Driving oxygen coordinated ligand exchange at nanocrystal surfaces using trialkylsilylated chalcogenides†‡
Marissa A.
Caldwell
b,
Aaron E.
Albers
a,
Seth C.
Levy
a,
Teresa E.
Pick
a,
Bruce E.
Cohen
a,
Brett A.
Helms
*a and
Delia J.
Milliron
*a
aThe Molecular Foundry, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, CA 94720, USA. E-mail: bahelms@lbl.gov; dmilliron@lbl.gov; Fax: +1 510 486 6166; Tel: +1 510 486 6723
bDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA
First published on 23rd November 2010
Abstract
A general, efficient method is demonstrated for exchanging native oxyanionic ligands on inorganic nanocrystals with functional trimethylsilylated (TMS) chalcogenido ligands. In addition, newly synthesized TMS mixed chalcogenides leverage preferential reactivity of TMS–S bonds over TMS–O bonds, enabling efficient transfer of luminescent nanocrystals into aqueous media with retention of their optical properties.
Advances in chemical synthesis have afforded high quality nanocrystals (NCs) of varying compositions with exceptional control over size and morphology.1 A central strategy used to achieve such control is the passivation of NC surfaces by hydrophobic ligands, such as oleic acid or octadecylphosphonic acid.2 However, for many applications it is necessary to replace the generic hydrophobic ligands with specific functional ligands, e.g., in order to facilitate aqueous dispersion,3 enable specific targeting4 or add complementary optoelectronic functionality.5 It remains a significant challenge to find general and efficient mechanisms for such ligand exchange reactions.
Conventionally, ligand exchange is carried out by mass action; excesses of several orders of magnitude of the new ligand are used to drive reactions. These reactions can nonetheless proceed slowly and the conditions can vary widely for specific ligand and inorganic core chemical compositions. For example, the commonly used exchange protocol for pyridine surface replacement involves hours of heating at elevated temperatures in neat pyridine,6 while amines or thiols are typically exchanged in concentrated solution at room temperature.7 Still other protocols involve phase transfer over hours or days.8
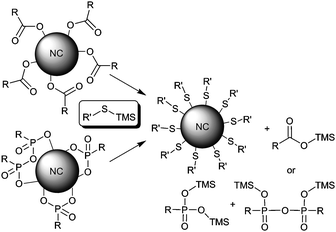
We hypothesized that broad applicability and greater efficiency might be achievable using a chemically-driven approach in which chemoselective reactions at the head groups of native ligands could render these ligands non-coordinating. Along this line, the Alivisatos group recently reported the use of various trimethylsilylated compounds (e.g., TMS-Cl or TMS-S-alkyl) to investigate the chemical nature of ligand binding sites at cadmium chalcogenide NC surfaces.9 In their scheme, native oxyanionic ligands at NC surfaces were chemically transformed by various TMS reagents to non-coordinating TMS phosphonate esters (and related species), with the reactive partner from the TMS-reagent replacing the oxyanionic ligand at the surface. We have investigated the extension of this chemoselective strategy to a diverse selection of NC compositions and common oxyanionic ligands (Scheme 1). We also report three novel TMS reagents derived from mercaptoalkanoic acids used for ligand exchange and quantitative transfer of quantum dots to aqueous buffers. We show conclusively that reactive ligand exchange by TMS-chalcogenides provides a general platform for efficient replacement of common native ligands on NCs of diverse inorganic composition with specifically designed functional ligands.
Proton Nuclear Magnetic Resonance (1H NMR) spectroscopy was used to evaluate the efficacy of oxygen coordinated ligand exchange using TMS reagents. For this purpose, trimethylsilylated 1-mercapto-3,6,9,12-tetraoxotridecane (TMS-S-TEG) was selected as the exchange ligand because of its distinct 1H NMR resonances between δ 3.2–4.0 ppm and was synthesized according to previously published procedures.10 By comparing the integration of 1H NMR resonances unique to the native and exchange ligands, respectively, we can approximate the exchange efficiency. In some cases native, unbound ligands persisted in the analyzed product, so the calculated exchange percentages are lower limits. These signals are readily distinguished in the post-exchange spectra where broad peaks sharpen, indicating a transition from a bound to an unbound state (Fig. 1).
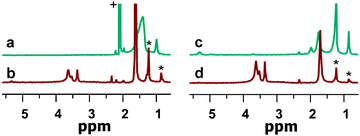 | ||
| Fig. 1 1H NMR spectra of phosphonate-capped CdSe NCs before (a) and after (b) ligand exchange and oleate-capped CdSe NCs before (c) and after (d) ligand exchange with TMS-S-TEG. (*) Denotes residual native ligand. (+) Denotes solvent impurity. | ||
To investigate generality of our reaction scheme with respect to both the native ligand functional head group and the inorganic core composition, several NCs were prepared. Alkylphosphonate- and alkylcarboxylate-capped CdSe and alkylcarboxylate-capped CdTe NCs were synthesized using an automated NC synthesis robot (the Workstation for Automated Nanomaterials Discovery and Analysis, or WANDA1a), and isolated using previously reported procedures.9,11Alkylcarboxylate-capped ZnO NCs were also synthesized and purified according to previously reported procedures.12 Reactive ligand exchange was performed by adding TMS-S-TEG (2–7 × 104 equivalents per NC, or approximately 8–100 per surface metal atom [ESI‡]) to a dispersion of NCs in chloroform and stirring for several hours at 25 °C. The TEG-S-bound NCs were isolated by several rounds of precipitation into hexanes from chloroform and subjected to NMR spectroscopic analysis in toluene-d8 to determine the extent of ligand exchange.
The impact of coordinating head groups on exchange efficiencies was evaluated by comparing alkylphosphonate and alkylcarboxylate-capped CdSe NCs. Despite differences in the chemistry of the native ligands, exchange occurred to approximately the same extent in both systems, affording 63% and 60% substitution, respectively. The composition of the inorganic core, however, was found to have a significant effect on ligand exchange efficiency. Alkylcarboxylate-capped CdTe NCs exhibited a higher extent of substitution (91%) compared to the analogous CdSe system, whereas similarly coordinated ZnO NCs showed markedly lower exchange (38%). This trend suggests that TMS-driven reactive ligand exchange is more effective on semiconducting NC compounds exhibiting more covalent character, and less effective on those which are more ionic (ionicity increases as CdTe < CdSe < ZnO), implying that the nanocrystal surface still exerts influence over the ligand exchange.
Encouraged by the general applicability of this reactive ligand exchange approach, we proceeded with novel trimethylsilylated reagents to transfer hydrophobic, highly luminescent semiconducting NCs to aqueous dispersions and evaluated the impact on NC optical characteristics. Ligand exchanges with hydrophilic ligands dihydrolipoic acid (DHLA), mercaptopropanoic acid (MPA), and mercaptoundecanoic acid (MUA) under mass action conditions have previously been reported as effective for preparing aqueous dispersions.13 We therefore synthesized TMS versions of these reagents (ESI‡) to evaluate the efficacy reactive ligand exchange for aqueous transfer.
Reactive ligand exchange was carried out on commercially available green-emitting (Qdot 545 ITK) or orange-emitting (Qdot 605 ITK) CdSe/ZnS core/shell hydrophobic quantum dots (Invitrogen Corporation) by adding the desired TMS reagent (5 × 105 equivalents per QD) to a dispersion of QDs in chloroform and stirring for several hours at 25 °C. An equal volume of sodium tetraborate buffer (50 mM, pH 10) was added, and the samples were allowed to stand for 1 h at 25 °C. Quantitative transfer of QDs to the aqueous phase was evidenced by the lack of detectable luminescence in the chloroform layer, with all green or orange luminescence observed in the aqueous phase.
To determine the hydrodynamic diameters of these aqueous QDs, dynamic light scattering (DLS) was performed before and after surface remodeling (Table 1). MPA-, DHLA-, and MUA-capped QDs followed a predictable size progression, with MPA producing the smallest QDs in each series. No evidence of aggregation was observed for any of the exchanged samples, in contrast to non-chemically driven exchange reactions under analogous conditions which resulted in severe aggregation [ESI‡]. In previous reports, longer reaction times and/or greater excesses were used to effect mass action exchange with this family of ligands.13,14
| Sample | d/nm | ΦI | ξ/mV |
|---|---|---|---|
| Qdot 545 ITK | 9 | 0.60 | NA |
| DHLA-QD545 | 10 | 0.07 | −34 |
| MPA-QD545 | 8 | 0.30 | −22 |
| MUA-QD545 | 13 | 0.52 | −40 |
| Qdot 605 ITK | 9 | 0.65 | NA |
| DHLA-QD605 | 13 | 0.03 | −23 |
| MPA-QD605 | 11 | 0.32 | −18 |
| MUA-QD605 | 20 | 0.35 | −56 |
NC optical absorption spectra were similar before and after exchange, indicating that these mild reaction conditions do not induce significant defects or etching of the NCs (Fig. 2). Before reaction, green-emitting QDs exhibit an absorbance maximum at 531 nm (Fig. 2a). Ligand exchange with bisTMS-DHLA, bisTMS-MPA, and bisTMS-MUA on these NCs proceeds with only slight variations in the resulting spectra, with the first exciton peak clearly resolved in all cases (Fig. 2a). Similar results were observed for orange-emitting NCs (Fig. 2b). Significantly enhanced absorbance below 450 nm, far from the excitonic features, was observed for MPA-capped QDs of both colors, though its origin is not yet known.
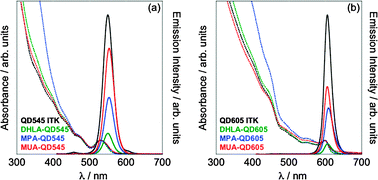 | ||
| Fig. 2 Absorption and emission spectra of variously capped (a) Qdot 545 NCs and (b) Qdot 605 NCs. Absorbance spectra, shown as dotted lines, are normalized at the first exciton peak. Emission spectra (solid lines) are normalized to reflect PL quantum yields. Spectra were acquired on 10 nM dispersions in CHCl3 or sodium tetraborate buffer (10 mM, pH 10) with excess exchange ligand at 1 mM at 25 °C (λex = 400 nm). | ||
Striking differences were observed in luminescence quantum yields before and after reaction with the three bisTMS mixed chalcogenides, though the spectral characteristics remained nearly unchanged in all cases (Fig. 2). Trends in PL quantum yields for both green- and orange-emitting QDs are similar; for both, reaction with bisTMS-DHLA proceeded with catastrophic losses in emission intensity (Fig. 2 and Table 1). In sharp contrast, bisTMS-MPA and bisTMS-MUA were much more effective in maintaining bright QDs in buffered aqueous environments. In the best case, the quantum yield of the MUA-capped QDs decreases by only 13% (7% absolute) following ligand exchange (Table 1). Similar decreases in quantum yield were also observed in MUA exchanged CdTe and CdSe cores (ESI‡).
Differences in ligand coverage, binding geometries, electronic passivation efficacy, and lability are known determinants of QD PL efficiency.15 While the contributions of these factors to the reported variations of quantum yield are challenging to disentangle, zeta potential measurements offer some insight (Table 1). All the QDs have negative zeta values, consistent with thiolate head group coordination to QD surfaces and carboxylate tail group presentation toward the aqueous environment. MPA consistently produced QDs with substantially smaller magnitude zeta values than MUA. Since these ligands each contain one thiolate and one carboxylate group per molecule, this trend likely reflects a higher coverage with MUA than with MPA. Furthermore, this zeta potential difference correlates with the differences in PL quantum yield between MPA- and MUA-capped QDs, suggesting that ligand coverage may be a major factor, particularly for the green QDs. However, the zeta potentials of DHLA-capped QDs lie between those of comparable MPA- and MUA-capped QDs, a result which does not correlate with their lowest PL quantum yield. In this case, however, zeta potential differences cannot be directly equated with differences in ligand coverage since DHLA contains an additional thiolate group per molecule. While further analyses will be required to fully unravel the combination of factors responsible, the empirical observations reported here are already useful in guiding ligand selection. Specifically, among ligands capable of facilitating aqueous dispersion of QDs, DHLA is particularly ill-suited for maintaining high PL quantum yield, while MUA introduced through our mild reactive exchange process is highly effective for smaller (green) QDs. MUA is also moderately effective for larger QDs, though the quantum yield of the orange QDs dropped from 65 to 35% following ligand exchange.
In summary, we have demonstrated the broad generality of oxyanionic ligand exchange at NC surfaces using TMS chalcogenido reagents. We have found that ligand exchange is largely independent of the native coordinating head group, but varies significantly with the inorganic core composition, correlating with the covalent character of the material. Among aqueous QD dispersions prepared using novel bisTMS ligands, MUA-capped QDs exhibit significantly higher quantum yield compared to QDs capped with DHLA or MPA. We anticipate that the mild reaction conditions and predictable functional group tolerance of this method may be leveraged to introduce designer ligands of interest for a wide variety of biological, electronic, and energy applications.
The authors gratefully acknowledge Dr Emory Chan for synthesis of CdSe and CdTe NCs using WANDA and Tracy Mattox for synthesis of ZnO NCs. Research was completed entirely at The Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA, USA and was supported by the Office of Science, Office of Basic Energy Sciences of the US Department of Energy under contract no. DE-AC02-05CH11231. M.A.C. is partially supported by the IBM Graduate Student Fellowship and the Stanford Non-Volatile Memory Technology Research Initiative (NMTRI) and its member companies. A.E.A. is partially supported by an Ernest Orlando Lawrence Postdoctoral Fellowship. S.C.L. was supported by the Office of Science, Department of Energy's Science Undergraduate Laboratory Internship (SULI) program.
Notes and references
- (a) E. M. Chan, C. Xu, A. W. Mao, G. Han, J. S. Owen, B. E. Cohen and D. J. Milliron, Nano Lett., 2010, 10, 1874 CrossRef CAS; (b) Y. Yin and A. P. Alivisatos, Science, 2005, 437, 664 CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630 CrossRef CAS.
- B. C. Mei, K. Susumu, I. Medintz and H. Mattoussi, Nat. Protoc., 2009, 4, 412 Search PubMed.
- (a) K. Susumu, B. C. Mei and H. Mattoussi, Nat. Protoc., 2009, 4, 424 Search PubMed; (b) M. Howarth, W. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397 CrossRef CAS.
- (a) D. J. Milliron, A. P. Alivisatos, C. Pitois, C. Edder and J. M. J. Fréchet, Adv. Mater., 2003, 15, 58 CrossRef CAS; (b) I. L. Medintz, T. Pons, S. A. Trammell, A. F. Grimes, D. S. English, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, J. Am. Chem. Soc., 2008, 130, 16745 CrossRef CAS; (c) E. J. McLaurin, A. B. Greytak, M. G. Bawendi and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 12994 CrossRef CAS; (d) D. Zheng, D. S. Seferos, D. A. Giljohann, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 3258 CrossRef CAS.
- (a) I. Lokteva, N. Radychev, F. Witt, H. Borchert, J. Parisi and J. Kolyny-Olesiak, J. Phys. Chem. C, 2010, 114, 12784 CrossRef CAS; (b) X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019 CrossRef CAS.
- F. Dubois, B. Mahler, B. Dubertret, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2007, 129, 482 CrossRef CAS.
- (a) B.-K. Pong, B. L. Trout and J.-Y. Lee, Langmuir, 2008, 24, 5270 CrossRef CAS; (b) W. Jiang, S. Mardyani, H. Fischer and W. C. W. Chan, Chem. Mater., 2006, 18, 872 CrossRef CAS; (c) A. M. Smith, H. Duan, M. N. Rhyner, G. Ruan and S. Nie, Phys. Chem. Chem. Phys., 2006, 8, 3895 RSC; (d) J. Aldana, Y. A. Wang and X. Peng, J. Am. Chem. Soc., 2001, 123, 8844 CrossRef CAS.
- J. S. Owen, J. Park, P.-E. Trudeau and A. P. Alivisatos, J. Am. Chem. Soc., 2008, 130, 12279 CrossRef CAS.
- (a) R. S. Glass, J. Organomet. Chem., 1973, 61, 83 CrossRef CAS; (b) A. W. Snow and E. E. Foos, Synthesis, 2003, 509 CrossRef CAS.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854 CrossRef CAS.
- Y. Chem, M. Kim, G. Lian, M. B. Johnson and X. Peng, J. Am. Chem. Soc., 2005, 127, 13331 CrossRef CAS.
- (a) G. P. Mitchell, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1999, 121, 8122 CrossRef CAS; (b) H. Mattoussi, J. M. Mauro, E. R. Goldman, G. P. Anderson, V. C. Sundar, F. V. Mikulec and M. G. Bawendi, J. Am. Chem. Soc., 2000, 122, 12142 CrossRef CAS.
- (a) W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS; (b) G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, 2nd edn, 2008 Search PubMed.
- (a) X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS; (b) F. Pinaud, X. Michalet, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Iver and S. Weiss, Biomaterials, 2006, 27, 1679 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Synthetic and experimental details, including ligand syntheses. See DOI: 10.1039/c0cc02220a |
| This journal is © The Royal Society of Chemistry 2011 |