A combinatorial approach to characterize the substrate specificity of protein arginine methyltransferase 1†‡
Kevin L.
Bicker
a,
Obiamaka
Obianyo
ab,
Heather L.
Rust
ab and
Paul R.
Thompson
*ab
aDepartment of Chemistry and Biochemistry, University of South Carolina, 631 Sumter St., Columbia, South Carolina, USA
bDepartment of Chemistry, The Scripps Research Institute, Scripps Florida, 120 Scripps Way, Jupiter, Florida, USA. E-mail: Pthompso@scripps.edu; Fax: +1 561-228-3050; Tel: +1 561-228-2860
First published on 6th July 2010
Abstract
The dysregulation of protein arginine methyltransferases (PRMTs) is implicated in a wide variety of disease states. Here we report the design, synthesis, and screening of a combinatorial peptide library used to characterize the substrate specificity of PRMT1. The information gained from this approach was used to develop a PRMT1 inhibitor with enhanced selectivity.
Over the last decade, protein methylation has been found to play an important role in numerous physiological processes, including DNA repair, RNA splicing, and, most predominantly, gene regulation.1 Although numerous residues are known to be methylated in vivo, the two most prominent are lysine and arginine; these residues are modified by the lysine methyltransferases and arginine methyltransferases (PRMTs) respectively.1 Of particular interest are the PRMTs, because the activity of several human members of this enzyme family is dysregulated in a variety of human diseases.2,3 For example, increased PRMT1 activity is associated with breast cancer (e.g., increased estrogen receptor α signaling) and heart disease (e.g., decreased nitric oxide production).2,3
Given their putative roles in human disease, our efforts have been focused on characterizing the molecular mechanism of arginine methylation, and using this information to guide the development of PRMT inhibitors.4–7 Recently, we reported that a chloroacetamidine containing 21 amino acid peptide, termed C21 (Fig. 1), could irreversibly inactivate PRMT1 with nanomolar to micromolar efficacy both in vitro and in cellulo.5 The structure of this inhibitor is based on the sequence of histone H4, a natural substrate for PRMT1, and Cl-amidine, a potent inhibitor of the Protein Arginine Deiminases (PADs).8,9
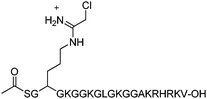 | ||
| Fig. 1 Structure of the PRMT1 inhibitor, C21. | ||
Given the covalent nature of this interaction, our experience developing synthetic lectins for cancer associated glycoproteins,10,11 and recently described on bead methods that have been used to characterize the peptide binding specificity of various proteins,12–15 we considered the possibility that a one-bead-one-compound (OBOC) combinatorial peptide library, incorporating the Cl-acetamidine warhead, could be used to explore the substrate specificity of arginine modifying enzymes. A major difference between these approaches, and our own, is that previous approaches have focused on the identification of non-covalent binders, whereas our approach is focused on identifying covalent binders. To our knowledge, this is the first description of such an approach; although we note that we and others have previously described the development of synthetic lectins that exploit the covalent but reversible interaction between a glycan and a boronic acid.10,11,16,17 For proof-of-concept, we focused our efforts on developing an OBOC Cl-acetamidine containing library that could be used to characterize the substrate specificity of PRMT1. We chose this enzyme for our initial studies because it is known to be the most promiscuous PRMT;18 therefore we expected that it would provide the most challenging test case for our methodology.
To develop the screening methodology, we initially synthesized a peptide library on amino terminated TentaGel resin via split-and-pool combinatorial methods using Fmoc strategies. The general sequence of this library is Ac-XXXO*XXXBBRM-resin, where X is a randomized amino acid, O* denotes the placement of the warhead modified ornithine, and B is β-alanine, which is used to project the peptide away from the resin (Fig. 2A).
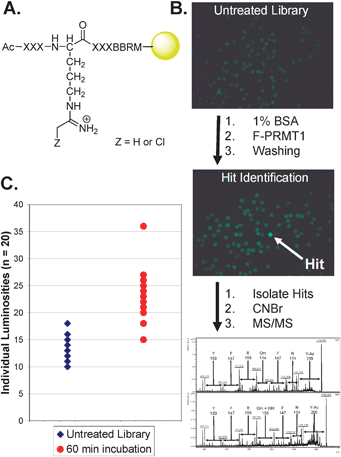 | ||
| Fig. 2 (A) The general library sequence. (B) The screening process used to identify and sequence F-PRMT1 hits, including the sequence of Hit34. Untreated library (upper) is preblocked with BSA, incubated with fluorescent enzyme, washed, and observed under a fluorescence microscope (middle) to identify hits, which are picked manually, cleaved from the resin, and sequenced using MS/MS methods (lower). (C) Binning chart of individual beads for untreated library and library incubated with F-PRMT1 for 60 min. Clustering of beads gives an indication of the level of background binding, while higher luminosity valued beads are recognized as hits. | ||
In total, 17 of the 20 natural amino acids were used in the library, generating a theoretical diversity of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 or roughly 1.4 × 106 unique peptide sequences. It is well known that in peptide library synthesis, coupling efficiency is affected by both the amino acid being attached and the one being attached to, and that this can lead to a biasing of the library for certain sequences.19 However, we are confident that by screening a significant number of beads (21000) this biasing is overcome. Excluded from the library were Cys (due to its potential reactivity with the Cl-acetamidine warhead), Met (due to its reactivity with cyanogen bromide), and Ile (because it has the same mass as Leu, making MS-based sequencing difficult). The Met and Arg residues on the C-terminus of the peptide are used for orthogonal cleavage from the resin with cyanogen bromide and to aid in MS ionization, respectively. Note that the chloroacetamidine warhead was not coupled exclusively to the ornithine, but rather a 1∶1 mixture of Cl-acetamidine and H-acetamidine was incorporated. Theoretically, this allows for up to half of the peptides on any given bead to react with PRMT1 while the other half will be available for sequence determination by MS-based methods. Incorporation of the mixed warhead system onto ivDde protected ornithine was achieved by first orthogonally removing the ivDde group with hydrazine followed by treatment with a 1∶1 mixture of ethyl acetimidate·HCl and ethyl chloroacetimidate·HCl (Scheme 1).
5 or roughly 1.4 × 106 unique peptide sequences. It is well known that in peptide library synthesis, coupling efficiency is affected by both the amino acid being attached and the one being attached to, and that this can lead to a biasing of the library for certain sequences.19 However, we are confident that by screening a significant number of beads (21000) this biasing is overcome. Excluded from the library were Cys (due to its potential reactivity with the Cl-acetamidine warhead), Met (due to its reactivity with cyanogen bromide), and Ile (because it has the same mass as Leu, making MS-based sequencing difficult). The Met and Arg residues on the C-terminus of the peptide are used for orthogonal cleavage from the resin with cyanogen bromide and to aid in MS ionization, respectively. Note that the chloroacetamidine warhead was not coupled exclusively to the ornithine, but rather a 1∶1 mixture of Cl-acetamidine and H-acetamidine was incorporated. Theoretically, this allows for up to half of the peptides on any given bead to react with PRMT1 while the other half will be available for sequence determination by MS-based methods. Incorporation of the mixed warhead system onto ivDde protected ornithine was achieved by first orthogonally removing the ivDde group with hydrazine followed by treatment with a 1∶1 mixture of ethyl acetimidate·HCl and ethyl chloroacetimidate·HCl (Scheme 1).
Fluorescein isothiocyanate labeled PRMT1 (F-PRMT1) was prepared analogously to previously described methods.10 To ensure that the fluorescent tag did not affect substrate binding or enzyme catalysis, the steady state kinetic parameters were determined for the AcH4-21 peptide. The AcH4-21 peptide is a histone H4 tail mimic that we have previously shown to be an excellent in vitro PRMT1 substrate.5 The results of these analyses (Table 1) indicate that the effects on Km and kcat are relatively minor (∼2-fold on both Km and kcat), indicating that the FITC tag has only limited effects on peptide substrate binding, thereby validating its use in screens.
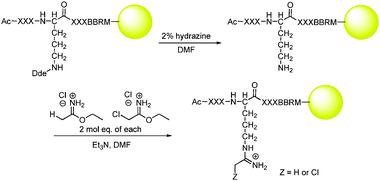 | ||
| Scheme 1 Deprotection of ornithine and subsequent attachment of the mixed warhead system. | ||
To screen the library, 10 mg aliquots of library resin were pre-swollen in 1 mL of Screening Buffer (see ESI‡), preblocked with 1 mg mL−1 bovine serum albumin (BSA) in Screening Buffer to prevent non-specific binding, and tumbled in 0.01 mg mL−1 F-PRMT1 in Screening Buffer containing 1 mg mL−1 BSA at 37 °C for the prescribed time. Resin was washed to remove unbound protein and fluorescent beads were then detected using a Leica MZ16F fluorescent stereoscope equipped with a GFP3 filter set (Ex. 450–490 nm; Em. 500–550 nm). Images were analyzed using Adobe Photoshop to acquire luminosity values for individual beads. The brightest beads, or ‘hits’, which are presumably the strongest binders, were manually picked, cleaved from the resin using cyanogen bromide, and sequenced using MS/MS based methodologies. Typical results from a screen are depicted in Fig. 2B and C.
Note that preliminary studies (see ESI‡) were used to identify both an optimal screening time (i.e., 60 min, Fig. S1, ESI‡) and concentration of F-PRMT1 (i.e., 0.01 mg mL−1, Fig. S2, ESI‡). Utilizing this optimized methodology, a total of 21000 beads were screened against F-PRMT1. These screens identified 57 hits§ yielding a hit rate of 0.3%. MS-based sequencing of the hits provided complete sequences for 45 hits, as well as 9 partial sequences, and 3 that were indeterminable The sequences of these hits are summarized in Table S2.‡ See Fig. S3 and S4 (ESI‡) for representative MALDI-TOF MS and MS/MS spectra, respectively.
Complete and partial sequences were entered into the WebLogo generator20 (http://weblogo.berkeley.edu/logo.cgi) to identify sequence homology (Fig. 3). Based on this analysis, PRMT1 appears to have an affinity for substrate peptides rich in Phe and Arg. In retrospect, this result is not altogether surprising, as PRMT1 is known to methylate RGG domain containing proteins (e.g., fibrillarin), which are rich in Arg, Gly, and Phe.21–23
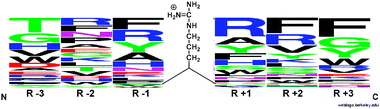 | ||
| Fig. 3 Determination of sequence homology for the identified F-PRMT1 hits. | ||
The preponderance of Phe and Arg residues at the R − 1 and R + 2 positions prompted us to determine whether the incorporation of these residues within the context of the AcH4-21 peptide could enhance the potency of C21. To examine this possibility, C21 and C21+2R were synthesized on TentaGel amino resin (Fig. 4A) and screened against F-PRMT1 as described above. Both of the compounds tested exhibited a higher average luminosity than the warhead library, which was used as a control in this study, indicating above average binding to F-PRMT1 (Fig. S5, ESI‡), and thereby validating our approach.
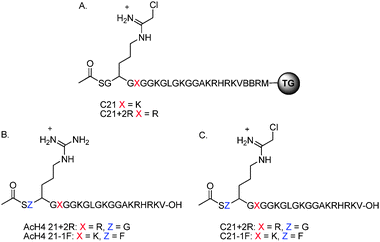 | ||
| Fig. 4 (A) Structure of the resin bound C21 derivatives. (B) Structure of AcH4-21+2R and AcH4−1F substrates. (C) Structure of C21+2R and C21−1F inhibitors. | ||
To further examine the contribution of these Phe and Arg residues, two AcH4-21 substrate analogues (Fig. 4B) and two C21 analogues (Fig. 4C) were synthesized on Wang polystyrene resin, cleaved from the resin, and purified by preparative-HPLC. To explore the effects of these mutations on substrate recognition, the steady state kinetic parameters were determined for the +2R and −1F mutant peptides. Note that we did not determine whether any of the hits were PRMT1 substrates because the hit peptides are only 7 residues in length and we have previously shown that peptides shorter than 13 residues are very poor PRMT1 substrates (Km ≥ 500 µM); PRMT1 is known to preferentially modify longer peptides with a preponderance of positively charged residues >10 amino acids C-terminal to the site of methylation.7 Additionally, for technical reasons (peptide solubility and quantities required), it is difficult to measure accurate Km values for short peptides. Finally, we have no baseline for comparison. Given these issues, we chose to validate our library data by generating ‘mutant’ peptides as opposed to testing hit sequences directly. This approach is advantageous because it provides a baseline with which to compare the contributions of residues present in the consensus sequence surrounding the site of methylation to substrate recognition.
The results of these ‘mutagenesis’ studies indicate that the Km and kcat values for AcH4-21+2R and AcH4-21−1F were very similar to the established AcH4-21 substrate (Table 2), showing that these modifications do not substantially alter the apparent affinity of the enzyme towards these analogues, as compared to AcH4-21. It is not surprising that little effect is seen when the Lys in the +2 position is replaced with an Arg, as these amino acids are not drastically different. However, the change from Gly to Phe in the −1 position is a substantial alteration in both amino acid side chain size and electronic properties. That this modification does not affect the affinity of the enzyme to the substrate, or its turnover, indicates that this methodology has successfully identified a significant alteration in the wild-type AcH4-21 substrate which the enzyme binds favorably.
| Substrate | Sequence | K m/µM | k cat a/min−1 | k cat/Kma/min−1 M−1 |
|---|---|---|---|---|
| a [SAM] = 15 µM; values for the AcH4-21 peptide are taken from Osborne et al.7 | ||||
| AcH4-21 | Ac-SGRGKGGKGLGKGGAKRHRKV | 1.23 ± 0.35 | 0.54 ± 0.01 | 4.38 × 105 |
| AcH4-21−1F | Ac-SFRGKGGKGLGKGGAKRHRKV | 1.68 ± 0.31 | 0.66 ± 0.01 | 3.93 × 105 |
| AcH421+2R | Ac-SGRGRGGKGLGKGGAKRHRKV | 0.83 ± 0.25 | 0.49 ± 0.01 | 5.94 × 105 |
To explore the effects of mutations in the peptide sequence on enzyme inhibition, IC50 values were determined for the C21+2R and C21−1F peptides (Table 3), using our gel-based methyltransferase assay, with a fixed concentration of AcH4-21, SAM, and PRMT in the presence of varying concentrations of inhibitor. Although AcH4-21+2R exhibited a slightly better kcat/Kmversus AcH4-21, the inhibitor derivative, C21+2R, has a markedly worse IC50 at greater than 500 µM. We theorize that this drastic decrease in potency is not due to a decrease in the affinity of the enzyme for the modified inhibitor, but rather the introduction of a new Arg that the enzyme can modify. C21−1F has an IC50 of 8.1 µM, which is higher (∼4-fold) than that established for C21 (1.8 µM).5 The reason for the decreased potency is not clear, but is likely due to the added steric bulk of the phenyl moiety. Nevertheless, we were encouraged by the finding that our method for determining PRMT1 substrate specificity had identified a considerable modification close to the warhead that minimally affects its inhibition of PRMT1. Given this finding we wished to explore the selectivity of C21−1F compared to C21, and therefore IC50 values (Table 3) were determined for each inhibitor against other major Type I PRMTs (i.e., 1, 3, 4, and 6). From these studies, we observed that although slightly less potent than C21, C21−1F is significantly more selective for PRMT1 compared with the established inhibitor. For example, whereas C21 is only ∼5-fold selective for PRMT1 over PRMT6, C21−1F is ∼17-fold selective, demonstrating that this methodology is capable of identifying residues around the substrate arginine that are important for PRMT1 recognition and selectivity.
| Inhibitor | Sequence | PRMT1/µM | PRMT3/µM | PRMT4/µM | PRMT6/µM |
|---|---|---|---|---|---|
| C21 | Ac-SGRGKGGKGLGKGGAKRHRKV | 1.8 ± 0.1 | >500 | >500 | 8.8 ± 0.5 |
| C21−1F | Ac-SFRGKGGKGLGKGGAKRHRKV | 8.1 ± 1.0 | >500 | >500 | 139.6 ± 31.6 |
The synthesis of an OBOC combinatorial peptide library incorporating a Cl-acetamidine warhead and the development of screening procedures for fluorescently labeled arginine modifying enzymes has provided a method for determining substrate specificity around a Cl-acetamidine warhead. We have used this proof-of-concept approach to explore the substrate specificity of PRMT1, the most promiscuous enzyme in the PRMT family. Several conserved amino acids were observed and implementation of these modifications into the established AcH4-21 substrate does not severely alter the kinetic parameters for these substrate analogues. The identification of a conserved Phe in the R − 1 position and the subsequent incorporation of this substantial modification into C21 produced an inhibitor (C21−1F), which, although slightly less potent than C21, is significantly more selective for PRMT1 over the other Type I PRMTs tested. Demonstrating the ability of this methodology to identify peptide specificity for PRMT1, the PRMT that should be most difficult to characterize, sets the stage for characterizing the substrate specificity of other members of the PRMT family of enzymes (e.g., CARM1), as well as other arginine modifying enzymes, such as the PADs, that react with the haloacetamidine warhead. This approach should also be amenable to screening for peptide, peptoid, and bead bound small molecule libraries to identify potent and selective inhibitors. More generally, the approach outlined herein can be readily adapted to identify selective and irreversible inactivators for a wide variety of enzymes by incorporating a particular warhead into a library.
The authors would like to acknowledge the support of the University of South Carolina and in part NIH grant GM079357.
Notes and references
- R. A. Copeland, M. E. Solomon and V. M. Richon, Nat. Rev. Drug Discovery, 2009, 8, 724–732 CrossRef CAS.
- M. T. Bedford and S. G. Clarke, Mol. Cell, 2009, 33, 1–13 CrossRef CAS.
- M. T. Bedford and S. Richard, Mol. Cell, 2005, 18, 263–272 CrossRef CAS.
- O. Obiamaka, T. C. Osborne and P. R. Thompson, Biochemistry, 2008, 47, 10420–10427 CrossRef.
- O. Obianyo, C. P. Causey, T. C. Osborne, J. E. Jones, Y.-H. Lee, M. R. Stallcup and P. R. Thompson, ChemBioChem, 2010, 11, 1219 CrossRef CAS.
- T. Osborne, R. L. W. Roska, S. R. Rajski and P. R. Thompson, J. Am. Chem. Soc., 2008, 130, 4574–4575 CrossRef CAS.
- T. C. Osborne, O. Obianyo, X. Zhang, X. Cheng and P. R. Thompson, Biochemistry, 2007, 46, 13370–13381 CrossRef CAS.
- Y. Luo, K. Arita, M. Bhatia, B. Knuckley, Y. H. Lee, M. R. Stallcup and P. R. Thompson, Biochemistry, 2006, 45, 11727–11736 CrossRef CAS.
- Y. Luo, B. Knuckley, Y. H. Lee, M. R. Stallcup and P. R. Thompson, J. Am. Chem. Soc., 2006, 128, 1092–1093 CrossRef CAS.
- Y. Zou, D. L. Broughton, K. L. Bicker, P. R. Thompson and J. J. Lavigne, ChemBioChem, 2007, 8, 2048–2051 CrossRef CAS.
- K. L. Bicker, J. Sun, P. R. Thompson and J. J. Lavigne, in preparation.
- Z. Songyang, S. E. Shoelson, M. Chaudhuri, G. Gish, T. Pawson, W. G. Haser, F. King, T. Roberts, S. Ratnofsky, R. J. Lechleider, B. G. Neel, R. B. Birge, J. E. Fajardo, M. M. Chou, H. Hanafusa, B. Schaffhausen and L. C. Cantley, Cell (Cambridge, Mass.), 1993, 72, 767–778 CrossRef CAS.
- G. Huyer, J. Kelly, J. Moffat, R. Zamboni, Z. Jia, M. J. Gresser and C. Ramachandran, Anal. Biochem., 1998, 258, 19–30 CrossRef CAS.
- S. H. Joo and D. Pei, Biochemistry, 2008, 47, 3061–3072 CrossRef CAS.
- Z. Songyang, A. S. Fanning, C. Fu, J. Xu, S. M. Marfatia, A. H. Chrishti, A. Crompton, A. C. Chan, J. M. Anderson and L. C. Cantley, Science, 1997, 275, 73–77 CrossRef CAS.
- N. Y. Edwards, T. W. Sager, J. T. McDevitt and E. Anslyn, J. Am. Chem. Soc., 2007, 129, 13575–13583 CrossRef CAS.
- M. Li, N. Lin, Z. Huang, L. Du, C. Altier, H. Fang and B. Wang, J. Am. Chem. Soc., 2008, 130, 12636–12638 CrossRef CAS.
- J. Tang, A. Frankel, R. J. Cook, S. Kim, W. K. Paik, K. R. Williams, S. Clarke and H. R. Herschman, J. Biol. Chem., 2000, 275, 7723–7730 CrossRef CAS.
- J. A. Boutin, I. Gesson, J.-M. Henlin, S. Bertin, P.-H. Lambert, J.-P. Volland and J.-L. Fauchere, Mol. Diversity, 1997, 3, 43–60 CrossRef CAS.
- G. E. Crooks, G. Hon, J. M. Chandonia and S. E. Brenner, Genome Res., 2004, 14, 1188–1190 CrossRef CAS.
- L.-S. Ai, C.-H. Lin, M. Hsieh and C. Li, Proc. Natl. Sci. Counc. ROC(B), 1999, 23, 175–180 Search PubMed.
- K. Fronz, S. Otto, K. Kolbel, U. Kuhn, H. Friedrch, A. Schierhorn, A. G. Beck-Sickinger, A. Ostareck-Lederer and E. Wahle, J. Biol. Chem., 2008, 283, 20408–20420 CrossRef CAS.
- K. Kolbel, C. Ihling, K. Bellmann-Sickert, I. Neundorf, A. G. Beck-Sickinger, A. Sinz, U. Kuhn and E. Wahle, J. Biol. Chem., 2009, 284, 8274–8282.
Footnotes |
| † This article is part of a themed issue of Molecular BioSystems on Post-translational modifications. |
| ‡ Electronic supplementary information (ESI) available: Methods, materials, equipment, and supplemental results. See DOI: 10.1039/c0mb00015a |
| § Hits are defined as beads with luminosity values 30% greater than the average library luminosity. |
| This journal is © The Royal Society of Chemistry 2011 |