Assembly of carbon nanotubes and alkylated fullerenes: nanocarbon hybrid towards photovoltaic applications†
Yanfei
Shen
*a,
Juan Sebastián
Reparaz
b,
Markus Raphael
Wagner
b,
Axel
Hoffmann
b,
Christian
Thomsen
b,
Jeong-O
Lee
c,
Sebastian
Heeg
d,
Benjamin
Hatting
d,
Stephanie
Reich
d,
Akinori
Saeki
e,
Shu
Seki
e,
Kaname
Yoshida
f,
Sukumaran Santhosh
Babu
a,
Helmuth
Möhwald
g and
Takashi
Nakanishi
*a
aNational Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, 305-0047, Japan. E-mail: shen.yanfei@nims.go.jp; nakanishi.takashi@nims.go.jp; Tel: +81-(0)29-860-4740
bTechnische Universität Berlin, 10623, Berlin, Germany
cKorea Research Institute of Chemical Technology, Daejeon, 305-343, Korea
dFreie Universität Berlin, 14195, Berlin, Germany
eGraduate School of Engineering, Osaka University, Osaka, Japan
fInstitute for Chemical Science, Kyoto University, Japan
gMax Planck Institute of Colloids and Interfaces, 14424, Potsdam, Germany
First published on 25th August 2011
Abstract
Taking advantage of the non-covalent interaction between alkyl chains and the sidewalls of a single-walled carbon nanotube (SWCNT), a nanocarbon hybrid of SWCNT and a fullerene (C60) derivative with long alkyl chains was constructed as a donor–acceptor pair for photovoltaics and nanodevice investigations. It was found that SWCNT could be mostly unbundled by the alkylated C60 (1) and was well-dispersed in organic solvents. As a photoactive material, the resultant nanocarbon hybrid, 1-SWCNT, performed well in light-energy harvesting applications in photoelectrochemical cells and nanoscale field-effect transistors (FET). Moreover, the 1-SWCNT assembly exhibited superhydrophobicity, providing an interesting opportunity to fabricate nanocarbon-based waterproof optoelectronic devices. In order to understand the photoexcitation process, the 1-SWCNT assembly was electrochemically and spectroscopically characterized. The electrochemical results showed that the SWCNT facilitated electronic communication between 1 and the electrode. The steady-state and time-resolved fluorescence and the photoluminescence excitation studies suggested efficient quenching of the singlet excited state of C60. Nanosecond transient absorption data revealed the one-electron reduction of fullerene, C60˙−, thereby demonstrating the photoinduced electron transfer from SWCNT to the C60 unit in the 1-SWCNT assembly.
1. Introduction
Since the discovery of carbon nanoclusters, such as single-walled carbon nanotubes (SWCNT) and fullerenes (e.g.C60), the use of nanocarbon science, including in organic synthesis,1–3 supramolecular material chemistry,4–10 surface nanotechnology11 and nanoelectronics,12,13 has rapidly developed. SWCNTs have emerged as attractive candidates for the development of light-energy harvesting and photovoltaic materials because of their unique structure and the presence of extended π-delocalization.14 Of these, SWCNT-based nanohybrids formed by electron donor–acceptor systems have been the focus of attention.15,16 The association with electron donors or acceptors can modulate the electronic properties of SWCNTs and results in new potential materials.On the other hand, fullerene (C60) has a highly delocalized 3D π-system and its properties have been intensively investigated in recent years. Among its most spectacular physical and chemical properties, C60 was found to be able to reversibly accept up to six electrons, behave as a strong electron acceptor and show exceptional electronic absorption bands expanding throughout the entire UV-vis wavelength range.17 The extraordinary electron acceptor properties have resulted in noteworthy advances in the research areas of light-induced electron transfer and solar energy conversion.18–21 Therefore, the hybridization of C60 with SWCNTs5,7,22–27 would couple the optical and electronic properties of SWCNTs together with the electron-acceptor feature of C60.
Covalent modification is one of the strategies for the attachment of C60 onto sidewall of SWCNT.28,29 However, this partly disrupts their electronic structure and carrier transport properties. As an alternative, non-covalent functionalization has been developed. The ability to fabricate nanocarbon assemblies, which are potentially exploitable for the bottom-up construction of nanodevices, demands the development of general and reliable approaches to control the self-assembly processes. To this end, applying π–π stacking interactions with C60 or polyaromatic substituents on C60 towards the periphery of SWCNTs has been well-established.5,7,30
Here, we report a facile, versatile and non-destructive way to construct nanocarbon hybrids by alkyl chain-assisted assembly between an alkylated C60 derivative 131,32 and SWCNTs (Scheme 1). The appropriate affinity of long aliphatic chains of 1 towards a SWCNT surface allows the solubilization of SWCNTs in organic solvents without disturbing their intrinsic electronic structures.33 The solubility of SWCNTs with the assistance of 1, maintaining electronic communication between them, makes it suitable for photovoltaic studies and field-effect transistor (FET) fabrication via wet processes. To grasp a deeper understanding of the photoenergy conversion process, we also performed a detailed spectroscopic study of the photoinduced electron transfer between the two components. In addition, the obtained nanocarbon assembly exhibits water repellency at its surface, which provides an extra benefit for the photovoltaic application, i.e., for the fabrication of waterproof devices.
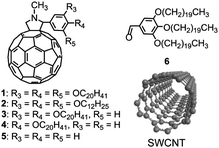 | ||
| Scheme 1 The molecular structures of 1–6 and SWCNT. | ||
2. Experimental
2.1 Materials
C60 derivatives 1–5 and 3,4,5-eicosyloxy benzaldehyde 6 were synthesized by following our previous synthetic procedure.34 Briefly, C60 derivatives were prepared by refluxing the corresponding benzaldehyde with N-methylglycine and C60 in dry monochlorobenzene. SWCNT (Super Purified grade, Unidym) produced by high-pressure decomposition of carbon monoxide (HiPco process) were used as received.2.2 Preparation of 1-SWCNT composite solution
The mixture of 1 (3.5 mg) and SWCNT (1 mg) was firstly grinded in a mortar, and then transferred to THF (8 mL) in a vial and followed by ultrasonication for about 6 h using the Branson 5510 ultrasonic equipment (40 kHz). The solution was subsequently centrifuged (Force Micro 1618) at 10000 rpm for 40 min, and two-thirds of the resultant supernatant liquid was decanted carefully from the settled solid. To get the ratio of SWCNT in the nanohybrids, 1 was firstly washed out from the 1-SWCNT nanohybrids through PTFE filter paper (Whatman, pore size 0.45 μm) with THF. The resulting SWCNT on filter paper was dried at 80 °C in a vacuum and weighed on an ultra-fine balance. The weight ratio of SWCNTS to 1-SWCNT nanohybrids was calculated to be about 18% from the obtained SWCNT weight and the amount of 1 added for preparing the nanohybrids. After evaporating the solvent from the as-obtained supernatant liquid, the solid 1-SWCNT was used for HR-TEM analysis. The supernatant liquid was used directly for spectroscopy characterizations, i.e., UV-vis–NIR, steady-state fluorescence, time-resolved fluorescence, photoluminenscence, transient absorption studies and photoelectrochemical measurements. UV-vis–NIR spectra were recorded with a Jasco V570 spectrophotometer.2.3 Electrochemical and photoelectrochemical measurements
Electrochemical experiments were conducted in a classic three-electrode system using a Garmy Reference 600 Potentiostat/Galvanostat/ZRA Instrument. The working electrode was a glassy carbon electrode, auxiliary and reference electrodes were a Pt ring and Ag/AgCl (saturated KCl), respectively. A thin film of 1-SWCNT on a glassy carbon electrode was prepared by depositing the 1-SWCNT supernatant liquid (10 μL) followed by drying in air for 24 h. Differential pulse (pulse amplitude 25 mV) voltammograms (DPV) of the cast film of 1-SWCNT were performed in 0.1 M aqueous n-Bu4NCl solution at 70 °C. Photoelectrochemical measurements were carried out in a two-electrode system with 1-SWCNT/FTO, 1/FTO or SWCNT/FTO as the working electrode and a Pt wire as the counter electrode. The aggregation of 1-SWCNT and 1 for photoelectrochemical measurements was obtained by rapid injection of an equivalent volume of methanol to a THF solution of 1-SWCNT and 1 (the concentration of 1 was 0.2 mM). The electrodes were prepared by casting methods. The short-circuit current was measured by immersing the working electrode and counter electrode into the electrolyte solution containing LiI (0.5 M) and I2 (0.01 M) in acetonitrile. A 150 W Xe lamp (Asahi Spectra, USA) equipped with a LAX-Cute Airmass 1.5 G filter was used as the light source. The incident light intensity was focused and calibrated to 1 Sun (1000 W m−2) by a 1 sun checker. The modified area of the working electrode (0.2 cm2) was illuminated from the back.2.4 FET device fabrication and measurements
SWCNTs were grown on Si wafers with 500 nm-thick SiO2. The liquid catalyst, consisting of Fe(NO3)2·9H2O, alumina nanopowder and Mo(acac)2 in methanol, was dispersed onto the substrates with patterned poly(methyl metacrylate). After lifting this off, the SWCNT was grown by heating to 900 °C in a furnace with CH4 as the carbon feedstock. Patterns for electrical leads were made with photolithography and the thermal evaporation of Ti and Au. The channel length between the source and drain electrodes was 5 or 7 μm. Electrical transport measurements were done in a custom-built probe station with Lab View-controlled electronics. Photoexcitation experiments were performed using an Osram halogen lamp (12 V, 100 W) equipped with a microscope in the probe station.2.5 Characterization
High-resolution transmission electron microscopy (HR-TEM) images were obtained by a JEOL transmission electron microscope, model JEM2200FS. Solid 1-SWCNT assemblies were dispersed in chloroform with ultrasonication. One drop of the solution was deposited on an electron microscopy microgrid and the sample was carefully rinsed with THF to show the clear visual state of the assembly, especially the debundled SWCNTs, before TEM measurements were carried out. All of the TEM images were recorded by a CCD camera with an accelerating voltage of 200 kV. Scanning electron microscopy (SEM) images were recorded by means of a Philips XL30 electron microscope at an accelerating voltage of 3 kV.Flash-photolysis time-resolved microwave conductivity (TRMC) measurements of 1-SWCNT and 1 were carried out using an X-band (9 GHz) microwave circuit at low power (approximately 3 mW) and a nanosecond laser irradiation at 355 nm with a photon density of 9.1 ×1015 cm−2.35 Samples for TRMC measurements were prepared by evaporating the THF solution of 1-SWCNT and 1 followed by drying the solid samples at 70 °C in a vacuum for about 5 h. The solid samples were pasted on a quartz plate with double-sided sticky tape (the tape does not disturb any TRMC signal). The smooth film was formed by physical smoothing with a spatula and used for TRMC measurements.
Photoluminescence excitation (PLE) data was acquired using a HORIBA Jobin Yvon Fluorolog-3 spectrofluorometer. A HeXe broadband lamp served as the excitation source. The PLE map was recorded in steps of 5 nm in excitation with an acquisition time of 20 s. The luminescence data was normalized with respect to the light source using a Si diode as the reference. The spectrometer response was taken into account by comparison with a known reference source. Photoluminescence (PL) spectra were collected using a HORIBA Jobin Yvon Fluorolog spectrometer equipped with 450 W of a Xenon arc lamp and 350 mm of focal point distance for the excitation monochromator. Slits for the excitation and emission monochromaters were fixed at 4 nm for the excitation and 4 nm for the emission monochromator and the integration time was 0.5 s. For the time-resolved PL measurements, the samples were excited using the second harmonic of a Ti sapphire laser with a pulse length of 2 ps. The excitation wavelength was tuned to 400 nm (3.1 eV). Transients were recorded by single photon counting using a Hamamatsu R3809U-52 microchannel plate (MCP). The emitted light was dispersed by a subtractive double monochromator with a spectral resolution of 0.1 nm. The instrumental time resolution was limited by the time walk of the MCP of about 30 ps, which allows the determination of lifetimes down to 10 ps using deconvolution techniques.
The nanosecond transient absorption measurement in the range 530–900 nm were performed by a monochromator, a streak camera and a continuous Xe lamp, while the one for 900–1600 nm was done by a monochromator, an InGaAs pin photodetector and a Xe flash lamp. The third harmonic generation (355 nm) of a nanosecond Nd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :YAG laser was used as an irradiation laser. The THF solutions were saturated with N2 before measurements were carried out.
:YAG laser was used as an irradiation laser. The THF solutions were saturated with N2 before measurements were carried out.
3. Results and discussion
3.1 Assembly of SWCNT with 1
The interaction between 1 and SWCNTs allows the unbundling and dispersion of SWCNTs in organic solvents, such as tetrahydrofuran (THF) (Fig. 1a) and chloroform (CHCl3) (Fig. S1†). As revealed in Fig. 1b, the well-resolved absorption peaks in the UV-vis–NIR spectrum of 1-SWCNT in THF suggest the presence of SWCNTs with small bundles8,9 and the preservation of the intrinsic electronic structure of the SWCNTs after the assembly with 1. The unbundling of SWCNTs was also supported by the high-resolution transmission electron microscopy (HR-TEM) analysis (Fig. S2†). In addition, as indicated by the arrows in the TEM image, several spherical objects with a diameter of approximately 1 nm close to the sidewall of the nanotubes are observed, which are likely to be C60 derivatives attached onto thin bundles of SWCNTs.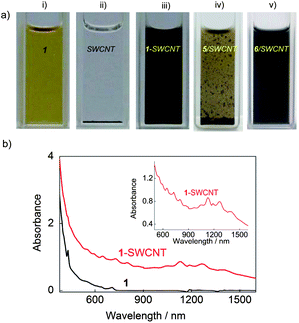 | ||
| Fig. 1 a) Photographs of 1 (i), SWCNT in the absence (ii) and presence (iii) of 1 and in the presence of 5 (iv) and 6 (v) in THF; b) UV-vis–NIR absorption spectra of 1 and 1-SWCNT in THF; the inset shows an enlarged area between 570–1500 nm showing the van Hove singularities. The concentration of 1 and SWCNTs are 0.25 mM and 0.125 mg ml−1, respectively. | ||
It is interesting to know which moiety (C60 or aliphatic chains) of 1 has the stronger affinity with the SWCNT sidewalls. To investigate this point, control experiments (Fig. 1a) were performed with 5 and 6 (Scheme 1), which are molecules containing the C60 unit and alkyl substituent moiety of 1, respectively. It was found that 6 has better performance than 5 in dispersing pristine SWCNTs, suggesting a more favorable affinity of long alkyl chains towards the sidewalls of SWCNTs in organic solvents. Apart from this control experiment, other C60 derivatives with a shorter alkyl chain length (2) or a lower number of alkyl chains (3, 4) can also disperse SWCNTs well (Fig. S3†), showing the capability of other various task-specific molecules to manipulate assemblies. This result is consistent with our previous report that alkylated C60 can align on the surface of graphite forming perfectly straight C60 nanowires,36,37 due to good lattice matching between the graphite and the all-trans conformation of the oligo-methylene units.38 Therefore, driven by the interaction between the long alkyl chains of C60 derivatives and SWCNTs, the nanocarbon assemblies of alkylated C60-SWCNT were successfully prepared. Alkylated C60-SWCNT assemblies are therefore expected to be a potential candidate for the wet-process fabrication of nanocarbon-based optoelectronic devices. While this non-covalent functionalization can more effectively preserve the intrinsic electronic features of SWCNT compared to covalent methodology, sonication during the sample preparation may cause shortened carbon nanotubes or defects along the sidewall (see the Raman spectra in Fig. S4†).
3.2 Electrochemical properties of 1-SWCNT assembly
It is known that SWCNTs have been widely utilized as a conducting channel to build a number of nanoscale electronic devices by chemical modification with various inorganic12 and organic39 semiconductors due to the unique electronic properties. As shown in Fig. 2, the cast film of 1 on a glassy carbon electrode in 0.1 M aqueous n-Bu4NCl solution at 70 °C exhibits two redox waves corresponding to the generation of C60 mono- and dianions at potentials of Ered,1 − 697 mV and Ered,2 − 865 mV vs.Ag/AgCl, respectively.401 is in a fluid-mesophase at 70 °C, which facilitates the diffusion of molecules (e.g. the supporting electrolyte, solvent and C60 molecules) throughout the system. In comparison, in the nanocarbon hybrid film of 1-SWCNT under the same conditions (see the differential scanning calorimetry (DSC) result in Fig. S5†), the first reductive peak positively shifts by approximately 50 mV and the peak intensity increases by a factor of 2. This result suggests that SWCNTs facilitate the electronic communication between 1 and the electrode surface. The enhanced electrochemical activity is attributed to the unique electronic properties of SWCNTs, which offers a better electron transfer pathway. This electrochemical result also provides additional positive evidence for the assembly of 1 and SWCNTs. In addition, the interaction between 1 and SWCNTs was also supported by the DSC result (See Fig. S5†).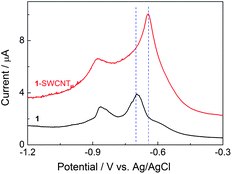 | ||
| Fig. 2 Differential pulse voltammograms (pulse amplitude 25 mV) of a cast film of 1-SWCNT and 1 on a glassy carbon electrode (0.1 M aqueous n-Bu4NCl solution, at 70 °C). | ||
3.3 Photoelectric conversion of 1-SWCNT assembly
Using the standard photoelectrochemical (PEC) cell configuration is a fundamental and facile method to evaluate the photoenergy conversion of semiconducting materials.41 Accordingly, photocurrent measurements were performed in a PEC cell (Fig. S6†) in acetonitrile containing LiI (0.5 M) and I2 (0.01 M), employing 1-SWCNT-coated FTO (1-SWCNT/FTO) as a working electrode and a Pt wire as a counter electrode. Fig. 3 shows the short-circuit photocurrent generation of 1-SWCNT/FTO upon excitation with white light. The anodic photocurrent response is prompt, steady and reproducible. Moreover, the photocurrent of 1-SWCNT/FTO is much larger than that of either 1/FTO (>160 fold) or SWCNT/FTO (>18 fold). The incident photon-to-electron conversion efficiency (IPCE) is calculated to be about 5% at 405 nm (see more discussion in Figs S7 and S8†). The direct interaction between 1 and the SWCNT could be a factor for driving away the charge carriers to the collecting surface, where the one-dimensional architecture of SWCNTs facilitates charge transport and charge collection.16 A particular advantage of our nanocarbon hybrid is the ease of nanodevice fabrication by simply cast/spin-coating from a volatile solution containing 1-SWCNT, which is desirable for industrial applications. Further improvements in the efficiency can be expected by varying the ratio of C60 derivative/SWCNT in the hybrids,16 increasing the C60 content in the derivative by reducing the aliphatic chain volume, photoelectrochemical cell optimization and/or the addition of a third component, such as an appropriate electron donor.42 For instance, the IPCE value was enhanced from 3% to 5% upon optimising the SWCNT content in the hybrid from 10 wt% to 18 wt%.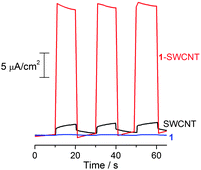 | ||
| Fig. 3 The short-circuit photocurrent response of 1/FTO, SWCNT/FTO and 1-SWCNT/FTO under white light (150 W Xe lamp). The electrolyte was 0.5 M LiI and 0.01 M I2 in acetonitrile and the counter electrode is a platinum wire. | ||
Single-SWCNT devices show excellent electronic properties, such as photoconductivity, and therefore have recently received much attention.43,44 Here, the photoinduced electron transport properties of the 1-SWCNT assembly were investigated by using a single-SWCNT-FET device (Fig. 4a). SWCNT-FETs were fabricated using patterned chemical vapour deposition (CVD) and photolithography (see details in the Experimental section). The decoration was performed by immersing SWCNT-FETs in a THF solution of 1 (1 mM) for about 1 s and rinsing with fresh THF, forming 40–120 nm-sized clusters of 1 along SWCNT surfaces (Fig. S9†). Fig. 4b shows the evolution of gate transfer characteristics upon decoration with 1, followed by photoexcitation. Without the decoration of 1, the SWCNT-FET (black squares) shows ambipolar characteristics (finite n-channel conductance). However, after assembly treatment with 1, the 1-decorated SWCNT-FET (red circles) shows unipolar p-type transport, with the gate threshold voltage positively shifting by ∼5 V. This p-type characteristic is attributed to the charge transfer between 1 and local parts of the SWCNTs, suggesting that 1 exerts a strong electron acceptor effect on the SWCNTs. The p-channel current decreases upon decoration of 1, which is probably due to the enhanced scattering. Moreover, upon illumination with light (11.3 mW), a further hole doping effect is observed and the device does not show complete depletion of conductance (empty red circles). In contrast, the bare SWCNT-FET (control experiment) shows almost no difference in the photoresponse (empty black squares). Fig. 4c shows the I–V characteristics of the device upon illumination. A gate voltage of 2 V was applied to the device to intentionally turn it off. In the dark state, the device was turned off and electronic conduction occurred only at high bias (>4 V) voltages (black circles). In contrast, upon illumination with light, a quasi-linear transport characteristic appears (open circles in Fig. 4c). The optical responsivity (R) of the device was estimated to be about 7 × 104 at −3 V bias according to the following formula:45
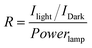 | (1) |
 | ||
| Fig. 4 a) A schematic illustration of the photoconductivity experiment using a FET equipped with a single SWCNT with the decoration of 1; b) the evolution of the gate transfer (Isd-Vg) characteristics of 1-decorated SWCNT-FET with and without light illumination and those of the SWCNT-FET with and without light illumination, bias voltages Vsd = 100 mV; c) the Isd-Vsd characteristics of the 1-decorated SWCNT-FET device with and without light illumination (at Vg = 2 V). | ||
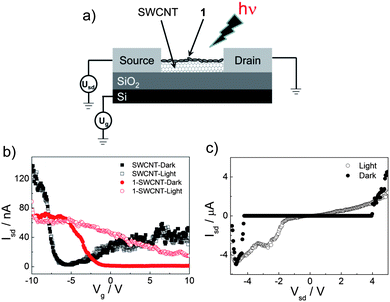
The photoelectric conversion performance of 1-SWCNT is supported by the photoconductivity evaluation via a flash-photolysis time-resolved microwave conductivity (TRMC) technique.35Fig. 5 shows the transient photoconductivity (ϕ∑μ, where ϕ represents the quantum efficiency of the charge carrier generation and ∑μ represents the sum of the nanometre-scale charge carrier mobilities) of 1-SWCNT and 1 upon 355 nm laser-pulse irradiation. In the presence of SWCNTs, the ϕ∑μ value increased by about 60% (1.6 × 10−4 cm2 V−1s−1) compared to that of 1. Therefore, the increased photoconductivity supports the electron communication in the 1-SWCNT assembly, which is consistent with the FET result.
 | ||
| Fig. 5 The TRMC kinetics of 1 and 1-SWCNT. | ||
Additionally, the 1-SWCNT assembly provides an anti-wetting surface, which is highly desirable for practical applications. The scanning electron microscopy (SEM) image (Fig. S11†) shows that the surface of the SWCNT is fully coated with the assembled 1 and exhibits a different thickness and roughness at both the nano- and micrometre scales. The hydrophobicity of 1, combined with the surface roughness of SWCNT, renders the 1-SWCNT assembly superhydrophobic, with a static water contact angle (CA) of 154° (Fig. S11a†). In contrast, the surface of the SWCNT film without 1 has a CA of only 104° (Fig. S11b†). The superhydrophobic films of 1-SWCNT possess high durability under a variety of environmental conditions, such as in various acidities, basicities, ionic strengths and polar solvents (Fig. S12†). Superhydrophobicity of nanocarbon materials8 would be beneficial for any applications in optoelectronic devices, by reducing the influence of water on their performance.
3.4 Steady-state fluorescence and photoluminescence excitation studies
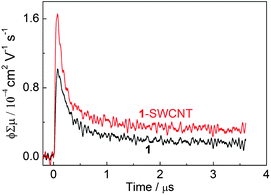
Since the photoexcitation process of 1-SWCNT is crucial in the photovoltaic system, steady-state fluorescence experiments were performed. Fig. 6a shows the steady-state fluorescence of 1 in the absence (curve i) and presence (curve ii) of the SWCNTs in CHCl3. In both cases, 1 exhibits a single transition at 712 nm. The fluorescence intensity of 1-SWCNT relative to that of 1 is reduced by 55%. Similar results were obtained in THF solution (Fig. S13a†), suggesting the occurrence of an interaction between the excited state of 1 and the SWCNTs. | ||
| Fig. 6 a) The steady-state fluorescence spectra of 1 (curve i) and 1-SWCNT (curve ii) in CHCl3, λex = 400 nm; PLE mapping of b) 1-SWCNT in THF solution and c) SWCNT in SDS aqueous solution with assignment.46 | ||
To obtain more information on the interaction between 1 and SWCNT in the excited state, photoluminescence excitation (PLE) mapping studies of SWCNTs in the near infrared (NIR) region were performed for the 1-SWCNT assembly in THF (Fig. 6b). For comparison, we also measured the PLE spectrum of the SWCNTs in SDS aqueous solution (Fig. 6c), where chiral indices of (9,4), (7,6), (8,4), (10,2), (7,5), (8,3), (9,2), (6,5) and (10,3) were observed.46 The emission from 1-SWCNT solution is relatively weaker than that of SWCNT-SDS. Since the PL intensity of the SWCNTs decreases with the externally applied potential,47 the weaker emission could be additional evidence for excited-stated events between 1 and SWCNTs. In addition, the absorption of 1 in the visible region and the small bundles of SWCNTs in solution might also weaken the emission of SWCNTs.
3.5 Time-resolved fluorescence and nanosecond transient absorption studies
The excited state dynamics of 1-SWCNT was further investigated by using picosecond time-resolved spectroscopy. As shown in Fig. 7a, the fluorescence decay of 1 in CHCl3 (red) can be represented by a single-exponential function with a lifetime of 1160 ps around 710 nm. Contrastingly, in the 1-SWCNT CHCl3 solution, a biexponential decay behavior was observed with lifetimes of 300 ps (40%) and 1430 ps (60%) around 710 nm. The different decays observed for 1 in both solutions indicate electronic coupling in the 1-SWCNT complex. While the larger component is related to the fundamental decay of unbound 1, the shorter component suggests the formation of the 1-SWCNT assembly and provides evidence for the energy/electron transfer process.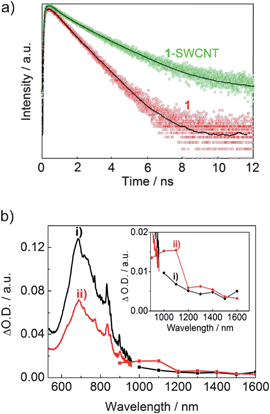 | ||
| Fig. 7 a) The time-resolved fluorescence spectra of 1 and 1-SWCNT (around 710 nm) and their fitting curves (solid lines) in CHCl3, λex = 400 nm, the y-axis is a logarithmic scale; b) the nanosecond transient absorption spectra of 1 (curve i) and 1-SWCNT (curve ii) in THF at 4 μs using 355 nm laser irradiation. Inset: enlarged absorption curve (900–1600 nm). | ||
To identify the transient intermediates involved in 1-SWCNT, transient absorption spectral studies were performed using a 355 nm laser,48 which corresponds to a wavelength of predominant C60 absorption (∼75%, see Fig. S14†) and, therefore, mainly excited the C60 moiety and formed a C60 triplet excited state (3C60*) via intersystem crossing from the singlet. In the absence of oxygen, transient absorption bands from the visible region to the near-IR region were observed for 1-SWCNT, as shown in Fig. 7b. The characteristic band of the triplet–triplet absorption of 1 centered at 680–700 nm was clearly observed, accompanying the transient absorption of one-electron reduced product (C60˙−) at around 1000–1100 nm for 1-SWCNT (red). However, a signature of radical anion was not observed for 1 (black). The spectroscopic data of the triplet and radical anion of 1 are in good agreement with those of pyrrolidinofullerene.49 The appearance of the transient absorption of the radical anion of 1 excludes the possibility that 1 acts as a donor in the assembly, since a C60 radical cation was not observed. Therefore, the observation of a C60 radical monoanion in the 1-SWCNT solution implies the occurrence of charge separation by the photoexcitation of 1, where 1 acts as an acceptor. Thus, in the photoelectrochemical cell, upon excitation, 1 transferred an electron to FTO and SWCNT transported a hole to iodine ion in the electrolyte.
According to the time-resolved fluorescence results, the charge separation rate κCS and the quantum yield ΦCS were calculated as follows:5
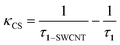 | (2) |
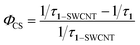 | (3) |
4. Conclusions
In summary, a nanocarbon hybrid of a C60 derivative and SWCNTs was fabricated. Taking advantage of the non-covalent interaction with alkyl chains in the C60 derivative, the SWCNTs were almost debundled and dispersed in organic solvents. The alkylated C60–SWCNT nanohybrid not only preserves the intrinsic optical and electronic properties of SWCNT, but couples this with the electron-accepting property of C60, facilitating device fabrication via classic wet processes. As examples, the nanocarbon assembly-modified transparent photoelectrodes and single-SWCNT device were fabricated and performed well in photovoltaic studies. In addition, a film of the same nanocarbon assembly exhibits superhydrophobicity, which provides an extra benefit for any photovoltaic application, i.e., towards the fabrication of waterproof devices. Photoinduced electron transfer within the 1-SWCNT assembly was probed by a series of spectroscopic techniques. Steady-state and time-resolved fluorescence studies suggest efficient quenching of singlet-excited C60 moieties in the assembly. More direct evidence for the photoinduced electron transfer was obtained from the nanosecond transient absorption studies, where the C60 radical anion was observed upon laser illumination. Therefore, the spectroscopic results, taken together with the light harvesting features of the 1-SWCNT assembly, provide clues for the future design of nanocarbon hybrid systems for photovoltaic applications.Acknowledgements
This work was supported, in part, by KAKENHI from the MEXT, Japan and PRESTO, JST, Japan, as well as the cluster of excellence ‘Unicat’, Germany. Y.S. gratefully acknowledges JSPS for the Postdoctoral Fellowships for Foreign Researchers. We thank the Kyoto Advanced Nanotechnology network for technical support with TEM measurements and Dr M. Takeuchi, Dr N. Shirahata and Dr S. Ishihara (NIMS) for useful discussion.References
- W. Zhang, J. K. Sprafke, M. Ma, E. Y. Tsui, S. A. Sydlik, G. C. Rutledge and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 8446–8454 CrossRef CAS.
- J.-F. Nierengarten, New J. Chem., 2004, 28, 1177–1191 RSC.
- S. Filippone, E. E. Maroto, Á. Martín-Domenech, M. Suarez and N. Martín, Nat. Chem., 2009, 1, 578–582 CrossRef CAS.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS.
- F. D'Souza, R. Chitta, A. S. D. Sandanayaka, N. K. Subbaiyan, L. D'Souza, Y. Araki and O. Ito, J. Am. Chem. Soc., 2007, 129, 15865–15871 CrossRef CAS.
- S.-Y. Ju, W. P. Kopcha and F. Papadimitrakopoulos, Science, 2009, 323, 1319–1323 CrossRef CAS.
- D. M. Guldi, E. Menna, M. Maggini, M. Marcaccio, D. Paolucci, F. Paolucci, S. Campidelli, M. Prato, G. M. Aminur Rahman and S. Schergna, Chem.–Eur. J., 2006, 12, 3975–3983 CrossRef CAS.
- S. Srinivasan, V. K. Praveen, R. Philip and A. Ajayaghosh, Angew. Chem. Int. Ed., 2008, 47, 5750–5754 CAS.
- C. Ehli, C. Oelsner, D. M. Guldi, A. Mateo-Alonso, M. Prato, C. Schmidt, C. Backes, F. Hauke and A. Hirsch, Nat. Chem., 2009, 1, 243–249 CrossRef CAS.
- T. Fujigaya, T. Morimoto, Y. Niidome and N. Nakashima, Adv. Mater., 2008, 20, 3610–3614 CrossRef CAS.
- D. Bonifazi, O. Enger and F. Diederich, Chem. Soc. Rev., 2007, 36, 390–414 RSC.
- S. Y. Jeong, S. C. Lim, D. J. Bae, Y. H. Lee, H. J. Shin, S. M. Yoon, J. Y. Choi, O. H. Cha, M. S. Jeong, D. Perello and M. Yun, Appl. Phys. Lett., 2008, 92, 243103 CrossRef.
- T. Kaneko, Y. Li, S. Nishigaki and R. Hatakeyama, J. Am. Chem. Soc., 2008, 130, 2714–2715 CrossRef CAS.
- T. Umeyama, N. Tezuka, F. Kawashima, S. Seki, Y. Matano, Y. Nakao, T. Shishido, M. Nishi, K. Hirao, H. Lehtivuori, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, Angew. Chem. Int. Ed., 2011, 50, 4615–4619 CAS.
- D. M. Guldi, G. M. A. Rahman, F. Zerbetto and M. Prato, Acc. Chem. Res., 2005, 38, 871–878 CrossRef CAS.
- A. Kongkanand, R. M. Domínguez and P. V. Kamat, Nano Lett., 2007, 7, 676–680 CrossRef CAS.
- F. Diederich and M. Gomez-Lopez, Chem. Soc. Rev., 1999, 28, 263–277 RSC.
- D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolskia and N. Martín, Chem. Soc. Rev., 2009, 38, 1587–1597 RSC.
- A. Takai, M. Chkounda, A. Eggenspiller, C. P. Gros, M. Lachkar, J. M. Barbe and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 4477–4489 CrossRef CAS.
- H. Imahori and S. Fukuzumi, Adv. Funct. Mater., 2004, 14, 525–536 CrossRef CAS.
- S. S. Babu, H. Möhwald and T. Nakanishi, Chem. Soc. Rev., 2010, 39, 4021–4035 RSC.
- T. Umeyama, N. Tezuka, M. Fujita, S. Hayashi, N. Kadota, Y. Matano and H. Imahori, Chem.–Eur. J., 2008, 14, 4875–4885 CrossRef CAS.
- J. K. J. van Duren, X. Yang, J. Loos, C. W. T. Bulle-Lieuwma, A. B. Sieval, J. C. Hummelen and R. A. J. Janssen, Adv. Funct. Mater., 2004, 14, 425–434 CrossRef CAS.
- Y. Yamamoto, G. Zhang, W. Jin, T. Fukushima, N. Ishii, A. Saeki, S. Seki, S. Tagawa, T. Minari, K. Tsukagoshi and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21051–21056 CrossRef CAS.
- Y. Shen, A. G. Skirtach, T. Seki, S. Yagai, H. Li, H. Möhwald and T. Nakanishi, J. Am. Chem. Soc., 2010, 132, 8566–8568 CrossRef CAS.
- M. S. Arnold, J. D. Zimmerman, C. K. Renshaw, X. Xu, R. R. Lunt, C. M. Austin and S. R. Forrest, Nano Lett., 2009, 9, 3354–3358 CrossRef CAS.
- A. S. D. Sandanayaka, Y. Takaguchi, Y. Sako, M. Tamura and O. Ito, Adv. Sci. Lett., 2010, 3, 353–357 CAS.
- J. L. Delgado, P. de la Cruz, A. Urbina, J. T. L. Navarrete, J. Casado and F. Langa, Carbon, 2007, 45, 2250–2252 CrossRef CAS.
- S. Giordani, J.-F. Colomer, F. Cattaruzza, J. Alfonsi, M. Meneghetti, M. Prato and D. Bonifazi, Carbon, 2009, 47, 578–588 CrossRef CAS.
- Y. Takaguchi, M. Tamura, Y. Sako, Y. Yanagimoto, S. Tsuboi, T. Uchida, K. Shimamura, S. Kimura, T. Wakahara, Y. Maeda and T. Akasaka, Chem. Lett., 2005, 34, 1608–1609 CrossRef CAS.
- H. Asanuma, H. G. Li, T. Nakanishi and H. Möhwald, Chem.–Eur. J., 2010, 16, 9330–9338 CrossRef CAS.
- T. Nakanishi, Chem. Commun., 2010, 46, 3425–3436 RSC.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593–596 CrossRef CAS.
- T. Nakanishi, T. Michinobu, K. Yoshida, N. Shirahata, K. Ariga, H. Möhwald and D. G. Kurth, Adv. Mater., 2008, 20, 443–446 CrossRef CAS.
- A. Saeki, S. Seki, T. Takenobu, Y. Iwasa and S. Tagawa, Adv. Mater., 2008, 20, 920–923 CrossRef CAS.
- T. Nakanishi, N. Miyashita, T. Michinobu, Y. Wakayama, T. Tsuruoka, K. Ariga and D. G. Kurth, J. Am. Chem. Soc., 2006, 128, 6328–6329 CrossRef CAS.
- T. Nakanishi, H. Takahashi, T. Michinobu, M. Takeuchi, T. Teranishi and K. Ariga, Colloids Surf., A, 2008, 321, 99–105 CrossRef CAS.
- J. P. Rabe and S. Buchholz, Science, 1991, 253, 424–427 CAS.
- H. Tanaka, T. Yajima, T. Matsumoto, Y. Otsuka and T. Ogawa, Adv. Mater., 2006, 18, 1411–1415 CrossRef CAS.
- T. Nakanishi, Y. Shen, J. Wang, S. Yagai, M. Funahashi, T. Kato, P. Fernandes, H. Möhwald and D. G. Kurth, J. Am. Chem. Soc., 2008, 130, 9236–9237 CrossRef CAS.
- M. Sharon, Wiley-VCH, Weinheim, Germany, 2002, p. 287.
- C. Li, Y. Chen, Y. Wang, Z. Iqbal, M. Chhowalla and S. Mitra, J. Mater. Chem., 2007, 17, 2406–2411 RSC.
- J. Kong, N. R. Franklin, C. W. Zhou, M. G. Chapline, S. Peng, K. J. Cho and H. J. Dai, Science, 2000, 287, 622–625 CrossRef CAS.
- H. S. Kim, E. K. Jeon, J. J. Kim, H. M. So, H. Chang, J. O. Lee and N. Park, Appl. Phys. Lett., 2008, 93, 123106 CrossRef.
- O. Bulteel, A. Afzalian and D. Flandre, Analog Integr. Circ. Sig. Process, 2010, 65, 399–405 CrossRef.
- S. M. Bachilo, M. S. Strano, C. Kittrell, R. H. Hauge, R. E. Smalley and R. B. Weisman, Science, 2002, 298, 2361–2366 CrossRef CAS.
- Y. Tanaka, Y. Hirana, Y. Niidome, K. Kato, S. Saito and N. Nakashima, Angew. Chem., Int. Ed., 2009, 48, 7655–7659 CrossRef CAS.
- A. Saeki, S. Seki, N. Satoh, K. Yamamoto and S. Tagawa, J. Phys. Chem. B, 2008, 112, 15540–15545 CrossRef CAS.
- K. G. Thomas, V. Biju, M. V. George, D. M. Guldi and P. V. Kamat, J. Phys. Chem. A, 1998, 102, 5341–5348 CrossRef CAS.
- S. Komamine, M. Fujitsuka, O. Ito, K. Moriwaki, T. Miyata and T. Ohno, J. Phys. Chem. A, 2000, 104, 11497–11504 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HR-TEM, Raman, DSC, AFM, SEM, solution photographs, IPCE, steady-state and time-resolved fluorescence spectra. See DOI: 10.1039/c1sc00360g |
| This journal is © The Royal Society of Chemistry 2011 |