Strategies for the intracellular delivery of nanoparticles
Leo Y. T.
Chou
,
Kevin
Ming
and
Warren C. W.
Chan
*
Institute of Biomaterials and Biomedical Engineering, Donnelly Center for Cellular and Biomolecular Research, Materials Science and Engineering, Chemical Engineering, Chemistry, University of Toronto, Toronto, ON M5S 3G9, Canada. E-mail: warren.chan@utoronto.ca
First published on 30th September 2010
Abstract
The ability to target contrast agents and therapeutics inside cells is becoming important as we strive to decipher the complex network of events that occur within living cells and design therapies that can modulate these processes. Nanotechnology researchers have generated a growing list of nanoparticles designed for such applications. These particles can be assembled from a variety of materials into desirable geometries and configurations and possess useful properties and functionalities. Undoubtedly, the effective delivery of these nanomaterials into cells will be critical to their applications. In this tutorial review, we discuss the fundamental challenges of delivering nanoparticles into cells and to the targeted organelles, and summarize strategies that have been developed to-date.
 Leo Y. T. Chou | Leo Chou is currently in his second year of PhD Studies in Biomedical Engineering at the University of Toronto. He received his BASc in Biomedical Engineering from the University of Toronto prior to joining Dr Chan's group. His current research is focused on the design and engineering of novel nanoparticles for applications in medical imaging and drug delivery. |
 Kevin Ming | Kevin Ming is currently in his second year of PhD studies in Biomedical Engineering at the University of Toronto. He received his BSc(Eng.) from Queen's University and MASc from the University of British Columbia, both in Electrical Engineering, prior to joining Dr Chan's group. His research interests are focused on developing a rapid, economical, and simple-to-use point-of-care diagnostic device for multiple disease detection. |
 Warren C. W. Chan | Professor Warren Chan received his PhD at Indiana University under Dr Shuming Nie in 2001 and was a post-doctoral fellow at University of California-San Diego under Dr Sangeeta Bhatia. He started at the University of Toronto in 2002 and since then, his research lab has been interested in developing nanoparticles for cancer and infectious disease applications. He is currently an Associate Professor at the Institute of Biomaterials and Biomedical Engineering (IBBME) and is the Canadian Research Chair in Bionanotechnology. |
1. Introduction
Advances in nanotechnology research have generated a growing list of contrast agents, therapeutics, and delivery vehicles. Many colloidal nanoparticles, which have at least one dimension in the 1 to 100 nm size range, are engineered for cellular biology and medical research and applications. In this size range, a wide variety of materials, including metals, metal oxides, and semiconductors, exhibit unique optical, electrical, and magnetic properties that can be tuned based on their size and shape;1–3 other types of materials, such as small molecules, lipids, polymers and other organic molecules, can be assembled into carriers for contrast agents and drugs to enhance payload and solubility. Collectively these materials can be synthesized, assembled into desirable geometries and configurations, and coated with targeting agents, and provide novel material properties for applications in molecular and cellular labeling, tracking, detection, drug delivery and medical imaging with high sensitivity and functionality.4 Conceptually, such modularity offers an infinite matrix of nanoparticles with different properties, making nanoparticle-based contrast agents and therapeutics more versatile than either small molecules or larger micron-sized particles in performing complex functions within physiological systems (Fig. 1).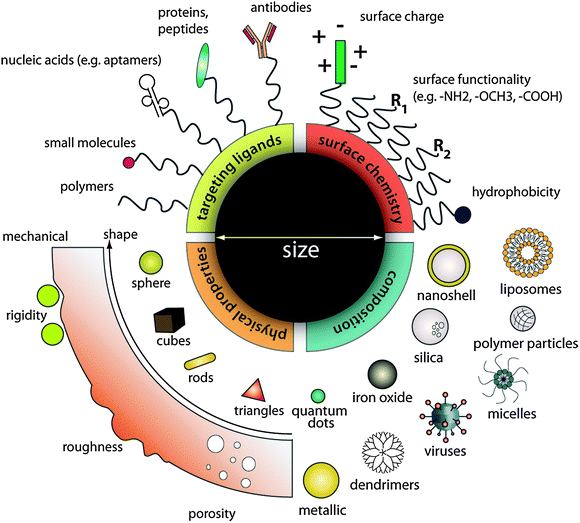 | ||
| Fig. 1 Designing nanoparticles for intracellular applications. Nanoparticles can be modularly assembled from different materials composition with different physical and chemical properties and functionalized with a myriad of ligands for biological targeting. Such flexibility in design freedom enables researchers to tailor nanoparticle for specific intracellular applications as contrast agents, drug delivery vehicles, and therapeutics. | ||
To fully realize the promise they hold, nanoparticles must be capable of reaching their biological targets with high efficiency and specificity. In particular, delivery and targeting at the subcellular level has recently become increasingly important as we strive to decipher complex events that occur within living cells such as gene regulation, signaling and transport, and to design therapies that can interject and modulate these processes (see Table 1). For example, fluorescent semiconductor quantum dots can image molecules over time scales of milliseconds to hours, providing researchers with a capability to monitor intracellular dynamics that cannot be accomplished using organic fluorophores. However, the delivery of quantum dots across the cell membrane remains challenging.5,6 Equally important is sub-cellular targeting for nanoparticle-based therapeutics: direct targeting of cytoplasmic organelles such as the mitochondria may improve photosensitizer-induced damage in photodynamic therapies7,8 while those carrying oligonucleotides for gene therapy must home in on the cytosol and nuclei of cells to be therapeutically effective.9,10
| Type of nanoparticle | Typical size range/nm | Structure and properties |
|---|---|---|
| Inorganic | ||
| Metals (Au, Ag, Cu) | 5–250 | ■ Easy to synthesize over a broad range of sizes and shapes (e.g. spheres, rods, core–shells); robust and functionalizable via thiol-metal chemistry |
| ■ Surface plasmon resonance; surface enhanced Raman scattering | ||
| Iron oxides | 5–200 | ■ Typically magnetite (MxFe3−xO4, M = Mn, Ni, Co, Fe) or maghemite (Fe2O3) |
| ■ Ferromagnetic or superparamagnetic properties | ||
| Quantum dots | 3–30 | ■ Typically II–VI or III–V chalcogenides synthesized as core–shell or alloy nanocrystalline colloids (e.g. CdSe/ZnS, CdTe1−xSex) |
| ■ Bright, photostable fluorophores with broad absorption and narrow emission; large two-photon cross section; FRET-donors | ||
| Silica | 3–100 | ■ Biodegradable; available also in micro- or mesoporous form for encapsulation of dyes and drugs; easily derivatizable with different surface chemistries using silanes |
| Layered double hydroxide | 50–200 | ■ Mg6Al2(CO3)(OH)16·4H2O |
| ■ Biocompatible and biodegradable in mildly acidic environments; high drug loading capacity | ||
| Calcium phosphate | 10–100 | ■ Ca5(PO4)3OH |
| ■ Biodegradable and biocompatible; can be doped with lanthanides or organic fluorophores | ||
| Organic | ||
| Liposomes | Multilayer: 500–5000 | ■ Spherical self-closed structures composed of one of more concentric phospholipid bi-layers |
| Unilayer: 100–500 | ■ Biocompatible, can entrap both hydrophobic and hydrophilic moieties; protects payload from external environment | |
| ■ Size and surface functionality can be tuned by adding new ingredients to the lipid mixture prior to synthesis | ||
| Polymer micelles | 20–200 | ■ Self-assembled spherical micelles composed of amphiphilic block co- or tri-polymers containing a hydrophobic core and a hydrophilic corona |
| ■ Hydrophobic payload can be entrapped in the core | ||
| ■ Geometry and functionality can be modularly controlled via the length and composition of the polymer blocks; can be biodegradable | ||
| Polymer nanoparticles | 50–300 | ■ Linear polymers with payload conjugated to the sidechain; precipitated into colloidal nanoparticles in solution |
| ■ Controllable size, surface functionality by adjusting polymer length, composition, and synthesis conditions; can be biodegradable | ||
| Dendrimers | 2–10 | ■ Radially hyperbranched polymers with regular repeat units |
| ■ High structural and chemical homogeneity; high ligand density and payload capacity per particle; controlled biodegradation | ||
| ■ Common dendrimers for biological applications: polyether, polyester, PAMAM | ||
| Carbon nanotubes | d = 0.5–3 | ■ Single or multi-layered graphene sheets rolled into concentric cylinders |
| l = 10 nm to several centimetres | ■ NIR-photoluminescence, strong resonance Raman scattering effects; directional conductivity, high tensile strength | |
| ■ Water-soluble through covalent chemical modification or non-covalent adsorption; ability to translocate cellular membranes via non-endocytosis mechanisms | ||
| Viral nanoparticles | 25–150 | ■ Self-assembled protein cages with multivalent surface functionalities |
| ■ Natural ability to internalize and unpack payload within cells | ||
Here we discuss the fundamental challenges of delivering nanoparticles into cells and to the targeted organelles, and summarize current strategies. We also describe some of the limitations of studying intracellular uptake of nanoparticles and provide a perspective on the development of this emerging research sub-theme in the field of nanotechnology.
2. Challenges in nanoparticle delivery and intracellular targeting
Portals of entry
The intracellular milieu is physically segregated from the environment by the plasma membrane, an elastic lipid bilayer embedded with domains of lipids, carbohydrates, and membrane proteins. In order to deliver nanoparticles into cells and to their subcellular targets, nanoparticles must first be able to traverse this plasma membrane. Nanoparticles may be internalized either by directly interacting with membrane-embedded receptors or indirectly by associating with the lipid bilayer. In the first approach, nanoparticles are functionalized with ligands that bind to receptors on the cell with high affinity and specificity. Ligands can be selected or engineered to target over-expressed receptors on healthy and diseased cells. Internalization of the resulting receptor–ligand complexes then leads to receptor-mediated endocytosis of the nanoparticles. Alternatively, nanoparticles can interact with the membrane via hydrophobic and electrostatic interactions and be taken into the cell through pinocytosis, a form of fluid-phase uptake where cell takes in the local extracellular environment by invaginating and pinching off pieces of the plasma membrane into vesicles containing the extracellular fluid.Controlling the route of nanoparticle uptake is important for mediating their intracellular fate and biological response. An increasing number of mechanistically distinct and highly regulated endocytic pathways are used by cells to traffic the extracellular cargoes to different intracellular locations and interactions11–13 (Fig. 2). Recent studies show that the endocytic processes not only function to internalize nutrients and membrane-associated molecules but orchestrate the spatiotemporal dynamics of cell signaling circuitry.14 An understanding of the cellular uptake mechanisms of nanoparticles would be important to determine what and how they modulate signaling and the subsequent molecular response of the cell.15–18
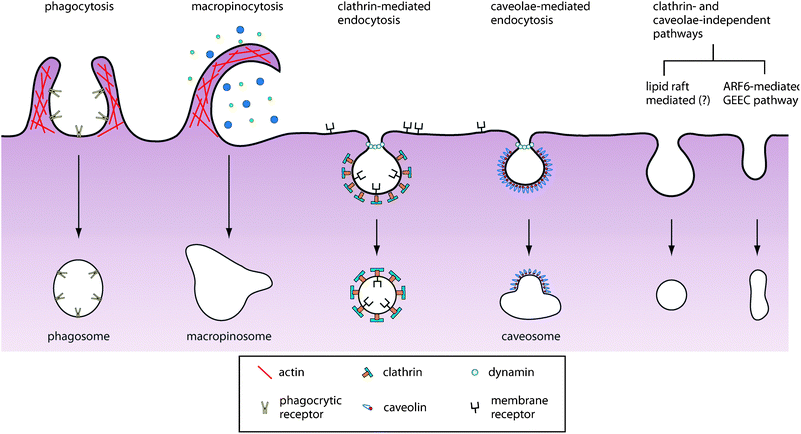 | ||
| Fig. 2 Pathways of entry into the cell. An increasing number of endocytic pathways are being defined, each mechanistically distinct and highly regulated at the molecular level. These pathways facilitate cellular signaling and cargo transport. Controlling the route of nanoparticle uptake is important for both mediating their intracellular fate as well as their biological response. | ||
Clathrin-mediated endocytosis is the most understood pathway of receptor-mediated endocytosis. In this pathway, ligand binding accelerates the recruitment of receptors to clathrin through adaptor proteins. Clathrin then polymerizes, driving the invagination of the pit, which is eventually released into the cytoplasm through the scission of the enzyme dynamin. This process is highly complex given that more than 50 different proteins can be found in clathrin-coated pits.19,20 Many other forms of endocytosis exist, including caveolae-mediated endocytosis and lipid-raft dependent endocytosis, though their mechanism is still poorly understood at the molecular level.
The endocytosis pathway is ligand dependent. Several pathogens, for example, use glycoproteins and lipid rafts on the plasma membrane as receptors to gain entry into the cell: anthrax toxin binds to cellular glyco-phosphatidylinositol-anchored proteins to enter the cell, whereas cholera and Shiga-like toxins use gangliosides.21 The iron-shuttling protein transferrin is well-known to bind its receptors and trigger endocytosis by the clathrin-dependent pathway, whereas caveolae-mediated endocytosis has emerged as an important entry mechanism for a number of viruses (e.g. SV40).22 However, an intrinsic cellular feature is that a particular receptor–ligand complex can internalize via multiple endocytic routes as a way to mediate net signaling output. For example, in the case of epidermal growth factor receptor (EGFR) and the transforming growth factor-b receptor (TGF-bR), internalization of the receptor–ligand complex can either lead to recycling if entered through clathrin-mediated endocytosis, or receptor degradation if entered via non-clathrin-mediated endocytosis.23 This redundancy makes it difficult to control nanoparticle route of entry solely on their ligand coating alone.
For a specialized set of cells, namely macrophages, monocytes and neutrophils, nanoparticles can also be internalized via phagocytosis.24 These cells function to clear off large particulates or debris such as remnants of dead cells and large pathogens such as bacteria or yeast. Targeted delivery to these cells can detect sites of inflammation and deliver drugs to malignant tumors. The process of phagocytosis occurs via several pathways, each mediated by a distinct receptor and can elicit biochemical cascades that result in different immune responses.25
Endosomal escape
Endocytosis transports nanoparticles into cells within vesicles and, depending on the mode of internalization, can either be recycled and exocytose out of the cell or trafficked to organelles including the lysosomes, golgi, and mitochondria. Endosomal trafficking is a complex intracellular process involving motor proteins that shuttle vesicles along microtubules within the cell. During this process, vesicles are sorted, fused or dissociated, as well as mature into endosomes and lysosomes (Fig. 3). For nanoparticles targeting the endolysosomal network, such as those used for treating lyosomal storage disorders, endocytic uptake provides immediate accessibility to their targets.26 However for many applications, the entrapment of nanoparticles within vesicles is undesirable. Moreover, the maturation of vesicles into late endosomes and lysosomes is characterized by rapid acidification from pH 6 to 4 within the vesicle and recruitment of degradative enzymes into the vesicle to digest vesicular content. For many applications, nanoparticles need to be engineered with mechanisms to escape from the endolysosomal network.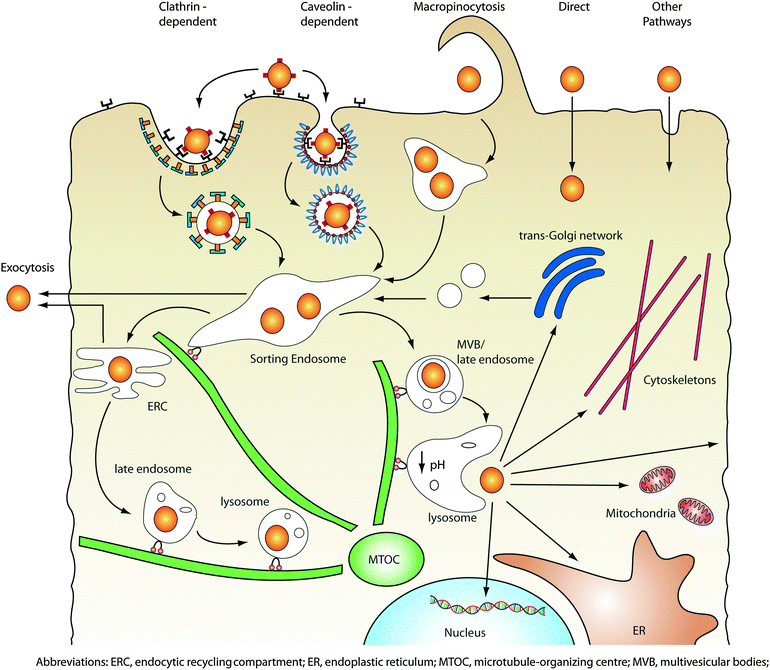 | ||
| Fig. 3 Intracellular transport of nanoparticles. After internalization via one or more of the endocytic pathways, nanoparticles are trafficked along the endolysosomal network within vesicles with the help of motor proteins and cytoskeletal structures. Vesicles can transport their contents into sorting endosomes, or excrete/recycle them back to the cell surface by fusing with the plasma membrane. Alternatively, endosomes can mature into lysosomes via luminal acidification and recruitment of degradative enzymes, which target the vesicle contents for degradation. In order to access cytoplasmic or nuclear targets, nanoparticles must be capable of escaping from the endolysosomal network as well as traverse through the crowded cytoplasm. | ||
Several strategies have been developed to overcome the endosomal barrier: viral capsids27 or a membrane-based envelope with the ability to fuse with endosomal membranes has been used to shuttle a wide variety of cargoes such as peptides, drugs, and nanoparticles from the endosome into the cytoplasm. This approach is limited by the complexity and cost of preparing viral vectors and the immunogenicity of the virus. Nanoparticles can also be coated with pH-sensitive synthetic peptides that undergo structural deformation under acidic pH to disrupt the vesicle membrane.28 Finally, polymers with a buffering capacity between pH 5.2 to 7.0 can be attached to nanoparticles to mediate their endosomal escape through the “proton-sponge effect”, where the proton-absorbing polymer induces osmotic swelling of the endosome and its eventual rupture (Fig. 4).29–31
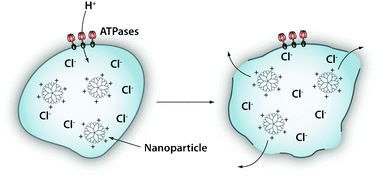 | ||
| Fig. 4 Endosomal escape by the “proton-sponge effect”. Acidification (pH decreases from 6 to 4) takes place during the process of endolysosomal maturation. The proton pumps (e.g. ATPases) pump H+ ions into the lumen of the endosome. Certain cationic polymers such as poly(ethyleneimine) are capable of sequestering these protons, thus sustaining the action of the proton pumps. To maintain electric neutrality, a parallel influx of Cl− ions and water accompanies the influx of protons. This swells and eventually ruptures the endosomal membrane allowing its contents to escape. | ||
Cytoplasmic transport and targeting
Once released from the endosomal compartments, nanoparticles must travel through the cytoplasm to bind with their targets. The cytoplasm is crowded with cytoskeletal structures, proteins, nutrients and small molecules. Diffusion mobility of the nanoparticle within the cytoplasm depends on its size and interactions with these biological entities. Binding to intracellular components may significantly retard the mobility of small particles while the geometry becomes important in the movement of larger nanoparticles.32 Within the cell, molecular motor proteins such as kinesin, dynein, and myosin typically transport vesicular cargoes using microtubule tracks or via actin filaments. The polymerization of actin can also propel a cargo particle in and out of the cell. Viruses hijack such systems upon cellular uptake in order to transport to the nucleus where they unload their genetic information.33 Similarly, these systems can be exploited by nanoparticles to move within the cytoplasm before reaching their targets of interest. Recently, it has been shown that, upon cellular internalization, semiconductor quantum dots functionalized with cell penetrating peptides are trapped within vesicles and transported towards the perinuclear region of the cell known as the microtubule organizing centre in a manner reminiscent of actin/kinesin mediated active transport.34However, very little is known about how nanoparticle design influences their transport in the cytoplasm. Researchers are attempting to answer this question by using a combination of fluorescent dyes and fluorescent proteins to study the intracellular nanoparticle transport process using multicolor fluorescence imaging but the limitation includes the requirements for analysis of large amounts of data and the inability to image single nanoparticles because of the diffraction-limited optical resolution. Hence, one can conclude that a major challenge for such studies is lack of available tools for tracking interactions between nanoparticles and large numbers of subcellular structures over space and time.
Circumventing the plasma membrane and endosomal barriers
The plasma membrane and endolysosomal vesicles present significant barriers for transporting nanoparticles into the cytoplasm or nucleus. For in vitro and ex vivo applications, the plasma membrane barrier can be bypassed by physically delivering nanoparticles into the cell. Two main approaches are currently being used: microinjection and electroporation. In the following section, we review the advantages and disadvantages of these two techniques, and highlight examples of their use in delivering nanoparticles into cells.3. Physical delivery strategies
Microinjection
Microinjection has been widely used in transgenic animal production, in vitro fertilization and in studies of non-permeable cells to mechanically transfer nucleic acids, peptides, proteins, and drugs into single cells.35 The samples are directly introduced into the cytoplasm or nucleus of a cell using a fine-tipped glass microcapillary under guidance of an optical microscope. This approach provides high payload efficiency and cell survival rate, and has few limitations in terms of cell or sample types. Microinjection is a purely mechanical method of delivery and thus a useful technique to evaluate the intracellular fate of nanoparticles beyond the endolysosomal compartment since the injected sample is the only variable in the experiment. For example, Derfus et al. microinjected quantum dots coated with poly(ethylene glycol) (PEG) and a nuclear localization signal, and observed the active transport and selective nuclear accumulation of the quantum dots within the nuclei of the majority of cells.36 Similarly, Medintz et al. microinjected quantum dot-fluorescent protein conjugates into different cell lines and compared the cytoplasmic distribution of quantum dots delivered using cell-penetrating peptides.37 Microinjected nanoparticle-conjugates distributed homogeneously (i.e. as monodisperse entities) across the entire cytoplasm rather than the more punctuated, localized patterns of endocytic uptake resulting from peptide-mediated delivery. In a similar approach, nickel-chelated quantum dots were microinjected into cells to label cytoplasmic proteins tagged with histidine residues via metal-affinity coordination.38The delivery of monodisperse nanoparticles into the cytoplasm is especially important for contrast agents used for tracking cell division and differentiation; tracers that distribute heterogeneously within the mother cell lead to uneven partitioning among daughter cells, making it very difficult to track lineage. Using microinjection, Dubertret et al. introduced phospholipid-coated quantum dots into early-stage Xenopus embryos in order to monitor biological activities and development.39 Injected quantum dots were cell autonomous, capable of labeling all embryonic cell types and remained monodisperse and photostable in vivo up to 4 days. Since microinjection delivers precise volumes into the cell, dose-dependent toxicity could be evaluated or avoided. It was found that up to 5 × 109 quantum dots could be introduced into the Xenopus embryo without inducing developmental abnormality. Similarly, Slotkin et al. microinjected neural stem and progenitor cells with quantum dots to study central nervous system development in the mouse embryo.40
Because of its serial nature, microinjection possesses obvious limitations: it is low throughput, technically demanding, can only be utilized in vitro or ex vivo, and expensive (∼$40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per device). Literature search of the past five years revealed very little use of microinjection for intracellular delivery of nanoparticles. Nevertheless, recent interests in single-cell analysis and single-molecule tracking with living cells may reinvigorate the use of microinjection, which provides reliable intracellular delivery and simplifies chemical modification of nanoparticles. Novel technologies are being developed to overcome inherent limitations of microinjection such as the development of smaller carbon-based nanometre-scale needles to improve delivery resolution and minimize cellular disruption;41 the integration of a fluid delivery component into atomic force microscopes to reduce manual operation and enable precision manipulation;42 high throughput nanosyringe arrays to eliminate the serial nature of the technique,43 and automatic delivery systems for controlling precisely the amount of volume injected into the cells.44
000 per device). Literature search of the past five years revealed very little use of microinjection for intracellular delivery of nanoparticles. Nevertheless, recent interests in single-cell analysis and single-molecule tracking with living cells may reinvigorate the use of microinjection, which provides reliable intracellular delivery and simplifies chemical modification of nanoparticles. Novel technologies are being developed to overcome inherent limitations of microinjection such as the development of smaller carbon-based nanometre-scale needles to improve delivery resolution and minimize cellular disruption;41 the integration of a fluid delivery component into atomic force microscopes to reduce manual operation and enable precision manipulation;42 high throughput nanosyringe arrays to eliminate the serial nature of the technique,43 and automatic delivery systems for controlling precisely the amount of volume injected into the cells.44
Electroporation
Electroporation is the technique of applying a rapid, high-voltage electric field impulse to temporarily generate hydrophilic pores in the cell plasma membrane. The pores then allow the passive transport of nanoparticles into the cell. This process is applicable to a large variety of cell types and delivers particles into many cells in a high throughput manner. However, this method requires specialized equipment and only works effectively when the cells are in suspension and can lead to cell death as a result of the electrical impulse.45Gold nanoparticles and oligonucleotide-modified gold nanoparticles were transported into mammalian cells by electroporation as an alternative to viral transfection of genes into cells for gene therapy.46 Microchips fabricated from poly(dimethylsiloxane) were used for cell culture and electroporation in situ. The electroporation was found to increase the delivery of both bare and oligonucleotide-coated gold nanoparticles into osteoblast cells as compared to no electroporation. Moreover electromigration—the gradual displacement of the metallic gold nanoparticles in the presence of a gradient electric field—can further increase delivery efficiency when used in conjunction with electroporation.
Chen and Gerion used electroporation to deliver quantum dots conjugated with nuclear localization sequence into the cytosol and subsequently into the nucleus.47 This was in contrast to quantum dots conjugated to random peptides that did not localize in any preferred region in the cell, showed near-homogenous distribution within the cytoplasm, and were excluded from the nucleus. The nuclear-localizing conjugates retained in the cell for a week without detectable negative cellular effects. Movement of conjugates from cytoplasm to nucleus was also visualized. Derfus et al. studied the effects of electroporation of quantum dots into live cells.36 It was observed that the quantum dots delivered via this method formed aggregates of up to 500 nm in diameter rather than being delivered individually. To test the hypothesis that the high-energy electric field caused polarization of quantum dot surfaces and the loss of electrostatically-adsorbed surface ligands, quantum dots cross-linked with bovine serum albumin (BSA) coat were delivered via electroporation. Aggregates were still observed both inside and outside the cell, suggesting that stabilization of surface ligand is not sufficient to prevent aggregation, and that the applied electric potential induced quantum dot aggregation.
Similarly, Slotkin et al. also investigated in utero electroporation of quantum dot-labeled neural stem and progenitor cells in viable mouse embryos. A mix of carboxyl-functionalized quantum dots and farnesylated enhanced green fluorescence protein (EGFP-F) plasmid were co-electroporated with the ventricular neuroepithelium of mouse embryos. Initially, GFP transfection efficiency was greater than the quantum dots', which the authors hypothesized to be due to the nanoparticles not carrying an adequate charge potential to be sufficiently propelled by the electroporation process. However, the labeling efficiency of neuroepithelial cells in a developing mouse nervous system increased dramatically when the quantum dots became more negatively charged when their surface is saturated with carboxyl functional groups.
4. Biochemical delivery strategies
Viruses represent an archetypal biological nanoparticle that exploits various cellular uptake and transport mechanisms to deliver their cargo either within the cytoplasm or nucleus of the cell.33,48 Similarly, synthetic nanoparticles can be engineered using biochemical approaches to facilitate their passage across cellular barriers such as the plasma membrane, the endolysosomal network, the cytoplasm and even the nuclear membrane. This can be achieved either by decorating nanoparticles with chemical functional groups and targeting biomolecules or by encapsulating nanoparticles within a carrier matrix. Biochemical strategies are high throughput and more versatile than the physical approaches—modular components made from different materials can be hierarchically assembled to create nanoparticles with multiple functionalities.Cationic coatings
Cationic molecules can interact strongly with negatively charged cell plasma membrane to induce membrane permeability. One explanation for the membrane disrupting effect is the formation of nanoscale holes on the plasma membrane upon interaction with cationic species. The extent of this effect is highly dependent on the charge density of the polycation where higher charge density is associated with greater potency.49Various types of cationic coatings have been used as transfection agents to deliver nanoparticles into cells, including cationic liposomes,50,51 polypeptides,52 and amine-containing polymers.53,54 For example, cationic liposomes such as lipofectamine are able to complex with negatively charged, water-soluble quantum dots through simple electrostatic interactions under co-incubation in media. The resulting liposome–nanoparticle complex is then delivered into cells via a second incubation step.36 Quantum dots internalized with the help of cationic liposomes have been found to form aggregates of several hundred nanometres in diameter in the cytoplasm, thus limiting their use in molecular imaging. On the other hand, cationic liposomes have been used to improve the delivery efficiency of plasmid DNA-coated gold nanoparticles into cells. Using plasmid DNA encoding for green fluorescent protein expression, researchers showed that liposome–DNA–gold nanoparticle construct had improved transfection efficiency over the DNA–gold nanoparticle construct alone. This was attributed to the neutralization of the negative charges on DNA–gold nanoparticle constructs, leading to improved electrostatic association with the plasma membrane.55
Nanoparticles can also be encapsulated within phospholipids in situ. Lipid carriers containing nanoparticles can be prepared by an established thin-film hydration approach, which allows the use of a cocktail of phospholipids to generate micelles or bilayer vesicles with mixed surface properties. For example, Kostarelos and co-workers demonstrated that quantum dots can be encapsulated within multilamellar lipid vesicles consisting of dioleoylphosphatidylcholine (DOPC) and DC-cholesterol. These constructs were able to penetrate human lung epithelial carcinoma A549 cells. The use of DC-cholesterol was critical to successful cell penetration as it changes the surface charge of DOPC vesicles from negative to positive. Bilayer lipid vesicles also offer the possibility of delivering both water-soluble and organically-functionalized nanoparticles by entrapping them either within the central aqueous pocket or the lipid bilayer compartment, respectively. Vogel et al. exploited this versatility to selectively label both the plasma membrane and the cytosolic components of human embryonic kidney HEK293 cells with hydrophobic and hydrophilic quantum dots, respectively.56 Other types of nanoparticles, such as magnetic iron oxides, have also been delivered into cells using cationic lipid-based transfection agents.
Certain amine-containing cationic polymers such as polyethyleneimine (PEI) can be attached as ligands to the surface of nanoparticles through standard conjugation chemistries. These cationic polymers are capable of inducing membrane permeability, facilitating nanoparticle entry into the cells. In particular, PEI has been widely investigated for gene delivery because of its ability to complex with and condense DNA, and transfect a broad range of cell lines with high efficiency. However, two major concerns for the use of cationic polymers are their cytotoxicity and poor stability in biological buffers or culture media. Zhang and co-workers addressed these challenges by designing a polymer coating that combines PEI with the biocompatible polymer PEG and the polysaccaride chitosan.57 This new polymer was then attached to the surface of iron oxide nanoparticles for in vivo gene delivery. These magnetic delivery vehicles were found to be non-toxic and demonstrated a high level of expression of the delivered plasmid DNA in an in vivo mouse model. Similarly, Duan and Nie grafted PEI with PEG to deliver quantum dots into cells with improved viability.58 Semiconductor quantum dots encapsulated within these PEG-modified amines were stable in biological buffer, capable of penetrating cell membranes, disrupt endosomal organelles, and access the cytosolic space in living cells. The endosomolytic effect arises from the so-called “proton-sponge effect”—the ability of multivalent amine-containing polymers to absorb protons and build up osmotic pressure in acidic organelles. This osmotic pressure destabilizes and eventually ruptures the endosomes, thus releasing the trapped materials into the cytoplasm. Nie and Gao et al. exploited the proton-sponge effect to deliver siRNA-wrapped quantum dots into MDA-MB-231 breast cancer cells.30 By coating quantum dots with a balanced composition of tertiary amine and carboxylic acid groups, the nanoparticle and its siRNA cargo were capable of performing the functions of penetrating cell membrane, endosomal release, carrier unpacking and intracellular transport.
Cell penetrating peptides
Cell-penetrating peptides (CPPs) have been used to facilitate translocation of cargoes across the plasma membrane and to specific organelles within the cell (see Table 2).59,60 Many CPP sequences are derived from natural sequences, such as the protein-transduction domains of viruses. For example, the Tat peptide derived from the HIV-1 virus has been popularly used to deliver a variety of nanoparticles into cells.61 In particular, Tat-conjugated fluorescent quantum dots were used as model systems to investigate the trajectory of Tat-functionalized nanoparticles within cells.34 These nanoparticles were confined within vesicles tethered to the inner vesicle surface. The quantum dot-loaded vesicles were trafficked along microtubule tracks and eventually localize at a perinuclear region known as the microtubule organizing center. This finding, however, cannot be generalized; some Tat-conjugated nanoparticles were observed to translocate to the nucleus62 while others remain in the cytosol or trapped in the endolysosomal compartment.63 This difference in intracellular localization may be dependent on the cell line studied and also on the properties of the engineered nanoparticle such as its size and surface chemistry. Results from various studies have suggested that the endocytic mechanism and intracellular fate of CPP mediated uptake into cells strongly depend on the attached cargo.| Name | Origin | Sequence | Cargo types |
|---|---|---|---|
| Peptides deriving from protein transduction domains | |||
| Tat | HIV-Tat protein | PGRKKRRQRRPPQ | Protein/peptide/siRNA/liposome/nanoparticle |
| Penetratin | Homeodomain | RQIKIWFQNRRMKWKK | Peptide/siRNA/liposome |
| Transportan | Galanin–mastoparan | GWTLNSAGYLLGKINLKALAALAKKIL | Protein/PNA/siRNA |
| VP-22 | HSV-1 structural protein | DAATATRGRSAASRPTERPRAPAR-SASRPRRPVD | Protein |
| Amphipathic peptides | |||
| MPG | HIV Gp41-SV40 NLS | GALFLGFLGAAGSTMGAWSQPKKKRKV | siRNA/ODN/plasmid/nanoparticle |
| Pep-1 | Trp-rich motif-SV40 NLS | KETWWETWWTEWSQPKKKRKV | Protein/peptide |
| MAP | Chimeric | KALAKALAKALA | Small molecule/plasmid |
| SAP | Proline-rich motif | VRLPPPVRLPPPVRLPPP | Protein/peptide |
| PPTG1 | Chimeric | GLFRALLRLLRSLWRLLLRA | Plasmid |
| Cationic peptides | |||
| Polyarginine | Chimeric | (R)n, n=8–10 | Protein/peptide/siRNA/ODN/plasmid |
| hCT (9-32) | Human calcitonin | LGTYTQDFNKTFPQTAIGVGAP | Protein/plasmid DNA |
| SynB | Protegrin | RGGRLSYSRRRFSTSTGR | Doxorubicin |
| Pvec | Murine VE-cadherin | LLIILRRRIRKQAHAHSK | Protein/peptide |
To facilitate endosomal escape Tat peptide can be combined with pH-sensitive proteins such as the influenza virus hemagglutinin protein HA2 to create fusogenic Tat-HA2 peptides.64 This fusion peptide is capable of disrupting the integrity of the endosomal lipid membrane at low pH without being cytotoxic like other endosomal membrane destabilizers such as PEI. Tat-HA2 peptides have been used to deliver gold nanoparticles into NIH3T3 fibroblast cells for intracellular imaging of actin filaments.64,65
Some peptide sequences also possess a nuclear localization sequence that interacts with the nuclear pore complex. By using a mixed cocktail of peptides containing either cell-penetrating or nuclear localization sequences, nanoparticles can be delivered into specific cells and then into the nucleus.63,66 Moreover, by optimizing the ligand density of CPPs (i.e. CALNN and Pntn) and PEG-stabilizer on the surface of the gold nanoparticle, single monodisperse gold nanoparticles can be delivered into the cytosol and nucleus of the cell.67
Ligand-mediated internalization
Attaching targeting ligands onto the nanoparticle scaffold imbue them with selectivity for specific cell types through association with cell surface antigens or membrane receptors that are overexpressed in these cells. Receptor–ligand binding can then facilitate internalization through receptor-mediated endocytosis. The strength of nanoparticle interactions with these antigens or membrane receptors can be controlled by the type of ligand (e.g. affinity) and by changing the ligand density (e.g. avidity) attached to the nanoparticle surface. For in vivo applications, targeting agents can be used to direct nanoparticles towards diseased tissues following systemic administration. These targeting moieties can be broadly classified as proteins (antibodies and their fragments), nucleic acids (e.g. aptamers), or other receptor ligands (e.g. small molecules, peptides, vitamins, and carbohydrates). Many of the targeting agents developed to-date has been for cancer applications.68 See Table 3 for a representative list of these targeting ligands and examples of their use in nanoparticle delivery.| Type | MW/kDa | Diameter/nm | Characteristics | Examples of use in nanoparticle delivery |
|---|---|---|---|---|
| Receptor ligands | ||||
| Proteins | 30–150 | Variable | Produced using recombinant DNA technologies, can be biologically active, susceptible to proteases | EGF,18 transferrin,69–72,113 NGF,73 cholera toxin B74,75 |
| Peptides | 0.5–10 | Variable | Facile synthesis and modification, diverse libraries and screening technologies, susceptible to peptidases, renal retention | YSA,76 chlorotoxin,77 LABL,78 RGD,79,80 F381 |
| Small molecules | 0.1–1.0 | 0.5–2.0 | Chemical synthesis, simple modification and coupling chemistries, can be biologically active, highly variable affinities | Folic acid,84–90 LHRH91 |
| Engineered antibody/antibody fragments (divalent) | ||||
| Whole antibodies | 150 | 15–20 | High affinity, divalent, many clinically approved examples, contains biologically active constant (Fc) region, long circulation | Cetuximab,93 HER2,95,96 and many others94 |
| (Fab′)2 | 100 | 10–15 | Improved affinity, can be engineered to a variety of sizes and arrangements of protein domains | |
| Diabody | 50–80 | 5–10 | Mono-specific or bi-specific dimer of ScFv | HER299 |
| Minibody | 80 | 10 | Can be produced genetically | |
| Engineered antibody fragments (monovalent) | ||||
| Fab′ | 50 | 5–10 | Can be produced genetically or enzymatically by cleavage of monoclonal antibodies | HER2100 |
| ScFv | 25 | 3–5 | Lowered affinity, rapid clearance from circulation, renal retention, reduced stability, reduced immunogenicity | CD19,101 A33,102 CEA,104 EGFR103,105 |
| Nanobody | 15 | 2–3 | Smallest antigen-binding fragment, single domain, can bind cryptic epitopes | |
| Aptamers | ||||
| DNA/RNA | 10–30 | 2–3 | Rapid clearance, automated chemical synthesis, susceptible to nucleases without chemical modification | AS1411,110 A10,109 PrPc,111 TD05,107 T-cell specific108 |
Many endogenous proteins with known membrane-bound receptors can be used for targeting via receptor-mediated endocytosis. For example, the endogenous iron-transporting protein transferrin has been used to deliver nanoparticles into a variety of cell types.69–72 The ligand is well-known to bind to its receptor and induce internalization via clathrin-dependent endocytosis. Transferrin receptors are also overexpressed in a variety of cancers, making transferrin a popular ligand of choice for cancer targeting. Several growth factors such as epidermal growth factor (EGF)18 and neural growth factor (NGF)73 attached to nanocarriers have been shown to enter cells and elicit specific molecular responses. Other biologically derived proteins such as plant and bacterial toxins have also been used as ligands to deliver nanoparticles into cells.74,75 As more compact alternatives, peptides have been used to facilitate nanoparticle entry into cells.76–78 Peptides with affinity and selectivity for specific cell types can be synthesized and screened using diverse libraries, and chemically modified with established chemistry making them versatile ligands for nanoparticle functionalization. For example, arginine–glycineaspartic acid (RGD) motifs have been used to target cell adhesion integrin αvβ3 on endothelial cells, resulting in increased intracellular drug delivery in different cell models.79,80 Nanoparticles functionalized with a tumor-homing peptide and siRNA cargo have been shown to internalize and knock down the fluorescence in GFP-expressing HeLa cells.81
Targeting of diseased cells can also be achieved using drugs, molecules and antibodies that bind receptors overexpressed on the surfaces of such cells. Folic acid is a widely used targeting molecule for delivering cargo to cells with folate receptors, which are overexpressed in ovarian and breast cancer.82–84 Folic acid is inexpensive, exhibits a high binding affinity for the folate receptor (i.e. Kd ≈ 10−10 M), and is efficiently internalized into cells via receptor-mediated endocytosis even when conjugated to other molecules. Folic acid has been demonstrated to deliver many types of nanoparticles such as polymer,85 gold,86–88 magnetic,89 and semiconductor nanocrystals90 selectively into cancer cells. Other small molecules such as luteinizing-hormone-releasing-hormone91 have also been investigated as nanoparticle targeting agents. In an effort to increase throughput and efficacy, a large library comprised of 146 different nanoparticles decorated with different small molecules was synthesized and screened against various cell lines where the hits were discovered to be effective targeting agents in vivo.92
Effective targeting of antigens overexpressed on diseased cells has been routinely achieved using monoclonal antibodies.93,94 For example, ErbB2/EGFR receptor overexpressing breast cancer cells has been targeted using nanoparticles functionalized with anti-EGFR antibodies such as Herceptin®.95,96 Antibodies exhibit the additional advantage in that their size, affinity, and selectivity can now be rationally engineered.97,98 Monoclonal antibody fragments can be engineered that retain the same targeting specificity as the original whole antibody but contribute minimally to the size of the attached cargo;99,100 fusion proteins can be created by combining two or more genes to produce a new protein with desired properties. For example, single-chain variable fragments (ScFv)—fusion protein consisting of antibody light- and heavy-chain variable domains connected via a flexible peptide linker—exhibit reduced immunogenicity and size while retaining the same cell type targeting ability. ScFv fragments have been attached to nanoparticles as smaller targeting ligands to direct nanoparticle entry into various cell types.101–105 Using molecular biology techniques, these engineered antibody fragments can be further forged into multivalent and multispecific reagents, for example by dimerization of two heterogeneous protein mimetics, and attached with therapeutic payloads. It is also possible to increase binding selectivity by engineering proteins to detect a specific structure or conformation of a target receptor. Application of these novel targeting ligands to nanoparticles provides another degree of freedom in dictating how nanoparticles bind cells and interact with intracellular components.
Finally, aptamers can also be coated onto the surface of nanoparticles for in vivo and in vitro targeting. Aptamers are short single-stranded DNA or RNA oligonucleotides selected in vitro to specific cellular targets and specific aptamers for specific targets can be selected from a large number of random sequences using a technique called Systematic Evolution of Ligands by Exponential Enrichment (SELEX).106 SELEX has been used to generate aptamers capable of binding a wide variety of targets including small molecules, peptides, proteins, and even whole cells.107,108 Aptamers with known sequences can be chemically synthesized in large scales and chemically modified to improve stability against nucleases.109–111
Sub-cellular targeting using biodegradable carriers
A major limitation in most nanoparticle delivery strategies has been the difficulty of achieving active targeting of specific subcellular organelles after entry into the cell. This is because the same nanoparticle surface that was modified to enable intracellular delivery is also needed for the attachment of subcellular targeting antibodies. To address this challenge, Chan et al. recently proposed using poly(D,L-lactide-co-glycolide) (PLGA) as an environmentally responsive carrier system to deliver protein-conjugated quantum dots into cells.112 Protein-functionalized quantum dots were encapsulated within PLGA nanospheres via a double microemulsion procedure and then selectively fractionated in a sucrose density gradient to yield a homogenous population of hybrid nanocomposites (d ≈ 104.5 ± 7.8 nm). The biodegradable carrier system was found to internalize within cells and were initially trapped in vesicles. A decrease in the pH caused a change in the surface charge of the PLGA nanosphere that induces the nanosphere to translocate from endolysosomal vesicles into the cytosol. Subsequent biodegradation of the carrier system allows release of the encapsulated active-targeting quantum dots. The use of nanospheres as a delivery vehicle did not induce significant cytotoxic effects. By utilizing the surface of the biodegradable polymer for cell penetration, the chemical and structural properties of protein-coated quantum dots were preserved and enable them to actively seek specific intracellular targets after the polymer has degraded. Aside from quantum dots, the polymer carrier system could also encapsulate other payloads such as organic dyes and drugs. The researchers used the PLGA nanospheres to deliver both antibody-conjugated quantum dots and MitoTrack Red for co-staining of actin filaments and mitochondria respectively.5. Technical limitations
One of the goals in designing nanoparticle-based contrast agents and drug delivery systems is to be able to correlate cellular responses to the physicochemical properties of the engineered nanoparticle and to determine the optimal material's properties of the particle for intracellular uptake and targeting. Establishment of such “first-principles” would enable biologists, pharmacologists, and biomedical engineers to rationally construct functional nanoparticles tailored for their particular applications. However, this task is quite challenging; multiple factors can simultaneously affect the interaction of the nanoparticle with cellular structures, including the intrinsic nanoparticle properties and extrinsic factors such as cell type and physiological state. Recent studies have begun to systematically investigate the causative effects of nanoparticle geometry113,114 and surface functionality115 on cellular behaviors such as uptake, intracellular localization, and toxicity, but to-date the picture is quite primitive and far from complete. Because of the differences in experimental design and nanomaterial properties, results from independent studies are difficult to interpret. Thus, successful targeting strategies must be determined for a particular nanoparticle system on a case-by-case basis.Secondly, particle-cell interactions at the nanoscale can be remarkably different from that of small molecules or large-micron sized particles.116 For example, the plasma membrane of a cell contains spatially distinct lipid domains, enzymes, carbohydrates, and protein receptors that function to mediate cellular functions such as signal transduction, immune surveillance, cell division, polarization, and maintaining cellular homeostasis. These domains can interact with exogenous materials that come in close proximity to the cell's surface membrane to establish various forces of interactions that ultimately determine the cellular response towards the material. Whereas a large, micron-sized particle will experience a surface-area averaged force of these interactions, nanoparticles are small enough to interact with only one or several domains at a time. Depending on their position on the membrane surface of the cell, their interactions can differ. This leads to divergent cellular responses such as different routes of uptake.
Finally, both the nanoparticle–cell interactions and the cellular structures themselves are inherently dynamic. Most analytical technologies only provide a snap shot at specific time point. Imaging technologies such as transmission electron microscopy offer highly sensitive ultrastructural information about the subcellular localization of nanoparticles but require sample fixation and can only detect electron dense materials such as metallic nanoparticles but not polymers, liposomes, and dendrimers. Optical imaging techniques such as fluorescence microscopy are capable of live cell analysis but provide limited resolution and sensitivity to resolve molecular interactions. In order to understand how nanoparticles interact with cellular structures during the processes of cellular internalization and trafficking, labeling cellular proteins with fluorescent proteins, organelle trackers, as well as selective use of cellular uptake inhibitors are currently used in conjunction with optical imaging. Correlative imaging technologies (i.e. electron and optical microscopy imaging) are also being developed.117 Nevertheless, substantial improvement in cost and accessibility, speed of image acquisition and data analysis will be required in order for these techniques to be widely used.
6. Conclusions and outlook
The ability to target nanoparticles to specific cells and subcellular components greatly impacts their performance as molecular contrast agents, detection probes and drug delivery vehicles for biomedical applications. While numerous exciting and novel applications of nanoparticles have been demonstrated in the last decade, our ability to deliver nanoparticles to their subcellular targets and to control their trajectory within the cell is still very primitive. In addition, quantitative descriptions on the kinetics, amount, mechanisms, and trajectories of nanoparticle uptake and trafficking are lacking. Such unanswered questions result in our inability to rationally design nanoparticles as well as fears of their toxicity. These barriers hamper the clinical utility and benefit of nanoparticle-based technologies. Thus, fundamental studies must continue to understand how nanoparticles interact with molecules, organelles, and cellular structures, and to be able to identify causative relationships between physicochemical properties of engineered nanoparticles with cellular responses. Further understanding of cellular and molecular biology is needed, as many of the mechanisms involved in cellular uptake and transport remain poorly understood at the molecular level, making it difficult to study nanoparticle behaviors therein.Secondly, the optimum design of the nanoparticle will be highly application specific. For example, controlling ligand valence on the surface of nanoparticles is very important for applications in single-molecule sensing, tracking, and labeling. For in vivo applications such as drug delivery and therapeutics, the engineered nanoparticle needs to overcome additional biological barriers before being taken up by the targeted cells, such as evading undesired immune responses, maintaining colloidal stability in blood circulation, extravasation into the targeted organs, and achieving effective tissue penetration.118 In lieu of the complexity of nanoparticle–cell interactions and the diverse and sequential presentation of biological barriers on the cellular and the tissue level, next generation nanoparticles may well be designed with functions or properties that can be dynamically triggered or activated.
Continuous advances in synthetic chemistry and materials chemistry will help realizing such novel classes of nanoparticles and nanoscaled “systems” with integrated functionalities and properties. For example, multifunctional nanoparticle probes are recently being developed to overcome limitations inherent in single component platforms such as multimodal contrast agents comprised of magnetic nanoparticles coupled with either optical or radiolabeled probes that allow medical imaging in two modes;119 carbon nanotubes as a longboat delivery platform and intracellular transporter decorated with quantum dot fluorophores for visualization.120 These developments are expected to bring unprecedented opportunities for nanotechnology to medicine and biological research. Understanding of how these particles interact with cells, and implement strategies to successfully and efficiently target them to their biological targets of interest will be important for their advance towards safe and effective use.
Acknowledgements
W. C. W. Chan acknowledges the Canadian Institute of Health Research, Natural Sciences and Engineering Research Council (NSERC), Canadian Foundation for Innovation, and Ministry of Research and Innovation for funding support. L. Y. Chou acknowledges NSERC for a student fellowship.References
- A. M. Smith and S. M. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS
.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS
- W. B. Cai and X. Y. Chen, Small, 2007, 3, 1840–1854 CrossRef CAS
- F. Pinaud, S. Clarke, A. Sittner and M. Dahan, Nat. Methods, 2010, 7, 275–285 CrossRef CAS
- J. B. Delehanty, H. Mattoussi and I. L. Medintz, Anal. Bioanal. Chem., 2009, 393, 1091–1105 CrossRef CAS
- E. Buytaert, M. Dewaele and P. Agostinis, Biochim. Biophy. Acta, Rev. Cancer, 2007, 1776, 86–107 Search PubMed
- S. W. Ryter, H. P. Kim, A. Hoetzel, J. W. Park, K. Nakahira, X. Wang and A. M. K. Choi, Antioxid. Redox Signaling, 2007, 9, 49–89 Search PubMed
- D. Putnam, Nat. Mater., 2006, 5, 439–451 CrossRef CAS
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Pharmacol. Rev., 2006, 58, 32–45 CrossRef CAS
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37–44 CrossRef CAS
- S. Mayor and R. E. Pagano, Nat. Rev. Mol. Cell Biol., 2007, 8, 603–612 CrossRef CAS
- S. Kumari, M. G. Swetha and S. Mayor, Cell Res., 2010, 20, 256–275 CrossRef CAS
- G. Scita and P. P. Di Fiore, Nature, 2010, 463, 464–473 CrossRef CAS
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS
- S. J. H. Soenen, N. Nuytten, S. F. De Meyer, S. C. De Smedt and M. De Cuyper, Small, 2010, 6, 832–842 CrossRef CAS
- Y. F. Huang, H. P. Liu, X. L. Xiong, Y. Chen and W. H. Tan, J. Am. Chem. Soc., 2009, 131, 17328–17334 CrossRef CAS
- T. P. Thomas, R. Shukla, A. Kotlyar, B. Liang, J. Y. Ye, T. B. Norris and J. R. Baker, Jr., Biomacromolecules, 2008, 9, 603–609 CrossRef CAS
- S. L. Schmid, Annu. Rev. Biochem., 1997, 66, 511–548 CrossRef CAS
- S. A. Mousavi, L. Malerod, T. Berg and R. Kjeken, Biochem. J., 2004, 377, 1–16 CrossRef CAS
- J. Gruenberg and F. G. van der Goot, Nat. Rev. Mol. Cell Biol., 2006, 7, 495–504 CrossRef CAS
- R. G. Parton and A. A. Richards, Traffic, 2003, 4, 724–738 CrossRef CAS
- G. M. Di Guglielmo, C. Le Roy, A. F. Goodfellow and J. L. Wrana, Nat. Cell Biol., 2003, 5, 410–421 CrossRef CAS
- L. M. Stuart and R. A. B. Ezekowitz, Immunity, 2005, 22, 539–550 CrossRef CAS
- P. R. Taylor, L. Martinez-Pomares, M. Stacey, H. H. Lin, G. D. Brown and S. Gordon, Annu. Rev. Immunol., 2005, 23, 901–944 CrossRef CAS
- L. A. Bareford and P. W. Swaan, Adv. Drug Delivery Rev., 2007, 59, 748–758 CrossRef CAS
- F. Li, Z. P. Zhang, J. Peng, Z. Q. Cui, D. W. Pang, K. Li, H. P. Wei, Y. F. Zhou, J. K. Wen and X. E. Zhang, Small, 2009, 5, 718–726 CrossRef CAS
- S. Kobayashi, I. Nakase, N. Kawabata, H. H. Yu, S. Pujals, M. Imanishi, E. Giralt and S. Futaki, Bioconjugate Chem., 2009, 20, 953–959 CrossRef CAS
- J. E. Fuller, G. T. Zugates, L. S. Ferreira, H. S. Ow, N. N. Nguyen, U. B. Wiesner and R. S. Langer, Biomaterials, 2008, 29, 1526–1532 CrossRef CAS
- M. V. Yezhelyev, L. Qi, R. M. O'Regan, S. Nie and X. Gao, J. Am. Chem. Soc., 2008, 130, 9006–9012 CrossRef CAS
- M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9138–9143 CrossRef CAS
- A. S. Verkman, Trends Biochem. Sci., 2002, 27, 27–33 CrossRef CAS
- U. F. Greber and M. Way, Cell (Cambridge, Mass.), 2006, 124, 741–754 CrossRef CAS
- G. Ruan, A. Agrawal, A. I. Marcus and S. Nie, J. Am. Chem. Soc., 2007, 129, 14759–14766 CrossRef CAS
- Y. Zhang and L. C. Yu, Curr. Opin. Biotechnol., 2008, 19, 506–510 CrossRef CAS
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Adv. Mater., 2004, 16, 961–966 CrossRef CAS
- I. L. Medintz, T. Pons, J. B. Delehanty, K. Susumu, F. M. Brunel, P. E. Dawson and H. Mattoussi, Bioconjugate Chem., 2008, 19, 1785–1795 CrossRef CAS
- K. Boeneman, J. B. Delehanty, K. Susumu, M. H. Stewart and I. L. Medintz, J. Am. Chem. Soc., 2010, 132, 5975–5977 CrossRef CAS
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759–1762 CrossRef CAS
- J. R. Slotkin, L. Chakrabarti, H. N. Dai, R. S. Carney, T. Hirata, B. S. Bregman, G. I. Gallicano, J. G. Corbin and T. F. Haydar, Dev. Dyn., 2007, 236, 3393–3401 CrossRef CAS
- K. Yum, N. Wang and M. F. Yu, Nanoscale, 2010, 2, 363–372 RSC
- A. Meister, M. Gabi, P. Behr, P. Studer, J. Voros, P. Niedermann, J. Bitterli, J. Polesel-Maris, M. Liley, H. Heinzelmann and T. Zambelli, Nano Lett., 2009, 9, 2501–2507 CrossRef CAS
- S. Park, Y. S. Kim, W. B. Kim and S. Jon, Nano Lett., 2009, 9, 1325–1329 CrossRef CAS
- F. O. Laforge, J. Carpino, S. A. Rotenberg and M. V. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11895–11900 CrossRef CAS
- D. J. Stephens and R. Pepperkok, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4295–4298 CrossRef CAS
- C. P. Jen, Y. H. Chen, C. S. Fan, C. S. Yeh, Y. C. Lin, D. B. Shieh, C. L. Wu, D. H. Chen and C. H. Chou, Langmuir, 2004, 20, 1369–1374 CrossRef CAS
- F. Q. Chen and D. Gerion, Nano Lett., 2004, 4, 1827–1832 CrossRef CAS
- D. Mudhakir and H. Harashima, AAPS J., 2009, 11, 65–77 Search PubMed
- P. R. Leroueil, S. Hong, A. Mecke, J. R. Baker, Jr., B. G. Orr and M. M. Banaszak Holl, Acc. Chem. Res., 2007, 40, 335–342 CrossRef CAS
- W. T. Al-Jamal, K. T. Al-Jamal, P. H. Bomans, P. M. Frederik and K. Kostarelos, Small, 2008, 4, 1406–1415 CrossRef CAS
- W. T. Al-Jamal, K. T. Al-Jamal, B. Tian, L. Lacerda, P. H. Bornans, P. M. Frederik and K. Kostarelos, ACS Nano, 2008, 2, 408–418 CrossRef CAS
- M. Babic, D. Horak, M. Trchova, P. Jendelova, K. Glogarova, P. Lesny, V. Herynek, M. Hajek and E. Sykova, Bioconjugate Chem., 2008, 19, 740–750 CrossRef CAS
- M. A. Herrero, F. M. Toma, K. T. Al-Jamal, K. Kostarelos, A. Bianco, T. Da Ros, F. Bano, L. Casalis, G. Scoles and M. Prato, J. Am. Chem. Soc., 2009, 131, 9843–9848 CrossRef CAS
- D. K. Chatteriee, A. J. Rufalhah and Y. Zhang, Biomaterials, 2008, 29, 937–943 CrossRef CAS
- W. K. Rhim, J. S. Kim and J. M. Nam, Small, 2008, 4, 1651–1655 CrossRef CAS
- G. Gopalakrishnan, C. Danelon, P. Izewska, M. Prummer, P. Y. Bolinger, I. Geissbuhler, D. Demurtas, J. Dubochet and H. Vogel, Angew. Chem., Int. Ed., 2006, 45, 5478–5483 CrossRef CAS
- F. M. Kievit, O. Veiseh, N. Bhattarai, C. Fang, J. W. Gunn, D. Lee, R. G. Ellenbogen, J. M. Olson and M. Zhang, Adv. Funct. Mater., 2009, 19, 2244–2251 CrossRef CAS
- H. Duan and S. Nie, J. Am. Chem. Soc., 2007, 129, 3333–3338 CrossRef CAS
- K. M. Stewart, K. L. Horton and S. O. Kelley, Org. Biomol. Chem., 2008, 6, 2242–2255 RSC
- F. Heitz, M. C. Morris and G. Divita, Br. J. Pharmacol., 2009, 157, 195–206 CrossRef CAS
- V. P. Torchilin, Adv. Drug Delivery Rev., 2008, 60, 548–558 CrossRef CAS
- J. M. de la Fuente and C. C. Berry, Bioconjugate Chem., 2005, 16, 1176–1180 CrossRef CAS
- A. G. Tkachenko, H. Xie, Y. L. Liu, D. Coleman, J. Ryan, W. R. Glomm, M. K. Shipton, S. Franzen and D. L. Feldheim, Bioconjugate Chem., 2004, 15, 482–490 CrossRef CAS
- S. Kumar, N. Harrison, R. Richards-Kortum and K. Sokolov, Nano Lett., 2007, 7, 1338–1343 CrossRef CAS
- S. Kumar, J. Aaron and K. Sokolov, Nat. Protocols, 2008, 3, 314–320 CrossRef CAS
- B. Kang, M. A. Mackey and M. A. El-Sayed, J. Am. Chem. Soc., 2010, 132, 1517–1519 CrossRef CAS
- P. Nativo, I. A. Prior and M. Brust, ACS Nano, 2008, 2, 1639–1644 CrossRef CAS
- D. A. Scheinberg, C. H. Villa, F. E. Escorcia and M. R. McDevitt, Nat. Rev. Clin. Oncol., 2010, 7, 266–276 Search PubMed
- R. Q. Huang, Y. H. Qu, W. L. Ke, J. H. Zhu, Y. Y. Pei and C. Jiang, FASEB J., 2007, 21, 1117–1125 CrossRef CAS
- C. H. Choi, C. A. Alabi, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1235–1240 CrossRef CAS
- T. Kakudo, S. Chaki, S. Futaki, I. Nakase, K. Akaji, T. Kawakami, K. Maruyama, H. Kamiya and H. Harashima, Biochemistry, 2004, 43, 5618–5628 CrossRef CAS
- R. Kumar, I. Roy, T. Y. Hulchanskyy, L. N. Goswami, A. C. Bonoiu, E. J. Bergey, K. M. Tramposch, A. Maitra and P. N. Prasad, ACS Nano, 2008, 2, 449–456 CrossRef CAS
- B. Cui, C. Wu, L. Chen, A. Ramirez, E. L. Bearer, W. P. Li, W. C. Mobley and S. Chu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13666–13671 CrossRef CAS
- C. Tekle, B. Deurs, K. Sandvig and T. G. Iversen, Nano Lett., 2008, 8, 1858–1865 CrossRef CAS
- S. K. Chakraborty, J. A. J. Fitzpatrick, J. A. Phillippi, S. Andreko, A. S. Waggoner, M. P. Bruchez and B. Ballou, Nano Lett., 2007, 7, 2618–2626 CrossRef CAS
- K. E. Scarberry, E. B. Dickerson, J. F. McDonald and Z. J. Zhang, J. Am. Chem. Soc., 2008, 130, 10258–10262 CrossRef CAS
- O. Veiseh, J. W. Gunn, F. M. Kievit, C. Sun, C. Fang, J. S. H. Lee and M. Q. Zhang, Small, 2009, 5, 256–264 CAS
- N. Zhang, C. Chittasupho, C. Duangrat, T. J. Siahaan and C. Berkland, Bioconjugate Chem., 2008, 19, 145–152 CrossRef CAS
- N. Nasongkla, E. Bey, J. M. Ren, H. Ai, C. Khemtong, J. S. Guthi, S. F. Chin, A. D. Sherry, D. A. Boothman and J. M. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS
- M. Oba, K. Aoyagi, K. Miyata, Y. Matsumoto, K. Itaka, N. Nishiyama, Y. Yarnasaki, H. Koyama and K. Kataoka, Mol. Pharmaceutics, 2008, 5, 1080–1092 CrossRef CAS
- A. M. Derfus, A. A. Chen, D. H. Min, E. Ruoslahti and S. N. Bhatia, Bioconjugate Chem., 2007, 18, 1391–1396 CrossRef CAS
- A. R. Hilgenbrink and P. S. Low, J. Pharm. Sci., 2005, 94, 2135–2146 CrossRef CAS
- C. P. Leamon and P. S. Low, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 5572–5576 CrossRef CAS
- Y. Bae, W. D. Jang, N. Nishiyama, S. Fukushima and K. Kataoka, Mol. BioSyst., 2005, 1, 242–250 RSC
- S. H. Kim, J. H. Jeong, K. W. Chun and T. G. Park, Langmuir, 2005, 21, 8852–8857 CrossRef CAS
- X. Shi, S. Wang, S. Meshinchi, M. E. Van Antwerp, X. Bi, I. Lee and J. R. Baker, Jr., Small, 2007, 3, 1245–1252 CrossRef CAS
- V. Dixit, J. Van den Bossche, D. M. Sherman, D. H. Thompson and R. P. Andres, Bioconjugate Chem., 2006, 17, 603–609 CrossRef CAS
- R. Bhattacharya, C. R. Patra, A. Earl, S. F. Wang, A. Katarya, L. Lu, J. N. Kizhakkedathu, M. J. Yaszemski, P. R. Greipp, D. Mukhopadhyay and P. Mukherjee, Nanomed. Nanotechnol. Biol. Med., 2007, 3, 224–238 CrossRef CAS
- C. Sun, R. Sze and M. Q. Zhang, J. Biomed. Mater. Res., Part A, 2006, 78, 550–557 CrossRef
- D. J. Bharali, D. W. Lucey, H. Jayakumar, H. E. Pudavar and P. N. Prasad, J. Am. Chem. Soc., 2005, 127, 11364–11371 CrossRef CAS
- C. Leuschner, C. S. Kumar, W. Hansel, W. Soboyejo, J. Zhou and J. Hormes, Breast Cancer Res. Treat., 2006, 99, 163–176 CrossRef CAS
- R. Weissleder, K. Kelly, E. Y. Sun, T. Shtatland and L. Josephson, Nat. Biotechnol., 2005, 23, 1418–1423 CrossRef CAS
- X. G. Pan, G. Wu, W. L. Yang, R. F. Barth, W. Tjarks and R. J. Lee, Bioconjugate Chem., 2007, 18, 101–108 CrossRef CAS
- G. P. Adams and L. M. Weiner, Nat. Biotechnol., 2005, 23, 1147–1157 CrossRef CAS
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS
- J. Yang, C. H. Lee, H. J. Ko, J. S. Suh, H. G. Yoon, K. Lee, Y. M. Huh and S. Haam, Angew. Chem., Int. Ed., 2007, 46, 8836–8839 CrossRef CAS
- A. M. Wu and P. D. Senter, Nat. Biotechnol., 2005, 23, 1137–1146 CrossRef CAS
- P. Holliger and P. J. Hudson, Nat. Biotechnol., 2005, 23, 1126–1136 CrossRef
- B. Barat, S. J. Sirk, K. E. McCabe, J. Q. Li, E. J. Lepin, R. Remenyi, A. L. Koh, T. Olafsen, S. S. Gambhir, S. Weiss and A. M. Wu, Bioconjugate Chem., 2009, 20, 1474–1481 CrossRef CAS
- H. W. Chen, J. Gao, Y. Lu, G. Kou, H. Zhang, L. Fan, Z. G. Sun, Y. J. Guo and Y. Q. Zhong, J. Controlled Release, 2008, 128, 209–216 CrossRef CAS
- W. W. Cheng and T. M. Allen, J. Control Release, 2008, 126, 50–58 CrossRef CAS
- D. K. Kirui, D. A. Rey and C. A. Batt, Nanotechnology, 2010, 21, 105105 CrossRef
- X. Qian, X. H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS
- K. L. Vigor, P. G. Kyrtatos, S. Minogue, K. T. Al-Jamal, H. Kogelberg, B. Tolner, K. Kostarelos, R. H. Begent, Q. A. Pankhurst, M. F. Lythgoe and K. A. Chester, Biomaterials, 2009, 31, 1307–1315
- L. Yang, H. Mao, Y. A. Wang, Z. Cao, X. Peng, X. Wang, H. Duan, C. Ni, Q. Yuan, G. Adams, M. Q. Smith, W. C. Wood, X. Gao and S. Nie, Small, 2009, 5, 235–243 CrossRef
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS
- Y. R. Wu, K. Sefah, H. P. Liu, R. W. Wang and W. H. Tan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5–10 CrossRef CAS
- J. E. Smith, C. D. Medley, Z. Tang, D. Shangguan, C. Lofton and W. Tan, Anal. Chem., 2007, 79, 3075–3082 CrossRef CAS
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS
- W. Hwang do, H. Y. Ko, J. H. Lee, H. Kang, S. H. Ryu, I. C. Song, D. S. Lee and S. Kim, J. Nucl. Med., 2010, 51, 98–105 CrossRef
- L. Q. Chen, S. J. Xiao, L. Peng, T. Wu, J. Ling, Y. F. Li and C. Z. Huang, J. Phys. Chem. B, 2010, 114, 3655–3659 CrossRef CAS
- B. Y. S. Kim, W. Jiang, J. Oreopoulos, C. M. Yip, J. T. Rutka and W. C. W. Chan, Nano Lett., 2008, 8, 3887–3892 CrossRef CAS
- B. D. Chithrani and W. C. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS
- B. D. Chithrani, A. A. Ghazani and W. C. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS
- T. S. Hauck, A. A. Ghazani and W. C. W. Chan, Small, 2008, 4, 153–159 CrossRef CAS
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS
- A. Sartori, R. Gatz, F. Beck, A. Rigort, W. Baumeister and J. M. Plitzko, J. Struct. Biol., 2007, 160, 135–145 CrossRef
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS
- F. Bertorelle, C. Wilhelm, J. Roger, F. Gazeau, C. Menager and V. Cabuil, Langmuir, 2006, 22, 5385–5391 CrossRef CAS
- N. Q. Jia, Q. Lian, H. B. Shen, C. Wang, X. Y. Li and Z. N. Yang, Nano Lett., 2007, 7, 2976–2980 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2011 |