Separation of enantiomers on diastereomeric right- and left-handed helical poly(phenyl isocyanide)s bearing L-alanine pendants immobilized on silica gel by HPLC
Kazumi
Tamura
,
Toshitaka
Miyabe
,
Hiroki
Iida
and
Eiji
Yashima
*
Department of Molecular Design and Engineering, Graduate School of Engineering, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: yashima@apchem.nagoya-u.ac.jp; Fax: +81-52-789-3185; Tel: +81-52-789-4495
First published on 23rd August 2010
Abstract
Diastereomeric left- and right-handed helical polyisocyanides composed of L-alanine-bound phenyl isocyanides were prepared. The helical polymers with a narrow molecular weight distribution were immobilized on a silica gel support via chemical bonding and their optical resolution abilities were evaluated as chiral stationary phases (CSPs) for high-performance liquid chromatography (HPLC). The CSP prepared from the left-handed helical polyisocyanide resolved racemic cyclic ether and carbonyl compounds and cyclic dianilides and dibenzamides, whereas the right-handed helical polyisocyanide-based CSP showed a rather complementary chiral recognition ability and specifically resolved racemic metal acetylacetonate complexes, which were not separated on the former CSP at all. Additionally, the elution order of some enantiomers was reversed on the CSPs, thus indicating that the macromolecular helicity of the L-alanine-bound-polyisocyanides played a critical role in the enantioselectivity and elution order of the enantiomers.
Introduction
The separation of enantiomers, in particular, those of chiral drugs has attracted great attention over the past few decades because a pair of enantiomers often shows quite different physiological activities. Therefore, the development of practical techniques for preparing enantiomers with a high enantiomeric excess has strongly been demanded in the pharmaceutical industry. Chromatographic enantioseparations, particularly resolution by high-performance liquid chromatography (HPLC), have significantly advanced during the past decades for determining their enantiomeric excess and also for obtaining pure enantiomers on analytical and industrial scales.1 The preparation of a chiral stationary phase (CSP) possessing an effective chiral recognition ability for a variety of enantiomers with different functionalities is the key to this separation technique. Currently, many commercial CSPs for HPLC are available, for example, polysaccharide-,2 protein-3 and synthetic helical polymer-based CSPs4 and chiral small molecule-based CSPs.5 Among them, the polysaccharide- and synthetic helical polymer-based CSPs, particularly, CSPs composed of cellulose and amylose derivatives2 and one-handed helical polymethacrylates prepared by helix-sense-selective polymerization6 have been recognized as the most popular and practical CSPs. Highly ordered helical structures appear to be responsible for their excellent resolving abilities. In contrast to the small-molecule CSPs, however, the chiral recognition mechanism of polymer-based CSPs is difficult to elucidate mostly because of the difficulty in determining their exact structures in both the solid state and in solution, although a plausible chiral recognition mechanism for polysaccharide-based CSPs has been proposed on the basis of chromatographic enantioseparation7 supported by spectroscopic and computational studies.8Recently, we found that both right (P)- and left (M)-handed helical polyisocyanides with a different molecular weight (MW) and a narrow molecular weight distribution (MWD) can be prepared by the living polymerization of an enantiomerically pure phenyl isocyanide bearing an L-alanine pendant with a long n-decyl chain (L-1) using the µ-ethynediyl Pt–Pd complex (2)9 as the catalyst, followed by facile fractionation with acetone (Fig. 1).10 The fractionated single-handed helical polyisocyanides (M- and P-poly-L-1s) maintained their living feature and can be used as an initiator (macroinitiator) for the further block copolymerizations of isocyanides.11 Most interestingly, the rodlike macroinitiators and resulting block copolymers self-assembled to form regular two-dimensional (2D) smectic crystals on a nanometre scale on a substrate, which makes it possible to determine their molecular lengths and helical structures including helical pitch, helical sense, and handedness excess by direct observation of the helical molecules by high-resolution atomic force microscopy (AFM).10,11
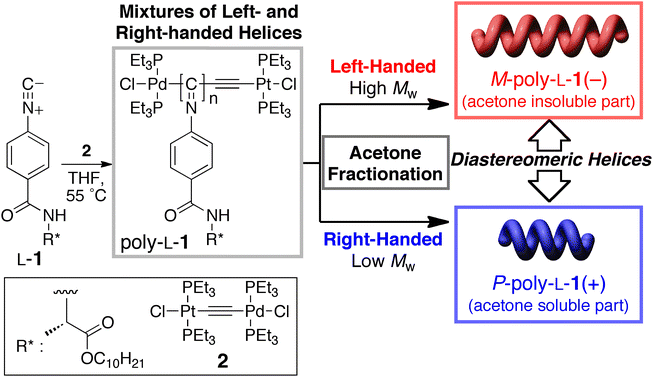 | ||
| Fig. 1 Schematic illustration of the helix-sense-selective living polymerization of L-1 with µ-ethynediyl Pt–Pd complex (2), yielding a mixture of diastereomeric, right- and left-handed helical poly-L-1's with different MWs and a narrow MWD, which can be further separated into the left-handed helical M-poly-L-1(−) and right-handed helical P-poly-L-1(+). | ||
Here we show the synthesis of M- and P-polyisocyanides with a narrow MWD and composed of a long L-1 block and a short achiral phenyl isocyanide block, immobilization on silica gel via chemical bonding, and their chiral recognition abilities as CSPs for HPLC (Fig. 2). We anticipated that the M- and P-poly-L-1s bearing the same L-alanine residues as the pendants, but with the opposite main-chain helical conformation might show different chiral recognition abilities toward enantiomers depending on their helical sense. The preferred-handed helicity induced by the L-alanine pendants would contribute to and/or enhance or reduce the enantiomer-selectivity during the chromatographic separation. These results would provide an insight into the importance of the helical chirality of the main chain and point chirality of the pendants toward chiral recognition events, and also fundamental understanding critical to the development of novel helical polymer-based CSPs.
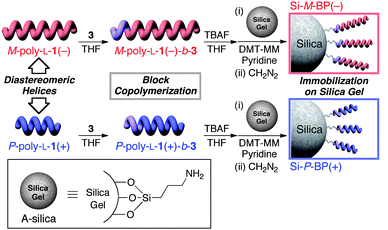 | ||
| Fig. 2 Schematic illustration of the helix-sense-selective living block copolymerization of 3 using diastereomeric left-handed M-poly-L-1(−) and right-handed P-poly-L-1(+) as the macroinitiators obtained by acetone fractionation of the as-prepared poly-L-1 and immobilization of the resulting block copolymers on silica gel through amidation reaction (Si-M-BP(−) and Si-P-BP(+)). | ||
Experimental
Materials
Anhydrous tetrahydrofuran (THF), pyridine and dimethylformamide (DMF) were purchased from Wako (Osaka, Japan) and stored under dry nitrogen. THF was further dried over LiAlH4 under nitrogen, and distilled under high vacuum just before use. tert-Butyldimethylchlorosilane (TBDMS-Cl), tetra-n-butylammonium bromide (TBAB) and tetra-n-butylammonium fluoride (TBAF, 1 M in THF) were purchased from Wako and imidazole was from Kishida (Osaka, Japan). 4-Isocyanobenzoic acid,12 4-isocyanobenzoyl-L-alanine decyl ester (L-1),13 the µ-ethynediyl Pt–Pd complex (2)9a and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM)14 were prepared according to the previously reported methods.The solvents used in the chromatographic experiments were of HPLC grade. The racemates were commercially available or were prepared by the usual methods.15 Porous spherical (3-aminopropyl)-triethoxy silanized silica gel (Daiso gel SP-1000-7-APSL, A-silica) with a mean particle size of 7 µm and a mean pore diameter of 100 nm was kindly supplied from Daicel Chemical Industries (Tokyo, Japan).
Measurements
The melting points were measured on a Yanako melting point apparatus and are uncorrected. The NMR spectra were measured using a Varian VXR-500S spectrometer (Varian, Palo Alto, CA) operating at 500 MHz for 1H using TMS as the internal standard. The IR spectra were recorded using a JASCO FT/IR-680 spectrometer (JASCO, Hachioji, Japan). The absorption and CD spectra were obtained in a 1.0 mm quartz cell at 25 °C using a JASCO V570 spectrophotometer and a JASCO J820 spectropolarimeter, respectively. The polymer concentration was calculated on the basis of the monomer units and was 0.2 mg mL−1. The optical rotations were measured in a 2 cm quartz cell on a JASCO P-1030 polarimeter. The size exclusion chromatography (SEC) was performed using a JASCO PU-2080 liquid chromatograph equipped with UV-visible (JASCO UV-2070) and CD (JASCO CD-2095) detectors. Two Tosoh TSKgel Multipore HXL-M SEC columns (Tosoh, Tokyo, Japan) were connected in series, and THF containing 0.1 wt% TBAB was used as the eluent at the flow rate of 1.0 mL min−1. The molecular weight calibration curve was obtained with standard polystyrenes (Tosoh). The SEC-MALS measurements were performed using an HLC-8220 GPC system (Tosoh) equipped with a differential refractometer coupled to a DAWN-HELEOS MALS device with a semiconductor laser (λ = 690 nm) (Wyatt Technology, Santa Barbara, CA) operated at 25 °C using two TSKgel Multipore HXL-M columns (Tosoh) in series. The scattered light intensities were measured by eighteen light scattering detectors at different angles. The differential refractive index increment, dn/dc, of the polymer with respect to the mobile phase at 25 °C was also measured by an Optilab rEX interferometric refractometer (Wyatt Technology). The thermogravimetric (TG) analyses were conducted on a SEIKO EXSTAR6000 TG/DTA 6200 (Seiko Instruments Inc., Chiba, Japan) under a heating rate of 10 °C min−1 in a nitrogen flow of 200 mL min−1 flow rate of nitrogen.The chromatographic separations of enantiomers were performed using a JASCO PU-2080 Plus liquid chromatograph equipped with Multi UV-Vis (JASCO MD-2010 Plus) and polarimetric (JASCO OR-2090 Plus, Hg–Xe without filter) or CD detectors at room temperature. A solution of racemate was injected into the chromatographic system using a Rheodyne Model 7725i injector (20 µL loop).
Synthesis of 4-tert-butyldimethylsiloxycarbonylphenyl isocyanide (3)
Imidazole (102 mg, 1.50 mmol) and TBDMS-Cl (225 mg, 1.50 mmol) were added to a solution of 4-isocyanobenzoic acid (200 mg, 1.36 mmol) in dry DMF (4.8 mL) (Scheme 1a). The reaction mixture was stirred at room temperature under a dry nitrogen atmosphere. After 22 h, water was added, and the solution was extracted with diethyl ether (3 × 20 mL). The organic extracts were dried over anhydrous Na2SO4. After filtration, the solvent was removed by evaporation, and the crude product was purified by column chromatography (SiO2) with CHCl3–methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as the eluent to give 3 as a pale yellow solid (98.0 mg, 28%).
:1, v/v) as the eluent to give 3 as a pale yellow solid (98.0 mg, 28%).
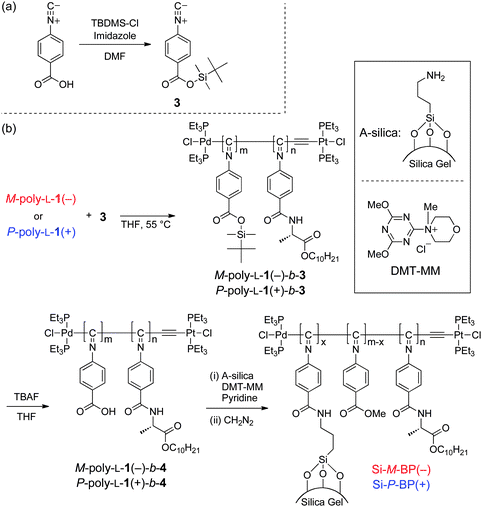 | ||
| Scheme 1 (a) Synthesis of 3. (b) Synthesis of M-poly-L-1(−)-b-4 and P-poly-L-1(+)-b-4 and immobilization on silica gel. | ||
Spectroscopic data of 3: mp: 101.8–102.2 °C; νmax(KBr)/cm−1 2122 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and 1693 (C
N) and 1693 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); δH (500 MHz, CDCl3, rt) 0.38 (s, CH3, 6H), 1.02 (s, CH3, 9H), 7.44 (d, J = 8.5 Hz, aromatic, 2H) and 8.06 (d, J = 8.5 Hz, aromatic, 2H). Found: C, 64.45; H, 7.38; N, 5.29. C14H19NO2Si requires C, 64.33; H, 7.33; N, 5.36%.
O); δH (500 MHz, CDCl3, rt) 0.38 (s, CH3, 6H), 1.02 (s, CH3, 9H), 7.44 (d, J = 8.5 Hz, aromatic, 2H) and 8.06 (d, J = 8.5 Hz, aromatic, 2H). Found: C, 64.45; H, 7.38; N, 5.29. C14H19NO2Si requires C, 64.33; H, 7.33; N, 5.36%.
Preparation of right- and left-handed helical poly(phenyl isocyanide) macroinitiators
The right- and left-handed helical poly(phenyl isocyanide) macroinitiators were prepared as outlined in Fig. 1 in a similar way to that previously reported.10 The polymerization of L-1 (2.00 g, 5.60 mmol) was carried out in a dry glass ampule under a dry nitrogen atmosphere in THF using 2 ([L-1]/[2] = 100) as the catalyst at 55 °C for 20 h. The obtained poly-L-1 (1.86 g, 93% yield) was then fractionated with acetone into left-handed helical M-poly-L-1(−) and right-handed helical P-poly-L-1(+) in 62 and 13% yield, respectively. The resulting M-poly-L-1(−) and P-poly-L-1(+) were further used as the macroinitiators.Block copolymerization of 3 with M-poly-L-1(−) and P-poly-L-1(+) as the macroinitiators
A typical experimental procedure is described below (see Scheme 1b and Fig. 2). M-Poly-L-1(−) (300 mg) and monomer 3 (22 mg, [L-1]/[3] = 10) were placed in a dry ampule, which were then evacuated on a vacuum line and flushed with dry nitrogen. After the evacuation–flush procedure had been repeated three times, a three-way stopcock was attached to the ampule, and dry THF (8.4 mL) was added by a syringe. The mixture was then stirred under a dry nitrogen atmosphere and heated to 55 °C. After 20 h, the resulting copolymer (M-poly-L-1(−)-b-3) was precipitated in a large amount of methanol, collected by centrifugation and dried overnight in vacuo at room temperature (288 mg, 90% yield). In the same way, P-poly-L-1(+)-b-3 was prepared using P-poly-L-1(+) as the macroinitiator (87% yield).Spectroscopic data of M-poly-L-1(−)-b-3: νmax(film)/cm−1 3279 (N–H), 1748 (CO ester), 1634 (amide I) and 1537 (amide II); δH (500 MHz, THF-d8, 55 °C) 0.20–0.80 (broad, SiCH3, 0.6H), 0.89 (broad, CH3 and SiC(CH3)3, 3.9H), 1.29 (broad, CH2, 14H), 1.55 (broad, CH3 and CH2, 5H), 4.09 (broad, CH2, 2H), 4.30–4.80 (broad, CH, 1H), 4.8–7.8 (broad, aromatic, 4.4H) and 8.0–9.0 (broad, NH, 1H); [α]D20 −1817 (c 0.1 in THF). Found: C, 69.51; H, 8.28; N, 7.60. (C21H30N2O3)10(C14H19NO2Si)1(H2O)1.4 requires C, 69.49; H, 8.38; N, 7.60%.
Spectroscopic data of P-poly-L-1(+)-b-3: νmax(film)/cm−1 3262 (N–H), 1749 (CO ester), 1634 (amide I) and 1537 (amide II); δH (500 MHz, CDCl3, 55 °C) 0.20–0.80 (broad, SiCH3, 0.6H), 0.91 (broad, CH3 and SiC(CH3)3, 3.9H), 1.31 (broad, CH2, 14H), 1.62 (broad, CH3 and CH2, 5H), 4.11 (broad, CH2, 2H), 4.52 (broad, CH, 1H), 4.8–7.8 (broad, aromatic, 4.4H) and 8.3–9.2 (broad, NH, 1H); [α]D20 +1633 (c 0.1 in THF). Found: C, 68.25; H, 7.97; N, 7.44. (C21H30N2O3)10(C14H19NO2Si)1(H2O)3.8 requires C, 68.73; H, 8.41; N, 7.51%.
Synthesis of M-poly-L-1(−)-b-4 and P-poly-L-1(+)-b-4
To a solution of M-poly-L-1(−)-b-3 (252 mg, 0.720 mmol) in THF (25 mL) was added a solution of TBAF in THF (1 M, 0.41 mL, 0.41 mmol) and the mixture was stirred at room temperature for 3 h. The solution was poured into a large amount of 1 M aqueous HCl–methanol (1:5, v/v) and the resulting yellow precipitate was collected by centrifugation, washed with methanol and dried in vacuo at room temperature overnight to give M-poly-L-1(−)-b-4 (216 mg, 88% yield). In the same way, P-poly-L-1(+)-b-4 was prepared in 98% yield from P-poly-L-1(+)-b-3.
Immobilization of M-poly-L-1(−)-b-4 and P-poly-L-1(+)-b-4 on silica gel
A-Silica (850 mg) was dispersed in a solution of M-poly-L-1(−)-b-4 (210 mg, 0.619 mmol) in pyridine (4.5 mL), and DMT-MM (57.6 mg, 0.161 mmol) was added to the suspended solution. After stirring at room temperature for 7 h, the resulting silica gel was collected by filtration, washed with pyridine, THF and methanol and dried in vacuo at room temperature overnight. The silica gel was then treated with diazomethane in diethyl ether to convert the remaining carboxylic acid groups of M-poly-L-1(−)-b-4 into the methyl esters at room temperature. The M-poly-L-1(−)-b-4-bound silica gel (Si-M-BP(−)) thus obtained was collected by filtration, washed with CHCl3, THF and methanol and dried in vacuo at room temperature overnight (927 mg). The content of M-poly-L-1(−)-b-4 chemically bonded to silica gel was estimated by TG analysis and was 9.9 wt%.In the same way, P-poly-L-1(+)-b-4 was chemically bonded to silica gel (Si-P-BP(+)), and its content was estimated to be 4.7 wt%.
Preparation of chiral columns
Each column packing material was packed into a stainless-steel tube (25 cm × 0.20 cm (i.d.)) by conventional high-pressure slurry packing technique using a Chemco Slurry-Packing Apparatus Model 124A (Chemco, Osaka, Japan).7a The plate numbers of the columns were 2000–2600 for benzene with hexane–2-propanol (90:10) as the eluent at a flow rate of 0.1 mL min−1. The dead time (t0) was estimated using 1,3,5-tri-tert-butylbenzene as the nonretained compound.16
Results and discussion
Synthesis of diastereomeric helical polyisocyanides and preparation of chiral stationary phases for HPLC
The left- and right-handed helical poly(phenyl isocyanide) macroinitiators (M- and P-poly-L-1s, respectively) were prepared by the living polymerization of L-1 with 2 as the catalyst in THF at 55 °C for 20 h, followed by fractionation with acetone into the acetone-insoluble (high MW M-poly-L-1(−), Mn = 6.0 × 104) and acetone-soluble (low MW P-poly-L-1(+), Mn = 2.2 × 104) fractions according to a previously reported method (Fig. 1).10 The helical sense (P or M) and helical sense excesses (>85%) of the M-poly-L-1(−) and P-poly-L-1(+) were determined by comparison of the sign and intensities of the Cotton effect at 364 nm to those of the same polymers previously prepared,10 whose helical senses and helical sense excesses had been directly determined by high-resolution AFM observations. As we recently reported, the M-poly-L-1(−) and P-poly-L-1(+) maintain their living feature and can be used as a macroinitiator for the further block copolymerization of phenyl isocyanides.11 In order to immobilize poly-L-1's onto an amino-functionalized silica gel surface through chemical linkages, reactive carboxyl pendant groups were introduced by the block copolymerization of 4-tert-butyldimethylsiloxycarbonylphenyl isocyanide (3) using the M-poly-L-1(−) and P-poly-L-1(+) as macroinitiators ([L-1]/[3] = 10) in THF at 55 °C (Fig. 2). The copolymerizations, the progress of which was monitored by IR, were terminated after 3 had been completely consumed. The obtained block copolymers (M-poly-L-1(−)-b-3 and P-poly-L-1(+)-b-3) were soluble in THF and mostly soluble in CHCl3. Because the block copolymers showed almost identical 1H-NMR spectra to those of the corresponding macroinitiators, the contents of the 3 residues of M-poly-L-1(−)-b-3 and P-poly-L-1(+)-b-3 were assumed to be equal to the amount of 3 used for the block copolymerizations. The Cotton effect intensities of M-poly-L-1(−)-b-3 (Δε364 = −19.2) and P-poly-L-1(+)-b-3 (Δε364 = +17.7) at 364 nm, which reflect that the helical sense excess of helical poly(aryl isocyanide)s, slightly decreased after the block copolymerization of 3 with M-poly-L-1(−) (Δε364 = −20.6) and P-poly-L-1(+) (Δε364 = +18.7) (Fig. 3). These results suggest that the achiral 3 monomer units incorporated into the main chain may not have a fully single-handed helical structure.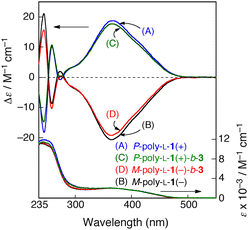 | ||
| Fig. 3 CD and absorption spectra of P-poly-L-1(+) (A) and M-poly-L-1(−) (B) in CHCl3 and P-poly-L-1(+)-b-3 (C) and M-poly-L-1(−)-b-3 (D) in THF at 25 °C (0.2 mg mL−1). | ||
After the deprotection of the tert-butyldimethylsilyl groups in M-poly-L-1(−)-b-3 and P-poly-L-1(+)-b-3 with TBAF, the condensation reaction between the deprotected carboxyl pendant groups of the copolymers and the free amino groups on the A-silica was then performed using DMT-MM14 as a condensing agent, giving the chiral packing materials having helical polymers on the surface chemically bonded through the amide linkage. The obtained silica gels were isolated by filtration and treated with diazomethane in diethyl ether to convert the remaining carboxylic acid groups of the polymers into the methyl esters. The resulting silica gels were then washed with CHCl3, THF and methanol to remove the unbound polymers, yielding the left- and right-handed helical poly(phenyl isocyanide)s chemically bonded to silica gel (Si-M-BP(−) and Si-P-BP(+), respectively). The contents of the polymers chemically bonded to the silica gel estimated by TG analyses were 9.9 and 4.7 wt%, respectively. The Si-M-BP(−) and Si-P-BP(+) were packed into stainless-steel columns (25 cm × 0.20 cm (i.d.)) by the conventional high-pressure slurry packing procedure.7a
Chromatographic enantioseparation
The chemically bonded-type CSPs allowed us to use diverse solvents17 including CHCl3 and THF, as the eluents, although these solvents dissolve or swell the poly(phenyl isocyanide)s. The results for the resolution of a variety of racemic compounds with different functional groups (5–12) and a series of cyclic amides (13–16) and metal complexes of acetylacetonate (17–20) on Si-M-BP(−) and Si-P-BP(+) under various chromatographic conditions are summarized in Table 1 and Fig. 4. The racemic trans-stilbene oxide (5) was almost completely separated on a column packed with the Si-M-BP(−) using hexane–2-propanol (98:2, v/v) as the eluent (Fig. 5a). The peaks were detected by a UV detector and identified by a polarimetric detector. The (+)- and (−)-enantiomers eluted at the retention times of t1 and t2, respectively, showing almost complete separation. The capacity factors, k1′ [= (t1 − t0)/t0] and k2′ [= (t2 − t0)/t0], were 0.19 and 0.32, respectively. The separation factor α [= k2′/k1′] and the resolution factor RS [= 2(t2 − t1)/(w1 + w2)] were estimated to be 1.70 and 1.44, respectively. The racemate 5 was also resolved on Si-M-BP(−) under various eluent systems with relatively high α values, but their RS values varied depending on the type of polar solvents in the hexane (Table 1). Interestingly, the Si-P-BP(+) composed of the same L-1 units, but possessing the opposite right-handed helical conformation in the main chain could not resolve the racemate 5 under the same chromatographic conditions, although the reversed elution order was detected by the polarimetric detector. A similar tendency was observed for the racemates 6 and 9, which could not be resolved on Si-M-BP(−) and Si-P-BP(+) in hexane–2-propanol (90:10, v/v), but was partially separated into enantiomers on Si-M-BP(−) (α = 1.17 and 1.10, respectively) in hexane–THF (98:2, v/v), indicating that the selection of a suitable eluent mixture is an important factor for the efficient separation of the racemates.17 In sharp contrast, the Si-P-BP(+) resolved a heterocyclic amide compound 11, which was hardly separated on the Si-M-BP(−).
| Polymer | Si-M-BP(−) | Si-P-BP(+) | ||||
|---|---|---|---|---|---|---|
| k 1′ | α | R S | k 1′ | α | R S | |
|
a
Conditions: column, 25 × 0.20 cm (i.d.); eluent, hexane–2-propanol (98:2, v/v); flow rate, 0.1 mL min−1. The signs in parentheses represent the optical rotation of the first-eluted enantiomers.
b Eluent: hexane–2-propanol (90:10, v/v).
c Eluent: hexane–THF (98:2, v/v).
d Eluent: hexane–THF (90:10, v/v).
e Eluent: hexane–CHCl3 (95:5, v/v).
f The signs in parentheses represent those of CD detection (254 nm) of the first-eluted enantiomer.
|
||||||
| 5 | 0.19 | 1.70 (+) | 1.44 | 0.02 | ca. 1 (−) | — |
| 5 | 0.15 | 1.63 (+) | 1.08 | 0.08 | ca. 1 (−) | — |
| 5 | 0.34 | 1.67 (+) | 1.71 | 0.35 | ca. 1 (−) | — |
| 5 | 0.15 | 1.68 (+) | 1.02 | — | — | — |
| 5 | 0.15 | 1.79 (+) | 0.72 | 0.03 | ca. 1 (−) | — |
| 6 | 0.70 | 1.17 (−) | 0.29 | 0.43 | ca. 1 (+) | — |
| 7 | 0.17 | ca. 1 (−) | — | 0.05 | ca. 1 (−) | — |
| 8 | 0.61 | ca. 1 | — | 0.25 | ca. 1 | — |
| 9 | 1.65 | 1.10 (−) | 0.74 | 0.35 | ca. 1 (+) | — |
| 10 | 0.39 | ca. 1 | — | 0.04 | ca. 1 | — |
| 11 | 2.19 | ca. 1 (−) | — | 1.65 | 1.13 (+) | 0.35 |
| 12 | 2.38 | 1.46 (+) | 2.52 | 1.95 | 1.08 (−) | 0.29 |
| 13 | 1.89 | ca. 1 | — | 1.68 | ca. 1 | — |
| 14 | 2.02 | 1.53 (+) | 1.82 | 1.54 | 1.10 (+) | 0.33 |
| 15 | 1.25 | 1.13 (−) | 0.48 | 1.10 | 1.25 (−) | 0.85 |
| 16 | 0.67 | 1.31 (+) | 0.50 | 0.42 | 1.80 (+) | 0.92 |
| 17 | 2.43 | ca. 1 (−)f | — | 1.66 | 1.22 (+)f | 0.47 |
| 18 | 1.54 | ca. 1 (+)f | — | 1.03 | 1.20 (−)f | 0.46 |
| 19 | 2.41 | ca. 1 (+)f | — | 1.81 | 1.16 (−)f | 0.39 |
| 20 | 0.70 | ca. 1 (+)f | — | 0.36 | ca. 1 (+)f | — |

|
||||||
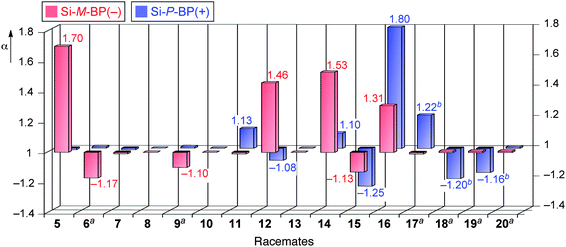 | ||
| Fig. 4 Histograms of the separation factors (α) on Si-M-BP(−) and Si-P-BP(+). Conditions: column, 25 × 0.20 cm (i.d.); eluent, hexane–2-propanol (98:2); flow rate, 0.1 mL min−1. The signs of the vertical axis (α value) represent the optical rotation of the first-eluted enantiomers. aEluent: hexane–THF (98:2, v/v). bThe signs of the vertical axis (α value) represent the CD detection (254 nm) of the first-eluted enantiomer. | ||
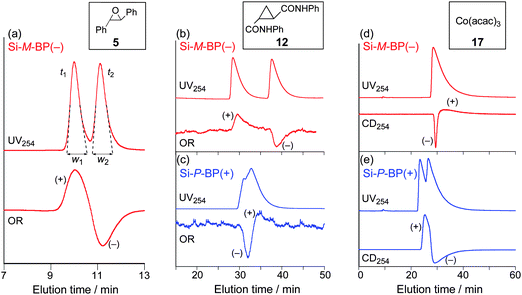 | ||
| Fig. 5 Chromatograms for the resolution of 5 (a), 12 (b and c) and 17 (d and e) on Si-M-BP(−) (red lines) and Si-P-BP(+) (blue lines). Eluent: (a–c) hexane–2-propanol (98:2, v/v); (d and e) hexane–THF (98:2, v/v). | ||
We noted that the complete base-line separation of the racemic trans-cyclopropanedicarboxylic acid dianilide (12) was achieved on the Si-M-BP(−) with the elution order of enantiomers such that the (+)-isomer eluted first followed by the (−)-isomer (α = 1.46) (Fig. 5b), while the reversed elution order was observed on the Si-P-BP(+) composed of the opposite right-handed helical conformation in the main chain (Fig. 5c), showing a partial separation (α = 1.08). These results clearly revealed the major contribution of the macromolecular helicity (helical chirality) of the polyisocyanides for the enantioseparation of specific enantiomers 12 rather than the pendant L-alanine residues (point chirality).
Analogous cyclic dianilide and dibenzamide racemates (13–16) were then employed to investigate such a synergistic effect of the helical chirality and point chirality of Si-M-BP(−) and Si-P-BP(+) during enantioseparation. The trans-cyclopropanediamine dibenzamide (13) was not resolved at all on both CSPs. However, the four- and six-membered cyclic dianilides (14 and 16) and dibenzamide (15) derivatives were partially or almost completely resolved on both CSPs with the same elution order. The enantioselectivity (α = 1.53) of Si-M-BP(−) for 14 was much greater than that of Si-P-BP(+) (α = 1.10). In contrast, the racemates 15 and 16 were better resolved on Si-P-BP(+) (α = 1.25 and 1.80, respectively). These results suggest that the point chirality that originated from the L-alanine pendant amide residues of the polymers as well as the macromolecular helicity of the polymer backbones influence the chiral recognition for these cyclic dianilides and dibenzamides.
We previously reported that the left- and right-handed helical poly-L-1s possess a similar 15/4 (a 15 units per 4 turns) helical structure assisted by four sets of intramolecular hydrogen-bonding networks based on the X-ray diffraction analyses of the oriented polymer films and high-resolution AFM observations of the two-dimensionally packed polymer crystals on a substrate.10 The dianilide and dibenzamide racemates probably interacted with the pendant amide residues of the diastereomerically helical poly(phenyl isocyanide)s through intermolecular hydrogen bond formation, in which the pendant amide residues of the poly-L-1s would also arrange in a helical array with an opposite screw-sense from each other, thus showing a different enantioselectivity toward the enantiomers of the racemic amide-containing compounds.
For the resolution of the metal acetylacetonate complexes, 17–19 were found to be resolved on Si-P-BP(+) with hexane–THF (98:2, v/v) as the eluent,18 and their first-eluted isomers possess the Λ-configuration judging from the Cotton effect signs at 254 nm.19 If the polymers used for the CSPs are enantiomeric, the racemates should be separated with the same α values with the reversed elution order. However, 17–19 were slightly separated (α = ca. 1) on the opposite left-handed helical poly-L-1-based Si-M-BP(−) column with the reversed elution order (Fig. 5d and e). These results clearly suggest that the macromolecular helicity (left- and right-handed helices of poly-L-1s) plays a crucial role for the resolution of the enantiomers, and the point chirality of the pendant groups (L-alanine residues) of the polymers is also involved with the chiral recognition during the enantiomer separation (positive or negative synergistic effect).
Conclusions
In summary, we have prepared two helical polyisocyanide-based CSPs covalently bonded to silica gel and investigated their chiral recognition abilities for HPLC. The polyisocyanides are composed of the same L-alanine repeating units, but they are completely different in their helical sense (right- and left-handed helices). The left-handed helical polyisocyanide-based CSP efficiently resolved the racemic cyclic ether and carbonyl compounds and cyclic dianilides and dibenzamides, while the right-handed helical polyisocyanide-based CSP showed a rather complementary chiral recognition ability and specifically separated the racemic metal acetylacetonate complexes, which could not be resolved on the former CSP at all. The enantioselectivity and elution order of the enantiomers are significantly influenced by the helical structures of the L-alanine-bound-polyisocyanides; the elution order of some enantiomers was reversed on the CSPs and the enantioselectivities of some enantiomers were enhanced or reduced depending on the CSPs. These results could be explained by the positive and negative synergistic effects of the chirality of the L-alanine pendants and helical chirality of the polymers. These results provided useful information both for understanding the chiral discrimination mechanism of the other helical polymer-based CSPs and for designing significantly better CSPs.Acknowledgements
We are deeply grateful to Professor K. Onitsuka (Osaka University) for his generous supply of the Pt–Pd catalyst. We thank Professor Y. Okamoto (Nagoya University and Harbin Engineering University) and Dr T. Ikai (Kanazawa University) for valuable discussions. This work was supported in part by a Grant-in-Aid for Scientific Research (S) from the Japan Society for the Promotion of Science (JSPS) and the Global COE Program “Elucidation and Design of Materials and Molecular Functions” of the Ministry of Education, Culture, Sports, Science, and Technology, Japan. K.T. expresses thanks for a JSPS Research Fellowship (No.2605) for Young Scientists.Notes and references
- For recent reviews on chromatographic enantioseparation, see: (a) N. M. Maier, P. Franco and W. Lindner, J. Chromatogr., A, 2001, 906, 3–33 CrossRef CAS; (b) E. R. Francotte, J. Chromatogr., A, 2001, 906, 379–397 CrossRef CAS; (c) S. Andersson and S. G. Allenmark, J. Biochem. Biophys. Methods, 2002, 54, 11–23 CrossRef CAS; (d) C. Roussel, A. D. Rio, J. Pierrot-Sanders, P. Piras and N. Vanthuyne, J. Chromatogr., A, 2004, 1037, 311–328 CrossRef CAS; (e) Y. Liu, A. W. Lantz and D. W. Armstrong, J. Liq. Chromatogr. Relat. Technol., 2004, 27, 1121–1178 CrossRef CAS; (f) Y. Zhang, D.-R. Wu, D. B. Wang-Iverson and A. A. Tymiak, Drug Discovery Today, 2005, 10, 571–577 CrossRef CAS; (g) Chiral Analysis, ed. K. W. Busch and M. A. Busch, Elsevier, Amsterdam, 2006 Search PubMed; (h) Chirality in Drug Research, ed. E. Francotte and W. Lindner, Wiley-VCH, Weinheim, 2006 Search PubMed; (i) G. Gübitz and M. G. Schmid, Molecular Biotechnology, 2006, 32, 159–179 Search PubMed; (j) Chiral Separation Techniques: a Practical Approach, ed. G. Subramanian, Wiley-VCH, Weinheim, 3rd edn, 2007 Search PubMed; (k) Y. Okamoto and T. Ikai, Chem. Soc. Rev., 2008, 37, 2593–2608 RSC.
- For reviews on polysaccharide-based CSPs, see: (a) K. Oguni, H. Oda and A. Ichida, J. Chromatogr., A, 1995, 694, 91–100 CrossRef CAS; (b) E. Yashima, C. Yamamoto and Y. Okamoto, Synlett, 1998, 344–360 CAS; (c) Y. Okamoto and E. Yashima, Angew. Chem., Int. Ed., 1998, 37, 1020–1043 CrossRef; (d) E. Yashima, J. Chromatogr., A, 2001, 906, 105–125 CrossRef CAS; (e) K. Tachibana and A. Ohnishi, J. Chromatogr., A, 2001, 906, 127–154 CrossRef CAS; (f) R. W. Stringham, Adv. Chromatogr., 2006, 44, 257–290 CAS; (g) T. Ikai and Y. Okamoto, Chem. Rev., 2009, 109, 6077–6101 CrossRef CAS.
- For recent reviews on protein-based CSPs, see: (a) J. Haginaka, J. Chromatogr., A, 2001, 906, 253–273 CrossRef CAS; (b) M. C. Millot, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2003, 797, 131–159 CrossRef CAS; (c) J. Haginaka, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 875, 12–19 CrossRef CAS.
- For reviews on synthetic helical polymer-based CSPs, see: (a) T. Nakano, J. Chromatogr., A, 2001, 906, 205–225 CrossRef CAS; (b) C. Yamamoto and Y. Okamoto, Bull. Chem. Soc. Jpn., 2004, 77, 227–257 CrossRef CAS For other leading references on synthetic helical polymer-based CSPs, see: Polyisocyanides:; (c) A. Yamagishi, I. Tanaka, M. Taniguchi and M. Takahashi, J. Chem. Soc., Chem. Commun., 1994, 1113–1114 RSC; (d) A. Tsuchida, T. Hasegawa, K. Kobayashi, C. Yamamoto and Y. Okamoto, Bull. Chem. Soc. Jpn., 2002, 75, 2681–2685 CrossRef CAS ; Polyacetylenes:; (e) E. Yashima, S. Huang and Y. Okamoto, J. Chem. Soc., Chem. Commun., 1994, 1811–1812 RSC; (f) E. Yashima, T. Matsushima, T. Nimura and Y. Okamoto, Korea Polym. J., 1996, 4, 139–146 Search PubMed ; Polyacrylamides and polymethacrylamides:; (g) G. Blaschke, Angew. Chem., Int. Ed. Engl., 1980, 19, 13–24 CrossRef; (h) G. Blaschke, W. Bröker and W. Fraenkel, Angew. Chem., Int. Ed. Engl., 1986, 25, 830–831 CrossRef; (i) K. Morioka, Y. Isobe, S. Habaue and Y. Okamoto, Polym. J. (Tokyo, Jpn.), 2005, 37, 299–308 CrossRef CAS ; Poly(maleimide)s:; (j) H. Gao, Y. Isobe, K. Onimura and T. Oishi, Polym. J. (Tokyo), 2007, 39, 764–776 Search PubMed; (k) T. Oishi, H. Gao, T. Nakamura, Y. Isobe and K. Onimura, Polym. J. (Tokyo), 2007, 39, 1047–1059 Search PubMed.
- For recent reviews on chiral small molecule-based CSPs, see: (a) F. Gasparrini, D. Misiti and C. Villani, J. Chromatogr., A, 2001, 906, 35–50 CrossRef CAS; (b) T. J. Ward and A. B. Farris, J. Chromatogr., A, 2001, 906, 73–89 CrossRef CAS; (c) V. A. Davankov, J. Chromatogr., A, 2003, 1000, 891–915 CrossRef CAS; (d) M. H. Hyun, Bull. Korean Chem. Soc., 2005, 26, 1153–1163 CrossRef CAS; (e) M. Lämmerhofer and W. Lindner, Adv. Chromatogr., 2008, 46, 1–107 CAS; (f) I. D'Acquarica, F. Gasparrini, D. Misiti, M. Pierini and C. Villani, Adv. Chromatogr., 2008, 46, 109–173 CAS.
- (a) Y. Okamoto, K. Suzuki, K. Ohta, K. Hatada and H. Yuki, J. Am. Chem. Soc., 1979, 101, 4763–4765 CrossRef CAS; (b) H. Yuki, Y. Okamoto and I. Okamoto, J. Am. Chem. Soc., 1980, 102, 6356–6358 CrossRef CAS; (c) Y. Okamoto, H. Mohri and K. Hatada, Polym. J. (Tokyo), 1989, 21, 439–445 Search PubMed.
- (a) Y. Okamoto, M. Kawashima and K. Hatada, J. Chromatogr., A, 1986, 363, 173–186 CrossRef CAS; (b) Y. Kaida and Y. Okamoto, Bull. Chem. Soc. Jpn., 1993, 66, 2225–2232 CrossRef CAS.
- (a) E. Yashima, M. Yamada, Y. Kaida and Y. Okamoto, J. Chromatogr., A, 1995, 694, 347–354 CrossRef CAS; (b) E. Yashima, C. Yamamoto and Y. Okamoto, J. Am. Chem. Soc., 1996, 118, 4036–4048 CrossRef CAS; (c) C. Yamamoto, E. Yashima and Y. Okamoto, Bull. Chem. Soc. Jpn., 1999, 72, 1815–1825 CrossRef CAS; (d) C. Yamamoto, E. Yashima and Y. Okamoto, J. Am. Chem. Soc., 2002, 124, 12583–12589 CrossRef CAS; (e) M. Lämmerhofer, J. Chromatogr., A, 2010, 1217, 814–856 CrossRef.
- (a) K. Onitsuka, T. Joh and S. Takahashi, Bull. Chem. Soc. Jpn., 1992, 65, 1179–1181 CAS; (b) K. Onitsuka, T. Joh and S. Takahashi, Angew. Chem., Int. Ed. Engl., 1992, 31, 851–852 CrossRef.
- H. Onouchi, K. Okoshi, T. Kajitani, S.-i. Sakurai, K. Nagai, J. Kumaki, K. Onitsuka and E. Yashima, J. Am. Chem. Soc., 2008, 130, 229–236 CrossRef CAS.
- Z. Q. Wu, K. Nagai, M. Banno, K. Okoshi, K. Onitsuka and E. Yashima, J. Am. Chem. Soc., 2009, 131, 6708–6718 CrossRef CAS.
- S. Coco, E. Espinet, P. Espinet and I. Palape, Dalton Trans., 2007, 3267–3272 RSC.
- T. Kajitani, K. Okoshi, S.-i. Sakurai, J. Kumaki and E. Yashima, J. Am. Chem. Soc., 2006, 128, 708–709 CrossRef.
- (a) M. Kunishima, C. Kawachi, J. Morita, K. Terao, F. Iwasaki and S. Tani, Tetrahedron, 1999, 55, 13159–13170 CrossRef CAS; (b) D. Son, A. Wolosiuk and P. V. Braun, Chem. Mater., 2009, 21, 628–634 CrossRef CAS.
- Y. Kaida and Y. Okamoto, Bull. Chem. Soc. Jpn., 1992, 65, 2286–2288 CrossRef CAS.
- H. Koller, K.-H. Rimböck and A. Mannschreck, J. Chromatogr., A, 1983, 282, 89–94 CrossRef CAS.
- (a) E. Yashima, H. Fukaya and Y. Okamoto, J. Chromatogr., A, 1994, 677, 11–19 CrossRef CAS; (b) C. Yamamoto, T. Hayashi, Y. Okamoto, S. Ohkubo and T. Kato, Chem. Commun., 2001, 925–926 RSC; (c) P. Franco, A. Senso, L. Oliveros and C. Minguillón, J. Chromatogr., A, 2001, 906, 155–170 CrossRef CAS; (d) T. Zhang, C. Kientzy, P. Franco, A. Ohnishi, Y. Kagamihara and H. Kurosawa, J. Chromatogr., A, 2005, 1075, 65–75 CrossRef CAS; (e) T. Ikai, C. Yamamoto, M. Kamigaito and Y. Okamoto, Chem. Rec., 2007, 7, 91–103 CrossRef CAS.
- The complexes 17–20 were hardly separated when hexane–2-propanol (98:2, v/v) and hexane–chloroform (95:5, v/v) were used as the eluent.
- (a) R. B. Von Dreele and R. C. Fay, J. Am. Chem. Soc., 1971, 93, 4936–4938 CrossRef; (b) B. Nordén, Inorg. Nucl. Chem. Lett., 1975, 11, 387–394 CrossRef CAS; (c) I. Jonás and B. Nordén, Inorg. Nucl. Chem. Lett., 1976, 12, 43–47 CrossRef CAS; (d) H. Kobayashi, H. Matsuzawa, Y. Kaizu and A. Ichida, Inorg. Chem., 1987, 26, 4318–4323 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |