Aqueous synthesis of color-tunable and stable Mn2+-doped ZnSe quantum dots
Pengtao
Shao
a,
Qinghong
Zhang
a,
Yaogang
Li
*b and
Hongzhi
Wang
*a
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University, Shanghai, 201620, China. E-mail: wanghz@dhu.edu.cn; Fax: +86-21-67792855
bEngineering Research Center of Advanced Glasses Manufacturing Technology, Ministry of Education, Donghua University, Shanghai, 201620, China. E-mail: yaogang_li@dhu.edu.cn
First published on 22nd October 2010
Abstract
The aqueous synthesis of color-tunable and stable Mn2+-doped ZnSe quantum dots (Mn:ZnSe d-dots) via a nucleation-doping strategy was realized in a three-necked flask and the photoluminescence (PL) peak position was easily tuned from 572 nm to 602 nm by prolonging the epitaxial-growthg time. The chemical stability of the resulting d-dots is much better than that of CdTevia an aqueous synthesis. Separation of nucleation and growth of crystallization, and multiple-step-injection of both zinc precursor and 3-mercaptopropionic acid (MPA) stabilizing reagent, were employed to realize a balanced, mutually diffused interface and a pure ZnSe shell. The resulting nanocrystals show excellent optical properties, implying that nanocrystals with high quality could also be achieved via aqueous synthesis.
Introduction
With the significant development of emissive materials for biomedical labeling,1 wavelength-tunable lasers2 and solar cells,3 semiconductor nanocrystals or quantum dots (QDs), have attracted more and more attention, with their exceptional optical properties (due to quantum confinement) and biological applications versus organic dyes.4 As classic representatives of the luminescent semiconductor nanocrystals, cadmium chalcogenides (e.g. of CdS, CdSe and CdTe) have been widely studied due to their outstanding properties, such as narrow and symmetric emission, color-tunable, broad and strong absorption, zero scattering, high stability, and solution processibility.5 Especially, continuous synthesis of high quality ZnS-coated CdSe composite particles in a microfluidic reactor improved the optical properties of QDs to the extreme, which can be directly analyzed online and adjusted to tailor the optical properties.6,7 Despite all these advantages mentioned above, the intrinsic toxicity of such QDs containing cadmium cannot be ignored no matter what we do, and any leakage of cadmium can cause devastating harm to the environment, especially to tissue.Doped semiconductor nanocrystals (d-dots), replacing the cadmium in the cadmium chalcogenide QDs with zinc as the absorption zone and introducing the transition ions (Mn or Cu) as the emissive centre,8 can, not only potentially retain most of the outstanding properties above, but also mitigate the toxicity problems. Impurities introduced intentionally can strongly modify the electronic, optical, and magnetic properties of bulk semiconductors.9 Since the position of the ZnS valence band-edge is located too far from the vacuum-level for direct injection of holes from presently available p-type conducting polymers,10 better results are expected for ZnSe, which has a valence band-edge at higher energy with respect to ZnS.11 So ZnSe has been regarded as an efficient host to dope different transition ions to modify the properties mentioned above.9 The nearly identical lattice constants of zinc blend ZnSe and MnSe allow MnSe existing stably in the zinc blend structures,8 and the energy from absorbed photons can be efficiently transferred to the impurity, quickly localizing the excitation and suppressing undesirable reactions on the nanocrystal surface.8 The unique magnetic spin characteristics of Mn2+ have the potential to make the Mn2+ doped ZnSe QDs become a class of mainstream emissive materials.5 For Mn:ZnSe, Mn2+ is a harder Lewis acid compared to Zn2+, so it is difficult to synthesize high quality QDs by traditional routes. The successful synthesis of Mn:ZnSe was not reported until 2000 by Suyver et al. for the first time,12 and later Norris et al. obtained high quality Mn:ZnSe QDs by a high-temperature organometallic method.13 The big breakthrough was achieved through new strategies called growth-doping strategy and nucleation-doping strategy reported by Peng's group.8
High quality Mn:ZnSe QDs reported by Peng's groupvia a nucleation-doping strategy were also carried out through a high-temperature organometallic method.8 The fundamental properties of the as-prepared QDs were greatly improved, such as high quantum yield (QY), controllable color tunability and high color purity (emission peak width).8 But, in order to conjugate with various bioprobes for biomedical labeling, complicated surface modification can dramatically reduce the properties mentioned above and the excessively large physical sizes of d-dots could also cast a doubtful future over this promising field. So, direct aqueous-phase synthesis of Mn:ZnSe d-dots without surface functionalization has been paid more attention in recent years.
In 2009, Wang's group prepared Mn:ZnSe d-dots via a nucleation-doping strategy with 3-mercaptopropionic acid (MPA) as the capping reagent in aqueous solution.14 Compared with organometallic synthesis, aqueous synthesis is cheaper, simpler, less toxic, and the Mn:ZnSe d-dots synthesized in aqueous solution are biologically compatible.14 But the Mn:ZnSe d-dots QY reported by Wang's group was only 2.4%, and the photoluminescence (PL) peak position was only at 570 nm. According to Peng's report, the Mn:ZnSe d-dots’ PL peak position can be tuned from about 575 nm to 595 nm,8 so the fundamental properties of Mn:ZnSe d-dots can be further improved.
In this article, we primarily focused on controlling the size, shape, and structure of d-dots and the formation of the mutually diffused core (Mn (Zn)Se core) and pure ZnSe shell to tailor the optical properties, and finally we successfully prepared high quality Mn:ZnSe d-dots. Through adjusting experimental parameters, the photoluminescence (PL) peak position of the as-prepared d-dots was controllably tuned from 572 nm to 602 nm, and the QY improved to 4.8%, and the chemical stability is much better than that of CdTe QDs via aqueous synthesis.
Experimental
Materials
All chemicals were of analytical reagent grade and were used without further purification. 3-mercaptopropionic acid (MPA, 98%) was a product of Aladdin, selenium powder (99.95%) was purchased from Mei Xing Chemical Co., Ltd. Shangha, China, Zn(OAc)2·2H2O (99.0%), MnCl2·4H2O (99.0%) was purchased from Sinopharm Chemical Reagent Co., Ltd. The ultrapure water used in all syntheses had a resistivity of 18.2 MΩ cm−1.Preparation of precursor solution
A literature method was used to synthesize NaHSe.15 The MPA stabilizing reagent solution was prepared by dissolving 1.064 g MPA in 5 mL water, and then the pH of the solution was adjusted to 11.0 by drop-wise addition of 2.0 M NaOH solution with stirring. MnCl2 solution was prepared by dissolving 0.0495 g MnCl2·4H2O in 10 mL water. Zn(OAc)2 solution was prepared by dissolving 0.1372g Zn(OAc)2·2H2O in 25 mL water. All precursor solutions were prepared freshly.Synthesis of Mn2+-doped ZnSe quantum dots
In a typical experiment, 1 mL MnCl2 solution was loaded into 50 mL three-necked flask, and then degassed for 30 min by nitrogen flow and subjected to reflux at 100 °C. Fresh NaHSe solution was injected swiftly into a 50 mL three-necked flask keeping for 40 min to obtain the ideal size of MnSe core. After that, a 1 mL Zn(OAc)2 solution was added through a syringe into a three-necked flask at 80 °C by 100 μL min−1, then the moderate MPA stabilizing reagent solution was diverted to the three-necked flask, and kept for 20 min, and then another 12 mL Zn(OAc)2 solution by 500 μL min−1, then moderate MPA stabilizing reagent solution was injected, kept for 15 min, and then the rest of the Zn(OAc)2 solution was added, and so was the rest of the MPA stabilizing reagent solution. From the second injection of Zn precursor solution, the reaction was monitored by PL spectroscopy. By varying the epitaxial-growth time, we can get Mn2+-doped ZnSe QDs with a different PL peak position. By this approach, one can obtain Mn:ZnSe d-dots whose emission wavelength was tunable from 572 nm to 602 nm.Characterization of Mn2+-doped ZnSe QDs
One of the key steps in the preparation of Mn2+-doped ZnSe d-dots was the removal of excess stabilizing reagent from the nanocrystals. For this to occur, the as-prepared colloids were precipitated by methanol addition and were redissolved in pure water at pH 9.0, which resulted in partial removal of the stabilizer.16 The obtained dispersion was allowed to take TEM measurements. Then the d-dots were precipitated with ethanol, and subsequently dried under vacuum to take the XRD measurements. The pH value of the MPA stabilizing reagent was measured by a Shiyi Co., Ltd. Shanghai, pHS-3CA pH meter. UV-vis spectra were obtained by a Perkin Elmer Co., USA Lambda 35 UV-vis spectrophotometer. PL, both excitation and emission, spectra of the QDs, were obtained by a JASCO, Japan, FP-6600 fluorescence spectrophotometer. The powder X-ray diffraction (XRD) was obtained by a Rigaku, Japan, D/MAX-2550X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) at 40 kV and 100 mA. Transmission electron microscopy (TEM) images and were obtained by JEOL, Japan, JEM-2100F HRTEM operated at 200 KV.Results and discussion
Results of Mn2+-doped ZnSe quantum dots
It is well-known that the optical properties of d-dots are the result of quantum confinement and the competition between radiative and non-radiative pathways mainly depending on the separation between the impurity, the surface, and other impurities.8,17,18 For Mn2+-doped ZnSe QDs particularly, the optical properties are mainly dependant on the size of the d-dots, and the unique, mutually diffused core—Mn and Zn distributed equally in the lattice of the entire core—as the emissive center, which was a result of the sp–d exchange interaction between the Zn and Mn ions, with the pure ZnSe shell acting as the absorption zone. The existence of a pure MnSe core could not occur with Mn–Mn emission centers (peak position: about 640 nm), which could broaden the PL, but also decrease the QY. So how the mutually diffused core and pure ZnSe shell at different sizes of d-dots could be obtained was considered throughout the entire synthesis process.According to the “self-purification” theory proposed by Erwin et al.,19 only if the size of nucleus reaches or exceeds that of so-called “magic clusters” produced at the nucleation stage, which are too stable to be doped,20–22 the doping will occur. So a higher temperature for nucleation and a lower one for shell epitaxial growth were necessary. For Mn2+-doped ZnSe quantum dots, the surface morphology of the core was critically important for diffusion, and aggregation of the final d-dots also needed to be avoided. The multiple-step-injection of 3- mercaptopropionic acid (MPA) cannot only modify the surface morphology of the nucleus to increase the interface (or Mn (Zn) Se core) and avoid the expulsion of Mn, but also reduce the surface energy, preventing the aggregation of the final d-dots.
Based on such a consideration above, the synthesis of high quality quasi core/shell Mn2+-doped ZnSe QDs in this report was a modification of nucleation-doping strategy.8 The separation of nucleation and growth of crystallization was realized by two oil baths at 100 °C and 80 °C respectively (once the nucleation at 100 °C was complete, the three-necked flask was swiftly transferred to an oil bath at 80 °C). Fig. 1 depicts the formation of quasi core/shell Mn2+-doped ZnSe QDs. Desirably small-sized MnSe nuclei were obtained through refluxing at 100 °C 40 min after the injection of anionic ions in the absence of MPA in the aqueous solution, and then the reaction mixture was cooled to 80 °C, a small amount of Zn precursor solution was injected to start the shell nucleation. As Mn2+ is a harder Lewis acid compared to Zn2+, Zn ions injected could undoubtedly gradually replace the Mn ones in the core. If more Zn precursor solution was injected, separated ZnSe particles could be observed by the appearance of ZnSe band gap emission.
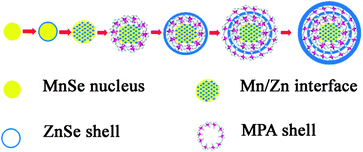 | ||
| Fig. 1 Schematics of the nucleation and growth mechanism of Mn2+-doped ZnSe QDs. | ||
10 min later, sufficient MPA stabilizing reagent solution was injected to form an efficient protective layer on the core surface. 5 min later, a stoichiometric amount of Zn precursor solution was added, and the reaction was monitored by UV-vis PL spectra until the appearance of the emission peak at 572 nm. Though a protective layer was formed, according to “Fick's first law”, the Zn ions from the shell could still more or less diffuse to the core zone, finally forming the ideally mutual diffused core as the emissive center. The outer ZnSe shell worked as an absorption zone, and prevented the Mn ions' exile to the surface, which could cause the self-quenching. The rest of the MPA and Zn precursor solutions were injected separately into the reaction mixture, and the significant PL red-shift from 572 nm to 602 nm was observed due to quantum confinement, consistent with the explanation by Peng and co-workers, which is as the ZnSe outer-shell grew thicker, the crystal field for each dopant ion became more symmetric over a long range, and consequently, the crystal field splitting of the d-orbital became smaller, which resulted in a PL red shift.8
In short, by controlling the injection volume of precursors in each step, the ideal core and shell were obtained; by varying the ZnSe shell epitaxial-growth time, the size of the as-prepared d-dots was controllable; by injecting the MPA stabilizing reagent solution, the growth of d-dots was adjusted and the undesirable emission was suppressed due to vanishing of the passivating dangling bonds.
The wide-angle X-ray diffraction patterns of the as-prepared power samples of Mn2+-doped ZnSe d-dots at different sizes are shown in Fig. 2. The three strong peaks with 2θ values of 27.47°, 45.63°, and 54.09° correspond to the (111), (220), and (311) planes respectively, which prove that Mn2+-doped ZnSe d-dots have a cubic zinc blend structure. The average particle sizes of Mn2+-doped ZnSe d-dots (PL peak position: 572 nm, and 598 nm), estimated from the diffraction line broadening, is 3.8 nm and 9.7 nm according to the Scherrer Formula, which could be supported by the TEM micrograph of the Mn:ZnSe d-dots (see Fig. 3). As shown in Fig. 3, the HRTEM image indicated the distances between the adjacent lattice fringes to be 0.3 nm, basically consistent with the literature value for the (111) d spacing, 0.324 nm (JCPDF No. 800021), meaning that the as-prepared d-dots are in the cubic phase, not wurtzite or rock-salt phase.
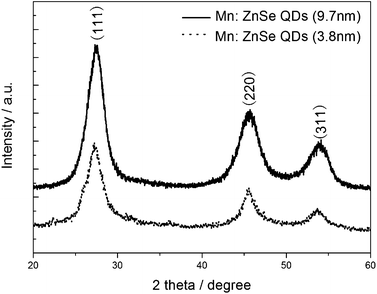 | ||
| Fig. 2 XRD patterns of the Mn:ZnSe QDs. | ||
 | ||
| Fig. 3 A TEM and a HRTEM image of Mn2+-doped ZnSe QDs. | ||
The UV-vis absorption spectra of the as-prepared d-dots at different reaction times are showed in Fig. 4. From the absorption spectra, an absorption band of Mn:ZnSe QDs at around 370 nm was observed, and a significant red-shift of absorption spectra of Mn2+-doped ZnSe d-dots, with prolonging reaction time, demonstrated the overcoating reaction of the ZnSe shell on the MnSe nucleus. This phenomenon is similar to that reported by Peng's group5 and Wang's group.14
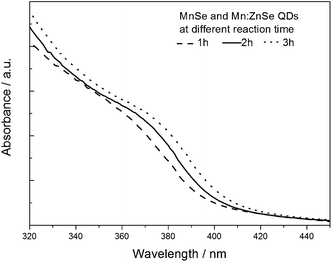 | ||
| Fig. 4 The UV-vis absorption spectra of Mn2+-doped ZnSe QDs. | ||
The PL emission evolution of Mn2+-doped ZnSe d-dots from 572 nm to 602 nm at different ZnSe shell epitaxial-growth times is shown in Fig. 5. These emissions are assigned to pseudo-tetrahedral (4T1 to 6A1) transitions of the Mn2+ ions incorporated into the ZnSe QDs. As is well-known, according to the different locations of dopant atoms, nanocrystals could be surface-doped, internal- or centre-doped, and the PL of the centre-doped and internal-doped nanocrystals were stable while the surface-doped ones were very fragile.8 In this article, the as-prepared QDs were centre-doped via a nucleation-doping strategy. For centre-doped QDs, the ZnSe shell could impact on the PL position of the resulting samples duo to quantum size effects. As ZnSe overcoating proceeded on the mutually diffused core, the thickness of the ZnSe shell increased, the lattice field around a given Mn centre became more symmetric, so the d-orbital of the Mn centre will experience a smaller electric field difference along different directions, resulting in a smaller splitting of the energy levels,5 and the significant red-shift of the PL spectra occurred.
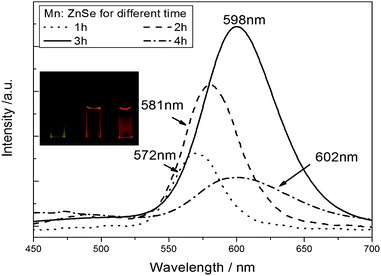 | ||
| Fig. 5 PL emission spectra of Mn2+-doped ZnSe QDs at different ZnSe shells. | ||
The influence of experimental parameters
The successful synthesis of color-tunable Mn2+-doped ZnSe QDs was built on the basis of optimal experimental conditions. In order to obtain high quality QDs, we systematically studied the influence of a series of experimental parameters, and made a corresponding explanation, including a pH value, the molar ratio of [MPA]/[Zn], and epitaxial-growth time on the PL performance of the obtained Mn2+-doped ZnSe QDs.Influence of pH Value
The pH value played an important role in nucleation and growth of crystallization. PL emission evolution of Mn2+-doped ZnSe QDs at different pH values is shown in Fig. 6. From the PL emission spectra, as the pH value increased from 9.0 to 11.0, the PL intensity gradually increased, but when the pH value reached 12.0, the PL intensity decreased. For this, we believed that the effect of the pH to the resulting d-dots was to activate Mn and Zn ions to form MnSe and ZnSe, according to Scheme 1, and the optimized parameter is 11.0, since sufficient OH ions did not only stimulate rapidly to form small-sized MnSe cores, which was critically important to enhance the PL QY, but also located the ZnSe on the surface of the mutually diffused core, preventing the oxidation of Mn ions. When pH values increased further, the PL intensity decreased, we believed that a large excess of OH ions may cause the formation of the manganese oxide or manganese hydroxide or zinc oxide, which were a disastrous influence on d-dots' QY.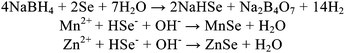 | ||
| Scheme 1 Equations for the formation of MnSe and ZnSe. | ||
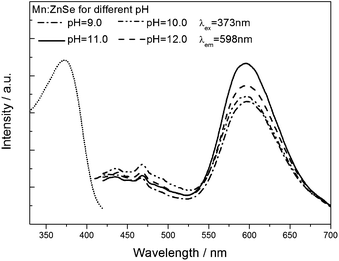 | ||
| Fig. 6 PL emission spectra of Mn2+-doped ZnSe QDs at different pHs. | ||
Influence of the molar ratio of [MPA]/[Zn]
The MPA stabilizing reagent played a determining role for the optical quality of the resulting QDs shown in Fig. 7. As we said above, Mn ions are harder Lewis acids compared to Zn ions, the Mn precursor was significantly less active than the Zn one, so even if the MnSe cores were formed already, the Mn sites would be continuously substituted by Zn, and finally the ideal core-shell structure changed into a single ZnSe spherical one, of which Mn ions embedded in the surface, and this could self-quench both the Mn ion 4T1 → 6A1 emission and the ZnSe band-edge emission.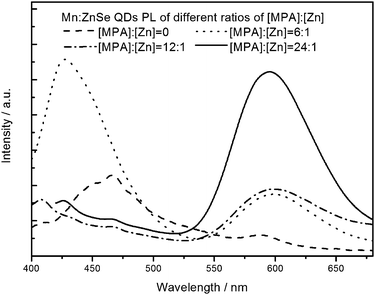 | ||
| Fig. 7 The PL emission spectra of Mn2+-doped ZnSe QDs at different MPA stabilizing. | ||
Thiolate ligands, especially deprotonated thiols are considered to be strong ligands for II–VI semiconductor nanocrystals, and, compared with thioglycolic acid (TGA), TGA-like 1-thioglycerol (TG), TG-like ligands, the longer chain and unique secondary coordination of MPA allow to form the most favorable hexagonal configuration with the ions of the core surface, in which the carbonyl oxygen and the thiol sulfur atoms coordinate with the same cation site,23 and more importantly, the hexagonal configuration would prohibit Mn substituted by Zn as an efficient protective layer on the core surface, which could significantly enhance the PL QY. The last injection of MPA also reduced the surface energy, preventing particle-aggregation.
Influence of ZnSe shell epitaxial-growth time
Influence of ZnSe shell epitaxial-growth time was not only indicated by the red-shift of the PL, but also in the change of PL intensity (see Fig. 5). The PL intensity evolution could be explained by the energy between the Zn and Mn atoms. When the stoichiometric amount of Zn precursor was injected into the three-necked flask, since Mn2+ is a harder Lewis acid compared to Zn2+, even in the presence of MPA, which is an efficient protective layer on the core surface, the Zn ions undoubtedly could still gradually replace the Mn ones in the core, forming the mutual diffused interface as the emissive center, so the shell was very thin, resulting in the energy absorbed by Zn minus the non-radiative relaxation is not adequate enough to excite all of the emitting Mn atoms. As the ZnSe shell got thicker, the PL intensity of the resulting d-dots increased, and when the epitaxial-growth time reached 3 h, the PL intensity reached its maximum (the QY is also at its maximum), because the thick ZnSe shell placed the emission centers–Mn ions deeper inside the QDs, making them more symmetrical, farther from the surface defects, and more importantly, there was sufficient energy from the Zn ions transferring to the Mn ions, causing the enhanced PL intensity. The quantum yield of Mn:ZnSe d-dots was 4.8%, calculated according to ref. 5. When the epitaxial-growth time was prolonged, Ostwald ripening occurred, and the ZnSe shell became thicker, so dislocations and low-angle grain boundaries appeared, relaxing the structure and causing the growth to proceed incoherently.24 These were the source of the non-radiative relaxation within the ZnSe overlayer, causing the significant PL decrease.PL stablity test
Compared that of traditional CdTe QDs,25 We studied the chemical stability of the as-prepared d-dots via chemioxidization by H2O2 according to a literature method.26 In the test, a 1 mL solution of 30% H2O2 diluted 1000 times was added to 10 mL of Mn2+-doped ZnSe d-dots samples under stirring with an optical density of 0.1 at room temperature, and the same treatment was done to traditional CdTe QDs (PL position at 600 nm). The PL emission spectra were obtained by taking aliquots at different time intervals, and the time dependence of the normalized PL intensities of both samples is shown in Fig. 8. Once H2O2 solution was introduced into the samples, a sharp decline in PL intensity for both the samples was observed, and after 10 min etching time, the PL intensity of CdTe QDs dropped down to about 2.5%, but that of the as-prepared d-dots still remained at about 52.5%, which strongly demonstrated the great chemical stability of the as-prepared d-dots.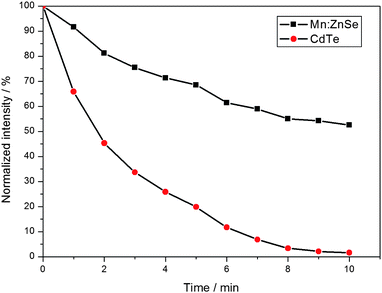 | ||
| Fig. 8 The time dependence of the normalized PL intensities of Mn2+-doped ZnSe and CdTe samples against H2O2 etching. | ||
Conclusions
By controlling the size of MnSe nucleus, mutual diffused interface, and varying the epitaxial-growth time, this article demonstrated that it is feasible to obtain color-tunable and stable Mn2+-doped ZnSe quantum dots (Mn:ZnSe d-dots) via nucleation-doping strategy in a facile aqueous route. The PL properties of the resulting QDs were optimized by adjusting the experimental parameters, and the highest PL QY of the resulting QDs could approach 4.8%. The stabilities of the resulting QDs to UV irradiation and chem-oxidation were also improved significantly in comparison with those of traditional CdTe QDs synthesized in aqueous solution. By rationally tailoring the experimental parameters, we can finally obtain color-tunable and stable Mn2+-doped ZnSe quantum dotsvia aqueous synthesis for application in fluorescent label fields.Acknowledgements
We gratefully acknowledge the financial support of Shanghai Municipal Education Commission (no.07SG37), National Natural Science Foundation of China (no. 51072034), Shanghai Leading Academic Discipline Project (B603), the Cultivation Fund of the Key Scientific and Technical Innovation Project (no. 708039), and the Program of Introducing Talents of Discipline to Universities (no. 111-2-04).Notes and references
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS.
- V. I. Klimov, S. A. Ivanov, J. Nanda, M. Achermann, I. Bezel, J. A. McGuire and A. Piryatinski, Nature, 2007, 447, 441 CrossRef CAS.
- I. Gur, N. A. Fromer, M. L. Geier and A. P. Alivisatos, Science, 2005, 310, 462 CrossRef CAS.
- M. BruchezJr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef CAS.
- N. Pradhan and X. Peng, J. Am. Chem. Soc., 2007, 129, 3339 CrossRef CAS.
- H. Z. Wang, H. Nakamura, M. Uehara, Y. Yamaguchi, M. Miyazaki and H. Maeda, Adv. Funct. Mater., 2005, 15, 603 CrossRef CAS.
- H. Z. Wang, H. Y. Li, M. Uehara, Y. Yamaguchi, H. Nakamura, M. Miyazaki, H. Shimizu and H. Maeda, Chem. Commun., 2004, 48 RSC.
- N. Pradhan, D. Goorskey, J. Thessing and X. Peng, J. Am. Chem. Soc., 2005, 127, 17586 CrossRef CAS.
- D. J. Norris, A. L. Efros and Steven C. Erwin, Science, 2008, 319, 1776 CrossRef CAS.
- J. F. Suyver, S. F. Wuister, J. J. Kelly and A. Meijerink, Phys. Chem. Chem. Phys., 2000, 2, 5445 RSC.
- G. H. Schoenmakers, E. P. A. M. Bakkers and J. J. Kelly, J. Electrochem. Soc., 1997, 144, 2329 CAS.
- J. F. Suyver, S. F. Wuister, J. J. Kelly and A. Meijerink, Phys. Chem. Chem. Phys., 2000, 2, 5445 RSC.
- D. J. Norris, Nan Yao, F. T. Charnock and T. A. Kennedy, Nano Lett., 2001, 1, 1 CrossRef.
- Chao Wang, Xue Gao, Qiang Ma and Xingguang Su, J. Mater. Chem., 2009, 19, 7016 RSC.
- P. V. Radovanovic and D. R. Gamelin, J. Am. Chem. Soc., 2001, 123, 12207 CrossRef CAS.
- Y. Yang, O. Chen, A. Angerhofer and Y. C. Cao, J. Am. Chem. Soc., 2006, 128, 12428 CrossRef CAS.
- H. Zhang, L. Wang, H. Xiong, L. Hu, B. Yang and W. Li, Adv. Mater., 2003, 15, 1712 CrossRef CAS.
- Zhiyong Tang, Nicholas A. Kotov and Michael Giersig, Science, 2002, 297, 237 CrossRef CAS.
- Steven C. Erwin, Lijun Zu, Michael I. Haftel, Alexander L. Efros, Thomas A. Kennedy and David J. Norris, Nature, 2005, 436, 91 CrossRef CAS.
- Jung Ho Yu, Xinyu Liu, Kyoung Eun Kweon, Jin Joo, Jiwon Park, Kyung-Tae Ko, Dong Won Lee, Shaoping Shen, Kritsanu Tivakornsasithorn, Jae Sung Son, Jae-Hoon Park, Young-Woon Kim, Gyeong S. Hwang, Margaret Dobrowolska, Jacek K. Furdyna and Taeghwan Hyeon, Nat. Mater., 2010, 9, 47 CrossRef CAS.
- V. N. Soloviev, A. Eichhöfer, D. Fenske and U. Banin, J. Am. Chem. Soc., 2001, 123, 2354 CrossRef CAS.
- H. Z. Wang, A. Tasiro, H. Nakamura, M. Uehara, M. Miyazaki, T. Watari and H. Maeda, J. Mater. Res., 2004, 19, 3157 CrossRef CAS.
- Hao Zhang, Dayang Wang and Helmuth Möhwald, Angew. Chem., Int. Ed., 2006, 45, 748 CrossRef CAS.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 46.
- C. Wang, Q. Ma and X. Su, J. Nanosci. Nanotechnol., 2008, 8, 4408 CrossRef CAS.
- Z. Fang, Y. Li, H. Zhang, X. Zhong and L. Zhu, J. Phys. Chem. C, 2009, 113, 14145 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |