-poly(OEGMA)) by in situATRP. | ||
In situ ATRP from tELP-Br
Next, in situATRP was carried out to grow a poly(OEGMA) brush from tELP-Br. The resulting ATRP solution was directly analyzed by SEC (Fig. 1). The SEC traces from UV detection at 280 nm (Fig. 1A) clearly show the appearance of a new peak with a lower elution time (10 min) and the disappearance of the elution peak at 16 min corresponding to tELP-Br, indicating that a higher molecular weight (MW) product, presumably tELP-g-poly(OEGMA), was formed and that the macroinitiator tELP-Br was completely converted to the conjugate. These results were also confirmed by RI detection (Fig. 1B). SDS-PAGE analysis provided visual confirmation of these results, as the band corresponding to tELP-Br with a mass of ∼86 kDa disappeared, and a smear with a much higher mass corresponding to the tELP-g-poly(OEGMA) conjugate was exclusively seen after in situATRP (Fig. S2†). In a control experiment, in which ATRP was performed from tELP in the same ATRP condition as from tELP-Br, a new peak at a lower elution time was not detected, indicating that the poly(OEGMA) brush could not be formed by in situATRP without modification of tELP with the ATRP initiator (Fig. S3†). This result was confirmed by SDS-PAGE as well (Fig. S2†).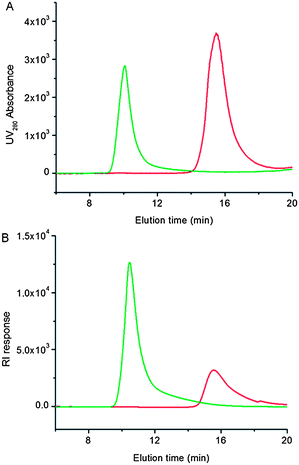 | ||
| Fig. 1 SEC traces of tELP-g-Br (red curve) and tELP-g-poly(OEGMA) (green curve). Panel A: UV traces at the absorbance of 280 nm; panel B: RI traces. | ||
Purification and characterization of tELP-g-poly(OEGMA)
The quantitative attachment of the ATRP initiator significantly eased the purification of tELP-g-poly(OEGMA), as there was no residual tELP left after polymerization. We simply dialyzed the resulting ATRP reaction mixture against water or PBS to remove low MW molecules including the ATRP catalyst and residual monomer. After dialysis, we determined the MW of the conjugate by static light scattering to be 600 kDa, which is nearly 7 times greater than that of tELP (86 kDa). The average MW of poly(OEGMA) graft chains was calculated to be 29 kDa. The polydispersity of the conjugate was determined to be 1.5. We further confirmed the formation of a poly(OEGMA) brush from the tELP-Br by 1H NMR (Fig. S4†). As expected, characteristic signals of poly(OEGMA) were clearly observed at 1, 2, 3.4, 3.6–3.8, and 4.2 ppm that correspond to CCH3, CH2C, OCH3, OCH2CH2, and C(O)OCH2, respectively.Inverse temperature phase transition
The effect of temperature on the turbidity of the conjugate and its precursors tELP-Br and tELP in PBS (10 μM) is shown in Fig. 2A. After attachment of the ATRP initiator to the (KV7F-72) blocks of tELP, a LCST phase transition25,26 was observed and the Tt shifted to 18 °C, compared to the ∼30 °C Tt of the parent block copolymer. This suggests that the ATRP initiator attachment increased the hydrophobicity of the (KV7F-72) blocks, thereby significantly changing the phase transition behavior of tELP. Interestingly, after in situgrowth of the poly(OEGMA) brush from tELP-Br, a similar LCST phase transition was observed, but with a significantly increased Tt of ∼58 °C. Poly(OEGMA) is also known to exhibit LCST behavior27 and has a Tt of 57 °C (Fig. S5†), which indicates that the poly(OEGMA) brush controls the LCST behavior of the tELP–poly(OEGMA) brush conjugate. These results were further confirmed by DLS analysis (Fig. 2B). Below their Tt, tELP and tELP-Br had a similar hydrodynamic radius (Rh) of ca. 8 nm. Consistent with the temperature-programmed turbidity measurements, the Rh of tELP started to increase at 25 °C, while the Rh of tELP-Br rapidly increased at 18 °C. The Rh of the conjugate was ca. 20 nm below 55 °C and dramatically increased at 58 °C. These data clearly demonstrate that in situgrowth of a poly(OEGMA) brush from tELP-Br changes its physicochemical properties and that the poly(OEGMA) dominates the temperature-dependent phase behavior of the biohybrid macromolecule.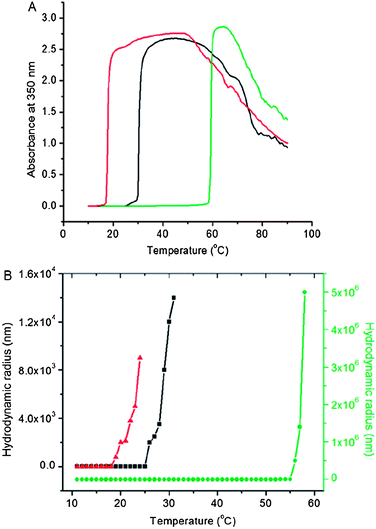 | ||
| Fig. 2 (A) Turbidity profiles of tELP (black), tELP-g-Br (red) and tELP-g-poly(OEGMA) (green) as a function of temperature. (B) DLS analysis of tELP (black), tELP-g-Br (red) and tELP-g-poly(OEGMA) (green) as a function of temperature. | ||
Thermoresponsive gelation
The viscoelastic properties of tELP and tELP-g-poly(OEGMA) were measured by oscillatory rheology as a function of temperature (Fig. 3). In Fig. 3A, the temperature sweep results of a 25 wt% aqueous solution of tELP are shown. As the temperature increased from 25 °C to 50 °C, the storage (G′) and loss (G′′) moduli both increased with temperature. G′′ in particular showed an inflection point at ∼33 °C, a value that is close to the Tt of 30 °C measured by temperature-programmed turbidimetry (Fig. 2A). With a further increase in temperature from 50 °C to 75 °C, neither G′ nor G′′ changed substantially. The value of G′′ is greater than that of G′ over the entire temperature range; the tELP is more viscous than elastic even in its coacervate phase at temperatures above Tt, indicating that either a fully cross-linked network has not been formed or that the physical network is weakly bonded so that it relaxes quickly on the time scale of the oscillatory experiment (10 rad s−1).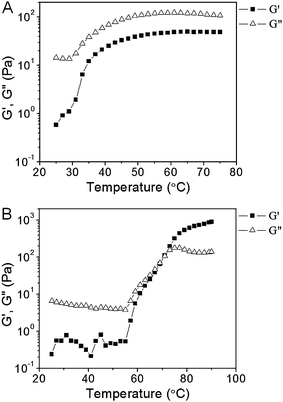 | ||
| Fig. 3 Oscillatory rheological profiles of tELP at 25 weight% in PBS (A), tELP-g-POEGMA at 25 weight% in PBS (B) as a function of temperature. | ||
In contrast, the tELP-g-poly(OEGMA) conjugate exhibited different rheological behavior (Fig. 3B). Between 25 °C and 55 °C, G′′ is greater than G′, corresponding to largely viscous behavior. Neither modulus changed significantly in this temperature range. Both G′ and G′′ increased dramatically, however, at temperatures greater than 55 °C, with a crossover point between G′ and G′′ at ∼70 °C. These results indicate the formation of an elastic network that is effectively solid on the time scale of the characterization, presumably due to the formation of a physically crosslinked hydrogel network. It is interesting to note that the evolution of the elastic network commenced at ∼55 °C, which is close to the Tt of the conjugate (Fig. 3B), but the conjugate does not fully attain its elastic properties until ∼70 °C. The origin of this thermal lag in the evolution of the elastic network, in particular the extent to which it is a kinetic rather than an equilibrium structural effect, is currently unclear and will be addressed in future studies.
The critical conclusion from the dynamic mechanical characterization is that the transitions in molecular structure and aggregation are coupled to changes in mechanical properties, offering promise for simultaneous tuning of all of these properties. Clearly, the dependencies of Tt and viscoelastic properties on the architecture of these biohybrid macromolecules are complex, as they involve both structural and dynamic components (e.g., the mesh size of the network vs. the relaxation dynamics of the physical associations). A full characterization will require systematic study of mechanical properties as a function of key macromolecular variables such as the composition and segment ratios of the ELP blocks and the poly(OEGMA) brush length and brush density, in order to unentangle their contributions to the phase behavior and mechanical properties of these complex biohybrid macromolecules.
In vitro enzymatic degradation
Next, we examined the enzymatic degradation of tELP-g-poly(OEGMA) to determine if the growth of the poly(OEGMA) brush affected the enzymatic degradability of the ELP segments in the conjugate. This study was motivated by the notion that a likely application of this class of biohybrid macromolecules would be as injectable materials to form s.c. depots for the sustained delivery of drugs for systemic therapy or as injectable depots within diseased tissues (e.g., arthritic knee joints, solid tumors, or resected tumor cavities) for local delivery of drugs. The in vivodegradation of the depot, once the drug is released, would be desirable to prevent long-term in vivo accumulation of the depot. We chose collagenase because it is an important matrix protease, and because we had previously shown that ELPs have a surprising—and unanticipated—sensitivity to degradation by collagenase.28 After 1 h incubation with collagenase at 37 °C, we analyzed the mixtures by SDS-PAGE. The band at 86 kDa corresponding to tELP disappeared after 1 h incubation, confirming that the unmodified ELP is facilely degraded by collagenase (Fig. 4A). The discrete bands in Lane 3 correspond to an aliquot of collagenase as received from the supplier, suggesting that the collagenase preparation is somewhat impure. In contrast, after 1 h incubation, the smear corresponding to tELP-g-poly(OEGMA) with a high molecular weight disappeared and a new broad smear appeared in the range of low molecular weights (Fig. 4B), indicating the conjugate is facilely degraded by collagenase, similar to the unmodified ELP.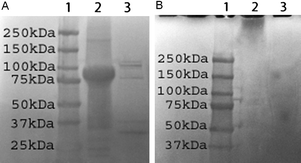 | ||
| Fig. 4 SDS-PAGE analysis of enzymatic degradation of tELP (A) and tELP-g-poly(OEGMA) (B). In panel A, lane 1: protein marker; lane 2: tELP; lane 3: tELP after digestion for 1 h. In panel B, lane 1: protein marker; lane 2: tELP-g-poly(OEGMA); lane 3: tELP-g-poly(OEGMA) after digestion for 1 h. | ||
Conclusions
We report the in situgrowth of poly(OEGMA) from a triblock ELP (tELP) by ATRP. The resulting tELP-g-poly(OEGMA) conjugate had a dumbbell-like structure with a poly(OEGMA) brush grafted from the first and third ELP blocks. The conjugate showed a LCST behavior, and the inverse transition temperature (Tt) of the conjugate was significantly different from that of tELP, but similar to that of poly(OEGMA). The conjugate also showed different rheological properties from those of tELP. These results indicate that in situATRP of OEGMA from tELP-Br significantly changes the physicochemical properties of tELP, and that the physicochemical properties of the conjugate are a complex function of their architecture. The conjugate was easily degraded by collagenase, which showed that the sensitivity of the ELP to proteolytic degradation was retained by the conjugate.In conclusion, although this paper is the first demonstration of the site-specific in situgrowth of a synthetic bottlebrush polymer from a genetically encoded polymer, the results reported herein are simply the first step in the design of useful biohybrid architectures. Much remains to be done to design the appropriate ELP macroinitiator in terms of its length and block design and to tune the chain length and density of the poly(OEGMA) segment to create biohybrid macromolecular architectures that exhibit interesting self-assembly behaviors, which may be useful as drug delivery carriers and as scaffolds for regenerative medicine. These studies are currently in progress.
Notes and references
- M. L. Nucci, R. Shorr and A. Abuchowski, Adv. Drug Delivery Rev., 1991, 6, 133 CrossRef CAS.
- J. M. Harris, N. E. Martin and M. Modi, Clin. Pharmacokinet., 2001, 40, 539 CrossRef CAS.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261 CrossRef CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Macromol. Symp., 2004, 207, 139 CrossRef CAS.
- G. W. M. Vandermeulen and H. A. Klok, Macromol. Biosci., 2004, 4, 383 CrossRef CAS.
- W. Gao, W. Liu, J. A. Mackay, M. R. Zalutsky, E. J. Toone and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15231 CrossRef CAS.
- W. Gao, W. Liu, T. Christensen, M. R. Zalutsky and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16432 CrossRef CAS.
- P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. H. Chen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472–474 CrossRef CAS.
- Z. L. Ding, R. B. Fong, C. J. Long, P. S. Stayton and A. S. Hoffman, Nature, 2001, 411, 59–62 CrossRef CAS.
- A. S. Hoffman, Clin. Chem., 2000, 46, 1478 CAS.
- P. S. Stayton, A. S. Hoffman, M. El-Sayed, S. Kulkarni, T. Shimoboji, N. Murthy, V. Bulmus and C. Lackey, Proc. IEEE, 2005, 93, 726 CrossRef CAS.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276 RSC.
- A. S. Hoffman, P. S. Stayton, V. Bulmus, G. H. Chen, J. P. Chen, C. Cheung, A. Chilkoti, Z. L. Ding, L. C. Dong, R. Fong, C. A. Lackey, C. J. Long, M. Miura, J. E. Morris, N. Murthy, Y. Nabeshima, T. G. Park, O. W. Press, T. Shimoboji, S. Shoemaker, H. J. Yang, N. Monji, R. C. Nowinski, C. A. Cole, J. H. Priest, J. M. Harris, K. Nakamae, T. Nishino and T. Miyata, J. Biomed. Mater. Res., 2000, 52, 577 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955 CrossRef CAS.
- A. J. Dirks, R. J. M. Nolte and J. J. L. M. Cornelissen, Adv. Mater., 2008, 20, 3953 CrossRef CAS.
- T. S. Burkoth, T. L. S. Benzinger, V. Urban, D. G. Lynn, S. C. Meredith and P. Thiyagrajan, J. Am. Chem. Soc., 1999, 121, 7429 CrossRef CAS.
- J. Hentschel, E. Krause and H. G. Borner, J. Am. Chem. Soc., 2006, 128, 7722 CrossRef CAS.
- S. Kessel, A. Thomas and H. G. Borner, Angew. Chem., Int. Ed., 2007, 119, 9181 CrossRef.
- P. Jing, J. S. Rudra, A. B. Herr and J. H. Collier, Biomacromolecules, 2008, 9, 2438 CrossRef CAS.
- A. P. Esser-Kahn and M. B. Francis, Angew. Chem., Int. Ed., 2008, 47, 3751 CrossRef CAS.
- D. W. Lim, D. L. Nettles, L. A. Setton and A. Chilkoti, Biomacromolecules, 2008, 9, 222 CrossRef CAS.
- H. W. Ma, J. H. Hyun, P. Stiller and A. Chilkoti, Adv. Mater., 2004, 16, 338 CrossRef CAS.
- A. Hucknall, D. H. Kim, S. Rangarajan, R. T. Hill, W. M. Reichert and A. Chilkoti, Adv. Mater., 2009, 21, 1968 CrossRef CAS.
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel, M. Schmidt and A. Chilkoti, J. Am. Chem. Soc., 2008, 130, 687 CrossRef CAS.
- A. J. Simnick, C. A. Valencia, R. Liu and A. Chilkoti, ACS Nano, 2010, 4, 2217 CrossRef CAS.
- J. F. Lutz, O. Akdemir and A. J. Hoth, J. Am. Chem. Soc., 2006, 128, 13046 CrossRef CAS.
- M. F. Shamji, H. Betre, V. B. Kraus, J. Chen, A. Chilkoti, R. Pichika, K. Masuda and L. A. Setton, Arthritis Rheum., 2007, 56, 3650 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 shows 1H NMR spectra of tELP and tELP-g-Br. Fig. S2 shows SEC analysis after 60 min ATRP using tELP as a macroinitiator. Fig. S3 shows SEC analysis after 60 min ATRP using unmodified tELP as a macroinitiator (negative control). Fig. S4 shows 1H NMR spectrum of tELP-g-poly(OEGMA). Fig. S5 shows turbidity profile of POEGMA as a function of temperature. See DOI: 10.1039/c1py00074h |
| This journal is © The Royal Society of Chemistry 2011 |