Chemical tools to discover and target bacterial glycoproteins
Danielle H.
Dube
*,
Kanokwan
Champasa
and
Bo
Wang
Bowdoin College, Department of Chemistry & Biochemistry, Brunswick, Maine, USA. E-mail: ddube@bowdoin.edu; Fax: +1 207 725 3017; Tel: +1 207 798 4326
First published on 23rd August 2010
Abstract
Bacterial protein glycosylation is an important post-translational modification that can distinguish pathogenic bacteria from human cells. This review discusses recent findings in the field of bacterial glycobiology, with a particular focus on the unusual structures of bacterial glycans and their link to pathogenesis. We then describe how chemical tools can augment the study of this class of biomolecules, offering the potential to unveil novel pathogen-associated targets. Finally, this article highlights recent advances in targeting bacteria with therapeutics based on their unique glycans.
 Danielle H. Dube | Danielle Dube is an Assistant Professor of Chemistry and Biochemistry at Bowdoin College. She received her PhD in Chemistry from the University of California, Berkeley under the guidance of Professor Carolyn Bertozzi in 2005. She then pursued post-doctoral training with Professor Jennifer Kohler at Stanford University. In 2007 Danielle joined the faculty at Bowdoin College. Her research interests are in the areas of chemical biology and glycobiology, including the development of chemical tools to target, alter, and understand glycosylation. Recent work in the lab has focused on using chemical tools to discover and target bacterial glycoproteins. |
 Kanokwan Champasa | Kanokwan (Paggard) Champasa was born in Thailand and was named a Royal Thai Scholar in 2006. She has conducted undergraduate research in the Dube lab since Spring 2009, where she has focused on metabolic profiling of the bacteria H. pylori's glycoproteins. Upon graduating from Bowdoin College with Honors in Biochemistry in May 2011, she plans to pursue her PhD. |
 Bo Wang | Bo Wang started conducting lab-based research as a high school student, when he worked in a lab studying the neuroprotective properties of erythropoietin. In Spring 2009 he joined the Dube lab, where he has focused on targeting the pathogen H. pylori based on its unique glycans. After he graduates from Bowdoin College with Honors in Biochemistry in May 2011, he intends to pursue his MD. |
1. Introduction
The effective treatment of infectious bacterial diseases is plagued by challenges associated with the emergence of bacterial strains that have evolved resistance to existing antibiotics. For example, 50% of Staphylococcus aureus isolates in US hospitals are methicillin-resistant,1 and extensively drug resistant tuberculosis poses a global threat.2 Therefore, there is an urgent need to identify more pathogen-associated targets that might be harnessed for selective treatment of bacterial infections.One long-overlooked class of molecular targets is bacterial glycoproteins. These biomolecules were once thought to be absent from bacteria, but recent work in the field of bacterial glycobiology has firmly established the synthesis of glycoproteins in bacteria.3–5 Two important themes have surfaced: (1) the ability of pathogenic bacteria to synthesize glycoproteins is often directly linked to their ability to cause disease;6 and (2) bacterial glycans have unique structures that distinguish them from their eukaryotic counterparts.7–10 Bacterial glycoproteins are therefore attractive targets for therapeutic intervention.
Despite their appeal as molecular targets, we know relatively little about the abundance, structure and function of bacterial glycoproteins. This paucity of knowledge is due in part to challenges associated with studying glycosylated proteins in any organism. Glycans are often complex, branched, heterogeneous structures, and their biosynthesis is not template directed.11 Moreover, diverse classes of enzymes are required for glycan biosynthesis and protein attachment. These characteristics render the use of genetic techniques to study glycoproteins difficult. The study of glycoproteins present in bacteria is further convoluted by their atypical structures, which differ from eukaryotic structures.7–10 While tools such as lectins (proteins that bind to glycans) can be used to globally profile bacterial glycoproteins,12 they do not reveal the details of glycan structures or their point of attachment to proteins.
Chemical tools can facilitate the detection and molecular characterization of glycoproteins and thus offer tremendous benefits for studying glycoproteins in a refined manner.13,14 For example, unnatural sugar analogs can label glycoproteins and ease enrichment, while mass spectrometry analysis can enable structural elucidation of glycoproteins. To date however, such tools have primarily focused on eukaryotic systems. The transfer of these tools from eukaryotic to prokaryotic systems to identify bacterial glycoproteins is not always straightforward.7 Hence, new tools are needed to study bacterial glycoproteins.
Here we provide an overview of the emerging toolset to study bacterial glycosylation. We begin with a brief review on bacterial glycoproteins, with a particular focus on their link to pathogenesis and unusual structures that have already been characterized. We then highlight chemical approaches to discover and characterize novel bacterial glycoproteins, offering the potential to unveil more pathogen-associated targets. Finally, we describe chemical approaches that could be used to target bacteria with therapeutics based on their distinctive glycans.
1.1 Bacterial glycoproteins are often linked to virulence
Protein glycosylation, the covalent addition of carbohydrates to proteins, is a ubiquitous post-translational modification that is most commonly associated with eukaryotic cells. Glycoproteins cover the surfaces of all eukaryotic cells, where they are crucial for mediating interactions between cells and their environment. Although the presence and importance of protein glycosylation in eukaryotes has been established for decades, protein glycosylation was long-believed to be absent from bacteria because bacterial cells lack the sophisticated compartmentalization that eukaryotic cells utilize during glycoprotein synthesis.3,6,67 For years, bacterial cell wall carbohydrates were thought to be the only sugar structures present in bacteria. However, recent work by Szymanski et al.,30Loganet al.,42 Comstock et al.68 and others has demonstrated that glycoproteins are in myriad bacterial species.Bacterial glycoproteins have been identified in a number of medically significant pathogens.3 Moreover, the ability of pathogenic bacteria to synthesize glycoproteins is in many cases directly linked to their ability to colonize the human host and cause disease (Table 1). For example, in the pathogens Campylobacter jejuni,24Escherichia coli H10407 and 2787,35,37Haemophilus influenzae,40 and Mycobacterium tuberculosis,45,46protein glycosylation appears to be directly involved in bacterial adherence and invasion. In C. jejuni17 and Helicobacter pylori,42glycosylation is crucial for proper flagella formation, motility, and colonization. In Neisseria sp.51 and Pseudomonas aeruginosa,60protein glycosylation is critical for the initiation and spread of infection. When the synthesis of bacterial glycoproteins is prevented through deletion of glycosylation genes, these pathogens lose their ability to adhere to and invade mammalian cells, initiate infection, and colonize their host.17,24,42 These observations suggest that inhibition of bacterial glycosylation enzymes could eliminate certain bacterial infections.
| Species | Protein(s) | Glycan structure (type)a | Glycosylation genes | Link to pathogenesis | Reference(s) |
|---|---|---|---|---|---|
| a Abbreviations: Glc = glucose; Gal = galactose; Man = mannose; Xyl = xylose; GalNAc = N-acetylgalactosamine; FucNAc = N-acetylfucosamine; DATDH = 2,4-diacetamido-2,4,6-trideoxyhexose; GATDH = glyceramido acetamido trideoxyhexose; Pse = pseudaminic acid; Bac = bacillosamine; Leg = legionaminic acid. | |||||
| C. jejuni | FlaA (flagellin) | Pse, Pse variants (O-linked) | PseB, C, G, H, I, F | Flagella assembly, motility, and colonization of host | 10, 15–22 |
| C. jejuni | Multiple, secreted | GalNAc2(Glc)GalNAc3Bac (N-linked) | Pgl glycosylation system | Adherence, invasion, animal colonization | 9, 23–30 |
| Campylobacter coli | FlaA (flagellin) | Pse, Pse variants, Leg (O-linked) | PseB, C, G, H, I, F | Flagella assembly, motility, and colonization of host | 17, 22, 31, 32 |
| E. coli H10407 | TibA adhesin, EtpA adhesin | Heptoses | TibC, Putative EtpC | Adherence, invasion, intestinal colonization; biofilm formation | 33–37 |
| E. coli 2787 | AIDA-I adhesin | Heptoses | Autotransporter adhesion heptosyltransferase (AAH) | Adherence, invasion | 36, 38, 39 |
| H. influenzae | HMW1 adhesin | Gal, Glc, Man; mono- and di-saccharides (N-linked) | HMW1C | Adherence to host epithelium | 40, 41 |
| H. pylori | FlaA, FlaB (flagellin) | Pse (O-linked) | PseB, C, G, H, I, F | Flagella assembly, motility, and colonization of host | 10, 19, 42–44 |
| M. tuberculosis | Multiple | Man; mono-, di- and tri-saccharides (O-linked) | Rv1002c protein mannosyltransferase | Adherence; protection from proteolysis; known and putative virulence factors | 45–50 |
| Neisseria meningitidis | Pilin | Gal2DATDH or Gal2GATDH (O-linked) | Pgl glycosylation system | Implicated in blocking immune response and spread of infection | 8, 51–55 |
| Neisseria gonorrhoeae | Pilin | Gal2DATDH (O-linked) | Pgl glycosylation system | Implicated in adherence, blocking immune response, and spread of infection | 51, 55, 56 |
| P. aeruginosa 1244 | Pilin | 5N-β-OH-C4-7NFm-Pse-Xyl-FucNAc (O-linked) | PilO | Aids in establishment of infection in mouse model | 57–61 |
| P. aeruginosa PAK | Flagellin (a-type) | Rhamnose core, with 4-amino-4,6-viosamine and other hexoses (O-linked) | Flagellin glycosylation island | Linked to virulence in mouse burn model | 62–66 |
The glycosylation enzymes responsible for the synthesis of bacterial glycoproteins have been elucidated in a number of cases. For example, Guerry, Logan and coworkers have characterized the enzymes responsible for the biosynthesis of the unusual sugar pseudaminic acid and its installment on flagellin proteins in C. jejuni17,19 and H. pylori,44 respectively. In addition, Jennings and coworkers have elucidated the glycosylation system responsible for the synthesis of glycans on the pili of Neisseria.52–55 Szymanski et al. and Wren et al. have reported the general glycosylation system responsible for the synthesis of glycans in C. jejuni.27,30 Finally, Comstock and colleagues have identified the general glycosylation system that installs glycans in the intestinal symbiont Bacteroides fragilis.68 These studies have revealed that, while several enzymatic steps are required for glycoprotein formation, bacteria appear to have only one enzyme encoded for each critical glycosylation reaction. This is in contrast to mammalian systems that have up to twenty glycosyltransferases that are capable of catalyzing the same reaction.69 Therefore, bacterial enzymes responsible for glycoprotein synthesis are particularly attractive disease targets.
1.2 Bacterial glycans have unique structures
The bacterial glycoproteins characterized to date largely fall into one of two classes, those with asparagine-linked (N-linked) glycans and those with serine/threonine-linked (O-linked) glycans. Although these two classes of glycoproteins are also found in eukaryotes,11 the structures of the glycans found on bacterial proteins differ markedly from their eukaryotic counterparts (Fig. 1). In contrast to the structure of N-linked glycans in eukaryotes, which contain an N-acetylglucosamine (GlcNAc)–Asn linkage70 (Fig. 1a, blue), N-linked glycans in C. jejuni contain a bacillosamine core9 (Fig. 1b, purple). Moreover, whereas O-linked glycans in eukaryotes contain GlcNAc, fucose, or N-acetylgalactosamine (GalNAc) linked to serine/threonine71 (Fig. 1a, maroon), O-linked glycans in bacteria have atypical monosaccharides. For example, 2,4-diacetamido-2,4,6-trideoxyhexose (DATDH),8N-acetylfucosamine (FucNAc),72pseudaminic acid,20 and legionaminic acid31 are at the core of O-linked bacterial glycans (Fig. 1b, orange, green, red, and blue, respectively). Most strikingly, several of the monosaccharide building blocks present in bacterial glycoproteins are amino- and deoxy-carbohydrates exclusively found in bacteria.8,9,73,74 In many cases these glycan structures are found on bacterial surfaces or secreted proteins involved in motility or adhesion (Table 1). Many of these distinctive monosaccharides are absent from human cells and have only been observed in a small number of pathogenic bacteria.8–10,42,74 Given their exclusive presence on bacterial cells, these glycan structures could serve as potential targets of therapeutic intervention.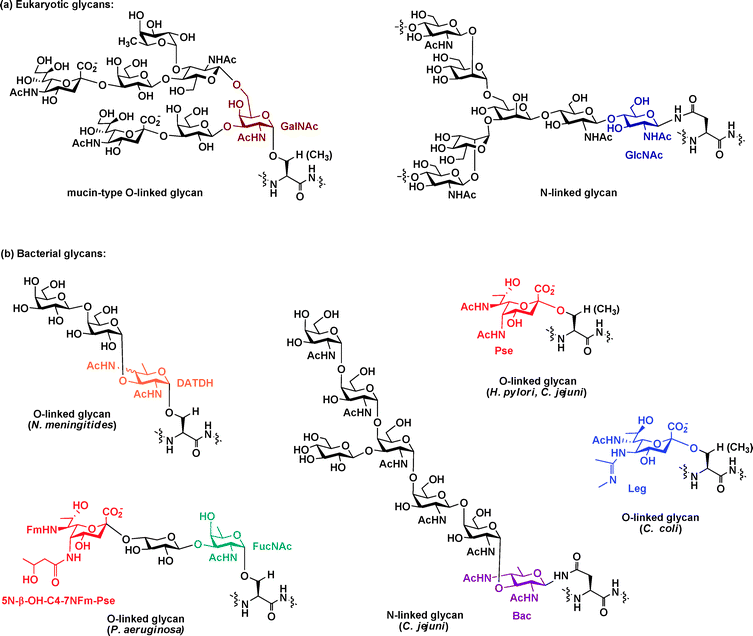 | ||
| Fig. 1 Bacterial glycan structures differ from their eukaryotic counterparts. (a) The two major classes of eukaryotic glycans, N-linked and O-linked, are depicted with GlcNAc and GalNAc, respectively, attached directly to the protein and highlighted. (b) Structures of some well-characterized bacterial glycans are depicted. The unusual amino- and deoxy-carbohydrates (DATDH, Pse, FucNAc, Bac, Leg) exclusively present in bacterial glycans are highlighted. Abbreviations: GlcNAc = N-acetylglucosamine; GalNAc = N-acetylgalactosamine; FucNAc = N-acetylfucosamine; DATDH = 2,4-diacetamido-2,4,6-trideoxyhexose; Pse = pseudaminic acid; Bac = bacillosamine; Leg = legionaminic acid. | ||
1.3 Bacterial glycoproteins remain underexplored
Even after Szymanski, Comstock and others30,42,68 firmly established the synthesis of glycoproteins in bacteria, bacterial glycoproteins have remained underexplored relative to their eukaryotic counterparts.7 With the exception of a small number of well-characterized examples, fundamental questions in bacterial glycobiology remain largely unanswered. Common questions for a range of bacterial species include: How many proteins are modified with glycans? What is the identity of these glycan-modified proteins? What are the structures of the glycans? What is their biological function? And finally, what is their evolutionary purpose? The process of answering these questions requires taking inventory of bacterial glycoproteins and developing tools for their selective manipulation.Most successful studies of bacterial glycoproteins have focused on certain features of the bacteria (e.g. flagellin16,42,62,65 or pili proteins57,58) and have not been performed at an organismal level. The global profiling of bacterial glycoproteins has been hampered by the relatively low abundance of these species in the bacterial milieu and the frequent inability to directly transfer biological methods that have been used to study eukaryotic glycoproteins to study their bacterial counterparts.7 One biological approach that is capable of detecting and globally profiling bacterial glycoproteins involves the use of carbohydrate-binding proteins called lectins7 (Fig. 2a).
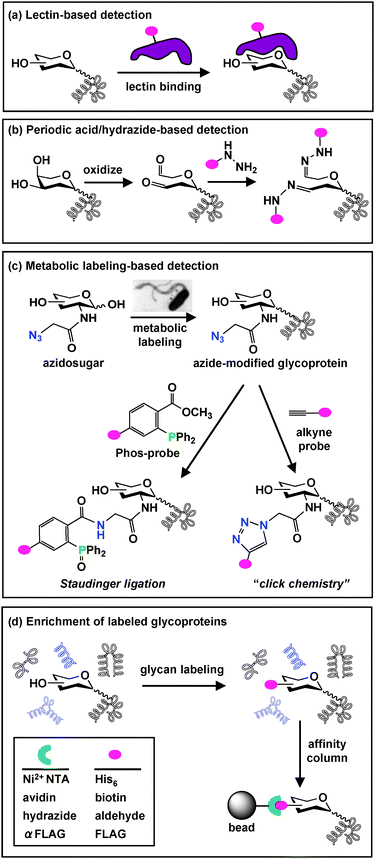 | ||
Fig. 2
Glycoprotein detection and enrichment strategies. (a) A common biological approach for glycan detection utilizes carbohydrate-binding lectins, which can be conjugated to an epitope tag ( ) for visualization. (b) A chemical approach oxidizes cis-diols of carbohydrates to aldehydes using periodic acid. Aldehydes are then reacted with hydrazide-conjugated tags to allow covalent attachment via a hydrazone linkage. (c) Azidosugars can be metabolically incorporated into cellular glycans to introduce azides into glycan structures. Azides can then be reacted with alkyne probesvia “click chemistry” (right) or phosphine probes (Phos-probe) via Staudinger ligation (left) to enable covalent attachment of probes to azide-labeled glycans. (d) Glycoproteins labeled using any of the described detection strategies (a–c) can be enriched from other cellular proteinsvia affinity column chromatography. ) for visualization. (b) A chemical approach oxidizes cis-diols of carbohydrates to aldehydes using periodic acid. Aldehydes are then reacted with hydrazide-conjugated tags to allow covalent attachment via a hydrazone linkage. (c) Azidosugars can be metabolically incorporated into cellular glycans to introduce azides into glycan structures. Azides can then be reacted with alkyne probesvia “click chemistry” (right) or phosphine probes (Phos-probe) via Staudinger ligation (left) to enable covalent attachment of probes to azide-labeled glycans. (d) Glycoproteins labeled using any of the described detection strategies (a–c) can be enriched from other cellular proteinsvia affinity column chromatography. | ||
The identification of >30 N-linked glycoproteins in C. jejuni is a successful example of lectin-based screening to globally profile bacterial glycoproteins. Linton et al. screened C. jejuniproteins with seven lectins and observed that soybean agglutinin (SBA), which has affinity for terminal GalNAc, bound to a large number of proteins.26 The specificity of SBA binding to GalNAc-modified proteins was confirmed by preincubating SBA with the monosaccharide GalNAc to compete away the interaction. Alternatively, specificity was confirmed by treating C. jejuni's proteins with a GalNAc-specific glycosidase that removes GalNAc residues prior to Western blot analysis. In both treatments, reduced SBA binding to C. jejuniproteins was observed relative to untreated controls. The authors then enriched GalNAc-binding proteinsvia immobilized SBA and identified two putative glycoproteins, PEB3 and CgpA, by mass spectrometry (MS) analysis.26 Follow-up glycan characterization experiments later confirmed that these proteins are modified with glycans containing terminal GalNAc.9 Therefore, with sufficient control experiments, lectin probing can detect bacterial glycoproteins and unveil some information about monosaccharide composition.
Lectin arrays are now available to speed the lectin screening process.75–77 Recently, up to 21 lectins have been covalently tethered on a glass slide to screen bacterial surface glycans.12 So far this microarray has enabled rapid detection of glycosylation patterns and dynamic changes in surface glycans of pathogenic E. coli during growth. This technology will greatly enhance our ability to track changes in glycoprotein expression and observe upregulation of unusual structures in response to environmental stimuli.
However, even when biological reagents such as lectins can be employed to detect and globally profile bacterial glycoproteins, they do not reveal the intricacies of glycan structures.7,78 Lectins display promiscuous binding specificities, rendering detailed structural characterization based on lectin binding alone impossible. To unambiguously identify the details of glycan structure, one must turn to MS or nuclear magnetic resonance (NMR) analysis.79 Moreover, prior knowledge of glycan structure required to select appropriate lectins is rarely available for the study of bacterial glycoproteins.7 Finally, not every glycan structure has a corresponding lectin partner.7,78 The limitations of lectin-based approaches for the study of bacterial glycoproteins contribute to our relative lack of understanding about this class of biomolecules. Therefore, there is a critical need for technologies that facilitate the analytical study of bacterial glycoproteins. Chemical tools have much to contribute in this area, as they have successfully enabled bacterial glycoprotein discovery78 and can provide detailed information that other methods cannot.79
2. Chemical approaches to discover bacterial glycoproteins
To globally profile bacterial glycoproteins, it is essential to find methods that are capable of detecting and identifying carbohydrates that are composed of atypical monosaccharide building blocks. A general approach to discovering bacterial glycoproteins requires the following steps, which are described in detail below: (1) detecting and enriching bacterial glycoproteins, (2) identifying modified proteins, and (3) characterizing the modifying glycans.2.1 Detecting and enriching bacterial glycoproteins
A combination of periodic acid/hydrazide labeling and lectin binding provided the first evidence of flagellin glycosylation in the pathogenic bacteria C. jejuni.16 Initial gel electrophoretic analysis of C. jejuni flagellin proteins demonstrated that the proteins were larger than predicted based on amino acid composition alone. The observed mass shift was confirmed to be the result of protein glycosylation using periodic acid oxidation and hydrazide-conjugated biotin.16 Western blot analysis with six different lectins revealed that the sialic acid-specific lectin, Limax flavus agglutinin (LFA), bound to flagellin proteins, suggesting that the proteins were modified by sialic acid. However, detailed structural analysis of C. jejuni's flagellin glycan ultimately revealed that these flagellin proteins are modified with the atypical nine-carbon monosaccharide pseudaminic acid, a structure that is related to but distinct from sialic acid.20
After glycan detection, affinity column chromatography is the most convenient method to enrich glycoproteins for identification7 (Fig. 2d). One purification option involves immobilizing glycan detection reagents on agarose beads to facilitate glycoprotein isolation. For example, lectin and hydrazide beads are commercially available and can be used to separate glycoproteins from their non-glycosylated counterparts. Alternatively, biotinylated reagents can be used for enrichment in conjunction with avidin beads (Fig. 2d). Once enriched, bacterial glycoproteins can be structurally characterized using a combination of MS and NMR-based analyses.79
The usefulness of lectin affinity column and periodic acid/hydrazide chemistry in profiling bacterial glycoproteins was recently demonstrated for the pathogenic bacteria Francisella tularensis.84 In this study, glycoproteins were enriched by one of two ways, using either lectin affinity columns or using a hydrazide-conjugated dye to probe all cellular proteins in a 2-dimensional (2D) gel format. Mass spectrometry analysis identified 104 putative glycoproteins in F. tularensis.
While periodic acid/hydrazide labeling and lectin binding can be used to globally profile bacterial glycoproteins, they provide only a static image of the bacterial glycome. These approaches are most powerful when used in conjunction with dynamic labeling approaches.
The most useful bioorthogonal moiety has proven to be the azide because of its small size, its absence in biological systems, its stability within cellular glycans, and its unique reactivity with phosphine and alkyne-based probes.87 Unnatural azide-containing sugar analogs (azidosugars) of sialic acid (Sia),92N-acetylgalactosamine (GalNAc),93N-acetylglucosamine (GlcNAc),94 and fucose (Fuc)95,97 have been incorporated into several classes of eukaryotic cellular glycans without the disruption of further glycan elaboration of the glycoproteins.13 Azide-labeled glycans can be covalently labeled with (1) phosphine probesvia Staudinger ligation,92 (2) alkyne probesviacopper-catalyzed azide–alkyne cycloaddition (click chemistry),98,99 or (3) cyclooctyne probesviacopper-free click chemistry100,101 (Fig. 2c). These modified glycoproteins can then be detected and enriched by virtue of their covalently attached epitope tags13,89 (Fig. 2d).
To enrich and identify azide-labeled glycoproteins, azides modified with probes containing epitope tags such as the FLAG peptide, biotin, or hexahistidine (His6) can be purified using anti-FLAG, avidin, or nickel–NTA conjugates, respectively88,89,102 (Fig. 2d). Although enrichment of azide-labeled glycoproteins has not yet been reported in a bacterial system, well-established protocols used in eukaryotic systems should be directly transferable. Using MOE and enrichment strategies, azide-modified glycoproteins have been globally identified in eukaryotic systems ranging from cancer cells102,103 to zebrafish104 and mice.105,106
Our laboratory recently demonstrated that MOE can similarly facilitate the study of bacterial glycoproteins.107 Based on the observation that GlcNAc is a common metabolic precursor to bacterial monosaccharides,19,44 we hypothesized that an azide-containing version of GlcNAc, N-azidoacetylglucosamine (GlcNAz),94 would be metabolically incorporated into bacterial glycoproteins and enable their global profiling. To test this hypothesis, we employed a peracetylated version of GlcNAz, Ac4GlcNAz, to metabolically label and profile H. pylori's glycoproteins. Peracetylation of this unnatural sugar facilitates its cellular entry, where it is converted to GlcNAz, processed by cellular machinery, and ultimately incorporated into glycoproteins.94 Treatment of H. pylori with Ac4GlcNAz resulted in robust azide-labeling of a large number of glycoproteins, far more than the two flagellin glycoproteins that had been previously characterized in H. pylori.107 Enzymatic treatment with peptide-N-glycosidase F (PNGase F), a glycosidase that removes N-linked glycans, or with a cocktail of O-glycosidases, which remove O-linked glycans, removed the azide label from proteins. These data provide strong evidence that the labeled bands observed upon metabolic labeling with Ac4GlcNAz are indeed glycoproteins. Efforts are currently underway in our laboratory to identify these glycoproteins and the nature of the modifying glycan. MOE should be broadly applicable in bacteria and will greatly facilitate the study of bacterial glycoproteins.
2.2 Identifying glycosylated bacterial proteins
The profiling/enrichment techniques interface nicely with traditional structural elucidation techniques. MS has been widely used to identify proteins and carbohydrate structures with high sensitivity and accuracy, and the availability of whole genome sequences allows rapid identification of proteins based on peptide segments detected during MS analysis.79 The strategy for identification of bacterial proteins is highly similar to that employed for identifying eukaryotic proteins and was reviewed in detail by Thiede et al.108 In principle, the enriched glycoproteins are separated via2D gel electrophoresis or liquid phase chromatography (LC) to yield a sample that contains a single protein. Following separation, proteins are enzymatically cleaved by in-gel digestion to generate peptides with suitable sizes for MS analysis. MS was successfully used to identify the first two non-flagellin glycoproteins, PEB3 and CgpA, in C. jejuni.26Additional sequence information of separated proteins can be generated by tandem MS (MS/MS). This approach was recently utilized to identify glycoproteins in the pathogen M. tuberculosis.45 Gonzalez-Zamorano and coworkers enriched mannosylated proteins from M. tuberculosis using the mannose-specific lectin ConA and then analyzed the enriched proteins by online LC-electron spray ionization (ES) MS.45Protein identification using MS/MS spectrum data was carried out using the MASCOT search algorithm109 with M. tuberculosisgenome sequence information. This study identified 41 putative mannose-containing proteins.45 Intriguingly, the majority of the identified glycoproteins correspond to known mycobacterial virulence factors, emphasizing the link between bacterial protein glycosylation and pathogenicity. These data suggest that identifying a group of similarly-modified proteins can provide insight into the crucial roles of specific glycans in bacteria.
2.3 Characterizing the glycans of bacterial glycoproteins
Unlike eukaryotic glycosylation, structural elucidation of bacterial glycans is even more challenging due to the presence of previously unidentified monosaccharide units, the lower abundance of bacterial glycoproteins, and the absence of common glycosylation pathways.79 Therefore, MS-based strategies for bacterial glycan characterization require further considerations and specialization.79 Successful MS-based strategies for characterizing bacterial glycans, including “bottom up” and “top down” approaches, are described below. The “bottom up” approach entails proteolyzing glycoproteins and structurally characterizing glycopeptides, whereas the “top down” approach involves directly characterizing the intact glycoprotein (Fig. 3).
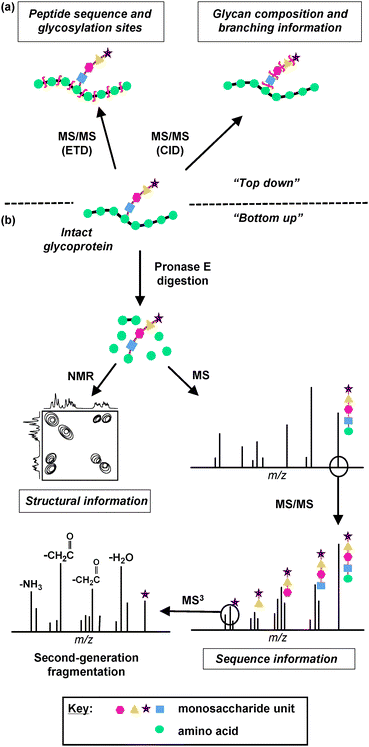 | ||
| Fig. 3 MS and NMR-based strategies for glycan characterization. Information about glycan structure can be derived by two MS approaches, (a) “top down” and (b) “bottom up”. (a) In the “top down” approach, an intact glycoprotein is analyzed directly using Electron Transfer Dissociation (ETD) or Collision Induced Dissociation (CID) MS techniques. ETD induces backbone fragmentation, resulting in a mass spectrum that reveals information about peptide sequence and glycosylation sites. CID favors fragmentation of glycosidic bonds, providing information about glycan composition and branching. (b) In the “bottom up” approach, the glycoprotein is first digested by Pronase E to yield the glycan moiety attached to a single amino acid. This glycan product is then analyzed by NMR to acquire structural information about the glycan and/or by MS and MS/MS to acquire sequence information. An ion peak of a monosaccharide can be subjected to second-generation fragmentation (MS3), which results in the loss of carbohydrate side chains and provides structural information about the monosaccharide unit. | ||
To characterize glycans with several monosaccharide units using the “bottom up” approach, glycopeptides are first generated from glycoproteins. Since PNGase F is unable to cleave certain carbohydrate–protein linkages, a non-specific protease, such as Pronase E, is used to generate single amino acid fragments that contain glycan moieties attached.113 The preferred amino acid–glycan product can be accessed by several rounds of Pronase E digestion with a high enzyme concentration and long incubation time. After digestion, the resulting glycans are subjected to MS and MS/MS, or NMR, analyses to obtain carbohydrate sequences and structures. This approach was successfully used to derive the structure and linkage of the glycan on C. jejuni's PEB3 protein.9 The Pronase E digested glycan product was characterized by NMR, leading to the identification of a novel monosaccharide building block, bacillosamine (Bac), on the Glc1GalNAc5Bac–Asn structure (Fig. 1b, purple). The site of glycan attachment was determined viaMS analysis by analyzing amide bond fragmentation ions to identify fragments that retain extra mass due to glycosylation.
The efficiency of the “top down” approach for characterization of bacterial glycoproteins was demonstrated in Schirm and coworker's analysis of flagellin glycosylation.10 In addition to the glycan structures and glycosylation sites reported in previous studies of C. jejuniglycosylation, this study found N-acetylglutamine linked to pseudaminic acidvia an ester bond, a novel glycan not observed in a “bottom up” analysis of the same C. jejuni flagellin.10 The elimination of the digestion and separation steps renders the “top down” approach highly sensitive. Therefore, it can be used to determine specific changes in modification patterns as a result of mutation or environmental conditions, changes which might be missed using the “bottom up” approach. The limitation of glycan structure characterization using a “top down” approach is that the MS-derived information is not enough to derive detailed structural information of novel glycans. Therefore, these experiments must be complemented with NMR analyses to obtain conformational and linkage information.116
Recent advances in our ability to structurally characterize bacterial glycans using MS and NMR-based analyses have greatly enhanced our understanding of bacterial glycobiology and have already unveiled unique structures that could serve as targets of therapeutic intervention. Chemical tools can facilitate the detection and molecular characterization of bacterial glycoproteins and thus offer tremendous potential for expanding our understanding of bacterial glycoproteins in the future.
3. Chemical approaches to target bacteria with therapeutics based on their unique glycans
The discovery of bacterial glycoproteins has revealed that bacteria utilize unusual monosaccharide building blocks that are absent from eukaryotes. As described above, in several cases the ability of bacteria to synthesize these atypical monosaccharides is directly tied to their virulence. These observations have stimulated the development of therapeutic strategies based on the presence of unique glycans on bacterial cells. In particular, three approaches are being applied to target bacterial glycans and glycosylation enzymes: (1) carbohydrate-based vaccination, (2) small-molecule inhibition of glycoprotein synthesis, and (3) metabolic disruption and covalent targeting of modified glycans. Below we describe these approaches and recent efforts in these areas.3.1 Carbohydrate-based vaccines
Efforts to improve upon glycan-targeting vaccines have focused on increasing the immunogenicity of glycans. One strategy that has achieved great success employs conjugate vaccines.121,123,124 In this approach, unique carbohydrate structures, either purified or synthetic, are linked to a carrier protein (Fig. 4a).124 In some cases the carrier protein, such as keyhole-limpet hemocyanin (KLH), possesses immune adjuvant characteristics that promote an immune response against the conjugate vaccine.125 When the conjugate vaccine circulates in the body, it elicits an immune response which leads to the production of antibodies against the glycan moiety (Fig. 4a). The adaptive immune system then mounts a defense against pathogenic cells covered with the carbohydrate antigen.126
 | ||
| Fig. 4 Conjugate carbohydrate-based anti-bacterial vaccines. (a) When an anti-bacterial conjugate vaccine circulates in the body, it elicits an immune response which leads to the production of antibodies against the glycan moiety of the vaccine. The immune system then mounts a defense against pathogenic cells covered with the carbohydrate antigen. This response in turn leads to eradication of pathogenic cells from the host. As shown, the conjugate vaccine is a therapeutic measure used after infection has occurred. Alternatively, the vaccine can be a prophylactic measure given before bacterial challenge. (b) Structures of synthetic conjugate vaccines containing a trisaccharide or tetrasaccharide moiety of Bacillus anthracis's spore glycoprotein BclA. These vaccines, which contain the unique sugar 2-O-methyl-4-(3-hydroxy-3-methylbutamido)-4,6-dideoxy-D-glucopyranose (anthrose), are antigenic in animal infection models and direct an immune response against the anthrose structure. | ||
The efficacy of conjugate vaccines was first demonstrated by Avery and Goebel in 1929.127 These authors established that conjugate vaccines increase the immunogenicity of polysaccharides and can initiate a long-term immune response in adults and infants against bacteria containing these glycan structures.127 Since that first demonstration, there has been a long history of utilizing anti-bacterial carbohydrate–protein conjugate vaccines to target glycans present on bacterial surfaces.123 In fact, a number of conjugate vaccines containing bacterial cell wall structures are used clinically. For example, Menactra MCV4 is a tetravalent vaccine consisting of an N. meningitidispolysaccharide core linked to diphtheria toxoid, and it demonstrates extremely high efficiency against several forms of meningitis.128 Prevenar is a licensed heptavalent vaccine consisting of Streptococcus pneumoniae's CPS linked to the carrier protein CRM197, and it affords protection in 97% of vaccinated infants.129 ActHib is one of a number of clinically available vaccines that protects against H. influenzae; like many of the others, it is a conjugate vaccine containing CPS purified from H. influenzae type b (Hib), and it has greatly decreased Hib infections in populations where it is used as a universal infant vaccine.130,131 Quimi-Hib, a fully synthetic conjugate vaccine containing components of Hib's CPS, provides long-term protection against Hib in infants and is now used routinely to immunize infants and children in Cuba.132 In addition to conjugate vaccines already in use in the clinic, myriad others are in the pipeline.133
Often, carbohydrate–protein conjugate vaccines fail to elicit a strong host immune response against bacterial glycans because the bacterial glycans are too similar to host glycans. For example, conjugate vaccines prepared with type V CPS isolated from Group B Streptococcus (GBS) produce only a weak host immune response. Kasper and coworkers hypothesized that this weak response was due to the presence of sialic acid, a monosaccharide also present on mammalian cells, on type V CPS.134 These authors demonstrated that a type V CPS conjugate vaccine that lacked sialic acid residues produced a robust host immune response.134 Similarly, Jennings and coworkers used a vaccine containing a chemically modified form of the surface antigen polysialic acid to elicit an immune response against N. meningitidis.135,136 Therefore, chemical modification of bacterial glycan structures that resemble host glycans promises to afford improved anti-bacterial carbohydrate-based vaccines in the future.
Bacterial glycoproteins need not be inherently immunogenic to form the basis of anti-bacterial vaccines. Boons and coworkers reported that a synthetic conjugate vaccine containing a trisaccharide moiety of Bacillus anthracis's spore glycoprotein BclA (Fig. 4b), which contains the unique sugar 2-O-methyl-4-(3-hydroxy-3-methylbutamido)-4,6-dideoxy-D-glucopyranose (anthrose), is antigenic in a rabbit infection model and directs an immune response against the anthrose structure.138 Seeberger and coworkers found complementary results that anthrax's anthrose tetrasaccharide conjugated to KLH (Fig. 4b) elicited antibodies in mice that recognize and bind to anthrax spores.139
These early results suggest that bacterial glycoproteins and conjugate vaccines containing their glycans will be important components of new anti-bacterial therapies. Advances in this area will be greatly aided by the continued discovery and characterization of bacterial glycoproteins, as well as efforts to chemically synthesize glycans containing distinctive bacterial monosaccharide building blocks.
Recent advances in carbohydrate synthesis technologies have enabled the utilization of automated techniques such as one-pot142 or solid phase oligosaccharide synthesis143 for the production of homogenous carbohydrates that can be tethered to protein carriers.141 The production of the first clinical synthetic conjugate vaccine against a bacterial pathogen in 2004 indicates that chemically synthesized glycans can elicit a powerful immune response.132 In their seminal study, Roy and coworkers synthesized a conjugate vaccine against Hib's CPS antigen polyribosephosphate (PRP) on large scale and clinically evaluated its ability to provide protection against bacterial meningitis.132 These authors synthesized PRP using a one-pot scheme, thus avoiding tedious purification steps required to isolate PRP from natural sources. Following conjugation to a macromolecule carrier, the vaccine induced a sufficient antibody response in infants for long-term protection against Hib.132 This vaccine is now commercially available under the trade name Quimi-Hib. For a recent review of synthetic anti-bacterial vaccines against other pathogens, see Pozsgay (2008).118
3.2 Small-molecule inhibitors of glycoprotein synthesis
Many small molecule inhibitors of enzymes involved in bacterial cell wall biosynthesis have proven clinical utility. Among the most prominent of these is the antibiotic penicillin, a small molecule inhibitor of peptidoglycan biosynthesis. Specifically, penicillin targets transpeptidases, enzymes responsible for crosslinking cell wall glycan chains viapeptide linkages.144 Other antibiotics, such as vancomycin, the lantibiotics, and moenomycin, disrupt peptidoglycan biosynthesis by inhibiting the peptidoglycan glycosyltransferases that catalyze the formation of cell wall glycan chains.145,146 Still other antibiotics function by preventing the formation of different cell wall glycopolymers, including cell wall teichoic acid.147,148 Although these antibiotics have saved countless lives, the emergence of bacterial strains that have evolved resistance to them has prompted the search for alternative treatments.One approach to circumvent antibiotic resistance involves modifying the structures of existing antibiotics. For example, once resistance to penicillin emerged, penicillin was chemically modified to produce a family of antibiotics.144,149 In addition, Kahne, Walker and others have identified novel inhibitors of peptidoglycan glycosyltransferase by altering moenomycin's lipid and truncating its core pentasaccharide.150–154 As an alternative to antibiotic scaffold modification, screens can be conducted to identify inhibitors of validated antibiotic targets. For example, Cheng and coworkers identified novel inhibitors of peptidoglycan glycosyltransferase by conducting a high-throughput screen of a library of 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds.155 A third approach involves identifying and inhibiting novel antibiotic targets, including glycosylation enzymes responsible for bacterial glycoprotein synthesis.
000 compounds.155 A third approach involves identifying and inhibiting novel antibiotic targets, including glycosylation enzymes responsible for bacterial glycoprotein synthesis.
Genetic deletion of glycosylation enzymes responsible for synthesizing and appending glycans onto bacterial proteins results in avirulent strains of certain bacteria (Table 1). Therefore, these enzymes are attractive targets for therapeutic intervention. To the best of our knowledge, there has only been one report of an inhibitor that interferes with bacterial glycoprotein synthesis.156 McNally et al. established that CMP–pseudaminic acid (CMP–Pse) is a potent inhibitor of PseB, the first enzyme in the pseudaminic acid biosynthetic pathway in C. jejuni and H. pylori.156CMP–Pse naturally provides feedback inhibition for Pse biosynthesis by binding directly to PseB's substrate binding site and acting as a competitive inhibitor. This study suggests that future efforts could focus on the rational design of a cell-permeable small molecule inhibitor based on CMP–Pse's interactions with PseB. Such an inhibitor would prevent C. jejuni and H. pylori from forming flagella and colonizing the host's gastrointestinal tract. Once such an inhibitor is created, follow up experiments in mammalian cells could be conducted to assure selectivity, lack of toxicity, and other desirable qualities.
Given that several bacterial glycoprotein synthesis pathways have been characterized (Table 1), the development of inhibitors that specifically interfere with these bacterial pathways should be possible. Several general strategies to inhibit glycosyltransferases and monosaccharide biosynthesis enzymes, including transition state analog design, bisubstrate analog design, structure-based design, fragment-based design, and library screening approaches, have been reviewed in detail elsewhere.157 Several of these strategies have already been employed to identify inhibitors of peptidoglycan glycosyltransferases.145,146,151,153–155,158,159 For instance, Walker and coworkers used structure-based design to produce a novel inhibitor of peptidoglycan glycosyltransferase that displayed potent antibacterial activity.158 These strategies should be directly transferable to bacterial glycosylation enzymes involved in bacterial glycoprotein synthesis. The development of small molecule inhibitors that interfere with these pathways would both provide leads for therapeutic interference and provide tools for the selective manipulation of bacterial glycoproteins.
3.3 Disrupting and covalently targeting bacterial glycans
As discussed earlier, MOE is a powerful technique that can enable bacterial glycoprotein discovery. The ability to metabolically introduce structural modifications into glycans also offers the opportunity to disrupt glycan function or covalently target unique glycans with therapeutics.85 Below we describe the reported examples in which MOE has been utilized to modify bacterial glycans and suggest the therapeutic potential of this approach in the future.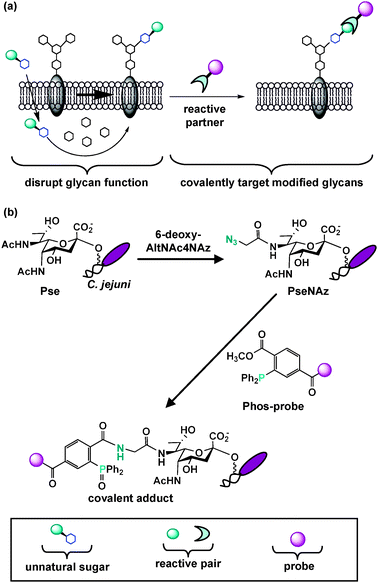 | ||
| Fig. 5 Metabolic oligosaccharide engineering introduces structural modifications into glycans, thus providing the opportunity to disrupt glycan function or covalently target unique glycans with therapeutics. (a) An unnatural sugar can be metabolically processed by the cell's carbohydrate biosynthetic enzymes and ultimately modify and potentially disrupt natural glycan structures. Modified cell surface glycans that contain a reactive functional group can be elaborated with a reactive partner to form a covalent adduct on the cell surface. This two-step process enables covalent targeting of modified glycans with probes for detection and therapeutic intervention. This general approach should be amenable to selectively disrupting and covalently targeting bacterial glycans. (b) Tanner, Logan and coworkers selectively replaced pseudaminic acid (Pse) residues on C. jejuni flagella with azido-pseudaminic acid (PseNAz) by feeding C. jejuni the azidosugar 6-deoxy-AltNAc4NAz, a dedicated metabolic precursor to PseNAz. They then covalently labeled and detected PseNAz residues on C. jejuni using Staudinger ligation with a phosphine probe (Phos-biotin). | ||
MOE can also disrupt glycan function in bacteria. Although sialic acid is primarily found in higher eukaryotes, this monosaccharide is also present within LOS and CPS of certain bacterial pathogens. Sialic acid on bacterial pathogens is believed to aid in binding to host surface receptors and in evading the host immune response. Thus, disrupting sialic acid structures on pathogenic bacteria may serve as a means to inactivate bacterial mechanisms required for initiation of infection and immunological tolerance within the host. Goon et al. metabolically introduced N-acyl variants of sialic acid into Haemophilus ducreyi's LOS by exploiting H. ducreyi's sialic acid salvage pathway.161 While some variants such as N-propanoyl sialic acid replaced natural sialic acid, others such as the 8-carbon analog N-octanoyl sialic acid inhibited LOS sialylation altogether.161 This result suggests that it may be possible to identify unnatural sialic acids that perturb sialylation of bacterial cell surfaces yet leave sialylation of eukaryotic cells unaffected. Exciting prospects for future work could focus on identifying analogs of sialic acid that are substrates or inhibitors of bacterial but not eukaryotic pathways. Moreover, efforts could focus on identifying analogs of monosaccharides that are exclusively found in bacteria (e.g.FucNAc, bacillosamine, Leg, DATDH). Such analogs would enable selective modulation of bacterial cell surface glycans, ultimately permitting us to disrupt their function or even covalently target them.
Ketone condensation reactions proceed relatively slowly outside of their pH optimum of 5–6, decreasing the utility of this ligation at physiological conditions.85 Moreover, ketones naturally present within cells can interfere with the reaction in complex tissue samples. Within the last decade, several other bioorthogonal reactions, all of which rely on the unique modes of reactivity of the azide,87 have emerged that proceed more favorably in physiological settings and are designed for application in vivo. Azides metabolically incorporated into cellular glycans via administration of azidosugars can be detected on live cellsvia several chemistries, including: (1) the Staudinger ligation between azides and triarylphosphines;92 (2) “copper-free click chemistry” between azides and cyclooctynes;100,168,169 and (3) the “phototriggered click reaction” between azides and cyclopropenones.101 To date, the Staudinger ligation has been demonstrated to proceed stably and efficiently in mice,105,106 and copper-free click chemistry employing difluorocyclooctyne (DIFO)-probes168 proceeds rapidly in zebrafish,104 nematodes,170 and mice.171 These seminal precedents suggest that covalent targeting of modified bacterial glycans using the Staudinger ligation or copper-free click chemistry within the context of an animal is possible. To translate this research to antimicrobial applications, all that is required is an azidosugar that can be exclusively incorporated into bacterial surface glycans yet leave mammalian surface glycans unmodified.
There are three reported examples describing metabolic incorporation of azides into bacterial glycans. In the first, Wang and coworkers incorporated and chemically elaborated azide-containing fucose monosaccharides, as well as other unnatural variants, into E. coli surface polysaccharides by introducing a permissive fucose salvage pathway from B. fragilis into E. coli.172 However, given the abundance of fucose on human cells and the need to genetically engineer E. coli to incorporate unnatural fucose analogs, selectively targeting pathogenic bacteria based on modified fucose residues is not an option at present. In another example, as described in section 2.1, our laboratory has metabolically labeled H. pylori's glycoproteins with azides by utilizing the azidosugar Ac4GlcNAz.107 Although Ac4GlcNAz is metabolically incorporated into mammalian surface glycans in cell culture, it is not detectably incorporated onto mammalian cell surfaces in mice.106 Therefore, we are exploring the possibility of using Ac4GlcNAz as a metabolic substrate to selectively label H. pylori's surface glycans with azides. Finally, Tanner, Logan and coworkers established that MOE can selectively label C. jejuni's pseudaminic acid residues with azides (Fig. 5b).160 These authors chemoenzymatically synthesized 6-deoxy-AltNAc4NAz, an azidoacetyl-containing variant of the pseudaminic acid metabolic precursor 6-deoxy-AltdiNAc. They then demonstrated that supplementing C. jejuni with 6-deoxy-AltNAc4NAz leads to selective metabolic replacement of PseNAc with PseNAz. Further, their work revealed that azide-covered C. jejuni can be tagged via Staudinger ligation with phosphine-based reactive partners to covalently attach biochemical probes (Fig. 5b).160 Their report is particularly exciting because it is the first example of labeling a sugar that is unique to pathogenic bacteria with an azide.
These MOE precedents demonstrate that it is possible to selectively modify glycans of pathogenic bacteria with azides. Modifying bacterial glycans sets the stage for labeling these glycans with chemical probesin vivo,105 providing a means to monitor and alter the bacterial coat, modulate immune recognition, and potentially render pathogenic bacteria harmless. For MOE to be successful in these pursuits, the efficiency of unnatural sugar incorporation and subsequent chemical labeling must be considered. Since a limited number of probes will be delivered to any particular pathogenic bacterium, probes with amplifiable effects will be the most valuable.
4. Conclusions
The past decade has witnessed a significant expansion in our understanding of bacterial glycoproteins. Although long-believed to be absent from bacteria, bacterial glycoproteins are now known to include unique structures that are often directly tied to the virulence of bacteria in human disease. Given their unusual structures and links to virulence, bacterial glycoproteins are intriguing targets of therapeutic intervention. Despite this realization, relatively little is known about bacterial protein glycosylation to date.Recent advances in our ability to detect bacterial glycoproteins and analyze them using mass spectrometry-based analyses will greatly ease the identification and characterization of these biomolecules in the future. Therefore, chemical approaches afford great promise for enhancing our basic understanding of bacterial glycobiology. Moreover, these approaches will surely unveil unique structures that could serve as novel pathogen-associated targets.
As the unique structures of bacterial glycans and the enzymes responsible for installing them have emerged, there has been tremendous excitement about using this information to eliminate certain bacterial infections. Taking a cue from successes in mammalian systems, the glycobiology community is now turning to carbohydrate-based vaccines, small molecule inhibitors of glycoprotein synthesis, and metabolic oligosaccharide engineering as approaches to target bacterial glycans and glycosylation enzymes. These approaches are already yielding promising leads and will no doubt garner further attention in the future.
Acknowledgements
We thank Jennifer Prescher and Peter Woodruff for comments on the manuscript. This work was supported by a Camille and Henry Dreyfus faculty startup award and a Research Corporation Cottrell College Science Award to D.D.References
- C. A. Arias and B. E. Murray, N. Engl. J. Med., 2009, 360, 439–443 CrossRef CAS.
- M. C. Raviglione and I. M. Smith, N. Engl. J. Med., 2007, 356, 656–659 CrossRef CAS.
- I. Benz and M. A. Schmidt, Mol. Microbiol., 2002, 45, 267–276 CrossRef CAS.
- P. M. Power and M. P. Jennings, FEMS Microbiol. Lett., 2003, 218, 211–222 CrossRef CAS.
- C. M. Szymanski and B. W. Wren, Nat. Rev. Microbiol., 2005, 3, 225–237 Search PubMed.
- M. A. Schmidt, L. W. Riley and I. Benz, Trends Microbiol., 2003, 11, 554–561 CrossRef CAS.
- L. Balonova, L. Hernychova and Z. Bilkova, Expert Rev. Proteomics, 2009, 6, 75–85 Search PubMed.
- E. Stimson, M. Virji, K. Makepeace, A. Dell, H. R. Morris, G. Payne, J. R. Saunder, M. P. Jennings, S. Barker and M. Panico, Mol. Microbiol., 1995, 17, 1201–1214 CrossRef CAS.
- N. M. Young, J. R. Brisson, J. Kelly, D. C. Watson, L. Tessier, P. H. Lanthier, H. C. Jarrell, N. Cadotte, F. St Michael, E. Aberg and C. M. Szymanski, J. Biol. Chem., 2002, 277, 42530–42539 CrossRef CAS.
- M. Schirm, I. C. Schoenhofen, S. M. Logan, K. C. Waldron and P. Thibault, Anal. Chem., 2005, 77, 7774–7782 CrossRef CAS.
- Essentials of glycobiology, ed. A. Varki, R. Cummings, J. D. Esko, H. Freeze, G. W. Hart and J. Marth, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2009 Search PubMed.
- K. L. Hsu, K. T. Pilobello and L. K. Mahal, Nat. Chem. Biol., 2006, 2, 153–157 CrossRef CAS.
- N. J. Agard and C. R. Bertozzi, Acc. Chem. Res., 2009, 42, 788–797 CrossRef CAS.
- J. A. Prescher and C. R. Bertozzi, Cell, 2006, 126, 851–854 CrossRef CAS.
- W. K. Chou, S. Dick, W. W. Wakarchuk and M. E. Tanner, J. Biol. Chem., 2005, 280, 35922–35928 CrossRef CAS.
- P. Doig, N. Kinsella, P. Guerry and T. J. Trust, Mol. Microbiol., 1996, 19, 379–387 CrossRef CAS.
- S. Goon, J. F. Kelly, S. M. Logan, C. P. Ewing and P. Guerry, Mol. Microbiol., 2003, 50, 659–671 CrossRef CAS.
- F. Liu and M. E. Tanner, J. Biol. Chem., 2006, 281, 20902–20909 CrossRef CAS.
- I. C. Schoenhofen, D. J. McNally, E. Vinogradov, D. Whitfield, N. M. Young, S. Dick, W. W. Wakarchuk, J. R. Brisson and S. M. Logan, J. Biol. Chem., 2006, 281, 723–732 CAS.
- P. Thibault, S. M. Logan, J. F. Kelly, J.-R. Brisson, C. P. Ewing, T. J. Trust and P. Guerry, J. Biol. Chem., 2001, 276, 34862–34870 CrossRef CAS.
- S. M. Logan, J. P. M. Hui, E. Vinogradov, A. J. Aubry, J. E. Melanson, J. F. Kelly, H. Nothaft and E. C. Soo, FEBS J., 2009, 276, 1014–1023 CrossRef CAS.
- S. M. Logan, J. P. M. Hui, E. Vinogradov, A. J. Aubry, J. E. Melanson, J. F. Kelly, H. Nothaft and E. C. Soo, FEBS J., 2009, 276, 1014–1023 CrossRef CAS.
- K. J. Glover, E. Weerapana, M. M. Chen and B. Imperiali, Biochemistry, 2006, 45, 5343–5350 CrossRef CAS.
- A. V. Karlyshev, P. Everest, D. Linton, S. Cawthraw, D. G. Newell and B. W. Wren, Microbiology, 2004, 150, 1957–1964 CrossRef CAS.
- J. C. Larsen, C. Szymanski and P. Guerry, J. Bacteriol., 2004, 186, 6508–6514 CrossRef CAS.
- D. Linton, E. Allan, A. V. Karlyshev, A. D. Cronshaw and B. W. Wren, Mol. Microbiol., 2002, 43, 497–508 CrossRef CAS.
- D. Linton, N. Dorrell, P. G. Hitchen, S. Amber, A. V. Karlyshev, H. R. Morris, A. Dell, M. A. Valvano, M. Aebi and B. W. Wren, Mol. Microbiol., 2005, 55, 1695–1703 CrossRef CAS.
- H. Nothaft, X. Liu, D. J. McNally and C. M. Szymanski, Methods Mol. Biol., 2010, 600, 227–243 CAS.
- N. B. Olivier, M. M. Chen, J. R. Behr and B. Imperiali, Biochemistry, 2006, 45, 13659–13669 CrossRef CAS.
- C. M. Szymanski, R. Yao, C. P. Ewing, T. J. Trust and P. Guerry, Mol. Microbiol., 1999, 32, 1022–1030 CrossRef CAS.
- D. J. McNally, A. J. Aubry, J. P. M. Hui, N. H. Khieu, D. Whitfield, C. P. Ewing, P. Guerry, J. R. Brisson, S. M. Logan and E. C. Soo, J. Biol. Chem., 2007, 282, 14463–14475 CrossRef CAS.
- I. C. Schoenhofen, E. Vinogradov, D. M. Whitfield, J.-R. Brisson and S. M. Logan, Glycobiology, 2009, 19, 715–725 CrossRef CAS.
- J. M. Fleckenstein, K. S. Roy, J. F. Fischer and M. Burkitt, Infect. Immun., 2006, 74, 2245–2258 CrossRef CAS.
- C. Lindenthal and E. A. Elsinghorst, Infect. Immun., 1999, 67, 4084–4091 CAS.
- C. Lindenthal and E. A. Elsinghorst, Infect. Immun., 2001, 69, 52–57 CrossRef CAS.
- C. Moormann, I. Benz and M. A. Schmidt, Infect. Immun., 2002, 70, 2264–2270 CrossRef CAS.
- O. Sherlock, R. M. Vejborg and P. Klemm, Infect. Immun., 2005, 73, 1954–1963 CrossRef CAS.
- I. Benz and M. A. Schmidt, Mol. Microbiol., 2001, 40, 1403–1413 CrossRef CAS.
- S. Laarmann and M. A. Schmidt, Microbiology, 2003, 149, 1871–1882 CrossRef CAS.
- S. Grass, A. Z. Buscher, W. E. Swords, M. A. Apicella, S. J. Barenkamp, N. Ozchlewski and J. W. St Geme, Mol. Microbiol., 2003, 48, 737–751 CrossRef CAS.
- J. Gross, S. Grass, A. E. Davis, P. Gilmore-Erdmann, R. R. Townsend and J. W. S. Geme, J. Biol. Chem., 2008, 283, 26010–26015 CrossRef CAS.
- M. Schirm, E. C. Soo, A. J. Aubry, J. Austin, P. Thibault and S. M. Logan, Mol. Microbiol., 2003, 48, 1579–1592 CrossRef CAS.
- I. C. Schoenhofen, V. V. Lunin, J.-P. Julien, Y. Li, E. Ajamian, A. Matte, M. Cygler, J.-R. Brisson, A. Aubry, S. M. Logan, S. Bhatia, W. W. Wakarchuk and N. M. Young, J. Biol. Chem., 2006, 281, 8907–8916 CrossRef CAS.
- I. C. Schoenhofen, D. J. McNally, J.-R. Brisson and S. M. Logan, Glycobiology, 2006, 16, 8C–14C CrossRef CAS.
- M. Gonzalez-Zamorano, G. Mendoza-Hernandez, W. Xolalpa, C. Parada, A. J. Vallecillo, F. Bigi and C. Espitia, J. Proteome Res., 2009, 8, 721–733 CrossRef CAS.
- S. L. Michell, A. O. Whelan, P. R. Wheeler, M. Partico, R. L. Easton, A. T. Etienne, S. M. Haslam, A. Dell, H. R. Morris, A. J. Reason, J. L. Herrmann, D. B. Young and R. G. Hewinson, J. Biol. Chem., 2003, 278, 16423–16432 CrossRef CAS.
- K. M. Dobos, K. Swiderek, K. H. Khoo, P. J. Brennan and J. T. Belisle, Infect. Immun., 1995, 63, 2846–2853 CAS.
- K. M. Dobos, K. H. Khoo, K. M. Swiderek, P. J. Brennan and J. T. Belisle, J. Bacteriol., 1996, 178, 2498–2506 CAS.
- M. J. Sartain and J. T. Belisle, Glycobiology, 2009, 19, 38–51 CAS.
- B. C. VanderVen, J. D. Harder, D. C. Crick and J. T. Belisle, Science, 2005, 309, 941–943 CAS.
- A. Banerjee and S. K. Ghosh, Mol. Cell. Biochem., 2003, 253, 179–190 CrossRef CAS.
- M. P. Jennings, M. Virji, D. Evans, V. Foster, Y. N. Srikhanta, L. Steeghs, P. van der Ley and E. R. Moxon, Mol. Microbiol., 1998, 29, 975–984 CrossRef CAS.
- P. M. Power, L. F. Roddam, M. Dieckelmann, Y. N. Srikhanta, Y. C. Tan, A. W. Berrington and M. P. Jennings, Microbiology, 2000, 146, 967–979 CAS.
- P. M. Power, L. F. Roddam, K. Rutter, S. Z. Fitzpatrick, Y. N. Srikhanta and M. P. Jennings, Mol. Microbiol., 2003, 49, 833–847 CAS.
- P. M. Power, K. L. Seib and M. P. Jennings, Biochem. Biophys. Res. Commun., 2006, 347, 904–908 CrossRef CAS.
- S. C. Ku, B. L. Schulz, P. M. Power and M. P. Jennings, Biochem. Biophys. Res. Commun., 2009, 378, 84–89 CAS.
- P. Castric, F. J. Cassels and R. W. Carlson, J. Biol. Chem., 2001, 276, 26479–26485 CrossRef CAS.
- J. Chamot-Rooke, B. Rousseau, F. Lanternier, G. Mikaty, E. Mairey, C. Malosse, G. Bouchoux, V. Pelicic, L. Camoin, X. Nassif and G. Dumenil, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14783–14788 CrossRef CAS.
- J. V. Kus, J. Kelly, L. Tessier, H. Harvey, D. G. Cvitkovitch and L. L. Burrows, J. Bacteriol., 2008, 190, 7464–7478 CrossRef CAS.
- J. G. Smedley, E. Jewell, J. Roguskie, J. Horzempa, A. Syboldt, D. B. Stolz and P. Castric, Infect. Immun., 2005, 73, 7922–7931 CrossRef.
- P. Castric, Microbiology, 1995, 141, 1247–1254 CrossRef CAS.
- S. K. Arora, M. Bangera, S. Lory and R. Ramphal, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9342–9347 CrossRef CAS.
- S. K. Arora, A. N. Neely, B. Blair, S. Lory and R. Ramphal, Infect. Immun., 2005, 73, 4395–4398 CrossRef CAS.
- W. L. Miller, M. J. Matewish, D. J. McNally, N. Ishiyama, E. M. Anderson, D. Brewer, J. R. Brisson, A. M. Berghuis and J. S. Lam, J. Biol. Chem., 2008, 283, 3507–3518 CAS.
- M. Schirm, S. K. Arora, A. Verma, E. Vinogradov, P. Thibault, R. Ramphal and S. M. Logan, J. Bacteriol., 2004, 186, 2523–2531 CrossRef CAS.
- A. Verma, M. Schirm, S. K. Arora, P. Thibault, S. M. Logan and R. Ramphal, J. Bacteriol., 2006, 188, 4395–4403 CrossRef CAS.
- P. Messner, J. Bacteriol., 2004, 186, 2517–2519 CrossRef CAS.
- C. M. Fletcher, M. J. Coyne, O. F. Villa, M. Chatzidaki-Livanis and L. E. Comstock, Cell, 2009, 137, 321–331 CrossRef CAS.
- E. Tian and K. Ten Hagen, Glycoconjugate J., 2009, 26, 325–334 CrossRef CAS.
- A. Helenius and M. Aebi, Science, 2001, 291, 2364–2369 CrossRef CAS.
- F. A. Hanisch, Biol. Chem., 2001, 382, 143–149 CrossRef CAS.
- J. Horzempa, T. K. Held, A. S. Cross, D. Furst, M. Qutyan, A. N. Neely and P. Castric, Clin. Vaccine Immunol., 2008, 15, 590–597 CrossRef CAS.
- M. Schirm, I. C. Schoenhofen, S. M. Logan, K. C. Waldron and P. Thibault, Anal. Chem., 2005, 77, 7774–7782 CrossRef CAS.
- R. K. Obhi and C. Creuzenet, J. Biol. Chem., 2005, 280, 20902–20908 CrossRef CAS.
- T. Zheng, D. Peelen and L. M. Smith, J. Am. Chem. Soc., 2005, 127, 9982–9983 CrossRef CAS.
- K. T. Pilobello, L. Krishnamoorthy, D. Slawek and L. K. Mahal, ChemBioChem, 2005, 6, 985–989 CrossRef CAS.
- A. Kuno, N. Uchiyama, S. Koseki-Kuno, Y. Ebe, S. Takashima, M. Yamada and J. Hirabayashi, Nat. Methods, 2005, 2, 851–856 CrossRef CAS.
- M. R. Bond and J. J. Kohler, Curr. Opin. Chem. Biol., 2007, 11, 52–58 CrossRef CAS.
- P. G. Hitchen and A. Dell, Microbiology, 2006, 152, 1575–1580 CrossRef CAS.
- J. M. Bobbitt, Adv. Carbohydr. Chem., 1956, 48, 1–41 CAS.
- E. A. Bayer, H. Ben-Hur and M. Wilchek, Anal. Biochem., 1988, 170, 271–281 CrossRef CAS.
- C. Hart, B. Schulenberg, T. H. Steinberg, W. Y. Leung and W. F. Patton, Electrophoresis, 2003, 24, 588–598 CrossRef CAS.
- T. H. Steinberg, K. P. O. Top, K. N. Berggren, C. Kemper, L. Jones, Z. J. Diwu, R. P. Haugland and W. F. Patton, Proteomics, 2001, 1, 841–855 CrossRef CAS.
- L. Balonova, L. Hernychova, B. F. Mann, M. Link, Z. Bilkova, M. V. Novotny and J. Stulik, J. Proteome Res., 2010, 9, 1995–2005 CrossRef CAS.
- D. H. Dube and C. R. Bertozzi, Curr. Opin. Chem. Biol., 2003, 7, 616–625 CrossRef CAS.
- O. T. Keppler, R. Horstkorte, M. Pawlita, C. Schmidts and W. Reutter, Glycobiology, 2001, 11, 11R–18R CrossRef CAS.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS.
- S. T. Laughlin, N. J. Agard, J. M. Baskin, I. S. Carrico, P. V. Chang, A. S. Ganguli, M. J. Hangauer, A. Lo, J. A. Prescher and C. R. Bertozzi, Methods Enzymol., 2006, 415, 230–250 CAS.
- S. T. Laughlin and C. R. Bertozzi, Nat. Protoc., 2007, 2, 2930–2944 Search PubMed.
- L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125–1128 CrossRef CAS.
- H. C. Hang and C. R. Bertozzi, J. Am. Chem. Soc., 2001, 123, 1242–1243 CrossRef CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846–14851 CrossRef CAS.
- D. J. Vocadlo, H. C. Hang, E.-J. Kim, J. A. Hanover and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9116–9121 CrossRef CAS.
- D. Rabuka, S. C. Hubbard, S. T. Laughlin, S. P. Argade and C. R. Bertozzi, J. Am. Chem. Soc., 2006, 128, 12078–12079 CrossRef CAS.
- S. G. Sampathkumar, M. B. Jones and K. J. Yarema, Nat. Protoc., 2006, 1, 1840–1851 Search PubMed.
- M. Sawa, T.-L. Hsu, T. Itoh, M. Sugiyama, S. R. Hanson, P. K. Vogt and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12371–12376 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS.
- R. Sprung, A. Nandi, Y. Chen, S. C. Kim, D. Barma, J. R. Falck and Y. M. Zhao, J. Proteome Res., 2005, 4, 950–957 CrossRef CAS.
- A. Nandi, R. Sprung, D. K. Barma, Y. Zhao, S. C. Kim and J. R. Falck, Anal. Chem., 2006, 78, 452–458 CrossRef CAS.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS.
- J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS.
- D. H. Dube, PhD thesis, University of California, Berkeley, 2005.
- M. B. Koenigs, E. A. Richardson and D. H. Dube, Mol. BioSyst., 2009, 5, 909–912 RSC.
- B. Thiede, W. Hhenwarter, A. Krah, J. Mattow, M. Schmid, F. Schmidt and P. R. Jungblut, Methods, 2005, 35, 237–247 CrossRef CAS.
- D. N. Perkins, D. J. C. Pappin, D. M. Creasy and J. S. Cottrell, Electrophoresis, 1999, 20, 3551–3567 CrossRef CAS.
- D. J. Harvey, Expert Rev. Proteomics, 2005, 2, 87–101 Search PubMed.
- J. Jang-Lee, S. J. North, M. Sutton-Smith, D. Goldberg, M. Panico, H. Morris, S. Haslam and A. Dell, Methods Enzymol., 2006, 415, 59–86 CAS.
- S. M. Haslam, S. J. North and A. Dell, Curr. Opin. Struct. Biol., 2006, 16, 584–591 CrossRef CAS.
- X. Liu, D. J. McNally, H. Nothaft, C. M. Szymanski, J. R. Brisson and J. Li, Anal. Chem., 2006, 78, 6081–6087 CrossRef CAS.
- N. Siuti and N. L. Kelleher, Nat. Methods, 2007, 4, 817–821 CrossRef CAS.
- J. Wiesner, T. Premsler and A. Sickmann, Proteomics, 2008, 8, 4466–4483 CrossRef CAS.
- S. M. Twine, C. J. Paul, E. Vinogradov, D. J. McNally, J. R. Brisson, J. A. Mullen, D. R. McMullin, H. C. Jarrell, J. W. Austin, J. F. Kelly and S. M. Logan, FEBS J., 2008, 275, 4428–4444 CrossRef CAS.
- S. H. Kaufmann, Nat. Rev. Microbiol., 2007, 5, 491–504 Search PubMed.
- V. Pozsgay, Curr. Top. Med. Chem., 2008, 8, 126–140 CrossRef CAS.
- T. J. Francis and W. S. Tillet, J. Exp. Med., 1930, 52, 573–585 CrossRef CAS.
- M. Heidelberger, M. M. Dilapi, M. Siegel and A. W. Walter, J. Immunol., 1950, 65, 535–541 CAS.
- A. H. Lucas, M. A. Apicella and C. E. Taylor, Clin. Infect. Dis., 2005, 41, 705–712 CrossRef CAS.
- P. Gardner, N. Engl. J. Med., 2006, 355, 1466–1473 CrossRef CAS.
- G. Ada and D. Isaacs, Clin. Microbiol. Infect., 2003, 9, 79–85 Search PubMed.
- M. L. Hecht, P. Stallforth, D. V. Silva, A. Adibekian and P. H. Seeberger, Curr. Opin. Chem. Biol., 2009, 13, 354–359 CrossRef CAS.
- J. Zhu, J. D. Warren and S. J. Danishefsky, Expert Rev. Vaccines, 2009, 8, 1399–1413 CrossRef CAS.
- S. H. E. Kaufmann, Nat. Rev. Microbiol., 2007, 5, 491–504 Search PubMed.
- O. T. Avery and W. F. Goebel, J. Exp. Med., 1929, 50, 533–550 CrossRef CAS.
- M. P. Girard, M. P. Preziosi, M. T. Aguado and M. P. Kieny, Vaccine, 2006, 24, 4692–4700 CrossRef CAS.
- S. Black, H. Shinefield, B. Fireman, E. Lewis, P. Ray, J. R. Hansen, L. Elvin, K. M. Ensor, J. Hackell, G. Siber, F. Malinoski, D. Madore, I. Chang, R. Kohberger, W. Watson, R. Austrian and K. Edwards, Pediatr. Infect. Dis. J., 2000, 19, 187–195 CrossRef CAS.
- W. G. Adams, K. A. Deaver, S. L. Cochi, B. D. Plikaytis, E. R. Zell, C. V. Broome and D. Wenger, J. Am. Med. Assoc., 1993, 269, 221–226 CrossRef CAS.
- A. Schuchat, K. Robinson, J. D. Wenger, L. H. Harrison, M. Farley, A. L. Reingold, L. Lefkowitz and B. A. Perkins, N. Engl. J. Med., 1997, 337, 970–976 CrossRef CAS.
- V. Verez-Bencomo, V. Fernandez-Santana, E. Hardy, M. E. Toledo, M. C. Rodriguez, L. Heynngnezz, A. Rodriguez, A. Baly, L. Herrera, M. Izquierdo, A. Villar, Y. Valdes, K. Cosme, M. L. Deler, M. Montane, E. Garcia, A. Ramos, A. Aguilar, E. Medina, G. Torano, I. Sosa, I. Hernandez, R. Martinez, A. Muzachio, A. Carmenates, L. Costa, F. Cardoso, C. Campa, M. Diaz and R. Roy, Science, 2004, 305, 522–525 CrossRef.
- C. Jones, An. Acad. Bras. Cienc., 2005, 77, 293–324 Search PubMed.
- H. K. Guttormsen, L. C. Paoletti, K. G. Mansfield, W. Jachymek, H. J. Jennings and D. L. Kasper, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5903–5908 CrossRef CAS.
- H. J. Jennings, A. Gamian and F. E. Ashton, J. Exp. Med., 1987, 165, 1207–1211 CrossRef CAS.
- R. A. Pon, M. Lussier, Q. L. Yang and H. J. Jennings, J. Exp. Med., 1997, 185, 1929–1938 CrossRef CAS.
- K. Roy, D. Hamilton, M. M. Ostmann and J. M. Fleckenstein, Vaccine, 2009, 27, 4601–4608 CrossRef CAS.
- A. S. Mehta, E. Saile, W. Zhong, T. Buskas, R. Carlson, E. Kannenberg, Y. Reed, C. P. Quinn and G. J. Boons, Chem.–Eur. J., 2006, 12, 9136–9149 CrossRef CAS.
- M. Tamborrini, D. B. Werz, J. Frey, G. Pluschke and P. H. Seeberger, Angew. Chem., Int. Ed., 2006, 45, 6581–6582 CrossRef CAS.
- C. E. Frasch, Vaccine, 2009, 27, 6468–6470 CrossRef CAS.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS.
- Z. Y. Zhang, I. R. Ollmann, X. S. Ye, R. Wischnat, T. Baasov and C. H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 CrossRef CAS.
- P. Seeberger and W. Haase, Chem. Rev., 2000, 100, 4349–4394 CrossRef CAS.
- T. Schneider and H. G. Sahl, Int. J. Med. Microbiol., 2010, 300, 161–169 CrossRef CAS.
- B. Ostash and S. Walker, Curr. Opin. Chem. Biol., 2005, 9, 459–466 CrossRef CAS.
- J. Halliday, D. McKeveney, C. Muldoon, P. Rajaratnam and W. Meutermans, Biochem. Pharmacol., 2006, 71, 957–967 CrossRef CAS.
- J. G. Swoboda, J. Campbell, T. C. Meredith and S. Walker, ChemBioChem, 2010, 11, 35–45 CAS.
- J. G. Swoboda, T. C. Meredith, J. Campbell, S. Brown, T. Suzuki, T. Bollenbach, A. J. Malhowski, R. Kishony, M. S. Gilmore and S. Walker, ACS Chem. Biol., 2009, 4, 875–883 CrossRef CAS.
- W. Sneader, Drug discovery: a history, John Wiley & Sons, West Sussex, England, 2005 Search PubMed.
- M. J. Sofia, N. Allanson, N. T. Hatzenbuhler, R. Jain, R. Kakarla, N. Kogan, R. Liang, D. S. Liu, D. J. Silva, H. M. Wang, D. Gange, J. Anderson, A. Chen, F. Chi, R. Dulina, B. W. Huang, M. Kamau, C. W. Wang, E. Baizman, A. Branstrom, N. Bristol, R. Goldman, K. H. Han, C. Longley, S. Midha and H. R. Axelrod, J. Med. Chem., 1999, 42, 3193–3198 CrossRef CAS.
- E. R. Baizman, A. A. Branstrom, C. B. Longley, N. Allanson, M. J. Sofia, D. Gange and R. C. Goldman, Microbiology, 2000, 146, 3129–3140 CAS.
- R. C. Goldman, E. R. Baizman, A. A. Branstrom and C. B. Longley, Bioorg. Med. Chem. Lett., 2000, 10, 2251–2254 CrossRef CAS.
- M. Adachi, Y. Zhang, C. Leimkuhler, B. Y. Sun, J. V. LaTour and D. E. Kahne, J. Am. Chem. Soc., 2006, 128, 14012–14013 CrossRef CAS.
- S. Fuse, H. Tsukamoto, Y. Yuan, T.-S. A. Wang, Y. Zhang, M. Bolla, S. Walker, P. Sliz and D. Kahne, ACS Chem. Biol., 2010, 5, 701–711 CrossRef CAS.
- T. J. R. Cheng, M. T. Sung, H. Y. Liao, Y. F. Chang, C. W. Chen, C. Y. Huang, L. Y. Chou, Y. D. Wu, Y. Chen, Y. S. E. Cheng, C. H. Wong, C. Ma and W. C. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 431–436 CrossRef CAS.
- D. J. McNally, I. C. Schoenhofen, R. S. Houliston, N. H. Khieu, D. M. Whitfield, S. M. Logan, H. C. Jarrell and J. R. Brisson, ChemMedChem, 2008, 3, 55–59 CrossRef CAS.
- J. D. Esko and C. R. Bertozzi, Chemical tools for inhibiting glycosylation, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2009 Search PubMed.
- Y. Q. Yuan, S. Fuse, B. Ostash, P. Sliz, D. Kahne and S. Walker, ACS Chem. Biol., 2008, 3, 429–436 CrossRef CAS.
- K. Lazar and S. Walker, Curr. Opin. Chem. Biol., 2002, 6, 786–793 CrossRef CAS.
- F. Liu, A. J. Aubry, I. C. Schoenhofen, S. M. Logan and M. E. Tanner, ChemBioChem, 2009, 10, 1317–1320 CrossRef CAS.
- S. Goon, B. Schilling, M. V. Tullius, B. W. Gibson and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3089–3094 CrossRef CAS.
- S. J. Luchansky, H. C. Hang, E. Saxon, J. R. Grunwell, C. Yu, D. H. Dube and C. R. Bertozzi, Methods Enzymol., 2003, 362, 249–272 CAS.
- O. T. Keppler, P. Stehling, M. Herrmann, H. Kayser, D. Grunow, W. Reutter and M. Pawlita, J. Biol. Chem., 1995, 270, 1308–1314 CrossRef CAS.
- L. K. Mahal, N. W. Charter, K. Angata, M. Fukuda, D. E. Koshland, Jr. and C. R. Bertozzi, Science, 2001, 294, 380–381 CrossRef CAS.
- N. W. Charter, L. K. Mahal, D. E. Koshland, Jr. and C. R. Bertozzi, J. Biol. Chem., 2002, 277, 9255–9261 CrossRef CAS.
- B. Buttner, C. Kannicht, C. Schmidt, K. Loster, W. Reutter, H. Y. Lee, S. Nohring and R. Horstkorte, J. Neurosci., 2002, 22, 8869–8875 CAS.
- R. Sadamoto, K. Niikura, P. S. Sears, H. T. Liu, C. H. Wong, A. Suksomcheep, F. Tomita, K. Monde and S. I. Nishimura, J. Am. Chem. Soc., 2002, 124, 9018–9019 CrossRef CAS.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzit, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS.
- S. T. Laughlin and C. R. Bertozzi, ACS Chem. Biol., 2009, 4, 1068–1072 CrossRef CAS.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS.
- W. Yi, X. W. Liu, Y. H. Li, J. J. Li, C. F. Xia, G. Y. Zhou, W. P. Zhang, W. Zhao, X. Chen and P. G. Wang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4207–4212 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |