Nuclear translocation of NF-κB in intact human gut tissue upon stimulation with coffee and roasting products
Tanja
Sauer
a,
Martin
Raithel
b,
Jürgen
Kressel
b,
Sonja
Muscat
a,
Gerald
Münch
c and
Monika
Pischetsrieder
*a
aDepartment of Chemistry and Pharmacy, Food Chemistry, Emil Fischer Center, Friedrich-Alexander University, Schuhstr. 19, 91052, Erlangen, Germany. E-mail: monika.pischetsrieder@lmchemie.uni-erlangen.de
bFunctional Tissue Diagnostics, Gastroenterology, Department of Medicine I, Friedrich-Alexander University, Ulmenweg 18, 91054, Erlangen, Germany
cDepartment of Pharmacology, School of Medicine, University of Western Sydney, Locked Bag 1797, Penrith, NSW 2751, Australia
First published on 9th September 2011
Abstract
In the healthy gut, NF-κB is a critical factor of the intestinal immune system, whereas inflammatory bowel diseases are associated with chronic activation of NF-κB. Previous studies indicated that coffee induces nuclear translocation of NF-κB in macrophages, an effect attributed to roasting products. In the present work, coffee extract or roasting products induced nuclear translocation of NF-κB in macrophages, Caco-2 cells, and primary human intestinal microvascular endothelial cells (up to fivefold, p < 0.001). Since the effect clearly depended on the cell type, ex vivo experiments were performed with intact human gut tissue from biopsies. The uniformity of the specimens and tissue viability during ex vivo incubation for up to 2 h were verified. Roasting products led to a concentration dependent significant increase of nuclear translocation of NF-κB in human gut tissue (up to 2.85 fold increase, p = 0.0321), whereas coffee extract induced a trend towards higher nuclear NF-κB concentration. NF-κB activation in macrophages and Caco-2 cells by roasting products was significantly blocked by co-incubation with catalase (p = 0.011 and p = 0.024) indicating involvement of H2O2-signaling. Monitoring of extracellular H2O2 indicated that roasting products in coffee constantly generate H2O2 by spontaneous oxygen reduction, which is only partially detoxified by cellular antioxidative systems. Thus, it can be concluded that ex vivo stimulation of intact human gut tissue is a valuable model to study nutritional effects on complex tissue systems. Furthermore, the consumption of coffee and roasting products may be able to induce nuclear NF-κB translocation in the human gut.
Introduction
The ubiquitous and inducible transcription factor nuclear factor (NF) κB is a key regulator of the intestinal immune system. Stimulation by bacteria, viruses or their metabolites leads to activation of NF-κB and, in consequence, to an immune response. The expression of more than 150 mostly immune-related proteins, such as cytokines, is regulated by NF-κB.1,2NF-κB consists of two subunits, most likely p50/p65. Without stimulation, the NF-κB dimer is localized in the cytoplasm bound to the inhibitory molecule IκB, which prevents the translocation of the NF-κB:IκB complex into the nucleus. Upon NF-κB activation, IκB kinase is induced causing the phosphorylation and degradation of IκB, which enables NF-κB to translocate into the nucleus. Once NF-κB accumulates in the nucleus, it binds to a specific κB site in the DNA coordinating the expression of genes that control diverse immune cell functions, including apoptosis, cytokine production, or inflammatory reactions.2,3 Knockout mice lacking functional expression of subunits of the NF-κB/IκB complex thus show diverse effects in cellular immune function such as suppression of antibody production or B cell proliferation upon lipopolysaccharide (LPS) challenge.4,5 Therefore, activation of NF-κB can be considered as a primary step of the intestinal immune response. Perpetuated activation of NF-κB, however, is associated with inflammatory bowel disease (IBD).2 In mice with experimental colitis, inflammation is promoted by Th1 cytokines, which are regulated by NF-κB.6 Likewise, nuclear NF-κB levels were elevated in macrophages and epithelial cells from patients with IBD, whereas it was almost completely absent in uninflamed mucosa. The number of NF-κB positive cells was associated to the degree of mucosal inflammation.7 Consequently, beneficial effects were observed in patients suffering from IBD upon blockage of NF-κB activation by drugs like corticosteroids, inhibitory cytokines, or antisense therapy.8 The diet seems to have a major influence on the development and symptoms of IBD.9 Although the control of the diet is an important component in the clinical treatment of IBD, experimental studies on the molecular mechanisms of defined food components are rare.It has been shown that coffee and coffee components considerably influence the activation of NF-κBin vitro and in vivo. In unstimulated macrophages, the presence of coffee induced strong activation of NF-κB. This effect was attributed to hydrogen peroxide (H2O2)-generating melanoidins and Maillard products formed during the roasting process.10 In LPS-stimulated monocytes as well as in the liver and the kidney of transgenic reporter mice, a high concentration of coffee inhibited LPS-induced activation of NF-κB.11 Although the diterpenoid lipids kahweol and cafestol are able to reduce LPS-induced NF-κB activationin vitro, this effect was rather related to melanoidins formed during the roasting process.11,12 The goal of the present study was, therefore, to investigate the intestinal activation of NF-κB by coffee. For this purpose, the time-, concentration- and cell-type-dependent NF-κB activation was examined upon cell stimulation with coffee and roasting products. Furthermore, a model was established to investigate the influence of food components on intact human gut mucosa tissue ex vivo. The model was then applied to analyze the effects of coffee and melanoidins on NF-κB activation.
Results
NF-κB activation by coffee extract in different cell types
NR8383 macrophages were incubated with 2 mg mL−1 coffee extract for 30 min to 6 h. Coffee extract up-regulated the nuclear NF-κB concentration in NR8383 macrophages in a time-dependent manner reaching the maximum of a threefold up-regulation after 2 h (p < 0.0001) (Fig. 1A). Thereafter, the activation slightly declined, but the nuclear NF-κB concentration was still significantly elevated after 6 h (p < 0.0001). The trypan blue dye exclusion test determined a cell viability of more than 90% during the entire incubation period. Nuclear NF-κB translocation did not only depend on time but also on the concentration (Fig. 1B). In fact, 1 mg mL−1 coffee extract did not activate NF-κB in macrophages after 2 h, while 4 mg mL−1 coffee extract caused a fourfold up-regulation (p < 0.0001). Cell viability measured by the trypan blue dye exclusion test exceeded 90%, independent from the coffee concentration. Since coffee extract activated NF-κB in macrophages, its impact on the NF-κB translocation in human epithelial colorectal adenocarcinoma (Caco-2) cells was also investigated. However, coffee extract did not induce NF-κB translocation in Caco-2 cells after an incubation period of 2 h, the time point of maximum NF-κB activation in macrophages (Fig. 1C).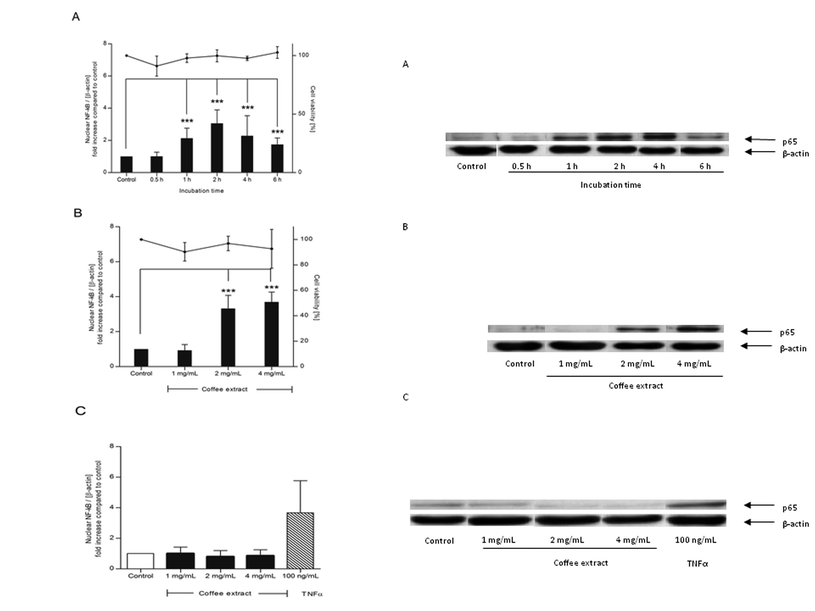 | ||
| Fig. 1 Nuclear translocation of NF-κB in NR8383 macrophages (A/B) and Caco-2 cells (C) induced by coffee extract. Cells were stimulated with coffee extract (1–4 mg mL−1) or TNF-α as positive control for 0.5–6 h. The intensity of the p65 signal (NF-κB subunit) was related to the loading control β-actin and expressed as n-fold increase compared to water-treated control cells (bars). Cell viability was assured by trypan blue dye exclusion test (points). Data is mean ± SD (A: n = 3–5; B: n = 3; C: n = 2). * p < 0.05, ** p < 0.01, *** p < 0.001. Representative Western blot of p65 and β-actin in NR8383 (A/B) and Caco-2 cells (C). | ||
NF-κB activation by roasting products in different cell types
Both NR8383 macrophages and Caco-2 cells were exposed to roasting products (Maillard reaction mixture (MRM), 25 mM) for 2 h. NF-κB was significantly activated in both NR8383 macrophages (p ≤ 0.0001) and Caco-2 cells (p = 0.0006) compared to the phosphate buffered saline (PBS)-treated control cells (Fig. 2). However, the level of NF-κB activation was considerably higher in NR8383 macrophages (4.9-fold) than in Caco-2 cells (1.6-fold). Moreover, it was shown that NF-κB activation in Caco-2 cells depended on the concentration. Next, NF-κB activation was determined in primary human intestinal microvascular endothelial cells (HIMEC). Compared to control cells, exposure to MRM 1.6-fold increased the nuclear translocation of NF-κB after 2 h (Fig. 2).Cytokine release as a result of stimulating NR8383 macrophages was analyzed using the Bio-Plex system including interleukin (IL)-1α, IL-1β, IL-6, IL-10, and tumor necrosis factor (TNF)-α. The results indicate that under the tested conditions, roasting products do not exert pro-inflammatory activity (data not shown).
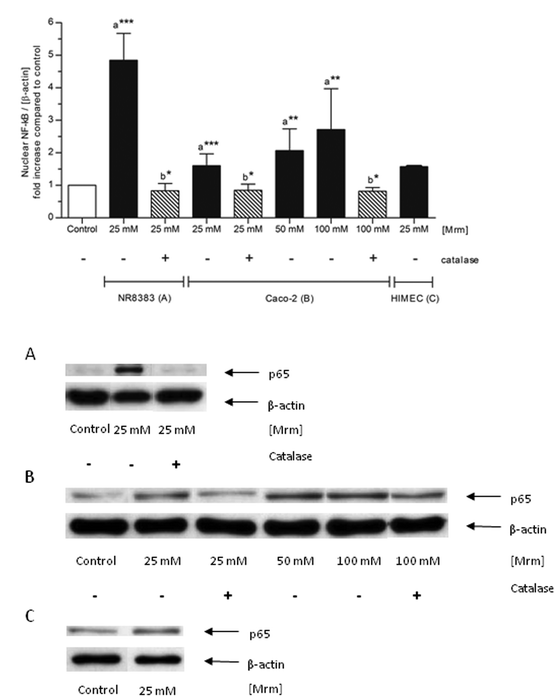 | ||
| Fig. 2 Nuclear translocation of NF-κB by roasting products in various cell types. NR8383 macrophages, Caco-2 cells, and HIMEC were stimulated with different concentrations of MRM for 2 h. In case of catalase co-treatment, catalase (150 U mL−1) was added ten minutes prior to stimulation. The intensity of the p65 signal (NF-κB subunit) was related to the loading control β-actin and expressed as n-fold increase compared to PBS-treated control cells. Data is mean ± SD (n = 2–8); * p < 0.05, ** p < 0.01, *** p < 0.001 compared to the control (a) or to the corresponding incubation without catalase addition (b). Representative Western blot of p65 and β-actin in NR8383 macrophages (A), Caco-2 cells (B) and HIMEC (C). | ||
Ex vivo stimulation of intact human gut tissue
Since the nuclear translocation of NF-κB induced by coffee and roasting products proved to be strongly dependent on the cell type, stimulation was repeated using intact human gut tissue. Thus, the actual cellular composition of the human gut as well as complex cellular interactions could be accounted for. To adapt ex vivo mucosa oxygenation for this nutritional study, the efficient nuclear protein extraction as well as the cell viability during ex vivo stimulation were studied. First, the nuclear protein concentration of the human gut tissue samples was measured. A correlation between the nuclear protein concentration and the wet weight of human gut tissue samples was calculated. The nuclear protein concentration increased with the wet weight of the tissue samples. The correlation coefficient r according to Pearson was 0.8185 for the nuclear protein concentration (p < 0.0001). Thus it could be confirmed that the nuclear NF-κB content, which was related to the nuclear protein concentration in the following experiments, represented nuclear NF-κB translocation in the tissue samples.Cell viability of the tissue cells during mucosa oxygenation ex vivo was investigated by the lactate dehydrogenase (LDH) assay. Intact human gut tissue samples were kept in modified PBS or Hanks buffer during mucosa oxygenation between 0.5 and 24 h. The amount of LDH secreted into the supernatant compared to overall LDH rose time-dependently in both media indicating a decrease in cell viability over time (Fig. 3). After 4.5 h, the cell viability still exceeded 75% in both media. However, after 6 and 24 h, cell viability decreased to about 50% and nearly 0%, respectively, independent from the cell culture medium. The difference in cell viability between both media was not significant. A similar LDH release was observed when a single human gut tissue sample was monitored between 0.5 and 24 h (data not shown). Therefore, a stimulation time of 2 h was chosen for further experiments to avoid notable cell death during the ex vivo incubation.
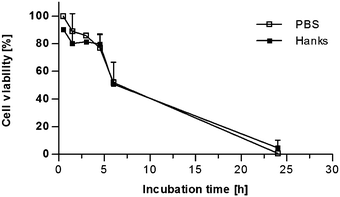 | ||
| Fig. 3 Time-dependent cell viability of human gut tissue samples during mucosa oxygenation ex vivo. The secretion of LDH assay into the supernatant was used as indicator of cell death and thus cell viability. For each time point a separate human gut tissue sample was incubated in modified PBS or Hank's balanced salt solution buffer (Hanks). LDH concentration was measured in the supernatant and in the tissue. The cell viability was expressed as the ratio between LDH concentration in the supernatant and the overall LDH concentration. Mean ± SD (n = 2–3) is shown. | ||
Stimulation of intact human gut tissue with coffee and roasting products
Human gut tissue samples were stimulated for 2 h with roasting products from MRM during mucosa oxygenation ex vivo. Indeed, MRM significantly increased nuclear translocation of NF-κB compared to the control tissue in a concentration-dependent manner (Fig. 4). A 2.85-fold increase in NF-κB translocation was detected in presence of the highest MRM concentration (100 mM).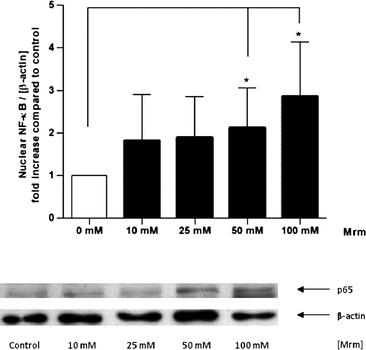 | ||
| Fig. 4 Nuclear translocation of NF-κB induced by roasting products in human gut tissue ex vivo. The tissue samples were stimulated with MRM (10–100 mM) in modified PBS for 2 h. The intensity of the p65 signal (NF-κB subunit) was related to the loading control β-actin and expressed as n-fold increase compared to PBS treated control. Data are mean ± SD (n = 5–7); * p < 0.05, ** p < 0.01, *** p < 0.001. Representative Western blot of p65 and β-actin in human gut tissue. | ||
The experiment was repeated with coffee extract as stimulant. Coffee extract induced a twofold higher NF-κB translocation compared to the control. The difference, however, was statistically not significant (Fig. 5).
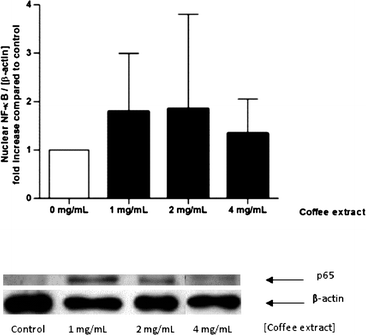 | ||
| Fig. 5 Nuclear translocation of NF-κB induced by coffee extracts in human gut tissue ex vivo. The specimens were stimulated with coffee extract (1–4 mg mL−1) in modified PBS for 2 h. The intensity of the p65 signal (NF-κB subunit) was related to the loading control β-actin and expressed as n-fold increase compared to control. Data are mean ± SD (n = 3–5). Representative Western blot of p65 and β-actin in human gut tissue. | ||
Generation of extracellular H2O2 during stimulation of NR8383 macrophages with roasting products
Co-treatment with catalase significantly blocked the MRM-induced activation in NR8383 macrophages (p = 0.0113) and in Caco-2 cells (p = 0.0242) independent from the MRM concentration (Fig. 2), indicating that NF-κB activation by roasting products is linked to H2O2-signaling. Incubation of cells with catalase, but without any stimulants did not show any effect on NF-κB and cell viability, ruling out any impact of catalase on cellular metabolism itself (data not shown).In order to clarify the role of H2O2 and of cellular H2O2-detoxification in MRM-induced NF-κB-translocation, NR8383 macrophages were incubated with MRM (10–100 mM) for up to 24 h and the extracellular H2O2 concentration was measured over time (Fig. 6A). In addition, H2O2 levels were analyzed in MRM stored at identical cell culture conditions, but in the absence of NR8383 macrophages (Fig. 6A). In the presence of NR8383, between 100 and 450 μM extracellular H2O2 was detected after 2 h depending on the MRM concentration. After 12 h, H2O2 levels were slightly decreased but not completely scavenged. In comparison, the overall H2O2 concentration was significantly higher in the absence of NR8383 macrophages than in the presence of cells. The H2O2 concentration in MRM without cells reached levels between 170 μM and 565 μM after 2 h dependent on the MRM concentration. Other than in the presence of cells, H2O2 generation was promoted with increasing incubation time.
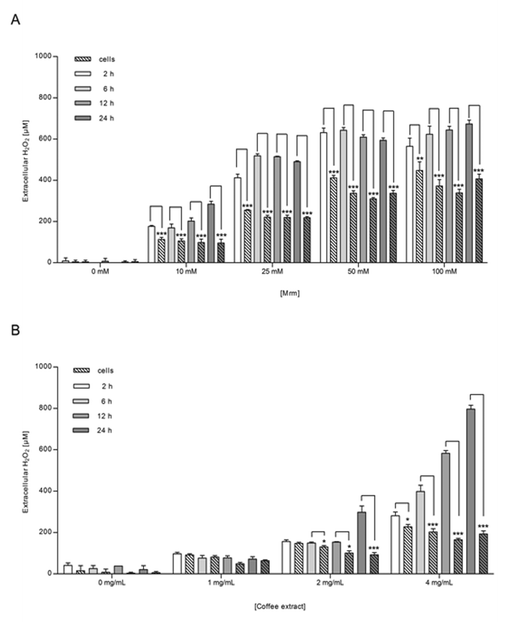 | ||
| Fig. 6 Extracellular H2O2 concentration after 2, 6, 12, and 24 h in NR8383 macrophages incubated with (A) MRM (10–100 mM) or (B) coffee extract (1–4 mg mL−1) for 24 h. Simultaneously, (A) MRM and (B) coffee extract were stored under similar cellular conditions but in the absence of NR8383 macrophages and H2O2 concentration was investigated likewise. Data is mean ± SD (A: n = 4; B: n = 3). * p < 0.05, ** p < 0.01, *** p < 0.001. | ||
Both experimental approaches were repeated under co-incubation of catalase (150 U mL−1). No significant amount of extracellular H2O2 was detected within the first 12 h (data not shown).
According to the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT) assay, the cell viability of NR8383 macrophages was not impaired, but cell growth was rather enhanced after incubation with MRM in low concentration. After exposure to 25 mM MRM for 24 h, the cell viability amounted to 173 ± 23%. In this case, the supplementation of catalase did not show any significant impact on the cell viability of MRM treated NR8383 macrophages. However, 50 mM and 100 mM MRM possessed cytotoxic potency (59 ± 21% and 23 ± 7% cell viability), which was reduced by co-treatment with catalase (121 ± 15% and 42 ± 5% cell viability).
Generation of extracellular H2O2 during stimulation of NR8383 macrophages with coffee extract
The generation of extracellular H2O2 after stimulation of NR8383 macrophages with coffee extract was in alignment with the results obtained for MRM; H2O2 generation depended on both coffee concentration and incubation time (Fig. 6B). In the presence of NR8383 macrophages, H2O2 levels were significantly lower than in the absence of cells especially for coffee extract applied in higher concentration. Under co-treatment with catalase, both experimental approaches did not result in significant amounts of H2O2 in any sample during 24 h (data not shown). Besides, it was confirmed that the cell viability of NR8383 macrophages did not decline to less than 90% after incubation with coffee extract (2 mg mL−1) for up to 6 h.Generation of extracellular H2O2 during mucosa oxygenation
Similarly, the extracellular H2O2 content was quantified during stimulation of human gut tissue ex vivo. As illustrated in Fig. 7, extracellular H2O2 could be detected in both samples, stimulated by MRM (Fig. 7A) and the coffee extract (Fig. 7B). The H2O2 concentration was always significantly higher in the absence of cells than in the presence of the tissue. An additional experiment clarified that air bubbling during mucosa oxygenation, which prevents ischaemia and supports the diffusion of the medium-dissolved stimuli into the tissue, did not have any impact on the H2O2 generating abilities of roasting products (data not shown).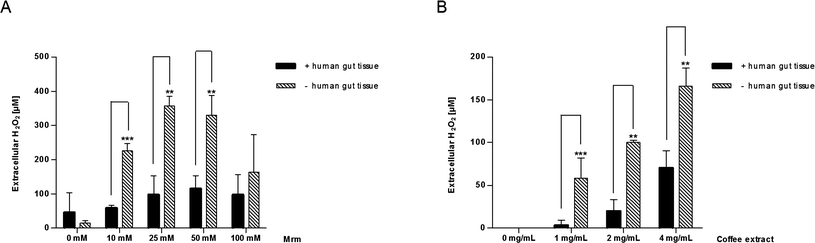 | ||
| Fig. 7 Extracellular H2O2 concentration after 2 h in human gut tissue samples incubated with (A) MRM (10–100 mM) or (B) coffee extract (1–4 mg mL−1) during mucosa oxygenation ex vivo. Simultaneously, MRM and coffee extract were cultivated under the same conditions as aforementioned but in the absence of the tissue samples and H2O2 concentrations were investigated likewise. Data are mean ± SD (A: n = 3; B: n = 2–5); * p < 0.05, ** p < 0.01, *** p < 0.001. | ||
Discussion
NF-κB is a multifunctional transcription factor which is involved in diverse cellular reactions such as inflammation, immune function, or cell survival.13 In the healthy gut, NF-κB is a critical factor of the intestinal immune system. On the other hand, inflammatory bowel diseases (IBD), such as Crohn's disease and ulcerative colitis, are associated with a chronic activation of NF-κB.2 Therefore, a balanced regulation of NF-κB is a crucial factor for gut health. Among food products, coffee seems to have a major influence on NF-κB regulation. In LPS-activated cell lines, coffee extract as well as the coffee component kahweol reversed NF-κB activation.11,12 A similar effect of coffee was also observed in transgenic NF-κB-luciferase mice.11 In the absence of co-stimulants, coffee extract activated NF-κB in macrophages.10 Both effects, NF-κB down-regulation of LPS-activated cells and NF-κB-activation in unstimulated macrophages seem to considerably depend on roasting products.10,11 These results are supported by a study on bread crust, another example of food rich in roasting products, that triggers the up-regulation of NF-κB in cardiac fibroblasts.14 Intestinal cell metabolism is characterized by diverse interaction of various cell types. Therefore, the present study focused on the influence of coffee and roasting products on the NF-κB regulation in several cell types and the role of reactive oxygen species herein. Furthermore, intact human gut tissue was proposed as a means to study the effect of coffee stimulation on complex cell structures ex vivo.Coffee extract as well as roasting products produced in a MRM significantly increased the nuclear translocation of NF-κB in macrophages in a concentration- and time-dependent manner. In contrast, stimulation of the colorectal epithelial cells Caco-2 by different concentrations of coffee did not lead to increased nuclear translocation of NF-κB. The incubation of Caco-2 cells with roasting products led to a significantly increased NF-κB response, which was, however, considerably lower than the effect on macrophages. Additionally, the primary human intestinal cells HIMEC were treated with MRM, since it could be expected that the metabolism of immortalized cell lines differs from cellsin vivo. As a matter of fact, NF-κB responded not only in multiple cell lines, but also in primary intestinal cells, which show closer similarities to cell metabolismin vivo. MRMs were already shown to induce NF-κB translocation in macrophages as well as in kidney cells.10 Besides, individual Maillard products such as aminoreductones15 and glycated ovalbumin16 elevated nuclear NF-κB amounts in macrophages and dendritic cells, respectively. Thus, it can be concluded that roasting products induce nuclear translocation of NF-κB in various cell types. The degree of NF-κB activation, however, varied depending on the cell type and showed the maximum effect in macrophages. The NF-κB activation by coffee was lower under the applied experimental conditions and could only be detected in macrophages. Furthermore, it was reported that coffee and coffee roasting products protect from hepatic inflammatory processes induced by a high fat diet in mice.17
The in vitro experiments raised the question how NF-κB responds in human intestinal tissue consisting of a wide range of cell types. The mucosa layer of the gut comprises a variety of cell types including immune cells such as macrophages, which showed the most explicit NF-κB response. Particularly in case of IBD such as Crohn's disease, macrophages are increasingly infiltrated in the mucosa of the intestine. Not only IBD, but also food allergies are triggered by specific reactions of the gastrointestinal immune system after food intake. Therefore, the immunomodulatory effect of coffee extract and roasting products was studied in the mucosa of the gut as the largest immunological organ of the body. Hence, a method was successfully established which allowed the stimulation of human gut tissue ex vivo.18,19 It was guaranteed that the tissue samples per se were uniform and comparable to each other and that cell viability of the tissue samples was not harmfully affected by the mucosa oxygenation ex vivo. Whereas roasting products formed in MRM elevated nuclear NF-κB levels in gut tissue significantly, coffee extract showed a trend towards activation.
In agreement with the in vitro studies, both coffee and roasting products showed similar effects on nuclear NF-κB translocation in human gut tissue, but with differences in the level of activation. These differences may be attributed to a different concentration of active roasting products in coffee and MRM, but also to coffee ingredients which may counteract NF-κB activation such as kahweol and 3-methyl-1,2-cyclopentanedione.12
Two milligrams per millilitre of coffee extract, which induced strong nuclear NF-κB translocation in macrophages, corresponds to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 diluted coffee beverage. Assuming a conversion rate for ribose into roasting products of 30%, 50 mM MRM would correspond to the melanoidin content of a coffee beverage. As far as we know, the concentration of coffee components in the gut after coffee consumption is not clear. Therefore, further studies are required to investigate the effect of coffee and roasting products on NF-κB activationin vivo.
:7 diluted coffee beverage. Assuming a conversion rate for ribose into roasting products of 30%, 50 mM MRM would correspond to the melanoidin content of a coffee beverage. As far as we know, the concentration of coffee components in the gut after coffee consumption is not clear. Therefore, further studies are required to investigate the effect of coffee and roasting products on NF-κB activationin vivo.
Furthermore, mechanisms were investigated which may explain how NF-κB activation in different cell types and complex human gut tissue can be differently affected by coffee and by roasting products. H2O2 seems to be a major factor of cell signaling induced by coffee and roasting products. H2O2 has been recognized as an important second messenger in redox signaling.20Co-treatment with catalase blocked the NF-κB activation in macrophages by the incubation with MRM or coffee extract, respectively.10 The present study revealed similar suppression for macrophages and Caco-2 cells. Since extracellular catalase cannot penetrate cellular membranes, its action is restricted to the decomposition of extracellular H2O2.21 It is well documented that coffee and roasting products are able to generate H2O2 in a cell-free system.10,22–24Extracellular H2O2 would then be able to penetrate the cell, activating H2O2-induced cell signaling including the activation of NF-κB. Indeed, supplements of pure H2O2 were shown to trigger the activation of NF-κB.25 Thus far, it is not fully clear to which extent coffee and roasting products are able to generate H2O2 in the anaerobic environment of the gastrointestinal tract, or if any H2O2 ingested with the coffee beverage reaches the gastrointestinal tract. For this purpose, studies are required to investigate the generation of H2O2 from roasting products in detail under different conditions. However, it has been shown that coffee drinking leads to increased urinary H2O2 concentrations in humans.26,27 These data strongly indicate continuous generation of H2O2 or high stability of coffee-derived H2O2in vivo. Furthermore, it has been demonstrated that other food components, such as green tea polyphenols, which are able to generate H2O2in vitro, are also able to generate reactive oxygen speciesin vivo. This prooxidative effect has been associated with the induction of apoptosis and the upregulation of antioxidative enzymes, eventually contributing to cancer preventive effects of tea polyphenols.28 The susceptibility of cells and tissue samples to nuclear NF-κB translocation depends on the activity of the stimulant to generate H2O2, but also on the ability of H2O2 to diffuse through cell membranes mediated by aquaporins.29 Furthermore, the cell-specific capacity of antioxidative enzymes such as glutathione peroxidase, peroxiredoxins, or catalase and numerous non-enzymatic antioxidants will have a major influence on NF-κB activation. The present study demonstrated that coffee and, particularly, roasting products are potent generators of H2O2 in different cell-free environments. In the presence of macrophages, H2O2 was clearly detectable in the extracellular space when exposed to coffee and roasting products, but the concentration was significantly lower than in the absence of cells. The decline of extracellular H2O2 can thus be attributed to diffusion across the cell membranes and also to cellular antioxidative systems. In C6 glioma cells, for instance, a bolus of H2O2 (≤100 μM) was completely detoxified.30 But in contrast to a one-time bolus of pure H2O2, roasting products in coffee and MRM generate H2O2 permanently, which could be responsible for the induction of nuclear NF-κB translocation in the different cell types.
In a similar way, the human gut tissue was able to significantly reduce extracellular H2O2 produced by coffee or roasting products. However, the detoxification rate of the tissue was higher compared to macrophages. The higher detoxification rate can be caused by the diversity of cells present in the tissue samples, but also by a higher cell density. Thus, the minor amount of residual H2O2 in the tissue samples may cause a reduced NF-κB response compared to macrophages. Finally, different gut sections may be differently affected by coffee roasting products. Whereas the upper gut section is directly exposed to roasting products, biotransformation processes and resorption may modulate the chemical structure or decrease the concentration of the roasting products reaching the lower gut resulting in changes of their bioactivity. The biological consequences of coffee-mediated NF-κB activation are still not fully clear. A screening assay for cytokine release indicated that NF-κB translocation induced by roasting products is not connected to pro-inflammatory activity, which is in agreement with previous studies.10 Active NF-κB regulated the expression of more than 150 target genes, including, for example, genes encoding immunoreceptors, cell adhesion molecule, stress response, cell-surface receptors, regulators of apoptosis, or enzymes.1 Therefore, large arrays for expression analysis have to be applied to delineate the full cellular response to intestinal NF-κB activation mediated by roasting products. Furthermore, the response of inflamed tissue, as present, for example, in IBD, may greatly differ from the response of unstimulated tissue.
It can be hypothesized that in a healthy gut, coffee-induced stimulation of NF-κB signaling supports the maintenance of immune homeostasis, particularly in the epithelial tissue, thus contributing to a balanced gut health.31 This assumption is in accordance with a moderate favorable effect of coffee drinking on colorectal cancer.32 In patients suffering from IBD, however, further activation of NF-κB by coffee may worsen inflammation processes associated with sustained elevated NF-κB activation.31
Experimental
Chemicals and cell culture
D-(−)-Ribose, L-lysine, phenylmethylsulfonyl fluoride (PMSF), dithiothreitol (DTT), NP-40, skim milk powder, sodium dodecyl sulfate (SDS), catalase (from bovine liver), albumin, di-sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O), sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O), ethylene glycol tetraacetic acid (EGTA), glycerol (anhydrous), bovine serum albumin (BSA), xylenol orange, ammonium ferrous sulfate, trypan blue, mouse anti-β-actin antibody (A5541), horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (A6154) and HRP-conjugated anti-mouse antibody (A6782) were purchased from Fluka-Sigma-Aldrich (Buchs, Switzerland). Rabbit polyclonal anti-p65 antibody (sc-109), anti-p65 antibody blocking peptide (sc-109p), and mouse monoclonal anti-p65 antibody (Sc-8008) were from Santa Cruz Biotechnology (Heidelberg, Germany). MTT, potassium chloride (KCl), magnesium chloride hexahydrate (MgCl2 × 6H2O), perchloric acid (PCA), magnesium chloride hexahydrate, potassium chloride, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and H2O2 (30%) were supplied by Merck KGaA (Darmstadt, Germany), lactate dehydrogenase (LDH) assay by Beckman Coulter (Krefeld, Germany), the bicinchoninic acid (BC) assay kit by Uptima, Interchim (Montluçon, France) and Dc-protein assay by BioRad Laboratories GmbH (Munich, Germany). Enhanced chemiluminescence (ECL) Western blotting detection reagents and Hyperfilm™ ECL were obtained from Amersham Biosciences (Munich, Germany) and chemiblot™ molecular weight marker from Chemicon (Schwalbach, Germany). NR8383 macrophages and Caco-2 cells were supplied by ATCC (Wesel, Germany), HIMEC, endothelial cell medium and endothelial cell growth supplement from ScienCell (Berlin, Germany). Other cell culture supplements and TNF-α were obtained from Biochrom AG (Berlin, Germany) and Invitrogen (Darmstadt, Germany). Endo-Paractol was purchased from Temmler Pharma GmbH & Co. KG (Marburg, Germany), roasted coffee beans (100% Coffea arabica) from a local retailer, dimethyl sulfoxide (DMSO) and sodium chloride (NaCl) from Acros Organics (Nidderau, Germany), protease inhibitor tablet (PIS) complete from Roche Diagnostics GmbH (Mannheim, Germany) and ethylenediaminetetraacetic acid (EDTA) from Carl Roth GmbH & Co. KG (Karlsruhe, Germany).Instruments
Universal grinder (Type A10) and Ultra Turrax homogenizer were obtained from IKA®-Werke GmbH & Co. KG (Staufen, Germany). The mucosa oxygenator was from Intestino-Diagnostics (Erlangen, Germany) and the VersaDoc™ imaging system from BioRad Laboratories GmbH (Munich, Germany).Preparation of coffee extract
Coffee extract was prepared using a standardized method. Fifteen grams of coffee beans (100% Coffea arabica) was ground for 30 s in a universal grinder. Next, 3.75 g ground coffee powder was filter-brewed with hot tap water (Ø 85 °C) to a total volume of 48 mL (coffee temperature: Ø 60 °C). The coffee was allowed to stand for 1 h in an ice-bath and pH was adjusted with NaOH (2 N) from pH 5.6 (Ø) to pH 7.4. Thereafter, the coffee solution was filled up with hot tap water to an overall volume of 50 mL followed by filter-sterilization (0.22 μm). In all experiments, coffee extract was used exactly 1 h after preparation. The coffee concentration used in the experiments refers to the dry weight, which was analyzed in at least three independent preparations per coffee batch.Preparation of roasting products in a MRM
The MRM was prepared from equimolar concentrated (0.5 M) D-(−)-ribose and L-lysine dissolved in PBS (137 mM NaCl, 8.1 mM Na2HPO4·2H2O, 2.7 mM KCl, 1.6 mM NaH2PO4·2H2O). The pH was adjusted from pH 9.7 to pH 9.4 by HCl (35%). Sterile aliquots of 20 mL were heated in capped screw neck bottles for 30 min at 120 °C in a drying cabinet. After cooling in an ice-bath, the pH was adjusted from pH 8.6 to pH 8.0 by HCl (35%). Finally, the solutions were filter-sterilized (0.22 μm) and aliquots were stored at −20 °C. Since the concentration of the reaction products in the final mixture is not known, the given concentration reflects the initial concentration of the reactants. Assuming that about 30% of ribose and lysine were converted into Maillard products under the applied conditions, the content of Maillard products in a 50 mM MRM would reflect, for example, the concentration of melanoidins in a coffee beverage (0.45 g melanoidins per 100 mL).33Cell culture
Cells were cultured at 37 °C in a humidified atmosphere with 5% CO2. Cell culture media were supplemented with penicillin (100 U mL−1) and streptomycin (100 μg mL−1). Rat macrophages (NR8383) were maintained in HAM's F12 medium supplemented with 15% (v/v) heat-inactivated fetal calf serum (FCS). Caco-2 cells were grown in minimum essential medium (MEM) supplemented with 20% (v/v) FCS and non-essential amino acid solution (0.1 mM). Primary HIMEC were cultivated in fibronectin-coated cell culture flasks in endothelial cell medium containing 5% (v/v) FCS and 1% (v/v) endothelial cell growth supplement. NR8383 macrophages were harvested by scraping; Caco-2 and HIMEC were detached by trypsin (0.25%)/EDTA (0.02%).Tissue culture
The study was approved by the ethics committee of the University Medical Center in Erlangen (Germany). Patients gave their informed consent to this local study. Mucosal gut biopsies (3–15 mg) were obtained from 17 patients during gastrointestinal endoscopies because of various gastrointestinal complaints to check for manifestations of inflammation, neoplasm, or food allergy. The specimens were taken from two localizations in the lower gastrointestinal tract, from the terminal ileum of the small intestine and from the ascending colon of the large intestine. Neither macroscopic nor microscopic investigation of all biopsies revealed any chronic inflammatory gut diseases such as Crohn's disease. Immediately after sampling, the biopsies were placed into hard plastic tubes containing PBS transport medium. The tubes were kept at 37 °C and bubbled constantly with room air providing an adequate oxygen supply at a pO2 of 85–95 mm Hg (mucosa oxygenation18). The wet weight was determined prior to stimulation.Analysis of NF-κB translocation in cultured cells: in vitro stimulation and nuclear protein extraction
Cells were stimulated by coffee extract, MRM, or TNF-α. In some experiments, the cells were co-incubated with catalase (150 U mL−1), which was added to the cells 10 min before stimulation. As negative control, cells were incubated with water/PBS instead of the stimulants.More specifically, NR8383 macrophages (3 × 106cells) were grown for 4 d. Thereafter, cells were incubated with the stimulants in PBS for up to 6 h. In order to replace cell culture medium by PBS, floating cells were collected by centrifugation (1500 rpm, 2 min, at room temperature) and both floating cells and adherent cells were washed separately with PBS. Finally, the floating cells were re-suspended in PBS and recombined with the adherent cells.
Caco-2 cells (1 × 106cells) and HIMEC (7.5 × 105cells) were grown for 5 d. In the case of HIMEC, the medium was refreshed on the 4th day. On the 5th day, the medium was removed for both, adherent cells were washed with PBS and cells were stimulated in PBS for 2 h.
After stimulation, the nuclear cell extracts were prepared on ice according to Andrews and Faller with slight modifications.34 Briefly, floating cells (NR8383) were collected by centrifugation (1500 rpm, 4 min, 4 °C); adherent cells by scraping. Floating and adherent cells were merged and washed three times with ice-cold PBS. Next, the cells were lyzed with 1 mL ice-cold hypotonic buffer Acell (10 mM HEPES, 10 mM KCl, 1 mM MgCl2·6H2O, 5% (v/v) glycerol, 0.5 mM EDTA, 0.1 mM EGTA), which was supplemented with protease inhibitor solution (Protease Inhibitor tablet complete (PIS) was dissolved in 0.5 mL PBS; 1% (v/v)), PMSF (2 mM in ethanol), and DTT (0.5 mM in water). After 15 min incubation on ice, 65 μL of 10% (v/v) NP-40 (in water) was added and the cells were mechanically lyzed by vortexing for 15 s. Cell nuclei were collected by centrifugation and washed with buffer Acell to ensure a complete removal of cytoplasmic proteins. Finally, nuclear proteins were extracted for 1 h with 52 μL ice-cold high salt extraction buffer B (20 mM HEPES, 1% (v/v) NP-40, 400 mM NaCl, 10 mM KCl, 1 mM MgCl2·6H2O, 20% (v/v) glycerol, 0.5 mM EDTA, 0.1 mM EGTA), which was supplemented with PIS (1%, v/v), PMSF (2 mM), and DTT (0.5 mM). The suspension was vortexed every 20 min. After 1 h, the supernatant nuclear extract was collected after centrifugation. The protein concentration of the nuclear extract was determined with the Dc-protein assay using BSA in high salt extraction buffer B as standard. Nuclear cell extracts were stored at −80 °C until use for Western blotting.
Analysis of NF-κB translocation in intact human gut tissue: ex vivo stimulation and nuclear protein extraction
Human gut tissue samples were stimulated according to a modified method of Raithel et al.19 Briefly, two biopsies, one from each intestinal segment, were stimulated in supplemented incubation medium (Dulbecco's PBS with 3% (w/v) albumin, 2.4% (v/v) HEPES buffer, and 0.002% (v/v) Endo-Paractol) with coffee extract or MRM for 2 h. The negative control biopsies were further analyzed without prior sample stimulation but exposed to water in case of the coffee experiments or PBS for the MRM experiments. During stimulation, human gut tissue samples were oxygenated at 37 °C by mucosa oxygenation.After stimulation, nuclear proteins were extracted according to a modified method of Thiele et al.35 In detail, intestinal human gut tissue samples were washed with PBS in a 50 mL Falcon tube for 2 min at 400 rpm. Cells were lyzed by vortexing in ice-cold hypotonic buffer Atissue (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl), which was freshly supplemented with DTT (0.5 mM), PMSF (0.2 mM), PIS (1% (v/v)), and NP-40 (0.56% (v/v)). To ensure lysis, the gut tissue was subjected to three freeze-thaw cycles in liquid nitrogen and finally mechanically disrupted in the Ultra Turrax homogenizer for 1 min on ice. After 30 min, the cell nuclei were collected by centrifugation and washed with buffer Atissue to assure the complete removal of cytoplasmic proteins. Subsequently, the nuclear proteins were extracted from the nuclei with 25 μL of high salt extraction buffer B.
NF-κB analysis by Western blotting
Nuclear amounts of NF-κB were detected immunochemically via Western blotting according to a modified method of Muscat et al.10 Analysis of nuclear NF-κB amounts has been described as a valid alternative to electrophoretic mobility shift assays.36,37 The nuclear protein amount in cells and human gut tissue sample was determined by a Dc-protein assay using BSA in high salt extraction buffer B as standard.Briefly, nuclear proteins were denatured in loading buffer (0.06 M Tris-HCl, 0.07 M SDS, 1.04 M urea, 0.064 M DTT, 7% (v/v) glycerol, bromophenol blue, adjusted to pH 7.4 with 2 N NaOH) for 7 min at 95 °C. Equal amounts of denatured protein (10 μg for cell experiments; 5 μg for tissue experiments) were separated by electrophoresis in a 12% SDS-polyacrylamide gel and afterwards transferred to a nitrocellulose membrane. The membranes were cut into two parts and the non-specific binding sites of the membrane were blocked with blocking buffer (0.15% (w/v) skim milk powder in PBS/Tween (0.1% (v/v) Tween 20 in PBS). Then the membranes were incubated overnight at 4 °C with the primary antibodies for p65, a subunit of NF-κB, or for β-actin as loading control. Primary antibodies included rabbit polyclonal anti-p65 (diluted 1:500 in blocking buffer) or, in the case of Caco-2 cells, mouse monoclonal anti-p65 (diluted 1:100 in blocking buffer) and mouse anti-β-actin (diluted 1:13333 in blocking buffer). Next, the membranes were incubated for 1 h with the HRP-conjugated second antibody (anti-rabbit 1:1500 diluted in blocking buffer; anti-mouse 1:2000 diluted in blocking buffer). The protein bands were visualized by ECL. The p65 band was identified by an anti-p65 antibody blocking peptide and a chemiblot™ molecular weight marker. The data was analyzed densitometrically by a VersaDoc™ imaging system. Besides the adjusted protein amount, the intensity of the p65 (NF-κB) signal was normalized to the loading control β-actin. Similar results were obtained when the NF-κB signal was not normalized to β-actin. NF-βB translocation was expressed as n-fold increase relative to the control exposed to the solvents PBS/water instead of the stimulants. The identity of p65 was verified by the use of a p65 blocking peptide.
Bicinchoninic assay (BC assay)
The total protein amount in human gut tissue samples was determined with the BC assay. Immediately after weighing, the tissue sample was homogenized in a defined volume of distilled water on ice for 2 × 20 s by an Ultra Turrax. The protein amount was examined according to the manufacturer's instructions. The protein concentration was calculated from an external standard curve using albumin as standard.Ferrous oxidation xylenol orange (FOX) assay
Extracellular H2O2 was measured with the FOX assay using PCA (FOXPCA assay) according to a modified method of Gay and Gebicki.38 For the assay, NR8383 macrophages (1 × 106cells) were grown for 4 d at 37 °C. Subsequently, the cells were stimulated with coffee extract or MRM in various concentrations for up to 24 h at 37 °C in serum-/phenol red-free medium. In some experiments catalase (150 U mL−1) was added to the cells 10 min before stimulation. Aliquots of 150 μL were taken after 2, 6, 12, and 24 h. Prior to the FOXPCA assay, floating cells were removed by centrifugation. According to the FOXPCA assay protocol, aliquots of 60 μL were mixed with 20 μL of solvent. As blank, catalase solution (120 U mL−1PBS) was added instead of solvent. After 15 min at room temperature, 20 μL of each sample was incubated in triplicate with 180 μL FOX reagent (0.5 M xylenol orange, 0.5 M ammonium Fe(II) sulfate in 0.11 M PCA) each. After shaking for 30 min, the absorbance was read at 550 nm. All readings were corrected for any interference from brown sample color. The H2O2 concentration was calculated from the difference in absorbance between sample and the catalase-blank via an external H2O2 calibration graph. The concentration of the H2O2 stock solution was determined by UV-spectroscopy, using the molar extinction coefficient (0.0394 cm2 μmol−1) at 240 nm.39 The experiment was repeated in the absence of cells but following the same experimental outline as described above. Similarly, extracellular H2O2 was measured during the stimulation of human gut tissue samples.MTT assay
The viability of NR8383 macrophages after stimulation with MRM was investigated according to a modified method of Mosmann.40 The method is based on the ability of active cells to metabolize the yellow MTT into dark blue insoluble formazan. Specifically, NR8383 cells (1 × 106cells) were allowed to settle overnight at 37 °C in a 12-well plate. Next, cells were stimulated with MRM (10 mM–100 mM) for 24 h at 37 °C in supplement-free medium. As positive control, cells were treated with 10% DMSO instead of MRM. Control cells were incubated with PBS without any stimulant. In some experiments, catalase (150 U mL−1) was added to the cells 10 min prior to stimulation. After stimulation, cells were incubated with MTT (1 mg mL−1 in PBS) for 3 h at 37 °C. Dark blue formazan crystals were formed which were dissolved by adding isopropanol/1 N HCl (25:1) solution. After shaking for 10 min, the absorbance was read at 595 nm. The absorbance of control cells was set to 100% viable cells. The cell viability of stimulated cells was expressed as percentage of control.
Trypan blue dye exclusion test
The cell viability of NR8383 macrophages after stimulation with coffee extract was determined by the trypan blue dye exclusion test. Depending on cellular membrane integrity, which is one main characteristic of vital cells, the anionic diazo dye trypan blue is absorbed into the cell and appears blue. Briefly, NR8383 macrophages (1 × 106cells) were grown for 4 d. Then the cells were exposed to coffee extract (1–4 mg mL−1) for up to 6 h in PBS. Control cells were incubated in PBS for 6 h. After stimulation, cells were treated with trypan blue dye solution (5 mg mL−1PBS). Viable as well as dead (blue) cells were counted under the microscopevia hemacytometer. The cell viability was calculated as the ratio between living cells and the total cell number. Values obtained for control cells were set to 100% cell viability. The cell viability of stimulated cells was expressed as percentage of control.LDH assay
A kinetic study of the cell viability of human gut tissue samples during mucosa oxygenation for ex vivo cultivation was measured using an LDH assay. LDH is a cytosolic enzyme, which is released into extracellular media due to cell membrane damage and, therefore, used as an indicator of cell viability.Human gut tissue samples were kept either in incubation media, modified Hank's balanced salt solution (Hanks) (3 g L−1 albumin, HEPES (1 M) 2.4% (v/v), FCS 1% (v/v)) or modified PBS (3 g L−1 albumin, HEPES (1 M) 2.4% (v/v)). LDH was measured after 0.5, 1.5, 3, 4.5, 6, and 24 h in the supernatant and in the tissue. A separate biopsy was used for each time point. Aliquots of the supernatant were used to determine the LDH release into the medium. In order to analyze the intracellular LDH amount, the human gut tissue samples were homogenized in an Ultra Turrax in a defined volume of bis-tris buffer (20 mM; pH 7) on ice for 30 s. The LDH concentration was quantified according to a protocol of Beckman Coulter. Cell death is defined by the LDH release, which is calculated as the percentage of extracellular LDH related to the total LDH amount. The cell death values were finally used to calculate the cell viability.
In order to rule out any influence of the human gut tissue samples themselves, the kinetic LDH release was analyzed for one single biopsy over time for 24 h. For that purpose a biopsy was incubated in a modified Hanks buffer for 24 h. Aliquots of the supernatant were taken after several time points and the extracellular LDH amount was determined as described above.
Cytokine analysis
NR8383 macrophages were stimulated with MRM (25 mM) as described above. The release of IL-1α, IL-1β, IL-6 IL-10 and TNF-α was determined by the Bio-Plex-Cytokine assay (Bio-Rad, Munich, Germany). One hundred nanograms per millilitre LPS was used as positive control.Statistical analysis
Data were analyzed statistically with Graphpad prism 5. The results were expressed as the mean ± SD from n independent experiments as indicated. Statistical significance of the data was calculated using unpaired, two-tailed Student's t-test. Levels of confidence were defined as * p < 0.05, ** p < 0.01, and *** p < 0.001.Conclusion
In summary, the present study showed that intact human gut tissue samples can be used to examine cellular effects of food components on complex cell systems. The impact of coffee extract and roasting products obtained from a MRM on the activation of NF-κB in cell lines, primary cell cultures, and human gut tissue was investigated. Thus, it was demonstrated that cellular effects of coffee and roasting products, which were observed in different cell lines, were also evident when intact human gut tissue was stimulated ex vivo. Moreover, this study provides growing evidence that the translocation of NF-κB is triggered by the roasting products in coffee extract. The roasting products generate extracellular H2O2, which escapes the cellular detoxification. After migration through the cell membrane, H2O2 might interfere with cellular signaling and eventually lead to the activation of NF-κB. Thus, coffee and roasting products may have to be considered as food items that affect the balance of NF-κB activation in the healthy and inflamed gut.Abbreviations
| NF | Nuclear factor |
| LPS | Lipopolysaccharide |
| IBD | Inflammatory bowel disease |
| Caco-2 cells | Human epithelial colorectal adenocarcinoma cells |
| MRM | Maillard reaction mixture |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| PBS | Phosphate buffered saline |
| HIMEC | Human intestinal microvascular endothelial cells |
| LDH | Lactate dehydrogenase |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide |
| PMSF | Phenylmethylsulfonyl fluoride |
| DTT | Dithiothreitol |
| SDS | Sodium dodecyl sulfate |
| BSA | Bovine serum albumin |
| HRP | Horseradish peroxidase |
| PCA | Perchloric acid |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| BC | Bicinchoninic acid |
| ECL | Enhanced chemiluminescence |
| DMSO | Dimethyl sulfoxide |
| FCS | Fetal calf serum |
| MEM | Minimum essential medium |
| PIS | Protease inhibitor solution |
| FOX | Ferrous oxidation xylenol orange |
References
- H. L. Pahl, Oncogene, 1999, 18, 6853–6866 CrossRef CAS.
- M. F. Neurath, C. Becker and K. Barbulescu, Gut, 1998, 43, 856–860 CrossRef CAS.
- Z. J. Chen, L. Parent and T. Maniatis, Cell, 1996, 84, 853–862 CrossRef CAS.
- W. C. Sha, H. C. Liou, E. I. Tuomanen and D. Baltimore, Cell, 1995, 80, 321–330 CrossRef CAS.
- F. Weih, D. Carrasco, S. K. Durham, D. S. Barton, C. A. Rizzo, R. P. Ryseck, S. A. Lira and R. Bravo, Cell, 1995, 80, 331–340 CrossRef CAS.
- W. Strober, B. Kelsall, I. Fuss, T. Marth, B. Ludviksson, R. Ehrhardt and M. Neurath, Immunol. Today, 1997, 18, 61–64 CrossRef CAS.
- G. Rogler, K. Brand, D. Vogl, S. Page, R. Hofmeister, T. Andus, R. Knuechel, P. A. Baeuerle, J. Scholmerich and V. Gross, Gastroenterology, 1998, 115, 357–369 CrossRef CAS.
- G. Dijkstra, A. J. Zandvoort, A. C. Kobold, A. de Jager-Krikken, P. Heeringa, H. van Goor, H. M. van Dullemen, J. W. Tervaert, A. van de Loosdrecht, H. Moshage and P. L. Jansen, Scand. J. Gastroenterol., 2002, 37, 546–554 CrossRef CAS.
- N. Rajendran and D. Kumar, World J. Gastroenterol., 2010, 16, 1442–1448 CrossRef CAS.
- S. Muscat, J. Pelka, J. Hegele, B. Weigle, G. Munch and M. Pischetsrieder, Mol. Nutr. Food Res., 2007, 51, 525–535 CAS.
- I. Paur, T. R. Balstad and R. Blomhoff, Free Radical Biol. Med., 2010, 48, 1218–1227 CrossRef CAS.
- J. Y. Kim, K. S. Jung, K. J. Lee, H. K. Na, H. K. Chun, Y. H. Kho and H. G. Jeong, Cancer Lett., 2004, 213, 147–154 CrossRef CAS.
- S. Ghosh and M. S. Hayden, Nat. Rev. Immunol., 2008, 8, 837–848 CrossRef CAS.
- S. Ruhs, N. Nass, V. Somoza, U. Friess, R. Schinzel, R. E. Silber and A. Simm, Mol. Nutr. Food Res., 2007, 51, 488–495 CAS.
- A. Wuhr, M. Deckert and M. Pischetsrieder, Mol. Nutr. Food Res., 2010, 54, 1021–1030 Search PubMed.
- T. Hilmenyuk, I. Bellinghausen, B. Heydenreich, A. Ilchmann, M. Toda, S. Grabbe and J. Saloga, Immunology, 2010, 129, 437–445 CrossRef CAS.
- P. Vitaglione, F. Morisco, G. Mazzone, D. C. Amoruso, M. T. Ribecco, A. Romano, V. Fogliano, N. Caporaso and G. D'Argenio, Hepatology, 2010, 52, 1652–1661 CrossRef CAS.
- M. Raithel, M. Weidenhiller, R. Abel, H. W. Baenkler and E. G. Hahn, World J. Gastroenterol., 2006, 12, 4699–4705 CAS.
- M. Raithel, M. Weidenhiller, M. Shaban, R. Abel, H. Tuchbreiter, B. Backhaus, N. Donhauser, H. W. Baenkler and E. G. Hahn, Inflammation Res., 2003, 52(Suppl. 1), s13–14 CrossRef CAS.
- H. J. Forman, M. Maiorino and F. Ursini, Biochemistry, 2010, 49, 835–842 CrossRef CAS.
- M. Schimmel and G. Bauer, Oncogene, 2002, 21, 5886–5896 CrossRef CAS.
- J. Hegele, G. Munch and M. Pischetsrieder, Mol. Nutr. Food Res., 2009, 53, 760–769 CAS.
- M. Akagawa, T. Shigemitsu and K. Suyama, Biosci., Biotechnol., Biochem., 2003, 67, 2632–2640 CrossRef CAS.
- B. Halliwell, M. V. Clement and L. H. Long, FEBS Lett., 2000, 486, 10–13 CrossRef CAS.
- J. Song, J. Li, J. Qiao, S. Jain, B. Mark Evers and D. H. Chung, Biochem. Biophys. Res. Commun., 2009, 378, 610–614 CrossRef CAS.
- K. Hiramoto, T. Kida and K. Kikugawa, Biol. Pharm. Bull., 2002, 25, 1467–1471 CAS.
- L. H. Long and B. Halliwell, Free Radical Res., 2000, 32, 463–467 CrossRef CAS.
- S. C. Forester and J. D. Lambert, Mol. Nutr. Food Res., 2011, 55, 844–854 CAS.
- G. P. Bienert, A. L. Moller, K. A. Kristiansen, A. Schulz, I. M. Moller, J. K. Schjoerring and T. P. Jahn, J. Biol. Chem., 2006, 282, 1183–1192 CrossRef.
- M. Gulden, A. Jess, J. Kammann, E. Maser and H. Seibert, Free Radical Biol. Med., 2010, 49, 1298–1305 CrossRef.
- A. Wullaert, M. C. Bonnet and M. Pasparakis, Cell Res., 2010, 21, 146–158 CrossRef.
- C. Galeone, F. Turati, C. La Vecchia and A. Tavani, Cancer, Causes Control, 2010, 21, 1949–1959 CrossRef.
- V. Fogliano and F. J. Morales, Food Funct., 2011, 2, 117–123 CAS.
- N. C. Andrews and D. V. Faller, Nucleic Acids Res., 1991, 19, 2499 CrossRef CAS.
- K. Thiele, A. Bierhaus, F. Autschbach, M. Hofmann, W. Stremmel, H. Thiele, R. Ziegler and P. P. Nawroth, Gut, 1999, 45, 693–704 CrossRef CAS.
- C. Kaltschmidt, B. Kaltschmidt, T. Henkel, H. Stockinger and P. A. Baeuerle, Biol. Chem. Hoppe-Seyler, 1995, 376, 9–16 CrossRef CAS.
- J. Korcok, L. N. Raimundo, X. Du, S. M. Sims and S. J. Dixon, J. Biol. Chem., 2005, 280, 16909–16915 CrossRef CAS.
- C. A. Gay and J. M. Gebicki, Anal. Biochem., 2002, 304, 42–46 CrossRef CAS.
- D. P. Nelson and L. A. Kiesow, Anal. Biochem., 1972, 49, 474–478 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |