Chemocatalytic conversion of cellulose: opportunities, advances and pitfalls
Jan A.
Geboers
,
Stijn
Van de Vyver
,
Roselinde
Ooms
,
Beau
Op de Beeck
,
Pierre A.
Jacobs
and
Bert F.
Sels
*
Centre for Surface Science and Catalysis, Catholic University of Leuven, Kasteelpark Arenberg 23, 3001 Heverlee, Belgium. E-mail: Bert.Sels@biw.kuleuven.be; Fax: (+32) 1632 1998; Tel: (+32) 1632 1593
First published on 30th June 2011
Abstract
In recent years, cellulose conversion to useful fuels and chemicals has become a focal point in academic and industrial research. This contribution highlights opportunities for cellulose valorization, summarizes recent advances in its chemocatalytic conversion and discusses the future of the field in the economic context of the first quarter of the 21st century.
Opportunities for cellulose conversion
Although the exact time frame for reaching the ‘peak oil’ moment remains a subject of much debate, it is clear that the supply of easily exploitable fossil fuels will run out eventually. Furthermore, CO2 emissions linked to the use of fossil fuels have been associated with global climate changes.1 The predicted growth of the world population and the increased demand for energy, fuels and chemicals due to an increased industrialisation will further exacerbate these issues,2 creating a great amount of academic and industrial interest in the valorization of alternative, renewable sources for fuels and chemicals.Although renewable energy from wind and water holds considerable potential, it is insufficient and incapable to fill the entire global energy demand. Whereas solar energy possibly provides a more final solution to the problem, it will only be able do so in the longer run. For a short and medium term solution to the deficit of both sustainable energy and renewable carbon, most experts look to biomass as a most viable alternative source.3
The first generation of biofuels, encompassing bioethanol and biodiesel, has already appeared on the market and shown both its potential and limitations. As bioethanol at present can only be used in a mixture with traditional gasoline,3b its potential as a long-term renewable fossil fuel alternative is severely limited. The same is largely true for biodiesel, although certain adapted engines have been shown to be capable of running on pure biodiesel. Moreover, concerns persist that a large production of the first generation biofuels might represent a threat for global food supply, already strained by an increasing population. Finally, although the environmental advantages of first generation biofuels produced in tropical climates are undeniable, the same is not necessarily true for biofuels produced in temperate climates, due to enhanced investment in fertilizers and lower returns in biomass production.
These problems could be avoided or mitigated by replacing the first generation biofuels with products derived from lignocellulosic biomass. Lignocellulose functions as the most important structural component in a majority of plants, making it widely available in much larger quantities than starch, oils and fats, the source materials of first generation biofuels. The global amount of terrestrial plant biomass produced annually through photosynthesis has been estimated to be about 56.8 × 109 tonnes of elemental carbon.4Lignocellulose is estimated to make up between 70 and 95% of this amount.5 Furthermore, the components of lignocellulose (40–50% cellulose, 16–33% hemicellulose and 15–30% lignin)6 are indigestible by humans and are readily available in several industrial waste streams, especially from the paper and agricultural industries. This severs the direct competition between lignocellulose valorisation and human food supply.
Cellulose is a homopolymer of glucose, while hemicellulose consists of several pentoses (mostly xylose [1] and arabinose [2]) and hexoses (mostly galactose [3], glucose and mannose). It often also contains non-sugar components such as acetyl groups (Fig. 1).7Lignin, on the other hand, is a (co-)polymer of aromatic lignols with monomers such as p-coumaryl [4], coniferyl [5] and sinapyl alcohols [6] (Fig. 2). Valorisation of (hemi)cellulose could therefore yield a diversity of platform chemicals such as the constituent sugars and their derivatives (levulinic acid [7], 5-hydroxymethylfurfural (5-HMF) [8], furfuryl alcohol [9], ethylene glycol [10], γ-valerolactone [11] and polyols [15–18] (Fig. 3)), while lignin could be a renewable source of aromatics.
 | ||
| Fig. 1 Some common hemicellulose monomers. | ||
 | ||
| Fig. 2 The aromatic monolignol monomers of lignin. | ||
 | ||
| Fig. 3 Examples of cellulose-derived platform chemicals. | ||
Since cellulose is the main component of lignocellulosic biomass, it has attracted much research interest. Its conversion in aqueous medium into chemicals and fuels is the main focus of this contribution. Because a certain knowledge of the structural chemistry of (ligno)cellulose is indispensable to fully appreciate the difficulties and recent developments in cellulose conversion, the next section is devoted to this topic. In another section a critical overview of recent literature will be made. Several extensive reviews on the subject have been published, also incorporating the extensive literature on cellulose conversion in ionic liquids.5,8,13,75 We therefore focus on the recent advances in aqueous cellulose conversion of the last 3 years. The final section of this review presents a critical outlook to the future of this domain.
Structure of lignocellulose
Cellulose [12] is one of the most abundant organic compounds on earth, since it is contained in almost all woody materials. Although cellulose has the same molecular formula as amylose [13], one of the main components of starch (C6H10O5), there is one fundamental difference among them: in amylose the glucose monomers are linked by α-1,4-glycosidic bonds, while in cellulose they are linked by β-1,4-glycosidic bonds (Fig. 4). This seemingly trivial difference has important physico-chemical consequences.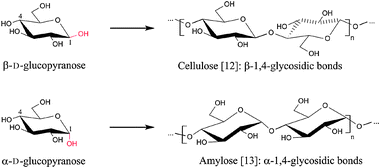
In cellulose, the glucose monomers are forced into a 4C1 chair conformation, while successive monomers are rotated over 180° around the polymer axis, to satisfy the bond angle of the bridging oxygen (O1; Fig 5). This forces the hydroxy and hydroxymethyl groups in an equatorial position, stabilizing the chair conformation and decreasing the flexibility of the glycosidic bond.9 As a result, the cellulose chain is a linear, rigid polymer, whereas amylose chains show more flexibility. Two intramolecular hydrogen bonds per anhydroglucopyranose unit further increase its rigidity. Every cellulose crystal phase exhibits a O3–H–O5′ bond (Fig 5). In addition, another crystal phase dependent hydrogen bond is formed. Finally, the β bond is intrinsically more stable than the α bond (in amylose).10
The hydroxy and hydroxymethyl groups in a cellulose chain protrude from the chain axis by virtue of their equatorial configuration. This makes them well suited for forming hydrogen bonds with neighboring cellulose chains and these interactions lie at the basis of the formation of planar cellulose sheets, which upon stacking form microfibrils. As recently revealed, an ordered multitude of weak CH–O bonds is at the basis of the affinity between the sheets,11 allowing formation of an ordered crystal network. The pattern of H-bonds in a cellulose crystal determines the internal cohesion, thereby determining its accessibility to catalysts and consequently its reactivity. Whereas an individual cellulose chain is not significantly more hydrophobic than an amylose chain, crystalline phase formation makes cellulose insoluble in water and most organic solvents. The functional groups and glycosidic bonds are well-protected by the microfibril structure, resulting in a half-life for non-catalyzed cellulose hydrolysis at 298 K between 5 and 8 million years.15Cellulose is not fully composed of crystalline domains, however. Between the crystalline domains of cellulose, amorphous areas/patches exist, which are much easier to hydrolyze. Molecular simulations estimated the existence of 8 hydrogen bonds per unit cell in cellulose crystals and up to 5.3 hydrogen bonds per unit cell in amorphous regions.13
Crystalline cellulose may exhibit several crystalline phases, differing in the alignment of the cellulose chains relative to each other and therefore in the pattern of the H-bonds. The natural form of cellulose is denoted as cellulose I and is the form present in microcrystalline Avicel PH-101 and α-cellulose, most often used in research. Cellulose I is a mixture of cellulose Iα (triclinic) and Iβ (monoclinic) in varying proportions depending on its origin.11,14 Another form of cellulose, which is thermodynamically most stable and denoted as cellulose II, is formed from cellulose I by mercerisation (alkali treatment) or regeneration (solubilisation and subsequent recrystallisation). When celluloses I and II are treated with liquid ammonia other forms of cellulose are obtained, denoted as IIII and IIIII, respectively. Celluloses IVI and IVII are formed by heating celluloses IIII and IIIII, respectively.12 More information on differences between allomorphs is available elsewhere.15
Compared to cellulose, hemicellulose has a very diverse composition containing several pentoses, hexoses and non-sugar compounds as monomers, the most abundant building block being xylose [1]. Furthermore, a typical hemicellulose chain is branched and shorter than that in cellulose, inhibiting crystal formation and making hemicellulose much easier to hydrolyse than cellulose.16
Lignin is another major component of lignocellulose, mainly present in woody biomass. It is a heavily branched and interconnected hydrophobic polymer consisting of the typical aromatic monomers [4], [5] and [6]. It is extremely resistant to degradation.
In cell walls, hemicellulose is associated with cellulose microfibrilsviahydrogen bonds, forming a rigid structure functioning as the structural backbone of the cell wall. It is surrounded by a very resistant lignin sheath to protect it against chemical and physical stress and degradation.16
The above discussion on the structure of lignocellulose illustrates the resistance of untreated woody biomass to degradation. In most cases, an intense pretreatment is a prerequisite for effective conversion. To increase its accessibility to catalysts, the protective lignin sheath must be removed, cellulose crystallinity must be reduced and the porosity and surface area of the biomass must be increased.17
Although the lignin fraction of biomass is usually burned to provide energy for the biorefinery, recently it has been advanced as a valuable source of renewable, aromatic platform chemicals, viz.benzene, toluene, xylene, phenolics, and alkane mixtures which are liquid at atmospheric pressure and ambient temperature.18
An economically viable hydrolysis procedure of cellulose still remains challenging, despite many years of research activity. Although cellulose hydrolysis can be both acid and base catalyzed, the former is more selective and therefore most intensively studied.19
Cellulose hydrolysis into glucose, followed by its conversion into degradation products, can be described kinetically by two consecutive first order reactions.20,21 Reported apparent activation energies vary between 172 and 189 kJ mol−1.22 Unfortunately, a reliable and unique kinetic model is not available for the following reasons:
(i) among the many acid degradation products, a family of solid by-products known as humins makes establishment of reliable kinetic data difficult;
(ii) the conversion of extensively degraded cellulose crystals, e.g. at high temperature (>488 K) in diluted acid or of decrystallised cellulose seems to obey different kinetic equations.23 Under these conditions, the temperature dependence of the hydrolysis reaction is enhanced, while its rate is increased by several orders of magnitude. Only under these conditions, i.e. after elimination of the physico-chemical barriers of which crystallinity is most important, cellulose hydrolysis can be considered to be under chemical control. This shows the immense influence of substrate crystallinity on cellulose hydrolysis. Therefore, any process in which cellulose hydrolysis is rate determining is to a large extent governed by the overall physico-chemical properties of the substrate, of which the crystallinity is dominating.
From cellulose to sugars
Presently, the hydrolysis of polysaccharides like cellulose into their constituent sugars with either biocatalysts or chemocatalysts is an active area of research.Biocatalysis utilizes cellulases, a complex mixture of enzymes, to convert cellulose to fermentable sugars. The cellulase complex is very specific for the cellulose substrate and produces glucose very selectively, in yields of typically about 75 till 85% under very mild conditions (pH 4–8, 318–353 K) and high glucose yields can be achieved with low amounts of enzyme.24 Contrary to reactions with mineral acids (vide infra), problems related to waste disposal, neutralisation and corrosion are non-existent. However, the use of cellulases requires a thorough pretreatment of the lignocellulosic biomass, not only to improve accessibility of the glycosidic bonds, but also to remove cellulase inhibitors like lignin. In the absence of this pretreatment, cellulose digestibility only is around 20%.17 Furthermore, hydrolysis with cellulases is slow and in most cases, cellulose feed concentration is limited by the need to keep the concentration of produced inhibitors low. All these factors significantly diminish the space time yield that can be achieved by enzymatic hydrolysis, necessitating the use of large reactors. The low space time yield, the need for pH regulating buffers, the high cost of enzymes and their troublesome separation from the product mixture all increase process costs.13
Chemocatalytic hydrolysis of cellulose has known several periods of revival and has occasionally been combined with biocatalytic fermentation steps to convert sugars to secondary chemicals. Industrially most important is the hydrolysis by concentrated or diluted mineral acids, predominantly by sulfuric acid. Typically, 1% of sulfuric acid is used at elevated pressure (>2.0 MPa) and temperature (>493 K). An advantage of this process over enzymatic hydrolysis is the high hydrolysis rate, with residence times of the order of several seconds.23 However, under the conditions mentioned, glucose is significantly degraded, reducing yields to about 60%.23,25,26 Moreover, some of the degradation products of glucose function as enzyme inhibitors in subsequent fermentation steps.27 Processes operating with concentrated (60–90%) sulfuric acid achieve glucose yields up to 87%. Indeed, under such conditions, the cellulose feed will be decrystallised, allowing the use of milder process conditions (>373 K).28,29 Large-scale application of this process is complicated by the recycling cost of the acid, reactor corrosion and formation of large amounts of neutralisation waste.
Recent research has been focusing on the development of solid acid catalytic systems for cellulose hydrolysis. Obviously, active solid catalysts could show advantages in process design, operation and cost reduction. Most significantly, after complete feed conversion, the solid nature of the catalysts will eliminate the need for catalyst separation from the liquid product solution. Furthermore, reactors will not be corroded, while acid neutralisation is not needed and neutralized waste is not produced, making the process more environmentally benign and sustainable.
Of course, solid acids have both their own advantages and disadvantages. Solid acids, e.g.zeolites, sulfonated zirconia and sulfonated carbons, can carry a high concentration of strong Brønsted acid sites, creating a very acidic environment in the catalyst pores or close to the catalyst surface. Provided the solids show high intragranular affinity for polysaccharides or related compounds, a very efficient catalyst system for cellulose hydrolysis will be available. As heterogeneous catalysts can be designed to carry multiple types of catalytic sites in close proximity, efficient one-pot multi-reaction pathways can be envisaged for cellulose conversion.
However, several obstacles must be overcome. Firstly, as water is the preferred solvent, several parameters affecting the acidity of the catalytic solid should be taken into account. Typically, hot water between 373 and 523 K can be very detrimental to many solid acids, leading to leaching of sites, loss of surface area or collapse of pore architecture. Most importantly, a critical hurdle when applying solid acids in cellulose hydrolysis consists in the insoluble nature of cellulose, excluding intensive contact between the β-1,4-glycosidic bonds and the intragranular catalyst acidity. Only acid sites at the external surface of a catalyst granule have a chance of occasional contact with the solid substrate. In the case of acid zeolites, most of the acid sites are located in a micropore architecture, inaccessible to cellulose chains. For this reason zeolites have generally shown inferior performance in cellulose hydrolysis,30,47 in contrast to their outstanding performance in the hydrolytic hydrogenation of starch (vide infra), which becomes soluble at elevated temperatures.31 This shows that besides acid strength and density, and glucan sorption affinity, textural features of the support critically determine catalyst success. On the other hand, the quality of the contact between cellulose and the solid catalyst can be improved by cellulose pretreatment to reduce particle size and/or crystallinity. Ball milling has proven to be an especially effective lab-scale pretreatment technique. It has the added advantage of avoiding possible contamination of the cellulose substrate, unlike cellulose pretreatment techniques using e.g. concentrated mineral acids. We also note that cellulose streams from biomass fractionation units and the paper industry are often much less crystalline than cellulose substrates used in research.
In Fig. 6 several recent reports from the vast domain of cellulose hydrolysis studies are summarised. As the figure shows, most successful recent systems are based on sulfonated carriers. This list is by no means exhaustive, but is meant to show the state of the art and the influence of the main factors in the efficient chemocatalytic hydrolysis of cellulose.
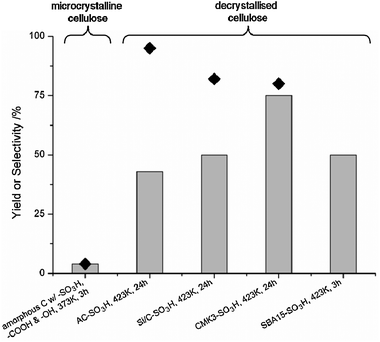 | ||
| Fig. 6 Examples of cellulose hydrolysis over solid acids. Bars: glucose yield, ◆: glucose selectivity. | ||
Onda et al. described the hydrolysis of ball-milled cellulose over a sulfonated activated carbon catalyst (AC-SO3H in Fig. 6).32 Despite its insoluble nature, the system compares favorably to a 0.01 M H2SO4 medium, which was attributed to an intimate affinity of the carbon substrate for glucans. The catalyst also showed excellent reusability. On the other hand, it was shown that even when using ball-milled cellulose, a cellulose fraction remains unconverted with the AC-SO3H catalyst at this relatively low temperature of 423 K. Other authors have further elaborated the use of glucan-sorbing carbon materials as efficient carriers for cellulose hydrolysis catalysts. Suganuma et al. (Fig. 6) were able to convert cellulose using an amorphous carbon, bearing acidic SO3H, COOH and OH groups, prepared by carbonisation of cellulose.33 This catalyst was capable of fully converting even microcrystalline cellulose at only 373 K. This extraordinary activity was ascribed to a high β-1,4-glucan affinity and a high hydration tolerance of the –SO3H acid groups.34 Although the system yielded 60–70% β-1,4-glucans, surprisingly no full depolymerisation to glucose was observed, perhaps due to a preference for the conversion of long cellulose chains over oligomers. A similar preference has been observed for inulin hydrolysis over an acidic carbon catalyst.35
Another recent catalytic system that takes advantage of glucan sorption effects is based on sulfonated silica-carbon composites (Fig. 6).30 The silica–carbon support was prepared from a co-carbonisation of TEOS, Pluronic F127 and sucrose and was then sulfonated. The catalyst was shown to be stable in sequential runs. A correlation was shown to exist between the turnover frequency (TOF) of the acid groups and the silica/carbon ratio of the catalyst, suggesting glucan sorption to occur primarily on the silica surface. The relatively high selectivity for glucose further supports this conclusion, as adsorbed glucans are expected to easily react further to glucose. A final promising carbon support for cellulose hydrolysis is the ordered mesoporous carbon CMK-3. It was recently shown to even exhibit hydrolysis activity without sulfonation, yielding over 20% glucose at 503 K.36 Pang et al. used a sulfonated CMK-3 catalyst, CMK3-SO3H (Fig. 6), with high surface area and high acid density, yielding over 75% glucose at full cellulose conversion.37
These results show that a highly active catalytic system can be developed by taking advantage of the glucan affinity of carbons. Furthermore, it is clear that strong acid sites like –SO3H can selectively form glucose when present in sufficiently high density. Finally, mesoporosity and a high surface area are needed to allow an efficient contact between the acid sites and the substrate, increasing the catalyst activity.
Recently, Kobayashi et al. proposed for the hydrolysis of ball-milled cellulose the use of transition metal-based catalysts, viz. Ru/CMK-3.36 A high reaction temperature (503 K) was applied, leading to a very fast conversion (68% after 1 min), at the expense however of glucose selectivity (50%). The CMK-3 carbon support was shown to hydrolyse cellulose mainly to oligosaccharides, while Ru catalysed hydrolysis of oligosaccharides to glucose. More research is needed to understand the nature of the active sites on the carbon support or the mechanism of the splitting of the glycosidic bond over a metal (cation) catalyst in the absence of H2.
Finally, attempts are made to separate via novel ways the catalyst from the reaction solution. For example, a sulfonated SBA-15 (mesoporous silica) was developed with magnetite (Fe3O4) nanoparticles incorporated during synthesis (Fig. 6, ‘SBA15-SO3H’).38 The nanoparticles made the resulting material magnetic enough to separate from solution with a permanent magnet. Unfortunately, as no conversion data were published, it is difficult to assess the system selectivity for glucose and compare it to the previously mentioned carbon-based catalyst supports.
Though it is beyond the scope of this article to provide an exhaustive overview of all recent research on chemocatalytic cellulose hydrolysis, it is noteworthy that several alternative approaches to this reaction are being explored. For example, impressive results have been achieved by the (soluble) heteropoly acids H4SiW12O40 and H3PW12O40 and transition metal salts thereof.39,40 Also, microwave radiation has been shown to be a powerful disruptor of cellulose crystals, resulting in a faster hydrolysis.41 Finally, by mechanically grinding cellulose and delaminated kaolinite together for 3 h, up to 84% solubilisation was achieved, though the soluble products were not fully analysed.42
In this section, we have tried to convince the reader of the great potential of chemocatalytic cellulose hydrolysis. However, though it can mitigate or eliminate many of the problems of biocatalytic and homogeneous chemocatalytic processes, it comes with its own set of problems and impediments. The most important one is the fundamental difficulty of contacting a solid substrate and a solid catalyst. By pretreating the cellulose feed and specifically designing a high-surface, water tolerant catalyst exhibiting high glucan affinity and high density of accessible strongly acidic sites, economic use of cellulose as an alternative energy and carbon source may be feasible.
From cellulose to platform chemicals
To accelerate cellulose conversion, increasing the reaction temperature (>423 K) is very effective. However, as mentioned before, this has the added effect of decreasing the process selectivity due to glucose degradation. Therefore, several researchers have combined cellulose hydrolysis with another reaction that converts glucose into more thermostable, value-added compounds. These processes are the subject of this section.An overview of most common pathways for cellulose conversion to platform chemicals is given in Scheme 1.
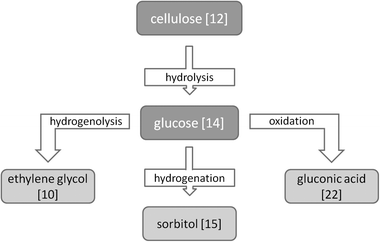 | ||
| Scheme 1 Overview of the most common pathways for cellulose conversion to platform chemicals. | ||
Cellulose to hexitolsvia hydrolytic hydrogenation
Out of the multifunctional catalytic pathways, hydrolytic hydrogenation has been most extensively studied (Scheme 1). Here, acid hydrolysis is coupled to a hydrogenation reaction under hydrogen pressure that converts glucose to sorbitol. Due to the elevated reaction temperature, isomerisation to mannitol [16] almost always occurs and in most systems, the acid groups also partially dehydrate the two alditols, resulting in sorbitan [17] isomers and sometimes even in isosorbide [18] (Fig. 7).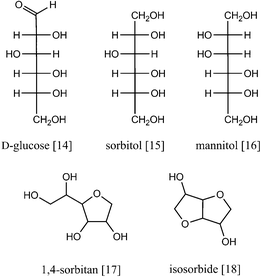 | ||
| Fig. 7 Hexoses and hexitols commonly formed in hydrolytic hydrogenation of cellulose. | ||
Almost all of the catalytic systems show similar components: (1) a dilute soluble acid or an acid support and (2) a transition metal catalyst, often Pt or Ru, that is capable of a fast hexose hydrogenation.31,35,43–45 To achieve a high overall selectivity to hexitols, obviously a high selectivity of each catalytic site for its specific reaction is imperative. In addition, the balance between the acid sites and the hydrogenation sites is of utmost importance. Should the acid sites dominate over the metal sites, then glucose will be formed faster than it can be hydrogenated. This free glucose will start to degrade into soluble and insoluble (polymeric) byproducts due to the high reaction temperature. When the metal sites dominate, metal-catalysed side reactions like hydrogenolysis will become important. Hydrogenation and hydrogenolysis are competitive reactions on the metal surface, so when the flow of glucose to the metal surface is insufficient, i.e. when hydrolysis is too slow, hydrogenolysis will take over and hexitols will be broken into smaller molecules through C–O and C–C bond breaking, resulting in ethylene glycol [10], propanediol [19], glycerol [20], erythritol [21], etc. (Fig. 8).46 Higher H2 pressures in general suppress hydrogenolysis and favor a high hexitol selectivity.
 | ||
| Fig. 8 Common hydrogenolysis byproducts in hydrolytic hydrogenation of cellulose. | ||
Despite the added difficulty of balancing multiple reactions on a catalyst surface, combining hydrolysis and hydrogenation can be a useful strategy since at higher reaction temperature the rate of cellulose conversion is enhanced very significantly. For example, it was shown that microcrystalline cellulose is solubilized up to 48% at 503 K in the absence of catalyst,53 whereas 35% solubilisation was observed at 463 K,48 in both cases almost completely to cellulose oligomers (cello-oligomers). This shows the important effect of heated water as a depolymerising factor in cellulose conversions. Fig. 9 lists some of the most interesting recent results in this field. As with the previous results for cellulose hydrolysis, a fair comparison of the results is sometimes complicated because of differences in substrate, crystallinity and pretreatment, as well as differences in process conditions.
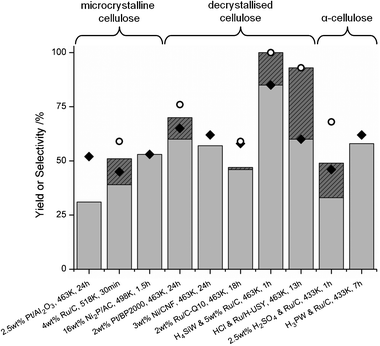 | ||
| Fig. 9 Hydrolytic hydrogenation of cellulose. Light grey bars show alditol yield, full bars show total hexitol yield, if published. ◆: alditol selectivity (for Pt/Al2O3 estimated with data from ref. 48), ○: hexitol selectivity. | ||
The recent revival of the hydrolytic hydrogenation of cellulose started with a contribution by Fukuoka and Dhepe (Fig. 9, ‘Pt/Al2O3’).47 It was established that Pt and Ru were the most interesting noble metals for this reaction, with Pt/γ-Al2O3 giving the best results. Although data on cellulose conversion were not included, Jollet et al. showed that the catalyst is active in the hydrolysis of cellulose, and that it is mainly responsible for converting the soluble cello-oligomers to glucose.48 However, about 30% microcrystalline cellulose is too resistant to be converted by the catalyst, even after prolonged reaction time. The nature of the hydrolysis sites on the catalyst remains unclear. The acid sites of γ-Al2O3 can be largely excluded, because cellulose solubilisation barely increased over pure γ-Al2O3 (41% vs. 35%) and no extra glucose was formed. Therefore, the metallic function must be responsible for the increased hydrolysis activity, either by contributing to the H+ pool by heterolytic cleavage of H2 with subsequent spillover onto the support, or by promotion of hydride transfer steps. Lastly, Fukuoka's work shows that classical solid acid supports, like H-USY and SiO2–Al2O3 loaded with Pt, are less performant than Pt/γ-Al2O3, even though these supports are all much more active in cellulose hydrolysis.
Based on the well-known superior activity and selectivity of Ru/C for glucose hydrogenation, its performance was also investigated in the hydrolytic hydrogenation of cellulose at high temperature (Fig. 9, ‘Ru/C’).49 Since conversion was not dependent on the amount of catalyst present, hydrolysis by solid acid sites on the carbon support was excluded. Instead, the hydrolysis activity was entirely ascribed to reversible generation of H+ ions in hot water (>440 K). In contrast to Pt, Ru seems to be exclusively responsible for hydrogenation of glucose. Similar results were obtained with Ru clusters supported on carbon nanotubes at lower temperature (458 K).50 The carbon nanotubes most likely exhibit some hydrolysis activity, enabling the use of a lower reaction temperature.
Recently, Kobayashi et al. investigated several metal catalysts on carbon and oxide supports (Fig. 9).51 A Pt/BP2000 (carbon black) catalyst proved reusable during three consecutive runs and showed better selectivity than comparable Ru, Rh, Ir and Pd catalysts. The BP2000 carbon support was shown to be superior to both other carbon supports and Al2O3, TiO2 and ZrO2 supports. In this system the presence of Pt also increases cellulose conversion, indicating that Pt is involved in reactions beyond the hydrogenation of glucose.
Although in the above examples noble metals have proven to be effective, cost considerations favor the use of cheaper metals such as nickel. Ding et al. (Fig. 9, ‘Ni2P/AC’) described the use of nickel phosphide catalysts, reaching full conversion with a 16 wt% Ni2P/AC catalyst.52 Unfortunately, recyclability of the catalyst is problematic due to leaching, a common problem with Ni under hydrothermal conditions. On the other hand, Van de Vyver et al. (Fig. 9, ‘Ni/CNF’) reported on a carbon nanofiber (CNF) supported Ni catalyst that was stable for at least three consecutive runs.53 In this catalyst, CNFs with Ni particles at their tip protrude from a central γ-Al2O3 particle. This design could enable the entanglement of cellulose particles and CNFs, thereby increasing the accessibility of the Ni particles. Furthermore, the Ni particles show an extraordinary selectivity towards glucose hydrogenation, whereas Ni is traditionally known to easily cleave C–C and C–O bonds.
In the hydrolytic hydrogenation of cellulose, the need for an external H2 pressure presents a clear disadvantage. The use of an alternative hydrogen source can therefore make the process more environmentally friendly. This issue was addressed in another recent contribution by the group of Fukuoka (Fig. 9, ‘Ru/C-Q10’).54 They investigated the use of metal catalysts on several carbon and oxide supports with 2-propanol as a hydrogen source using transfer hydrogenation, producing acetone as a byproduct. Although all the Ru-based catalysts showed an increased conversion relative to a blank reaction, especially carbon supported catalysts also exhibited a good glucose hydrogenation activity. Ru proved superior performance to other carbon supported metals (Ir, Pd, Pt and Au). The active site for the transfer hydrogenation seems to be located on Ru, its presence being dependent on the support. Unfortunately, the Ru/C catalysts were not stable in more than two consecutive catalytic runs. In view of these results other concepts, like the combined aqueous reforming and hydrogenation that was successfully applied to the conversion of glycerol to propanediol without external H2 pressure, are waiting to be explored.72
Also in the hydrolytic hydrogenation of cellulose, the use of soluble acids has been studied. Although this sacrifices the heterogeneous nature of the process it could be industrially interesting, since the high process temperature enables complete conversion with a very low acid concentration. A very low acid concentration diminishes many of the technical difficulties associated with the use of soluble acids, while a fast hydrolysis has significant selectivity benefits. Palkovits et al. (Fig. 9, ‘H2SO4 and Ru/C’) provide an interesting study on the use of a mineral acid and a heterogeneous hydrogenation catalyst for α-cellulose conversion.55aα-Cellulose, the fibrous product of a strong alkali extraction of wood or straw, contains significant amounts of hemicellulose (10–20%) and is significantly less crystalline than microcrystalline cellulose. The authors studied combinations of H2SO4 or H3PO4 with Pt/C, Pd/C and Ru/C. Like Fukuoka and Jollet, they also observed a beneficial influence of the metal catalysts on cellulose conversion and advanced an explanation different from the hydrogen spillover hypothesis. They noted that the reforming activity of the metals follows the sequence Pt > Pd > Ru, which is the same sequence as for the conversion increase. At the same time, the product spectrum shows a shift from C5–C6 products (Ru/C) towards smaller and gaseous products that are the result of C–C and C–O bond breaking (Pt/C). This supports the notion that the conversion increase upon addition of noble metals is not due to increased cellulose hydrolysis but rather direct (hydrogenolytic) cleavage of C–C and C–O bonds in the cellulose chain. Furthermore, Ru exhibited a greatly superior hydrogenation activity whereas reactions with Pt and Pd form mainly glucose as a C6 product. It is worth noting that by substituting H2SO4 in this system for HCl and increasing the reaction temperature to 488 K the double dehydration of sorbitol to isosorbide [18] is stimulated, leading to 50% isosorbide yield from cellulose at full conversion in 6 h.55b The high selectivity for isosorbide formation in aqueous conditions was ascribed to the previously reported promoting effect of Cl− on the dehydration of sorbitol to 1,4-sorbitan [17].51
By using the very acidic heteropoly acids (HPAs) H4SiW12O40 or H3PW12O40 in combination with Ru/C, Geboers et al. were the first to achieve quantitative conversion of cellulose to hexitols (Fig. 9, ‘H4SiW and Ru/C).46 The high activity of the HPAs for cellulose hydrolysis compared to mineral acids leads to a very fast conversion, minimizing the effect of hydrogenolysis reactions over Ru/C. It was suggested that this high hydrolysis activity might be due to a progressive hydrolysis mechanism by the HPAs, for which one glucose unit after another is released from the cellulose chain. By exploiting the dependency of hydrolysis rate on the water concentration reported by Yamaguchi,56 full conversion of 10 wt% aqueous cellulose was shown in 20 min.
More recently, Palkovits et al. applied the same catalytic system to the conversion of α-cellulose under milder conditions (Fig. 9, ‘H3PW and Ru/C’), reaching similar conclusions.57 Care must be taken when surveying their results in Fig. 9: the figure only lists C6 yield and therefore underestimates the true performance of the catalytic system. Using a combination of H4SiW12O40 and Ru/C, an 81% C4–C6 yield was achieved at 99% conversion, showing the contribution of the hemicellulose fraction of α-cellulose. H3PW12O40–Ru/C proved less selective, yielding only 66% C4–C6 at 94% conversion. Additionally, the authors reported the conversion of spruce and achieved 65% C4–C6 yield.
Finally, a system was shown combining bifunctional Ru/H-zeolites and trace amounts of mineral acids for the hydrolytic hydrogenation of cellulose at moderate temperatures (Fig. 9, ‘HCl and Ru/H-USY’).58 Both H-mordenite and H-USY zeolites were shown to be effective for this conversion. The low amount of mineral acid (typically 35 ppm) depolymerizes cellulose into soluble oligomers which are rapidly adsorbed into the zeolite pores, where they are further depolymerized into glucose. This results in a high intraporous glucose concentration, in close proximity to the intraporous Ru particles, suppressing hydrogenolysis on the Ru surface. This leads to a highly selective reaction, yielding up to 93% hexitols in 13 h when converting a 10 wt% feed.
These results show that hydrolytic hydrogenation of cellulose by soluble acids and a solid hydrogenation catalyst can be very fast and selective, though the economic feasibility of such a process remains unproven, and depends on the cost and effectiveness of acid recovery and recirculation.
Cellulose to ethylene glycolvia hydrolytic hydrogenolysis
Besides hydrolytic hydrogenation, several other interesting conversions of cellulose into valuable chemicals have been proposed. Ji et al. reported a remarkable system that converts cellulose in a one-pot aqueous reaction of 30 min at 518 K into 61% ethylene glycol (EG) [10] (Scheme 1).59,60 The catalyst consists of a nickel promoted tungsten carbide on activated carbon (Ni-W2C/AC). Later the work was expanded by using an SBA-15 support61 and a three-dimensional mesoporous carbon support.62 The greater pore accessibility and diffusion in the support enhanced the EG yield up to 75%. This beneficial influence of a mesoporous support structure implies partial cellulose depolymerisation before catalysis on the sites inside the pores occurs, since cellulose particles (about 50 μm) cannot enter mesopores. In view of the high reaction temperature, this degradation is most likely carried out by hot water.Cellulose to gluconic acidvia hydrolytic oxidation
Just as hydrolysis can be combined with a hydrogenation reaction, it could also be combined with an oxidation reaction to produce gluconic acid [22] (Fig. 10).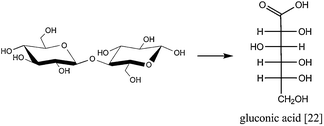 | ||
| Fig. 10 One-pot aqueous hydrolytic oxidation of cellobiose yields gluconic acid.63 | ||
Gluconic acid is an important food and pharmaceutical industry intermediate that is usually produced by enzymatic or chemical oxidation of glucose.63,64 Recently, Tan et al. showed the selective aqueous hydrolytic oxidation of cellobiose, the dimer of cellulose, over a Au/CNT catalyst, achieving 68% yield of gluconic acid at 81% conversion.63 Although chemical glucose oxidation usually occurs in a basic medium, the authors found the reaction to occur without pH control. Cellobiose hydrolysis was effected by acid groups on a HNO3 pretreated CNT support. To extend this process to cellulose conversion, the use of higher reaction temperatures is inevitable, with Au activity and selectivity possibly becoming an issue in these slightly acidic media.
From cellulose to biofuels
In the present petroleum-based economy, a range of fuels and base chemicals can be produced out of a single feedstock. If future technology is to be cellulose-based, the possibility of converting it into a series of commodities would greatly facilitate the transition. Therefore, it is important to note that cellulose conversion is not limited to base chemicals for the chemical industry. In the following paragraphs, recent developments in the conversion of cellulose into biofuels and fine chemicals are discussed.Biphasic cellulose conversion into furanic biofuels
The conversion of cellulose into furanics provides 5-hydroxymethylfurfural and its derivatives, a versatile feedstock not only for the production of polyesters and other plastics, but also diesel-like fuels and pharmaceuticals (Scheme 2). A key point in this conversion is the use of biphasic reaction media to extract the unstable 5-hydroxymethylfurfural before its facile rehydration into levulinic acid and formic acid. | ||
| Scheme 2 Conversion of cellulose into furanic biofuels. | ||
The first step towards furanics production from cellulose was published by Mascal and Nikitin. Cellulose reacted in an aqueous HCl/LiCl solution in a biphasic reactor with 1,2-dichloroethane to extract the products, producing mainly 5-chloromethylfurfural [23] (CMF; Fig. 11).65
CMF itself is not a biofuel candidate because of its Cl content. Therefore, the authors demonstrated the efficient conversion of CMF into ethoxymethylfurfural [24] (EMF) by stirring in ethanol or into 5-methylfurfural [25] (MF) by hydrogenation over PdCl2. EMF, a liquid at room temperature with an energy density comparable to that of gasoline, could be a promising biofuel.66 Since then, Mascal and Nikitin have shown the robustness of their system by expanding feedstocks to waste biomass sources such as wood, cotton, corn stover, newspapers and straw.67,68 The pentose fraction in these substrates yields mainly furfural [26] (Fig. 11). The presence of other components such as lignin did not hinder the process. However, this process would benefit from the use of a less corrosive acid instead of concentrated HCl and from the transition of 1,2-dichloroethane to a less toxic compound.69
Yang and Sen converted several hexoses, pentoses, cellulose and lignocellulose into 2,5-dimethyltetrahydrofuran [27] (DMTHF; from hexoses) and 2-methyltetrahydrofuran [28] (MTHF; from pentoses) for biofuels (Fig. 12).70a
 | ||
| Fig. 12 THF derivatives produced from cellulose.70a | ||
They used an aqueous solution of HI containing a soluble Rh catalyst under H2 pressure. By adding an organic solvent to the mixture, viz.benzene, selectivity was improved, resulting in 54% yield. The process has the advantage of being performed under relatively mild conditions (433 K, 2 MPa H2, 16 h). As a biofuel, DMTHF has a good energy density (8.8 kWh L−1) and a boiling point of 363–365 K. Very recently, the authors also showed the conversion of cellulose to MF, a useful intermediate, over a Pd/C catalyst in aqueous HI under H2 pressure, again with benzene as the organic phase, with 42% yield.70b
Obviously, large-scale application of these processes would benefit from the development of a heterogeneous variant and finding an alternative organic phase instead of benzene.
Cellulose conversion into valeric biofuels
Derivatives of γ-valerolactone (GVL) [11] are an alternative class of promising biofuels that can be produced from cellulose (Scheme 3).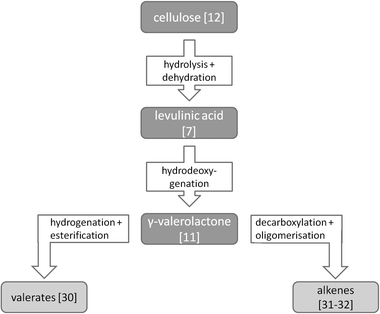 | ||
| Scheme 3 Cellulose conversion into valeric biofuels. | ||
Lange et al. proposed a process comprising an acid hydrolysis of cellulose to glucose and subsequent dehydration to levulinic acid [7], which is then converted to GVL and further hydrogenated to valeric acid [29].71 This can then be esterified to alkyl mono- and divalerates [30] (Fig. 13). Using Pt-loaded SiO2-bound H-ZSM-5 as a catalyst, the authors could not only convert GVL into valeric acid, but also directly into pentyl valerate, a renewable diesel additive. Higher energy density additives, such as di- and tri-valerates, can be produced by valeric acid esterification with EG [10], propylene glycol [19] or glycerol [20], products that can also be produced from cellulose and glycerol.72,59–62
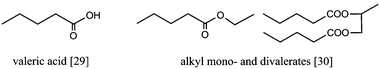 | ||
| Fig. 13 Hydrogenation of GVL yields valeric acid, which can be esterified to promising biofuels.71 | ||
It is noteworthy that the conversion of cellulose into levulinic acid can be performed with H2SO4, but would benefit from the development of highly acidic, stable heterogeneous catalysts. At present, the conversion of cellulose into levulinic acid over heterogeneous catalysts produces only about 10% levulinic acid.73
Further reinforcing the potential of GVL as a biofuel platform chemical, Bond et al. reported its conversion to liquid C8+alkenes for fuel applications.74,75GVL was decarboxylated to butenes [31] over SiO2–Al2O3. In the following step, these butenes were oligomerised to longer alkenes [32] over acid catalysts like H-ZSM-5 or Amberlyst 70 (Fig. 14). The fact that a CO2 molecule is expelled for every butene molecule produced sets the maximum attainable carbon efficiency at 83%. A major advantage is that the process has no need for external H2.
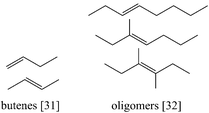 | ||
| Fig. 14 Butenes and their oligomers can be produced from GVL in an integrated process without the need for external H2.73 | ||
From cellulose to fine chemicals
As a last process showing the broad potential of cellulose as a renewable source of chemicals and fuels, we discuss the one-pot transformation of cellulose into alkyl glycosides. Methyl glycosides [33], important intermediates for the production of fine chemicals, can be produced by simply reacting cellulose over acid catalysts in methanol at 468 K (Fig. 15).76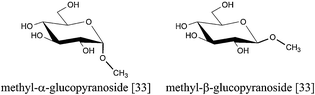 | ||
| Fig. 15 Catalytic conversion of cellulose into methyl glycosides. | ||
Especially H2SO4, H4SiW12O40 and H3PW12O40 were successful catalysts, yielding up to 50–60% methyl glycosides. Remarkably, the authors observed full conversion of microcrystalline cellulose after 1 h, showing the potential of small alcohols as an alternative solvent to water in chemocatalytic cellulose conversions.
Pitfalls, summary and outlook
So far in this contribution, we have dedicated some space to the physico-chemical structure of cellulose and to a brief literature overview of various chemocatalytic cellulose conversions, allowing the reader to understand the challenges in developing an active and selective catalytic process and to convey the economic opportunities of such processes in the near future. Many new strategies have been explored to address the mass transfer limitations and difficult solid–solid interactions that govern cellulose conversion. However, typical timescales for many heterogeneous systems are still in the order of days, leading to limited space time yields. To allow the large-scale use of cellulose as an industrial feedstock, issues concerning cellulose reactivity and catalyst effectivity will need to be addressed in the following years.In our opinion, the field of chemocatalytic cellulose conversion is at a turning point. In the past years, researchers have been focused mainly on developing new reaction pathways and processes and on gaining insights in the main parameters that govern catalyst performance. Among the main results of this period are (1) the demonstration by Fukuoka and Dhepe that fully heterogeneous conversion of cellulose is possible, (2) realising the hydrolysing power of heated water, (3) showing that noble metals can influence cellulose conversion, even if the mechanism is not yet fully understood, (4) synthesis of cheap, non-noble metal catalysts such as Ni that selectively hydrogenate sugars, (5) depolymerisation of highly crystalline cellulose feeds at low reaction temperature over acidic carbon surfaces and (6) the expansion of catalytic systems to produce a myriad of chemicals and fuels from cellulose. However, as fruitful as this period has been, it suffered from several growing pains, such as incomplete carbon balances, especially in multi-reaction pathways, and an inadequate characterisation of the substrates. Concerning the problem of incomplete carbon balances, much has already improved in the last few years. The earliest studies only reported yields of desired products, without any mention of byproducts or cellulose conversion. Since then, researchers have started reporting conversion based on gravimetrical analysis of the solids in the reaction mixture or on analysis of the dissolved organic carbon (DOC) in the reaction supernatants. In our own work, we have found a close agreement between the results of both techniques, but the following points should be considered: (1) the gravimetric method prevents any sampling during the reaction, because this results in the loss of solids, meaning that kinetic information is lost, (2) if the catalyst is unstable and partly dissolves, the gravimetric method becomes unusable, (3) if gas-phase products are formed, DOC analysis of the liquid phase will underestimate conversion, (4) it is known that in some reactions, the products undergo acid-catalysed condensation and dehydration reactions, forming insoluble humins. If this is the case, both methods underestimate conversion. Finally, both methods actually measure cellulose solubilisation. Cellulose depolymerisation to soluble oligomers thus registers as full conversion, though no glucose is formed.
On the other end of the carbon balance are reported products and byproducts. Since the chemical and physical properties of the products of cellulose reactions are very similar, full separation and identification of the product spectrum is a complicated matter. Often, several techniques have to be combined and even then MS identification is indispensable. However, even MS identification is not always reliable because of the similarity of the compounds. Nevertheless, a better understanding of the relation between the properties of the catalytic system and the obtained products is of high importance. We would like to note here recent work that shows a very detailed product analysis.55a A final remark concerns the definition of product yields. Yields have been reported on a mass basis, dividing the mass of a formed product by the total mass of cellulose introduced in the reactor. This can be misleading for many products, differences for instance arising from water molecules added during the reaction. It is preferable to report yields on a carbon basis, calculating the molar amount of carbon represented in a product and dividing by the total molar amount of carbon introduced in the reactor.
The problem of inadequate cellulose characterisation is more difficult to address. To allow a fair comparison between catalysts, it is crucial that information is provided on the substrate regarding its degradability. The main parameter to consider here is the ‘accessibility’ of the glycosidic bonds. It is however not clear which measurable parameters would provide a good indication for accessibility. Some parameters are clearly crucial, like crystallinity, crystal morphology, and particle porosity and size. Their relative importance is much more difficult to judge. Does crystallinity govern degradability, or is particle size equally important? It has been proposed that some catalysts degrade cellulose in an ablative manner, hydrolysing the less ordered outer surface of the particles. Internal chains become surface chains and partly lose their ordering, so they can be hydrolysed.77 In this case, particle surface area is much more important than crystallinity. This shows that the relative importance of the parameters likely depends on the nature of the catalyst, viz. an enzyme, a solid acid or a water-soluble acid. For chemocatalysts working on pure cellulose in hot water, crystallinity is likely to be the main characteristic, but when using lignocellulosic biomass as a substrate, the lignin and hemicellulose content also becomes important, since their presence brings with it added forms of protection against cellulose degradation. The matter is greatly complicated by the fact that the different parameters are very difficult to examine independently. For example, removal of lignin and hemicellulose influences the crystallinity and particle dimensions of the cellulose that remains, cellulose milling reduces its crystallinity but also its particle size, ammonia explosion influences porosity… This has been sidestepped by the use of microcrystalline Avicel PH-101 as an implicit benchmark substrate in many publications. However, therein lies an important problem. Avicel PH-101 is highly crystalline and poses a significant challenge to catalysts. On the other hand, other factors besides crystallinity become important when converting industrial lignocellulose feeds, which are usually significantly less crystalline than Avicel PH-101, but are also less pure. This means that potentially very powerful systems are discarded because they are unable to degrade the highly crystalline Avicel, which is ultimately not the goal. This is a strong argument to include crude lignocellulosic biomass in catalytic and characterisation studies. Unfortunately the physicochemical characterisation of these substrates is even more complicated than that of pure cellulose.
In fact, with cellulose, as with any biomass substrate, even the measurement of a single, seemingly well defined property such as crystallinity lacks standardisation. The problem has been described in a contribution by Park et al.77 They calculated a cellulose crystallinity index (CI) by several methods, three based on XRD data and one based on 13C CP MAS NMR data. The first method, the ‘XRD peak height method’, is the easiest to use and is most popular (Fig. 16).
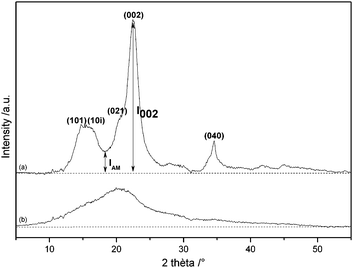 | ||
| Fig. 16 X-Ray diffractogram of (a) Avicel PH-101 and (b) Avicel after 24 h ball milling.46,53 Annotations after ref. 77. | ||
The CI is calculated from the difference in intensity between the crystalline (002) reflection and that of amorphous material (I002-IAM), divided by the intensity of the crystalline (002) reflection. In another approach, the XRD spectrum is deconvoluted into crystalline peaks and one amorphous peak. In the third method, an additional diffractogram of purely amorphous cellulose is measured and subtracted from the original one. The differential diffractogram is then regarded as the contribution of crystallinity and is divided by the area of the original diffractogram.
In the last method, based on NMR data (Fig. 17), the CI is calculated by separating the C4 region of the spectrum into crystalline (C4c) and amorphous (C4a) peaks and dividing the area of the crystalline peak by the total area of the C4 region. Using these techniques on 8 cellulose samples and a survey of literature data on Avicel PH-101, the authors showed that (1) the XRD peak height method gives a much higher CI than the other methods, (2) CI values based on the same technique for the same type of cellulose published by different authors differ by as much 25% and (3) the techniques all classify the cellulose samples in generally the same order. Furthermore, all of the XRD-based techniques lose their reliability for more amorphous samples. The main problems are (1) the absence of a completely amorphous reference sample, (2) the fact that cellulose reflections are broad and ill defined, even for crystalline cellulose forms and (3) peak broadening and shifting with decreasing particle size, as illustrated in Fig. 16. Therefore, crystallinity indices should be used with caution. Indeed, CI values for Avicel varied between 70 and 92% when performed by different authors. We argue that cellulose characterisation should at least consist of XRD, 13C NMR and FTIR data and that CI values should only be used to relatively classify samples within a study. When reported together, these techniques give the reader an indication about the properties of the used substrate.
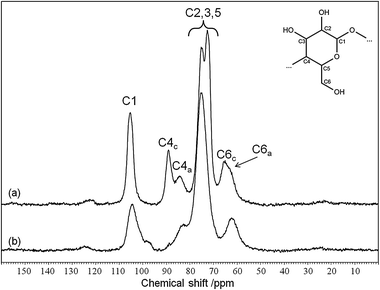 | ||
| Fig. 17 13C CP MAS NMR spectrum of (a) Avicel PH-101 and (b) Avicel after 24 h ball milling.46,53 Annotations after ref. 77. | ||
We believe that, now that the groundwork for successful aqueous cellulose conversion has been laid, the field might benefit most from systematic studies to further elucidate the fundamental factors that govern catalyst performance. For example, the rate determining step in most cellulose conversion processes is solubilisation. Conceptually, it is clear that a high acid density, β-glucan affinity, good accessibility of strong acid sites and a high surface area should increase the performance of a hydrolysis catalyst. However, beyond this conceptual image, not much is known. Systematic studies to investigate the individual importance of the different factors would be highly important in this field.
We would also like to note that the aqueous hydrolysis of cellulose can still be enigmatic at times. For example, Jollet et al. showed that the dissolution of cellulose into oligomers in hot water (423–463 K) reaches a plateau after about 100 h reaction, instead of proceeding to full solubilisation.48 Furthermore, these oligomers are not further depolymerised into glucose regardless of reaction time, and solubilisation seems to occur homogeneously throughout the cellulose particle, instead of preferentially on the amorphous regions. Finally and most remarkably, cellulose solubilisation seems to not only be dependent on water temperature but also on cellulose concentration: the higher the initial amount of cellulose loaded into the reactor, the higher the absolute amount of cellulose converted to soluble oligomers, excluding the possibility of saturation of the reaction mixture as explanation for the plateau. No explanation for this phenomenon has been given. In another example, Yamaguchi et al. showed that the speed of cellulose hydrolysis over a solid acid catalyst shows an optimum for a certain, presumably catalyst-dependent amount of water.56 Less water slows hydrolysis because water is a reactant, but too much water also slows hydrolysis, supposedly because of hydration of the acid sites, which remains to be proven. These examples show that the role of water in cellulose hydrolysis is also not fully understood. This should be further investigated and if possible exploited to develop highly active and selective catalysts for large-scale industrial applications.
Besides the investigations suggested above, there are some key areas that could trigger significant progress in the field in the next few years. The first one is the further development of biomass pretreatment techniques. Ball milling has proven extremely successful on a lab scale,30,32,36,37 allowing reduction of cellulose crystallinity and crystal size without introducing chemical contaminants into the substrate. However, it is too energy intensive for industrial application. A range of other techniques exist, each with their own advantages and drawbacks.3a Pretreatment remains one of the most important cost contributors in lignocellulosic conversion. A cheap, effective and environmentally benign pretreatment procedure would be an important breakthrough. Ionic liquids show considerable potential in this regard. Though still rather expensive, some ionic liquids are able to dissolve up to 25 wt% of cellulose,78 allowing rapid and selective conversion. Unfortunately, polar molecules such as glucose and polyols are difficult to separate from the solvent. A solution is to stop depolymerisation at the formation of cello-oligomers. These can be easily separated by precipitation by adding water and can be further converted in aqueous medium.79 However, cost-effective integration of ionic liquids in a lignocellulose fractionation strategy is not trivial.
Advancements in the deoxygenation of biomass-derived compounds such as sugars, hexitols and lignin monomers will allow the production of an even broader product spectrum from biomass. An exciting example was recently reported by Li and Huber, who showed the conversion of sorbitol into hexane over a Pt/SiO2–Al2O3 catalyst.80 Coupling this chemistry to the aqueous phase reforming of sorbitol as described by Huber et al. to produce H2 for the process could result in a truly green production of alkanes.81 With a similar strategy, glycerol was selectively converted to propane diol over Pt/NaY without the need for external H2.72
Another advancement could be the synthesis of a heterogeneous heteropoly acid-based catalyst for cellulose conversion. Heteropoly acids have proven to be extremely active and selective in several cellulose conversions,39,40,46,57 but their water solubility prevents them from being an industrially feasible catalyst. Several authors reported the incorporation of HPAs into materials such as zirconia, activated carbon, MCM-41, silica or MOFs.82 In water however, these materials exhibit significant leaching of the HPA, even at modest temperatures. Another approach might be the use of insoluble salts of HPAs.83 If this issue can be solved, heteropoly acids could be very powerful hydrolysis catalysts.
Finally, it is interesting to note that the range of possible cellulose-derived products is still expanding. For example, an interesting contribution by Chheda et al. shows a system that is capable of converting glucose and cellobiose with high selectivity to HMF [8], which was previously mainly done starting from fructose.84 This breakthrough might open up the way for the eventual production of HMF from cellulose and shows that the role that cellulose could play as a renewable feedstock for chemicals and fuels is still growing rapidly.
We have shown the effort that is being devoted to converting cellulosic biomass to base and intermediate chemicals and biofuels. Despite the omission of several other examples in the interest of space, we believe that the vast scope of the conversions that have been discussed shows that cellulose could indeed be part of the renewable feedstock of the future. On the other hand, we have tried to be both faithful and fair when discussing literature, not only showing what is possible in the field of chemocatalytic cellulose conversion, but also what is not. Cellulose is still a recalcitrant molecule and its aqueous conversion is no simple matter, requiring the development of a whole new form of solid–solid catalysis. It is for this reason that the recent progress in the field is both astonishing and highly encouraging. In the next decade, we expect to see several processes that employ highly advanced catalytic systems and pretreatment methods to convert lignocellulosic biomass from a wide range of sources into a whole array of fuels, base chemicals and fine chemicals. A renewable and more sustainable economy might be happening right now.
Notes and references
- Climate Change 2007, Synthesis Report, ed. R. Pachauri and A. Reisinger, IPCC, Geneva, Switzerland Search PubMed.
- S. Herrera, Nat. Biotechnol., 2006, 24(7), 755–760 CrossRef CAS.
- (a) D. Hayes, Catal. Today, 2009, 145, 138–151 CrossRef CAS; (b) M. Balat, Energy Convers. Manage., 2011, 52, 858–875 CrossRef CAS.
- M. Imhoff, L. Bounoua, T. Ricketts, C. Loucks, R. Harriss and W. Lawrence, Nature, 2004, 429, 870–873 CrossRef CAS.
- G. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS; R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610–626 Search PubMed.
- P. Mäki-Arvela, B. Holmbom, T. Salmi and D. Murzin, Catal. Rev. Sci. Eng., 2007, 49(3), 197–340 Search PubMed.
- L. Lynd, C. Wyman and T. Gerngross, Biotechnol. Prog., 1999, 15(5), 777–793 CrossRef CAS.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- M. Jarvis, Nature, 2003, 426, 611–612 CrossRef CAS.
- P. Dais and A. Perlin, Carbohydr. Res., 1982, 100, 103–116 CrossRef CAS.
- Y. Nishiyama, H. Chanzy and P. Langan, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS; Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan, J. Am. Chem. Soc., 2003, 125, 14300–14306 CrossRef.
- A. Gardiner and A. Sarko, Can. J. Chem., 1985, 63, 173–180 CrossRef CAS; J. Hayashi, A. Sufoka, J. Ohkita and S. Watanabe, J. Polym. Sci., Polym. Lett. Ed., 1975, 13, 23–27 CrossRef.
- P. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS.
- K. Mazeau, Cellulose, 2005, 12, 339–349 CrossRef CAS.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1110 CrossRef CAS; A. O’Sullivan, Cellulose, 1997, 4, 173–207 CrossRef.
- G. Huber and J. Dumesic, Catal. Today, 2006, 111(1–2), 119–132 CrossRef CAS.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS.
- J. Shabtai, W. Zmierczak and E. Chornet, US, 5959167, 1999 CrossRef CAS; N. Yan, C. Zhao, P. Dyson, C. Wang, L. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS; J. Zakzeski, P. Bruijnincx, A. Jongerius and B. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef.
- O. Bobleter, in Polysaccharides, ed. S. Dumitriu, 2nd edn, 2005, Marcel Dekker, New York, pp. 893–937 Search PubMed.
- J. Saeman, Ind. Eng. Chem. Res., 1945, 37, 43–52 CrossRef CAS.
- N. Mosier, C. Ladisch and M. Ladisch, Biotechnol. Bioeng., 2002, 79, 610–618 CrossRef CAS.
- B. Girisuta, L. Janssen and H. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- Q. Xiang and Y. Lee, Appl. Biochem. Biotechnol., 2003, 107, 505–514 CrossRef.
- M. Balat, Energy Convers. Manage., 2010, 52(2), 858–875 CrossRef.
- C. Wyman, R. Decker, M. Himmel, W. Brady, C. Skopec and L. Viikari, in Polysaccharides, ed. S. Dumitriu, 2nd Edn, 2005, Marcel Dekker, New York, pp. 995–1033 Search PubMed.
- H. Zhao, J. Hollady, Y. Wang, J. White and Z. Zhang, J. Biobased Mater. Bioenergy, 2007, 1, 210–214 CrossRef.
- J. Lange, Biofuels, Bioprod. Biorefin., 2007, 1, 39–48 CrossRef CAS.
- W. Farone and J. Cuzens, WO, 96/40970, Arkenol Inc., 1996 Search PubMed.
- D. Hayes, Catal. Today, 2009, 145(1–2), 138–151 CrossRef CAS.
- S. Van de Vyver, P. Li, J. Geboers, H. Schepers, F. de Clippel, C. Gommes, B. Goderis, P. Jacobs and B. Sels, Green Chem., 2010, 12(9), 1560–1563 RSC.
- P. Jacobs and H. Hinnekens, EP, 0329923, Synfina-Oleofina, 1989 Search PubMed.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC; A. Onda, T. Ochi and K. Yanagisawa, Top. Catal., 2009, 52, 801–807 CrossRef CAS.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS.
- M. Kitano, D. Yamaguchi, S. Suganuma, K. Nakajima, H. Kato, S. Hayashi and M. Hara, Langmuir, 2009, 25, 5068–5075 CrossRef CAS.
- A. Heinen, J. Peters and H. van Bekkum, Carbohydr. Res., 2001, 330, 381–390 CrossRef CAS.
- H. Kobayashi, T. Komanoya, K. Hara and A. Fukuoka, ChemSusChem, 2010, 3, 440–443 CrossRef CAS.
- J. Pang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2010, 46, 6935–6937 RSC.
- D. Lai, L. Deng, J. Li, B. Liao, Q. Guo and Y. Fu, ChemSusChem, 2011, 4, 55–58 CrossRef CAS.
- K. Shimizu, H. Furukawa, N. Kobayashi, Y. Itaya and A. Satsuma, Green Chem., 2009, 11, 1627–1632 RSC.
- J. Tian, J. Wang, S. Zhao, C. Jiang, X. Zhang and X. Wang, Cellulose, 2010, 17, 587–594 CrossRef CAS.
- Y. Wu, Z. Fu, D. Yin, Q. Xu, F. Liu, C. Lu and L. Mao, Green Chem., 2010, 12, 696–700 RSC.
- S. Hick, C. Griebel, D. Restrepo, J. Truitt, E. Buker, C. Bylda and R. Blair, Green Chem., 2010, 12, 468–474 RSC.
- A. Balandin, N. Vasyunina, S. Chepigo and G. Barysheva, Dokl. Akad. Nauk SSSR, 1959, 128, 941–944 CAS.
- V. Sharkov, Angew. Chem., Int. Ed., 1963, 2, 405–409 CrossRef.
- J. Robinson, C. Burgess, M. Bently, C. Brasher, B. Horne, D. Lillard, J. Macias, H. Mandal, S. Mills, K. O’Hara, J. Pon, A. Raigoza, A. Sanchez and J. Villareal, Biomass Bioenergy, 2004, 26, 473–483 CrossRef CAS.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46(20), 3577–3579 RSC.
- A. Fukuoka and P. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS.
- V. Jollet, F. Chambon, F. Rataboul, A. Cabiac, C. Pinel, E. Guillon and N. Essayem, Green Chem., 2009, 11, 2052–2060 RSC.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- W. Deng, X. Tan, W. Fang, Q. Zhang and Y. Wang, Catal. Lett., 2009, 133, 167–174 CrossRef CAS.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- L. Ding, A. Wang, M. Zheng and T. Zhang, ChemSusChem, 2010, 3, 818–821 CrossRef CAS.
- S. Van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. Van Tendeloo, P. Jacobs and B. Sels, ChemSusChem, 2010, 12, 1560–1563 CAS.
- H. Kobayashi, H. Matsuhashi, T. Komanoya, K. Hara and A. Fukuoka, Chem. Commun., 2011, 47, 2366–2368 RSC.
- (a) R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978 RSC; (b) G. Liang, C. Wu, L. He, J. Ming, H. Cheng, L. Zhuo and F. Zhao, Green Chem., 2011, 13, 839 RSC.
- D. Yamaguchi, M. Kitano, S. Suganuma, K. Nakajima, H. Kato and M. Hara, J. Phys. Chem. C, 2009, 113, 3181–3188 CAS.
- R. Palkovits, K. Tajvidi, A. Ruppert and K. Procelewska, Chem. Commun., 2011, 47(1), 576–578 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- N. Ji, T. Zhang, M. Zheng, A. Wang, X. Wang and J. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS.
- N. Ji, T. Zhang, M. Zheng, A. Wang, X. Wang, Y. Shu, A. Stottlemyer and J. Chen, Catal. Today, 2009, 147, 77–85 CrossRef CAS.
- M. Zheng, A. Wang, N. Ji, J. Pang, X. Wang and T. Zhang, ChemSusChem, 2010, 3, 63–66 CrossRef CAS.
- Y. Zhang, A. Wang and T. Zhang, Chem. Commun., 2010, 46, 862–864 RSC.
- X. Tan, W. Deng, M. Liu, Q. Zhang and Y. Wang, Chem. Commun., 2009, 7179–7181 RSC.
- P. Gallezot and A. Kiennemann, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VHC, Weinheim, vol. 5, 2008, pp. 2447-2476 Search PubMed.
- M. Mascal and E. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924–7926 CrossRef CAS.
- G. Gruter and F. Dautzenberg, EP, 1834950 A1, Avantium Int. B.V, 2007 Search PubMed.
- M. Mascal and E. Nikitin, ChemSusChem, 2009, 2, 423–426 CrossRef CAS.
- M. Mascal and E. Nikitin, ChemSusChem, 2009, 2, 859–861 CrossRef CAS.
- M. Rüsch gen. Klaas and H. Schöne, ChemSusChem, 2009, 2, 127–128 CrossRef.
- (a) W. Yang and A. Sen, ChemSusChem, 2010, 3(5), 597–603 CrossRef CAS; (b) W. Yang and A. Sen, ChemSusChem, 2011, 4, 349–352 CrossRef CAS.
- J.-P. Lange, R. Price, P. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS.
- E. D’Hondt, S. Van de Vyver, B. Sels and P. Jacobs, Chem. Commun., 2008, 6011–6012 RSC; S. Van de Vyver, E. D’Hondt, B. Sels and P. Jacobs, Stud. Surf. Sci. Catal., 2010, 175, 771–774 CrossRef CAS.
- D. Rackemann and W. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198 CrossRef CAS; J. Hegner, K. Pereira, B. DeBoef and B. Lucht, Tetrahedron Lett., 2010, 51, 2356–2358 CrossRef.
- J. Bond, D. Alonso, D. Wang, R. West and J. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS.
- D. Alonso, J. Bond and J. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC; R. Palkovits, Angew. Chem., Int. Ed., 2010, 49, 4336–4338 CrossRef CAS.
- W. Deng, M. Liu, Q. Zhang, X. Tan and Y. Wang, Chem. Commun., 2010, 46, 2668–2670 RSC.
- S. Park, J. Baker, M. Himmel, P. Parilla and D. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef.
- R. Swatloski, S. Spear, J. Holbrey and R. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47(42), 8047–8050 CrossRef CAS.
- N. Li and G. Huber, J. Catal., 2010, 270, 48–59 CrossRef CAS.
- G. Huber, R. Cortright and J. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS.
- B. Devassy and S. Halligudi, J. Catal., 2005, 236, 313–323 CrossRef CAS; Y. Izumi and K. Urabe, Chem. Lett., 1981, 663–666 CrossRef; C. Kresge, D. Marler, G. Rav and B. Rose, US Pat., 5366945, Mobil Oil, 1994 CrossRef; C. Caetano, I. Fonseca, A. Ramos, J. Vital and J. Castanheiro, Catal. Commun., 2008, 9, 1996–1999 CrossRef; P. Ferreira, I. Fonseca, A. Ramos, J. Vital and J. Castanheiro, Appl. Catal., B, 2010, 98(1–2), 94–99 CrossRef; S. Bajpe, C. Kirschhock, A. Aerts, E. Breynaert, G. Absillis, T. Parac-Vogt, L. Giebeler and J. Martens, Chem.–Eur. J., 2010, 16(13), 3926–3932 Search PubMed.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem. 10.1039/c1gc15350a.
- J. Chheda, Y. Romàn-Leshkov and J. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
| This journal is © The Royal Society of Chemistry 2011 |