Covalently incorporated protein–nanogels using AGET ATRP in an inverse miniemulsion†
Saadyah E.
Averick
a,
Andrew J. D.
Magenau
a,
Antonina
Simakova
a,
Bradley F.
Woodman
b,
Andrew
Seong
a,
Ryan A.
Mehl
b and
Krzysztof
Matyjaszewski
*a
aDepartment of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA 15213, USA. E-mail: km3b@andrew.cmu.edu; Fax: +1 412-268-6897; Tel: +1 412-268-3209
bDepartments of Chemistry, Franklin & Marshall College, Lancaster, PA 17604-3003, USA. E-mail: ryan.mehl@fandm.edu; Fax: +1 717-291-4343; Tel: +1 717-291-4125
First published on 24th March 2011
Abstract
Using a genetically engineered protein, containing a non-natural amino acid bearing an atom transfer radical polymerization (ATRP) initiator, protein–nanogel hybrids (PNHs) were synthesized by activator generated by electron transfer (AGET) ATRP in an inverse miniemulsion. The route presented is an appropriate synthetic strategy to covalently and site specifically incorporate green fluorescent protein (GFP) into well-defined nanogels. These PNHs were analyzed using dynamic light scattering (DLS), UV-visible fluorescence spectroscopy and confocal microscopy to confirm the successful integration of GFP proteins into each nanogel (NG), while preserving its native tertiary structure.
Protein–polymer hybrids (PPHs), first described in 1970,1,2 have revolutionized disease treatment3,4 and catalytic processes.5–8 PPHs are typically comprised of linear or branched polymers “grafted to” or “grafted from” a protein.3,9–13 The advantages of PPHs over native proteins, from a therapeutic perspective, include an increased in vivo stability, minimized immune recognition due to steric effects, enhanced in vivo circulation and therapeutic effect.1,14–16 Catalytically, PPHs further expand enzymatic processes to organic synthesis and media owing to their improved solubility in non-aqueous solvents.5,6
Recently, the concept of PNHs has been introduced in order to overcome some of the long-term stability issues associated with PPHs.5,8,17–19 PPHs exposed to organic solvents for long durations or reactions with harsh conditions are susceptible to deactivation, rendering them functionally inactive. However, encapsulation of proteins into nanogel matrices has demonstrated improved protein activity and stability for several systems, such as carbonic anhydrase,19lipase,5 and horseradish peroxidase20 among others.5,8 It has been found that proteins encapsulated in nanogels show superior temperature and organic solvent stability. Both of these characteristics are paramount to expanding the catalytic potential of enzymatic systems and their eventual use in therapeutic applications.
Typically, PNHs are synthesized in a two-step process. The proteins are first functionalized using N-hydroxysuccinimide (NHS) acrylate and subsequently copolymerized with acrylamides and crosslinkers using REDOX initiated free radical polymerization (FRP).5,8,17–19 Because NHS chemistry modifies lysine residues in a non-specific manner,16,21protein activity within PNHs has limited reproducibility between reaction batches. Moreover, non-specific functionalization potentially leads to deactivation of the protein active sites and denaturing. Although these systems show better stability relative to native proteins under harsh reaction conditions, they are also limited by several factors. Some of these factors include: monomer, particle size, protein loading capacity, limited potential for controlled release, and lower activity due to non-specific protein incorporation.
We envisioned a system where well-defined PNHs could be synthesized by combining controlled radical polymerization with a genetically engineered protein containing a site-specific initiator. These PNHs would therefore have controlled size distributions, potential to be combined with a wide range of monomers, minimal loss of protein activity due to nanogel encapsulation and control over protein loading per nanogel. Recently, the synthesis of a non-natural amino acid bearing an ATRP22–24 initiating group, 4-(2′-bromoisobutyramido) phenylalanine, and its genetic expression into the GFP was reported.25 This was accomplished using an engineered M. jannaschiityrosyl tRNA/aminoacyl-tRNA-synthetase pair vector in E. coli. By using genetic engineering, specific placement of the initiating amino acid was selected to be expressed at the 134 position of GFP (referred in the text as GFP1), thereby, protecting the protein's active sites and structurally weak regions. Furthermore, genetic engineering allows precise control over the number of chains attached (i.e. the number of initiation sites) to the protein, overcoming a primary drawback of traditional PPHs and PPNs. Thus, genetically engineered proteins have unique capabilities to solve some of the traditional limitations of PPH and PNH materials.
GFP–NGs were prepared through a water-in-oil inverse miniemulsion utilizing AGET ATRP to generated nanogels of ca. 200 nm in diameter (Scheme 1).26,27 The inverse miniemulsion was composed of a water phase consisting of Cu(II)Br2/tris(2-pyridylmethyl)amine (TPMA) as the catalytic species, 4% (w/w total solids) GFP1 and poly(ethylene glycol)isobutyryl bromide (PEG2000MI, Mn = 2000) coinitiator, oligo(ethylene oxide)methacrylate (PEG300MA, Mn = 300) monomer, and a poly(ethylene glycol) dimethacrylate (PEG4000DM, Mn = 4000) crosslinking agent. These reagents were dissolved in 1.46 ml of 0.1 M phosphate buffered saline (PBS) solution (pH 7.4) and emulsified with a 0.05% (w/w) of Span-80 in cyclohexane using ultrasonication to form stable droplets.28,29 After degassing, ascorbic acid was injected to initiate AGET ATRP which was stopped after 15 hours at 30 °C. The nanogels were purified by precipitation into THF followed by dialysis (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MWCO membrane) with water to remove all unreacted reagents and proteins.30,31
000 MWCO membrane) with water to remove all unreacted reagents and proteins.30,31
 | ||
| Scheme 1 Covalent attachment of GFP1 into a nanogel matrix using AGET ATRP in an inverse miniemulsion. | ||
The size distribution and uniformity of the resulting PNHs were characterized by DLS. PNHs created with GFP1 resulted in monomodal particles with a diameter of ca. 240 nm (Fig. 1A). The monomodal and narrow size distribution demonstrates that inverse miniemulsions are an effective method to produce well-defined PNHs. Confocal microscopy was then used to ascertain the structural characteristics of the hybrid material in aqueous solution at a concentration of 0.01 mg ml−1 and to confirm the nanogel particle size determined with DLS. The confocal microscopy image shown in Fig. 1B revealed well-defined spherical particles with a size of ca. 300 nm, similar to those determined by DLS. Furthermore, this image confirms the encapsulation and covalent incorporation of GFP1 into the nanogel matrix as indicated by its inherent green fluorescent response. Any unreacted GFP1 would have been removed from the resulting nanogel scaffold during dialysis.
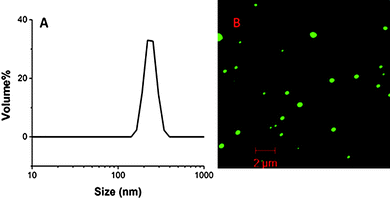 | ||
| Fig. 1 (A) Dynamic light scattering of GFP1–PNH (d = 240 nm, PDI = 0.25). (B) Confocal microscopy of GFP1–PNH. | ||
GFP1 was rationalized to be an excellent model system to efficiently evaluate if any potential damage or denaturing occurs to the protein during the AGET ATRP inverse miniemulsion process. Damage to the GFP's tertiary structure would result in a loss of its absorption and fluorescent properties.32 Therefore, GFP1–PNHs were studied using UV-vis and fluorescence spectroscopy and their spectra were compared to those of GFP wild type (GFP-wt). Fig. 2A shows the absorption spectra of GFP1–PNH to have a maximum absorption at 485 nm, the same maximum absorption wavelength as GFP-wt. In the emissions spectra (Fig. 2B) of GFP1–PNG and GFP-wt, the same maximum emission wavelength at 510 nm was observed for both samples. By comparing the emission and absorption spectra of GFP1–PNG and GFP-wt, it can be concluded that covalent encapsulation of GFP1 into the nanogel effectively maintained the GFP's tertiary structure.
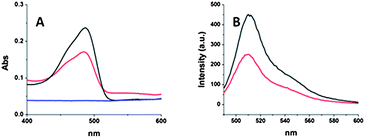 | ||
| Fig. 2 (A) UV-vis absorption spectra GFP-wt (black), GFP1–PNH (red), and GFP-wt–PNH (blue). (B) Emission spectra of GFP-wt (black) and GFP1–PNH (red). | ||
Due to the retention of GFP1's tertiary structure, the molar extinction coefficient of GFP can be used to estimate the loading of GFP1 into the nanogel. The concentration of GFP1 in a 0.42 mg ml−1 solution of nanogel was ∼0.09 mg ml−1 (i.e. a 2.1% (w/w) GFP1 within the nanogel scaffold). This value can be compared with the 4% targeted loading used in the inverse miniemulsion formulation, therefore preserving and covalently incorporating nearly 50% of the initially added GFP protein.
Previous literature has shown that proteins can be entrapped within a NG matrix,31 although limited evidence supporting retention of the proteins native structure was given. In order to examine the effect of covalent incorporation versus physical entrapment, a control experiment was conducted to explore the difference between our previously described covalently incorporated GFP1–PNH and an analogous nanogel utilizing a GFP-wt without a genetically engineered ATRP initiator. Identical reaction conditions were used, as in our former inverse miniemulsion, although GFP-wt was incorporated instead of GFP1. As shown in Fig. 2A, nanogels prepared with GFP-wt did not retain fluorescent properties upon purification, demonstrating the necessity of a covalent linkage to avoid protein leaching from the NG scaffold and denaturing.
AGET ATRP in inverse miniemulsion as a method to produce nanogels with covalent incorporation of proteins offers several advantages over commonly employed two-step FRP methodologies. Most notably, ATRP nanogels have been reported to have more homogenous and controlled structures in comparison to other methods.11 Also, a versatile array of nanogel properties can be finely tuned using ATRP, including particle size, loading capacity, and degree of swelling. In traditional two step PNH synthesis, proteins are anchored to the nanogel matrix by multiple linkage points stemming from the numerous acrylamide moieties on the protein surface. These multiple and random anchoring points to the matrix prevent controlled release of the protein for therapeutic applications. The GFP1 protein used in this work was covalently incorporated into the nanogel, with only one genetically encoded junction point. Therefore, GFP1 tethered to the nanogel matrix may provide an improved methodology for tuneable and controlled release in therapeutic applications.
Conclusion
A new efficient route to well-defined PNHs has been shown. By employing a genetically engineered protein, bearing a single ATRP initiator, AGET ATRP in an inverse miniemulsion could be used to produce covalently linked PNHs with a diameter of 240 nm. In addition, the protein's tertiary structure was maintained as demonstrated by retention of its fluorescent activity. PNGs with physically entrapped wild type GFP were synthesized, although no fluorescent activity was maintained. These results suggest either protein leaching from the nanogel or loss of the protein's tertiary structure. PNHs with a single covalent linkage are expected to be suitable for potential controlled release applications.Acknowledgements
Financial support from the NSF (Grant DMR 09-69301) and the CRP Consortium at Carnegie Mellon University is gratefully appreciated. The authors would also like to thank Dr Sidi A. Bencherif for giving insight into the synthesis of these nanogels.Notes and references
- A. Abuchowski, T. van Es, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578–3581 CAS.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS.
- J. Ge, D. Lu, J. Wang and Z. Liu, Biomacromolecules, 2009, 10, 1612–1618 CrossRef CAS.
- Y. Ito, H. Fujii and Y. Imanishi, Biotechnol. Prog., 1994, 10, 398–402 CrossRef CAS.
- K. Renggli and N. Bruns, in Green Polymer Chemistry: Biocatalysis and Biomaterials, American Chemical Society, 2010, vol. 1043, ch. 2, pp. 17–34 Search PubMed.
- M. Yan, J. Ge, Z. Liu and P. Ouyang, J. Am. Chem. Soc., 2006, 128, 11008–11009 CrossRef CAS.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508–6509 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- H. Li, A. P. Bapat, M. Li and B. S. Sumerlin, Polym. Chem., 2011, 2, 323–327 RSC.
- M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854–859 RSC.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657–7663 CrossRef CAS.
- J. P. Magnusson, S. Bersani, S. Salmaso, C. Alexander and P. Caliceti, Bioconjugate Chem., 2010, 21, 671–678 CrossRef CAS.
- K. Velonia, Polym. Chem., 2010, 1, 944–952 RSC.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS.
- J. Ge, D. Lu, J. Wang, M. Yan, Y. Lu and Z. Liu, J. Phys. Chem. B, 2008, 112, 14319–14324 CrossRef CAS.
- J. Hong, P. Gong, D. Xu, L. Dong and S. Yao, J. Biotechnol., 2007, 128, 597–605 CrossRef CAS.
- M. Yan, Z. Liu, D. Lu and Z. Liu, Biomacromolecules, 2007, 8, 560–565 CrossRef CAS.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263–6266 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- J. C. Peeler, B. F. Woodman, S. Averick, S. J. Miyake-Stoner, A. L. Stokes, K. R. Hess, K. Matyjaszewski and R. A. Mehl, J. Am. Chem. Soc., 2010, 132, 13575–13577 CrossRef CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- J. K. Oh, D. J. Siegwart, H.-i. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef CAS.
- J. K. Oh, D. J. Siegwart and K. Matyjaszewski, Biomacromolecules, 2007, 8, 3326–3331 CrossRef CAS.
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 5578–5584 CrossRef CAS.
- S. A. Bencherif, N. R. Washburn and K. Matyjaszewski, Biomacromolecules, 2009, 10, 2499–2507 CrossRef CAS.
- D. J. Siegwart, A. Srinivasan, S. A. Bencherif, A. Karunanidhi, J. K. Oh, S. Vaidya, R. Jin, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2009, 10, 2300–2309 CrossRef.
- W. Gao, W. Liu, T. Christensen, M. R. Zalutsky and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16432–16437 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00050k |
| This journal is © The Royal Society of Chemistry 2011 |