Design, synthesis and biological evaluation of novel benzothiazole and triazole analogs as falcipain inhibitors†
Falgun
Shah
a,
Yunshan
Wu
a,
Jiri
Gut
b,
Yakambram
Pedduri
a,
Jennifer
Legac
b,
Philip J.
Rosenthal
b and
Mitchell A.
Avery
*ac
aDepartment of Medicinal Chemistry, School of Pharmacy, University of Mississippi, University, MS 38677, USA. E-mail: mavery@olemiss.edu; Fax: +662-915-5638; Tel: +662-915-5879
bDepartment of Medicine, San Francisco General Hospital, University of California, San Francisco, CA 94143, USA
cNational Center for Natural Products Research, University of Mississippi, University, MS 38677, USA
First published on 27th September 2011
Abstract
We describe the design, combinatorial library synthesis and biological evaluation of compounds with benzothiazole and triazole cores as inhibitors of falcipain, cysteine proteases of the malaria parasite Plasmodium falciparum. These classes were originally discovered by structure-based virtual screening of a focused cysteine protease inhibitor library. Fifteen structural analogs of both series showed moderate inhibition of falcipain-2. Two compounds, 41 and 42, were predicted by docking studies to interact with polar residues buried in the S2 pockets of falcipain-2 and -3, and these compounds inhibited both enzymes. Compound 41 also demonstrated activity against chloroquine-resistant cultured P. falciparum parasites at the lower micromolar concentration. Evaluation of 41 and 42 against mammalian cysteine proteases of papain family suggest these polar residues of the S2 pocket may not be important for the design of selective inhibitors against falcipain.
Malaria, in particular infection with Plasmodium falciparum, is a severe disease causing hundreds of millions of illnesses and about one million deaths each year.1 The disease is prevalent throughout the tropics, mostly affecting children under the age of five.2 The efficacy of available antimalarial compounds is increasingly compromised due to the wide spread of resistant malaria parasites.3 Of particular concern are early signs of resistance to new artemisinin-based combination therapy (ACT) regimens, which are first-line treatments for uncomplicated falciparum malaria.4–6 These observations have triggered a devoted search for potential leads intervening in crucial biochemical pathways to facilitate development of next generation antimalarial drugs.3,7,8 Among these leads are inhibitors of falcipain cysteine proteases.9 The cysteine proteases of the P. falciparum are collectively known as falcipains (FPs), among which falcipain-2 (FP-2) and falcipain-3 (FP-3) are major hemoglobinases, present in the food vacuole of P. falciparum. They are responsible for host hemoglobin hydrolysis providing building blocks for parasites protein synthesis. Specific inhibitors of both enzymes blocked hemoglobin hydrolysis, formed hemoglobin filled food vacuoles in P. falciparum trophozoites and displayed antiplasomodial acivity.10–14 These findings suggest FPs as validated targets for the development of antimalarial chemotherapy.
Most of the potent FP-2 inhibitors described in the literature (see ref. 10, 11, 13, 15 and reviews ref. 16,17) were peptide analogs, displaying a covalent irreversible binding mode with the active site cysteine, and they were relatively non-selective. It is desirable to develop non-peptidic, non-covalent inhibitors of FPs to minimize toxicity while retaining in vivo activity and selectivity. As a part of our ongoing efforts to develop novel non-peptidic inhibitors of FPs,18–20 we previously carried out structure-based virtual screening (SBVS) of the focused cysteine protease inhibitor (FCPI) library built based on soft-electrophiles against the X-ray structure of FP-2.21 The study identified 21 diverse micromolar inhibitors of FP-2. The current efforts were initiated with the goal of optimizing the potency of lower micromolar FP-2 hits 1 and 2 with a tetrazole and a benzothiazole scaffold, respectively (Fig. 1). The aim was to establish the structure–activity relationships around these scaffolds. First, we carried out core-hopping in the benzothiazole series for the potency optimization of these hits. Then, a total of 35 structural analogs comprising both series were made and evaluated against the enzyme FP-2, and FP-3 and for the activity against cultured P. falciparum parasites. Twelve compounds from the benzothiazole series and three compounds from the triazole series showed moderate activity against FP-2. Two compounds from the benzothiazole series and one from the triazole series showed dual inhibition of FP-2 and FP-3. One particular compound from benzothiazole series showed corresponding inhibition of the cultured parasites. Docking of these inhibitors in the FP-2 active site provided insights about structure activity relationships and interactions of the inhibitors with the active site of FP-2.
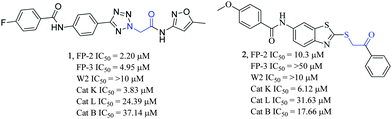 | ||
| Fig. 1 2D chemical structures of compound 1 and 2. The activities of 1 and 2 against the cysteine proteases: FP-2 and FP-3, the chloroquine-resistant (W2) strain of P. falciparum, and the mammalian cysteine proteases cathepsins (Cat) K, L, and B are shown. | ||
Core or scaffold hopping is a technique frequently used by medicinal chemists to discover novel compounds by mutating the molecule core or scaffold starting from known active molecules.22 Core hopping gives medicinal chemists an opportunity to pursue drug design in a different chemical space to improve potency, to improve adsorption, distribution, metabolism, excretion and toxicity liabilities of a given scaffold, to circumvent intellectual property rights associated with a parent scaffold or to speed up the lead optimization process.22
Our primary goal was to improve the potency of virtual screening hits 1 and 2. For the initial optimization of 2 with core-hoping strategy, we selected analogs which were commercially available, affordable and had an α-thioketone to act as a soft-electrophile prior to initiating extensive synthetic efforts. From the docking studies of 2, we envisioned that the replacement of the benzothiazole core with benzimidazole and thioacetamide moieties may improve the overall affinity of 2 by forming strong H-bond networks with the backbone Gly83 and Asn173. Therefore, our selection was limited to commercially available compounds (3–10) containing these cores with the goal of improving potency. While selecting compounds in these series, compounds with electron withdrawing groupspara to the phenyl ring (for example, 3, 4, 7) were preferred to enhance the reactivity of the carbonyl carbon with the catalytic Cys42 of the FP-2 active site. It is worthwhile to mention that compounds 9 and 10 can be reasonably enolic and may predominantly behave as α, β-unsaturated ketones in the FP-2 active site. This enolic behavior is in equilibrium, thus availability of the carbonyl tautomer should not be problematical. Selected compounds were docked in the crystal structure of FP-2 using Glide extra precision (XP) mode and a previously validated docking protocol.21 Although thioacetamide and benzimidazole compounds showed improved interaction profiles in the binding site (Figure S1), they were inactive against FP-2 at concentration up to 50 μM. These findings, although surprising, suggested the importance of the benzothiazole core for FP-2 inhibition and prompted us to design a library around this core.
From the docking pose of compound 2 shown in Fig. 2, it was evident that 2 could be modified to better occupy the S1′ and, S2 pockets of FP-2. For example, hydrophobic substituents can be placed on the 6-aminobenzothiazole scaffold to interact with hydrophobic residues such as Ala235, Ile85 and Leu172 lining the border of the S2 pocket. Also, the flat S1′ pocket of FP-2 is composed of hydrophobic residues such as Trp206 and Val152. Therefore, two regions of the benzothiazole core spanning the S1′ and S2 sites were selected to perform chemical transformations and, thereby to develop SAR information. In addition, α-thioketones of 2 were modified to α-thioesters and α-thioacetamide electrophiles to investigate the chemical reactivity of these electrophiles with the active site cysteine sulfur and determine its effects on FP-2 inhibition. Thus, three series were designed (Table 1).
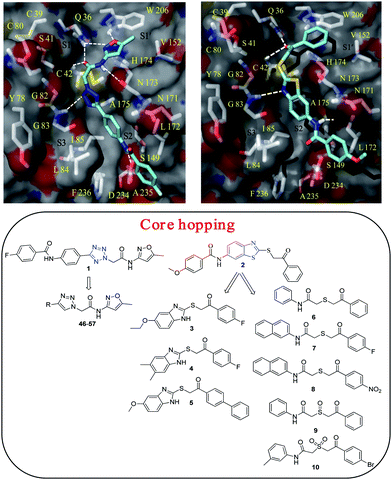 | ||
| Fig. 2 (top) docking pose of compounds 1 and 2 in the FP-2 binding site; (bottom) core hopping of compound 1 and 2 with tetrazole and benzothiazole cores (highlighted in blue), respectively are shown. The tetrazole core of 1 was modified to triazole core to take an advantage of click chemistry. The benzothiazole core of 2 was modified to commercially available benzimidazole and thioacetamide core containing compounds (3–10) with anticipated gain in potency. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Sr. No | R 1 | R 2 | FP-2 IC50 (μM) | Sr. No | R 1 | R 2 | FP-2 IC50 (μM) |
| a IC50 of compound 41 and 42 against FP-3 were 13.77 μM and 14.94 μM, respectively; compound 24 & 41 also inhibited the growth of W2 strain of P. falciparum with IC50 = 2.08 μM and 4.65 μM, respectively. IC50 of positive controls, artemisinin and chloroquine, against W2 strain were 0.012 μM and 0.062 μM, respectively. | |||||||
| 16 |

|
H | >50 | 27 |

|

|
>50 |
| 17 |

|
H | >50 | 28 |

|

|
>50 |
| 18 |

|
H | >50 | 29 |

|

|
31.34 |
| 19 |

|
H | >50 | 30 |

|

|
24.33 |
| 20 |

|

|
11.14 | 31 |

|

|
13.89 |
| 21 |

|

|
35.70 | 32 |

|

|
13.96 |
| 22 |

|

|
>50 | 33 |

|

|
16.48 |
| 23 |

|

|
13.18 | 34 |

|

|
16.87 |
| 24 |

|

|
>50 | 39 |

|

|
>50 |
| 25 |

|

|
43.25 | 40 |
|

|
>50 |
| 26 |

|

|
>50 | a 41 |

|

|
12.22 |
| E-64 | — | — | 0.05 | a 42 |

|

|
12.75 |

The deep hydrophobic S2 pocket in FPs harbors polar residues such as Asp234 and, Ser149 in FP-2 and Glu243 and, Ser158 in FP-3. Electrostatic interactions of compounds with the polar residues of the S2 pocket might lead to an overall boost in potency. In addition, sequence alignments of FPs with homologous mammalian protease of papain family (Fig. S2, ESI†) suggested that polar residues of the S2 pocket are replaced by hydrophobic residues in cathepsin K, and L and can be targeted to design selective inhibitors of FPs. Therefore, in the α-thioketones and α-thioacetamide series, compounds with protonated amino groups were designed (compounds 39–42) to exhibit ionic interactions with Asp234 of the S2 subsite. The preliminary docking studies suggested a preference for R over S isomers of designed compounds, and, hence, Boc-(D) phenyl glycine or Boc-(D) valine were used in the synthesis of 39–42.
In the tetrazole series, our aim was to investigate the effects of different substitutions on the activity of 1 that would be specifically interacting with the residues of the critical S2 pocket. Docking studies of 1 (Fig. 2) indicated that the tetrazole core could be replaced by a triazole ring without loss of key interaction with Gly83, thus optimization of 1 could be enhanced by click chemistry via classic Huisgen cycloaddition.23–26 Therefore, initial modification of 1 was focused on establishing an SAR using a 1, 2, 3-triazole core without altering the N-(5-methylisoxazol-3-yl)acetamide side chain of 1 which showed extensive H-bond networks with the key residues of the S1–S1′ pockets of the FP-2 active site. The alkyl or aralkyl groups with varying hydrophobicity and electronic properties were thus appended only at the fourth position of the 1,2,3-triazole moiety.
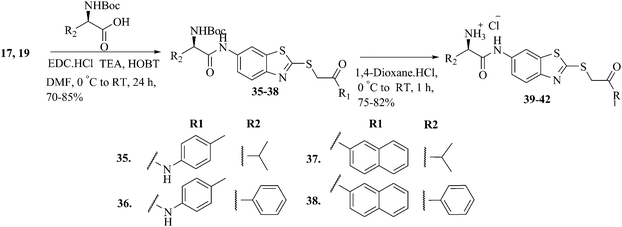
The synthetic strategies adopted to obtain various benzothiazole derivatives are highlighted in Schemes 1 and 2. Scheme 1 depicts condensation of appropriate α-haloketones (12, 13), α-haloacetate (14), and α-haloamides (15) with the commercially available 6-aminobenzothiazole-2-thione (11) in the presence of triethylamine to give respective S-alkylated α-thioketones (16, 17), α-thioesters (18) or α-thioacetamide (19). In the next step of N-acylation, intermediate 16–19 provided corresponding N-acylated derivatives 20–34 by treatment with various acid chlorides. Synthesis of designed analogs targeting polar residues of the S2 pocket was accomplished as depicted in Scheme 2. Accordingly, compounds 17 and 19 were coupled with (D)-N-Boc phenyl glycine and (D)-N-Boc valine using standard 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/hydroxybenzotriazole (EDCI/HOBT) peptide coupling conditions and triethylamine as a base to yield Boc-protected compounds 35–38. Deprotection of Boc with 4 M HCl in 1, 4-dioxane provided the target compounds 39–42 as HCl salts.
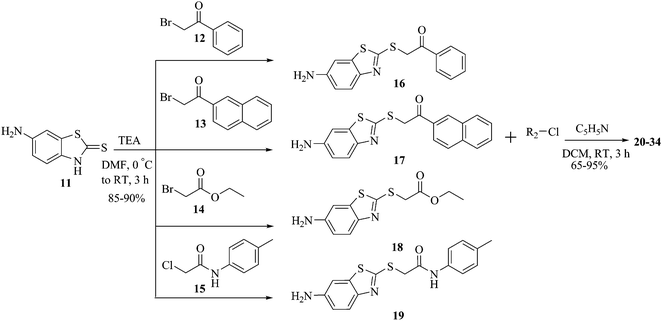 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
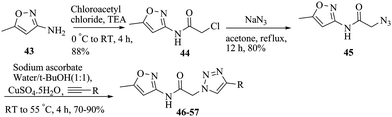
The synthesis of the triazole series is summarized in Scheme 3. The acylchloride intermediate 44 was readily synthesized by coupling commercially available 5-methylisoxazol-3-amine (43) with chloroacetyl chloride, which, upon substitution with sodium azide in acetone, gave an azide intermediate 45. The triazole derivatives 46–57 were then obtained through classic Huisgen 1, 3-dipolar cycloaddition between organic azide 45 and a series of terminal alkynes27–29 (Table 2).
 | ||
| Scheme 3 | ||










The synthesized compounds were evaluated for inhibition of recombinant FP-2 and, FP-3 and, the in vitrogrowth of the chloroquine resistant W2 strain of P. falciparum according to previously described procedures.21 The epoxysuccinate E-64 was used as a positive control for the enzyme assays whereas artemisinin and chloroquine were used as positive controls for testing against W2 strain of P. falciparum.
In the benzothiazole series, compounds lacking R2 substituents (for example, compounds 16–19) were devoid of activity against FP-2 (Table 1). A few analogs showed similar log order activity as that of parent compound 2, whereas others were inactive. Interestingly, no direct correlation was observed between the chemical reactivity of electrophiles such as α-thioketones (20–25, and 41, 42), α-thioesters (26–29) and α-thioacetamide (30–34, and 39, 40) towards Cys-42 and inhibition of FP-2. Overall, the compounds with α-thioacetamide and α-thioketones electrophiles showed moderate activity against FP-2 compared to those possessing α-thioesters electrophiles. Moreover, compounds 41 and 42 with protonated amino groups were the only compounds from the benzothiazole series displaying dual inhibition of FP-2 and FP-3. In the triazole series (Table 2), compounds with bulkier R group (48, 53 and 54) showed modest activities against FPs. However, as opposed to compound 1, none of the compounds from triazole series, except 48, inhibited FP-3.
Among the evaluated compounds, compound 24 and 41 showed activity against the W2 strain of P. falciparum. However, compound 24 was inactive against FP-2, suggesting action against targets other than FP-2. The majority of identified compounds specifically inhibited FP-2 but not FP-3, which may account for their inability to block parasite development, as deletion of only FP-2 by gene knockout was not lethal to parasites.30 Activity of compound 41 can be attributed to its dual inhibition against FP-2 and FP-3. However, compound 42 was not active up to 10 μM concentration in culture parasites, despite of having same log order of activity against FPs as 41. This suggests the obvious involvement of other physicochemical properties for inactivity of 42 in cultured parasites development such as the solubility, lipophilicity, polar-surface area etc.
To gain insight into the binding mode of test compounds, docking of compounds into the FP-2 binding site using the Glide docking program was performed. Previously, we compared abilities of two scoring functions implemented in Glide: Gscore and Emodel, to enrich known FP-2 actives from decoys downloaded from the DUD database.21 In our study, Emodel outperformed Gscore in retrieving actives from decoys and proved more appropriate for the apolar FP-2 binding site.21 Thus, we applied our validated Glide XP protocol with an Emodel scoring function to rank order the analogs from both series. Here, the distance of the electrophilic carbonyl carbon present in the putative hits to the cysteine sulfur (typically <3.5 Å) was used as a measure of covalent adduct formation. Docking studies revealed some important trends which are discussed below.
Among the benzothiazole series, compounds with α-thioacetamide and α-thioketones electrophiles were ranked higher (Emodel scores from −61 to −84), whereas compounds with α-thioesters showed Emodel scores of greater than −56, except compound 29 (Emodel score −61). This trend was consistent with the experimental affinity of compounds from the benzothiazole series. For example, most of the compounds from the α-thioesters were inactive up to 50 μM concentration, and, thus, ranked lower, except 29, which showed moderate activity (IC50 = 31.34 μM) against FP-2. In addition, in α-thioesters containing compounds, the distance of reactive carbonyl carbon from the catalytic Cys 42 of the FP-2 active site was always >4.0 Å. On the other hand, the majority of compounds in the α-thioacetamide series showed a similar log order of activity against FP-2, and they were always more active than corresponding α-thioesters. These observations suggest the predictive nature of the scoring scheme used here to separate actives from inactives in congeneric series of compounds. However, correlation of docking scores with activity was poor, perhaps due to similar log orders of activity for identified hits and the parent compound 2. In the benzothiazole series, bulkier R1 and R2 substituents affected the placement of the reactive carbonyl carbon in the vicinity of the catalytic Cys42 of the FP-2 active site. For example, compounds 24 and 25 with naphthalene in the S1′ pocket and quinoline or substituted phenyl in the S2 pocket, showed modest or no activity against FP-2.
The compounds with the α-thioacetamide electrophile (30–39) formed an additional H-bond with the Asn173 of FP-2, placing the reactive carbonyl carbon in an appropriate position for nucleophilic attack by Cys42. The docking pose of representative compound 31 from thioacetamide series is shown in Fig. 3. The p-toluene moiety of 31 showed hydrophobic interactions with Tyr 206 of the S1′ pocket. The thioacetamide linker exhibited interactions with key catalytic residues such as Cys 42 and Gln36 in addition to H-bond formation with Asn173. The anilinic –NH of 31 formed an H-bond with Asp234 of the S2 site.
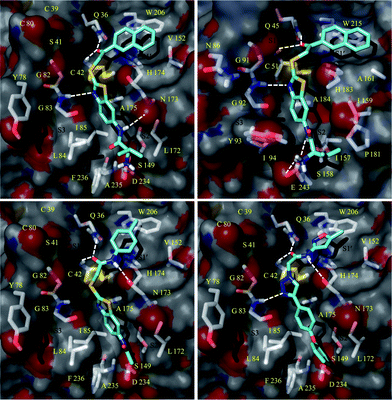 | ||
| Fig. 3 Docking pose of 42 (a), 31 (c), and 45 (d) in FP-2 and of 42 in FP-3 (b). | ||
FP-2 and FP-3 share 65% sequence identity.31 Both of these enzymes have a preference for substrate with a hydrophobic residue at the P2 position.32 The major difference between these two enzymes is in their S2 pockets. The Asp234, Leu84 and Leu172 of the S2 pocket in FP-2 is replaced by the bulkier Glu243, Tyr93 and Pro181 in FP-3, making the S2 pocket more narrow in FP-3.31 These differences are likely major contributing factors for distinct biochemical specificity and ligand recognition among these two homologous hemoglobinases. In the present study, compounds 41 and 42 showed inhibition of FP-2 and FP-3. The binding modes of 41 in FP-2 and FP-3 explain their inhibition, as follows (Fig. 3). First, as expected, the protonated amino functionality of 41 forms charge-charge interactions with Asp234 and Glu243 in the S2 pockets of FP-2 and FP-3, respectively. Second, the isopropyl side chain of 41 forms van der Waals interactions with Leu172 in FP-2 and Pro181 in FP-3. Third, the thiazole nitrogen of 41 interacts with the main chains of glycine residue (Gly83 in FP-2 and Gly92 in FP-3) that is highly conserved in papain-like family of cysteine proteases and considered important for inhibition of these enzymes. Fourth, the carbonyl carbon of α-thioketone linker interacts with the residues of an oxyanion hole formed by the backbone of Cys42 and side chain of Gln36. Finally, the naphthalene moiety exhibits van der Waals interactions with Tyr206 and Val152 in S1′ pocket of FP-2 and Tyr215 and Ala161 in S1′ pocket of FP-3. Collectively, the above interaction profiles of 41 contribute to its activity against both parasitic cysteine proteases.
Compounds (46–57) from the triazole series displayed similar binding modes as shown for the parent compound 1 (Fig. 2). However, very few compounds displayed inhibition of FP-2. Bulkier substituents at the R2 position, for example, compound 48, 53 and 54, afforded modest activity against FP-2. Additional hydrophobic interactions with Leu172 which is buried deep in the S2 pocket of FP-2 might account for the modest activity of these compounds against FP-2. The inactivity of other triazole analogs might be due to unsuitable or shorter hydrophobic R2 groups as compared to the parent compound 1.
Next, we screened compound 41 and 42 with protonated amino group against selected human papain-family of cysteine proteases, cathepsins K, L and B (Table 3). Although, cathepsins K and L1 lack corresponding polar residues in the S2 pocket (Leu in Cat K and Ala in Cat L1, see Figure S2) as present in FPs (Asp in FP-2, Glu in FP-3), compound 41 and 42 also inhibited these enzymes, whereas both compounds were inactive against cathepsin B having polar glutamate residue at the same position. These suggest the polar residues of S2 pocket, although important for improving potency, may not be crucial for design of selective inhibitors against falcipains.
| Compound No. | Cat K IC50 (μM) | Cat L1 IC50 (μM) | Cat B IC50 (μM) |
|---|---|---|---|
| 41 | 14.52 | 5.87 | >50 |
| 42 | 5.62 | 4.39 | >50 |
| E-64 | 0.004 | 0.016 | 0.005 |
The compounds reported in this study are ketone-based inhibitors and are likely to bind in a covalent but reversible fashion with the active site cysteine. However, extensive efforts would be required to actually prove that these compounds undergo covalent adduct formation with the catalytic cysteine moiety. The proposed mechanism of action of these compounds (shown in Fig. 4) which involves formation of a tetrahedral intermediate (hemithioacetal) after nucleophilic attack by the catalytic cysteine (stabilized by the Gln 36, Cys42 residues of the oxyanion hole) is supported by SAR and recent studies by Ellman and co-workers.33,34
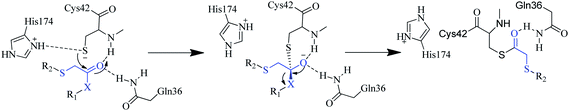 | ||
| Fig. 4 The proposed mechanisms of action for the α-thioketone soft-electrophiles containing compounds. | ||
In summary, the present study reports novel, non-peptidic active analogs of the benzothiazole and triazole series. Fifteen compounds showed moderate activity against FP-2 and, three were also active against FP-3. In the benzothiazole series, compounds 41 and 42, with protonated amines inhibited both hemoglobinases. However, these compounds also displayed activity against homologous mammalian cysteine proteases lacking corresponding polar residues, suggesting less significance of these residues in the design of selective inhibitors against FPs. The relative inactivity of synthesized analogs against FP-3 and cultured P. falciparum needs to be addressed in the subsequent lead optimization process.
References
- World Malarial Report, http://www.who.int/malaria/world_malaria_report_2009/en/, Accessed 17th May, 2011.
- J. Sachs and P. Malaney, Nature, 2002, 415, 680–685 CrossRef CAS.
- D. A. Fidock, Nature, 2010, 465, 297–298 CrossRef CAS.
- A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. Day, N. Lindegardh, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455–467 CrossRef CAS.
- H. Noedl, Y. Se, K. Schaecher, B. L. Smith, D. Socheat and M. M. Fukuda, N. Engl. J. Med., 2008, 359, 2619–2620 CrossRef CAS.
- N. J. White, Science, 2008, 320, 330–334 CrossRef CAS.
- W. A. Guiguemde, A. A. Shelat, D. Bouck, S. Duffy, G. J. Crowther, P. H. Davis, D. C. Smithson, M. Connelly, J. Clark, F. Zhu, M. B. Jimenez-Diaz, M. S. Martinez, E. B. Wilson, A. K. Tripathi, J. Gut, E. R. Sharlow, I. Bathurst, F. El Mazouni, J. W. Fowble, I. Forquer, P. L. McGinley, S. Castro, I. Angulo-Barturen, S. Ferrer, P. J. Rosenthal, J. L. Derisi, D. J. Sullivan, J. S. Lazo, D. S. Roos, M. K. Riscoe, M. A. Phillips, P. K. Rathod, W. C. Van Voorhis, V. M. Avery and R. K. Guy, Nature, 2010, 465, 311–315 CrossRef CAS.
- F. J. Gamo, L. M. Sanz, J. Vidal, C. de Cozar, E. Alvarez, J. L. Lavandera, D. E. Vanderwall, D. V. Green, V. Kumar, S. Hasan, J. R. Brown, C. E. Peishoff, L. R. Cardon and J. F. Garcia-Bustos, Nature, 2010, 465, 305–310 CrossRef CAS.
- P. J. Rosenthal, Int. J. Parasitol., 2004, 34, 1489–1499 CrossRef CAS.
- B. J. Lee, A. Singh, P. Chiang, S. J. Kemp, E. A. Goldman, M. I. Weinhouse, G. P. Vlasuk and P. J. Rosenthal, Antimicrob. Agents Chemother., 2003, 47, 3810–3814 CrossRef CAS.
- P. J. Rosenthal, J. E. Olson, G. K. Lee, J. T. Palmer, J. L. Klaus and D. Rasnick, Antimicrob. Agents Chemother., 1996, 40, 1600–1603 CAS.
- J. T. Palmer, D. Rasnick, J. L. Klaus and D. Bromme, J. Med. Chem., 1995, 38, 3193–3196 CrossRef CAS.
- P. J. Rosenthal, W. S. Wollish, J. T. Palmer and D. Rasnick, J. Clin. Invest., 1991, 88, 1467–1472 CrossRef CAS.
- P. J. Rosenthal, J. H. McKerrow, M. Aikawa, H. Nagasawa and J. H. Leech, J. Clin. Invest., 1988, 82, 1560–1566 CrossRef CAS.
- B. R. Shenai, B. J. Lee, A. Alvarez-Hernandez, P. Y. Chong, C. D. Emal, R. J. Neitz, W. R. Roush and P. J. Rosenthal, Antimicrob. Agents Chemother., 2003, 47, 154–160 CrossRef CAS.
- F. Shah, P. Mukherjee, P. Desai and M. Avery, Curr. Comput.-Aided Drug Des., 2010, 6, 1–23 CrossRef CAS.
- R. Ettari, F. Bova, M. Zappala, S. Grasso and N. Micale, Med. Chem. Res., 2010, 30, 136–167 CAS.
- P. V. Desai, A. Patny, J. Gut, P. J. Rosenthal, B. Tekwani, A. Srivastava and M. Avery, J. Med. Chem., 2006, 49, 1576–1584 CrossRef CAS.
- P. V. Desai, A. Patny, Y. Sabnis, B. Tekwani, J. Gut, P. Rosenthal, A. Srivastava and M. Avery, J. Med. Chem., 2004, 47, 6609–6615 CrossRef CAS.
- Y. Sabnis, P. J. Rosenthal, P. Desai and M. A. Avery, J. Biomol. Struct. Dyn., 2002, 19, 765–774 CAS.
- F. Shah, P. Mukherjee, J. Gut, J. Legac, P. J. Rosenthal, B. L. Tekwani and M. A. Avery, J. Chem. Inf. Model., 2011, 51, 852–864 CrossRef CAS.
- P. Ciapetti and B. Giethlen, Molecular Variation Based on Isosteric Replacements, Third edn, Academic Press, 2008 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- R. Breinbauer and M. Köhn, ChemBioChem, 2003, 4, 1147–1149 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- K. Barral, A. D. Moorhouse and J. E. Moses, Org. Lett., 2007, 9, 1809–1811 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- P. S. Sijwali, J. Koo, N. Singh and P. J. Rosenthal, Mol. Biochem. Parasitol., 2006, 150, 96–106 CrossRef CAS.
- I. D. Kerr, J. H. Lee, K. C. Pandey, A. Harrison, M. Sajid, P. J. Rosenthal and L. S. Brinen, J. Med. Chem., 2009, 52, 852–857 CrossRef CAS.
- S. Subramanian, M. Hardt, Y. Choe, R. K. Niles, E. B. Johansen, J. Legac, J. Gut, I. D. Kerr, C. S. Craik and P. J. Rosenthal, PLoS One, 2009, 4, e5156 Search PubMed.
- L. Huang, A. Lee and J. A. Ellman, J. Med. Chem., 2002, 45, 676–684 CrossRef CAS.
- K. Brak, P. S. Doyle, J. H. McKerrow and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6404–6410 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Docking poses of representative compounds from benzimidazole and thioacetamide series, multiple sequence alignment, 1H and 13C NMR of synthesized compounds. See DOI: 10.1039/c1md00129a |
| This journal is © The Royal Society of Chemistry 2011 |