Improved visible light photocatalytic activity of titania doped with tin and nitrogen
Enjun
Wang
a,
Tao
He
*b,
Lusong
Zhao
a,
Yongmei
Chen
c and
Yaan
Cao
*a
aTeda Applied Physics School, Institute of Physics, Nankai University, Tianjin, 300071, China. E-mail: caoyaan@yahoo.com; Fax: +86-22-66229310; Tel: +86-22-66229598
bNational Center for Nanoscience and Technology, Beijing, 100190, China. E-mail: het@nanoctr.cn
cSchool of Science, Beijing University of Chemical Technology, Beijing, 100029, China
First published on 15th October 2010
Abstract
Tin and nitrogen co-doped titania has been prepared by a hydrolysis precipitation method and studied by X-ray diffraction, X-ray photoelectron spectroscopy, diffuse reflectance UV-vis absorption spectra, and photoluminescence. The surface area has been determined by using the BET method. Tin is incorporated into the TiO2 crystal lattice in substitutional mode, while nitrogen is present as surface species. The resultant energy levels of tin doping and nitrogen surface states are located inside the bandgap, which are close to the conduction and valence bands, respectively. Hence, co-doping of tin and nitrogen can greatly enhance the absorption in the visible light region and inhibit the recombination of photogenerated charge carriers, leading to a higher photocatalytic activity for the co-doped catalyst than pure TiO2 and solely doped TiO2 with nitrogen or tin for degradation of 4-chlorophenol under both visible and UV-light irradiation. This indicates that co-doping simultaneously with two foreign elements is a feasible way to improve the photocatalytic activity of TiO2.
Introduction
Photocatalysis using titania (TiO2) offers a viable approach for degradation of organic and inorganic pollutants in air and water due to its high oxidizing capacity, photostability, and nontoxicity.1–4 However, TiO2 exhibits photoresponse only to UV light because it is a wide-gap semiconductor (3.2 eV), which impedes the effective usage of solar illumination in practical applications. To efficiently use solar energy, recent research interest has been mainly focused on extending its response to visible light.5–18 So far many efforts have been made in this field, such as surface modification,5,6 doping TiO2 with foreign ions,7–12 combining TiO2 with other semiconductors.13–15 A breakthrough was made in 2001 with a visible-light photocatalyst via doping TiO2 with nitrogen.7 From then on, many experiments and theoretical work have been done on nitrogen-doped TiO2,16–27 indicating that this new catalyst exhibits a relatively high visible-light photocatalytic activity due to the creation of energy levels of doping and/or surface states above the valence band,18–20 and/or bandgap narrowing.7,21In addition, pristine TiO2 exhibits a low quantum yield due to its relatively high recombination rate of photogenerated electron–hole pairs.28,29 In an aqueous suspension system, only a few electrons in the conduction band have high enough energy that can overcome the potential barrier to reach the surface and be captured by the surface adsorbed O2 since the energy bands of TiO2 bend upward at the solid–liquid interface.30 The majority of electrons will migrate to an opposite direction, the bulk of TiO2, leading to an increase in the recombination probability of electron–hole pairs during the photocatalytic reaction. If a doping energy level can be introduced below the conduction band of TiO2, therefore, the photogenerated electrons in the conduction band can transfer to the surface via this doping energy level, without crossing over the potential barrier, and take part in the photocatalytic process. This will facilitate the separation of photogenerated electron–hole pairs. Thus more photogenerated electrons and holes can contribute to the photocatalytic reaction, resulting in an enhancement of photocatalytic activity of the catalyst. This is confirmed by our previous work, in which Sn4+ ions were incorporated into the TiO2 lattice in substitutional mode.31,32 The doping energy level of Sn4+ ions is located at 0.4 eV below the conduction band of TiO2, which allows an efficient transfer of photogenerated electrons to the surface and suppresses the recombination of photogenerated charge carriers. Furthermore, Sn4+ doping can also improve the photocatalytic performance of TiO2 under visible light irradiation since it can lead to the visible-light response.31,33
Furthermore, the visible-light response and photocatalytic activity can be further improved for a photocatalyst with multi-dopants since the contribution can come from all the dopants. This has been demonstrated in several systems, such as (B,Ni),14 (N,Fe),25 (N,W),27 (N,B),34 (N,F)35,36 and (C,F,N).37 In this work, we present that a photocatalyst co-doped with nitrogen and tin can be prepared via a simple sol–gel method. Tin is incorporated into the TiO2 crystal lattice in substitutional mode, while nitrogen exists as surface species. Moreover, as expected, it exhibits a higher visible and UV-light photocatalytic activity than pure TiO2, tin-doped TiO2, and nitrogen-doped TiO2. The mechanism is also discussed in this contribution.
Experimental
Catalyst preparation
All chemicals were of analytical grade. Milli-Q water (> 18.2 MΩ cm) was used for all experiments. Co-doped photocatalysts were prepared by using the following protocol. First, deionized water (1 mL) was mixed with anhydrous ethanol (40 mL) at room temperature, followed by the dropwise addition of tetrabutyltitanate (Ti(OC4H9)4, 12 mL) to the solution under vigorous stirring. Concentrated hydrochloric acid (12 M) was used to adjust the pH to 0.5. Then 0.2 mL SnCl4, 1 mL water, and 3 mL ammonia were added in sequence under stirring. A white precipitate formed immediately upon addition of ammonia. After aging at room temperature for 24 h, the resultant precipitate was dried at 100 °C, followed by annealing at 450 °C for 2.5 h. The obtained samples were denoted as N/Sn-TiO2. Tin-doped TiO2 (Sn-TiO2), nitrogen-doped TiO2 (N-TiO2), and pure TiO2 were prepared using the same procedure, but with the addition of the corresponding doping reagent.Catalyst characterization
X-Ray diffraction (XRD) patterns were acquired on a Rigaku D/max 2500 X-ray diffraction spectrometer (Cu Kα, λ = 1.54056 Å) at a scan rate of 0.02° 2θ s−1. The average crystal size was calculated using the Scherrer equation (D = kλ/βcosθ). After degassing at 180 °C, the BET surface area was determined via the measurement of nitrogen adsorption–desorption isotherms at 77 K (Micromeritics Automatic Surface Area Analyzer Gemini 2360, Shimadzu). X-Ray photoelectron spectroscopy (XPS) measurements were carried out with an ESCA Lab 220i-XL spectrometer by using an unmonochromated Al Kα X-ray source (1486.6 eV). All spectra were calibrated using the binding energy (BE) of the adventitious C1s peak at 284.6 eV. Diffuse reflectance UV-vis absorption spectra (UV-Vis DRS) were collected with a UV-vis spectrometer (U-4100, Hitachi). Photoluminescence (PL) spectra were acquired by using the 340 nm line of a nanosecond Nd:YAG laser (NL303G) as excitation source at room temperature.38Photocatalytic reactivity test
Photocatalytic decomposition of 4-chlorophenol (4-CP) was used to evaluate the photocatalytic activity of the resultant catalyst, which was carried out using 10 mg of catalyst suspended in a 4-CP aqueous solution (40 mL, 5 × 10−5 mol L−1) under visible and UV-light irradiation. A sunlamp (Philips HPA 400/30S, Belgium) was used directly for the UV-light photocatalytic reaction, while for visible-light photocatalysis a 400 nm cutoff filter was employed to remove the UV light. The reactor was perpendicular to the light beam and located 10 cm away from the light source. The 4-CP solution was continuously bubbled using O2 gas at a flux of 5 mL min−1 under stirring at 25 ± 2 °C. The change in concentration of 4-CP was monitored by a UV-visible spectrometer (UV-1061PC, SHIMADZU) using 4-aminoantipyrine as the chromogenic reagent. The suspension was magnetically stirred in the dark for 30 min before photocatalytic reaction, which was long enough to reach the adsorption equilibrium of 4-CP. The control experiment was performed under identical conditions, but without a photocatalyst.Results
XRD patterns
Fig. 1 shows the XRD patterns of TiO2 (curve a), N-TiO2 (curve b), Sn-TiO2 (curve c), and N/Sn-TiO2 (curve d) powders. It is found that the majority of the crystal phase is anatase for all of the samples.27 The shape of the diffraction peaks of N-TiO2 and N/Sn-TiO2 is consistent with that of pure TiO2. For Sn-TiO2 sample (curve c), besides the anatase peaks, diffraction peaks at 26.6°, 33.9°, and 51.7° can also be observed, corresponding to the crystal planes of (110), (101), and (211) of SnO2, respectively.39 This indicates that a small amount of SnO2 was formed in the Sn-TiO2 sample during the doping process. However, the diffraction peaks of SnO2 are not observed in the N/Sn-TiO2 sample, suggesting that the addition of ammonia can inhibit the formation of SnO2.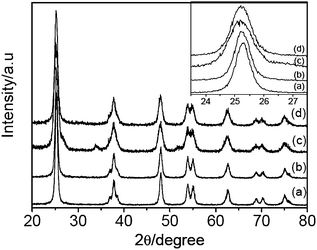 | ||
| Fig. 1 XRD patterns of TiO2 (a), N-TiO2 (b), Sn-TiO2 (c), and N/Sn-TiO2 (d). Inset is the enlarged XRD peaks of crystal plane (101). | ||
The inset in Fig. 1 is the enlarged XRD peaks of crystal plane (101) for all of the samples. Compared with the pure TiO2, no shift of the peak position is observable for N-TiO2 (curves b), while the peak of Sn-TiO2 (curves c) shifts to lower diffraction angles (Δ ≅ 0.08°). It is noted that N/Sn-TiO2 exhibits almost the same diffraction angle as that of Sn-TiO2 (inset of Fig.1). The diffraction peaks of crystal planes (101), (200), and (105) in the curves are used to determine the lattice parameter and crystal size of the samples, which are summarized in Table 1. It is found that the cell volume of N/Sn-TiO2 and Sn-TiO2 is much larger than that of pure TiO2, while the cell volume of N-TiO2 is almost the same as that of pure TiO2. The detailed mechanism is discussed in the following sections.
| Sample | a = b (Å) | c (Å) | Cell volume (Å3) | Crystal size (nm) | Specific surface area (m2 g−1) |
|---|---|---|---|---|---|
| TiO2 | 3.790 | 9.573 | 137.53 | 12.9 | 68.6 |
| N-TiO2 | 3.792 | 9.590 | 137.92 | 12.5 | 73.2 |
| Sn-TiO2 | 3.801 | 9.614 | 138.91 | 7.7 | 97.8 |
| N/Sn-TiO2 | 3.794 | 9.654 | 138.93 | 10.8 | 91.2 |
The doping mode is primarily determined by two factors: electronegativity and ionic radius of the doping ions.31 If the electronegativity and ionic radius of doping ions match those of the lattice ions in the oxide, the doping ions can substitute the lattice ions during the doping process.31 Because the electronegativity and ionic radius of Sn4+ ions (1.8, 69 pm) are close to those of Ti4+ ions (1.5, 53 pm) in TiO2,31,40 it is feasible for Sn4+ ions to replace and occupy lattice Ti4+ ions. Since the ionic radius of Sn4+ ions is larger than that of lattice Ti4+ ions, the cell volume and lattice parameters of Sn-TiO2 are larger than those of pure TiO2. For the doping mode of nitrogen in N-TiO2, since the ionic radius of the nitrogen ion (171 pm) is much larger than that of the oxygen ion (140 pm), a dramatic change in lattice parameter and cell volume would be expected if nitrogen were substituted into the lattice. However, an obvious increase in lattice parameter and cell volume is not observed after nitrogen doping according to XRD results. The doping of nitrogen into TiO2 through the substitutional mode can thus be excluded. Hence, it is proposed here that nitrogen dopants exist as surface species. In the case of N/Sn-TiO2, it seems the change in cell volume comes mainly from Sn dopants since the cell volume of N/Sn-TiO2 is almost the same as that of Sn-TiO2. Therefore, according to the results of XRD, it is reasonable to speculate that tin can incorporate into the TiO2 matrix in substitutional mode and nitrogen exists as surface species, which is confirmed by XPS results shown below.
In addition, it is noted that the size of N-TiO2 nanoparticles is similar to that of pure TiO2 (Table 1), indicating that nitrogen doping has almost no influence on the grain growth of N-TiO2. Tin doping can remarkably inhibit the grain growth of Sn-TiO2 since the crystal size of Sn-TiO2 is smaller than that of pure TiO2. The specific surface area ranks in the order of TiO2 < N-TiO2 < N/Sn-TiO2 < Sn-TiO2 (Table 1), which agrees with the trend for the change in crystal size of the samples. The fact that Sn-TiO2 exhibits a lower photocatalytic activity than N/Sn-TiO2 and N-TiO2 although it has the largest specific surface area indicates that here the doping of N and Sn, rather than the increase in surface area, plays a major role in the enhancement of photocatalytic activity.
XPS analysis
XPS measurements are used to investigate the chemical states of N and Sn in N-TiO2, Sn-TiO2, and N/Sn-TiO2 samples. Fig. 2 shows the N1s peaks of N-TiO2 and N/Sn-TiO2, in which broad peaks centered at about 399.4 eV (N-TiO2) and 400.4 eV (N/Sn-TiO2) are observed. These two peaks are much higher than the typical binding energy (BE) of 396.9 eV in TiN,41 indicating that the N atoms in N-TiO2 and N/Sn-TiO2 interact strongly with O atoms.18 Therefore the BE of 399.4 and 400.4 eV here are attributed to the oxidized nitrogen similar to NOx species, meaning that Ti–N–O linkage possibly formed on the surface of N-TiO2 and N/Sn-TiO2.42–46 Furthermore, it is highly possible that some N–O species coordinate with tin to form the Sn–N–O linkage on the surface of N/Sn-TiO2. For the latter case, the electron density of N atoms in Sn–N–O is lower than that in Ti–N–O since the electronegativity of Sn is larger than that of Ti,31 which may explain why the BE of nitrogen surface species for N/Sn-TiO2 (400.4 eV) is higher than that for N-TiO2 (399.4 eV).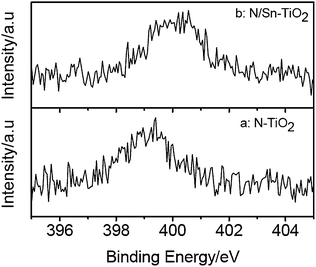 | ||
| Fig. 2 N1s XPS spectra of N-TiO2 (a) and N/Sn-TiO2 (b). | ||
Fig. 3 presents the Sn3d spectra of Sn-TiO2 and N/Sn-TiO2. The doublet peak around 486.4 eV and 494.9 eV is ascribed to Sn3d5/2 and Sn3d3/2, respectively, which arises from the doping Sn4+ ions that substitute lattice Ti in Sn-TiO2 and N/Sn-TiO2 samples because the Sn3d5/2 peak falls between that for SnO2 (486.7 eV) and metallic Sn (485.0 eV).31,47 According to XPS and XRD results, therefore, nitrogen is present as surface species in N/Sn-TiO2 and tin incorporates into TiO2 lattice in substitutional mode. According to XPS spectra, moreover, the Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn and Ti:N ratio in N/Sn-TiO2 sample is 1:0.034 and 1:0.059, respectively.
:Sn and Ti:N ratio in N/Sn-TiO2 sample is 1:0.034 and 1:0.059, respectively.
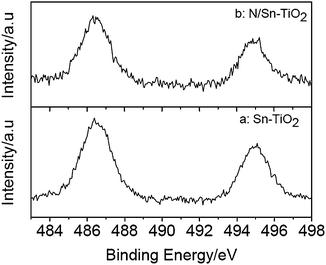 | ||
| Fig. 3 Sn3d XPS spectra of Sn-TiO2 (a) and N/Sn-TiO2 (b). | ||
UV-Vis DRS
Fig. 4 gives UV-Vis DRS absorption spectra of TiO2, N-TiO2, Sn-TiO2, and N/Sn-TiO2 samples. Pure TiO2 only exhibits a strong absorption in the UV region that is attributed to the band-to-band transition.27 Compared with the pure TiO2, N-TiO2 presents a small hump at around 450 nm tailing the visible-light region, which is a typical absorption feature of nitrogen-doped TiO2 and arises from the electron transition from surface state of NOx species to the conduction band of TiO2.48,49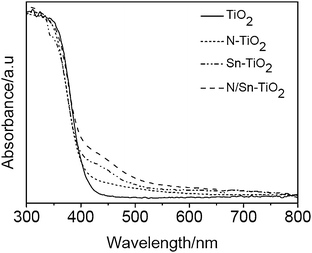 | ||
| Fig. 4 UV-Vis DRS absorption spectra of TiO2, N-TiO2, Sn-TiO2, and N/Sn-TiO2. | ||
As shown in Fig. 4, Sn-TiO2 shows a distinct hump at 450 nm, which agrees with our previous work.31 Since the doping energy level of Sn4+ ions is located at 0.4 eV below the conduction band, it is suggested that the visible-light absorption of Sn-TiO2 arises from the electronic transition from valance band to this doping energy level.
The N/Sn-TiO2 samples show a strong absorption in the visible range from 400 to 600 nm (Fig. 4), which is stronger than N-TiO2 and Sn-TiO2 because the contribution is from both the nitrogen surface species and substitutionally doped tin. This means that nitrogen and tin co-doped TiO2 catalysts are more sensitive to the visible light than TiO2, Sn-TiO2, and N-TiO2.
Photoluminescence spectra
Fig. 5 shows PL emission spectra of pure TiO2, N-TiO2, Sn-TiO2, and N/Sn-TiO2. Two peaks around 480 and 525 nm are observed for TiO2, which is attributed respectively to the transition from oxygen vacancies with two-trapped electrons and one-trapped electron to the valence band of TiO2.36,50–52 The energy levels relate to these two types of oxygen vacancies are located at 0.51 eV and 0.82 eV below the conduction band of TiO2, respectively.50,52 Thus, the origin of PL is explained as follows.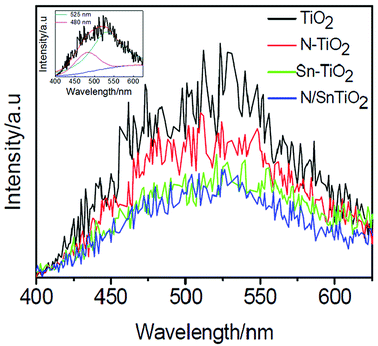 | ||
| Fig. 5 Photoluminescence emission spectra of TiO2 (black), N-TiO2 (red), Sn-TiO2 (green), and N/Sn-TiO2 (blue). Inset shows the fitting results for the TiO2 sample, from which two peaks can clearly be observed. | ||
First, the photogenerated electrons in the conduction band can fall into the oxygen vacancies through a non-irradiative process, and then recombine with the photogenerated holes in the valence band, accompanied by the fluorescence emission. Compared with pure TiO2, the emission intensity is weakened significantly for N-TiO2 and Sn-TiO2, while further weakened for N/Sn-TiO2. This implies that the recombination of charge carriers is effectively suppressed upon doping of N and/or Sn.
A lower PL intensity of N-TiO2 can be attributed to the capture of photogenerated holes by surface states (surface nitrogen species). The effective quenching of PL for Sn-TiO2 is due to the formation of the doping energy level of Sn4+ ions, located at 0.4 eV below the conduction band in the Sn-TiO2 sample. Since the doping energy level of Sn4+ ions is higher than those of oxygen vacancies (0.51 and 0.82 eV below the conduction band), the photogenerated electrons in the conduction band are inclined to move from the conduction band to the doping energy level of Sn4+ ions, rather than to the oxygen vacancies. As a result, the emission intensity originated from the transition from oxygen vacancies to the valence band decreases greatly after doping of Sn4+ ions. For N/Sn-TiO2 samples, the separation of photogenerated holes and electrons is enhanced due to the contribution from both the nitrogen surface species and substitutionally doped tin, resulting in a much stronger suppressed recombination of photogenerated carriers and, thus, the lowest luminescence intensity for N/Sn-TiO2 among all of the samples.
Photocatalytic activity
The photodegradation of 4-chlorophenol (4-CP) is used to evaluate the photocatalytic activity of TiO2, N-TiO2, Sn-TiO2, and N/Sn-TiO2. The adsorption curves in the dark indicate that the adsorption for all the samples can reach equilibrium after 30 min (Fig. 6c). The adsorption equilibrium amount for 10 mg of TiO2, N-TiO2 and Sn-TiO2 samples is almost the same (4 × 10−8 mol) by coincidence, and it is 8 × 10−8 mol for 10 mg N/Sn-TiO2. For all the samples, the ln(c0/c) value of 4-CP shows a linear relationship versus irradiation time, suggesting that the photodegradation may be a pseudo-first-order reaction. The photocatalytic results under both visible and UV light are given in Fig. 6 and Tables 2 and 3. The 4-CP is hardly photo-degraded in the control experiment (photolysis of 4-chlorophenol), regardless of visible or UV-light irradiation. The pure TiO2 photocatalyst shows a very low photocatalytic activity under visible-light irradiation (λ > 400 nm), while N-TiO2 and Sn-TiO2 exhibit a relatively high visible-light activity. N/Sn-TiO2 has an even higher visible-light activity than N-TiO2 and Sn-TiO2, for which 49.1% of 4-CP can be degraded after 8 h of visible-light irradiation. The photodegradation rate and specific photocatalytic activity of N/Sn-TiO2 are about four times those of pure TiO2, and about two times those of N-TiO2, indicating that co-doping TiO2 with nitrogen and tin is an effective way for preparing TiO2-based catalysts with a high visible-light photoactivity. The photocatalytic activity under UV-light irradiation exhibits the same order as that under visible-light irradiation, i.e., N/Sn-TiO2 > N-TiO2 > Sn- TiO2 > TiO2 (Tables 2 and 3).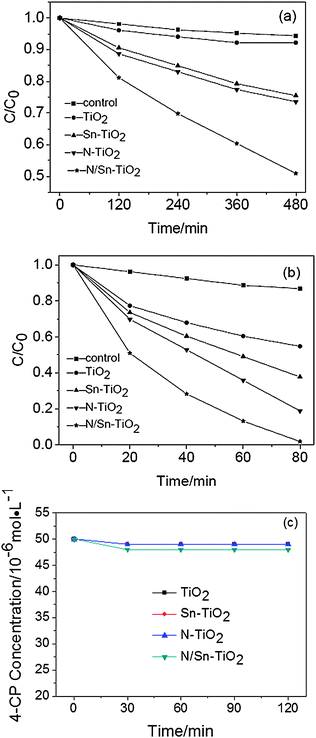 | ||
| Fig. 6 Photodegradation of 4-CP under visible (a), UV-light irradiation (b), and adsorption curve under dark for 10 mg sample (c). | ||
| Sample | 4-CP Degraded a (c0 − c)/c0 (%) | k (min−1) b | t 1/2 (min) | Specific photocatalytic activity (mol g−1 h−1) |
|---|---|---|---|---|
| a After reaction for 8 h. b Apparent rate constant deduced from linear fitting of ln(c0/c) versus reaction time. c The blank was the photolysis of 4-chlorophenol. | ||||
| Controlc | 5.7 | 1.14 × 10−4 | 6063 | — |
| TiO2 | 15.1 | 3.44 × 10−4 | 1987 | 5.45 × 10−6 |
| Sn-TiO2 | 24.5 | 5.83 × 10−4 | 1161 | 9.08 × 10−6 |
| N-TiO2 | 26.4 | 6.28 × 10−4 | 1066 | 1.09 × 10−5 |
| N/Sn-TiO2 | 49.1 | 1.38 × 10−3 | 487 | 1.82 × 10−5 |
| Sample | 4-CP degraded a (c0 − c)/c0 (%) | k (min−1)b | t 1/2 (min) | Specific photocatalytic activity (mol g−1 h−1) |
|---|---|---|---|---|
| a After reaction for 80 min. b Apparent rate constant deduced from linear fitting of ln(c0/c) versus reaction time. | ||||
| Control | 13.2 | 1.80 × 10−3 | 377 | — |
| TiO2 | 45.3 | 7.30 × 10−3 | 86 | 7.62 × 10−5 |
| Sn-TiO2 | 62.2 | 1.18 × 10−2 | 56 | 9.80 × 10−5 |
| N-TiO2 | 81.0 | 2.02 × 10−2 | 37 | 1.23 × 10−4 |
| N/SnTiO2 | 98.1 | 4.86 × 10−2 | 20 | 1.67 × 10−4 |
Discussion
Based on the aforementioned results, the reason that N/Sn-TiO2 exhibits a high photodegradation capacity of 4-CP under both visible and UV light can be explained using the scheme shown in Fig. 7. Pure TiO2 has a very low visible-light (λ > 400 nm) photodegradation capacity due to its large bandgap (3.2 eV, process A). After co-doping with tin and nitrogen, electrons can be excited simultaneously from valence band to Sn4+ doping energy level (process B), and from the surface states energy level (formed due to nitrogen surface species) to the conduction band (process C). Meanwhile, it is possible for photogenerated electrons at the conduction band to fall into the doping energy level of Sn4+, located at 0.4 eV below the conduction band.31 The photogenerated electrons can also transfer from the conduction band and Sn4+ doping energy level to the surface of nanoparticles.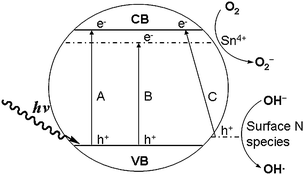 | ||
| Fig. 7 Scheme of photocatalytic mechanism of nitrogen and tin co-doped TiO2. | ||
Because the energy level of O2/O2− is lower than that of the doping energy level of Sn4+ ions and the conduction band (Fig. 7), photogenerated electrons are directly captured by the adsorbed O2 molecules on the surface of N/Sn-TiO2 to form O2− active species during the photocatalytic process (eqn (1)).2,31 The resultant O2− can react with the photogenerated electrons to form H2O2 (eqn (2)), which can further react with photogenerated electrons to produce OH− and the hydroxyl free radical OH• (eqn (3)). The obtained OH− can react with photogenerated holes to generate active species OH• (eqn (4)). Both O2− and OH• can oxidize 4-CP molecules absorbed on the catalyst surface.1 In addition, photogenerated holes at both the valance band and surface states can directly oxidize 4-CP, or be trapped by surface hydroxyl to form active species OH•. Eventually, 4-CP molecules are photodegraded into CO2 and H2O. For solely doped TiO2, however, only process B exists for Sn-TiO2 under visible-light irradiation, and process C for N-TiO2. Therefore, co-doping with nitrogen and tin can increase the quantity of photoinduced electrons and holes, compared with pure and solely doped TiO2. As discussed above (Fig. 5), moreover, N/Sn-TiO2 has the highest efficiency for the separation of photogenerated electrons and holes among all of the samples and, accordingly, the lowest recombination rate. As a result, more photogenerated electrons and holes will contribute to the photocatalytic process, resulting in a higher photocatalytic activity than that of pure TiO2, N-TiO2, and Sn-TiO2.
| O2 + e → O2− | (1) |
| O2 + 2H+ + 2e → H2O2 | (2) |
| H2O2 + e→ OH− + HO• | (3) |
| OH− + h+ → HO• | (4) |
In the case of UV light, process A is allowed for all of the samples. For pure TiO2, due to the existence of the potential barrier of TiO2 surface at the liquid–solid interface, the photogenerated electrons at the conduction band would migrate to TiO2 bulk along the band-bending direction rather than transfer to the TiO2 surface over the potential barrier, which increases the probability of electron–hole recombination. Compared with pure TiO2, N-TiO2 and Sn-TiO2, the photocatalytic activity of N/Sn-TiO2 under UV-light irradiation is improved due to existence of the energy levels of both doping Sn+4 ions and surface state of NOx species. Since the doping energy level of Sn+4 is located at 0.4 eV below the conduction band, the photogenerated electrons at the conduction band will directly transfer to the catalyst surface via this Sn+4 doping energy level, and then captured by the adsorbed O2 molecules on the surface of N/Sn-TiO2 to form O2− active species, which will further degrade 4-CP molecules adsorbed on the surface. Meanwhile, the photogenerated holes at the valence band can transfer to the surface state (surface NOx species), and then oxidize 4-CP molecules adsorbed on the N/Sn-TiO2 surface during the photocatalytic process. This effectively facilitates the separation of photogenerated electrons and holes and thereby inhibits their recombination, resulting in more electrons and holes that can contribute to the photocatalytic reaction. Compared with pure TiO2, N-TiO2, and Sn-TiO2, as a result, the photocatalytic activity of N/Sn-TiO2 is also improved under UV-light irradiation.
Conclusions
In summary, a new type of TiO2-based photocatalyst (N/Sn-TiO2) has been successfully prepared by co-doping TiO2 with tin and nitrogen using a simple sol–gel technique, which exhibits a higher photocatalytic activity than pure TiO2, nitrogen-doped TiO2, and tin-doped TiO2 under both visible and UV-light irradiation due to the contribution from both nitrogen surface species and substitutionally doped tin. This implies that co-doping with two foreign ions, metal and nonmetal, is a more efficient way to improve the photocatalytic activity of TiO2 than doping with just one type of ion. It is expected that detailed studies on the co-doped TiO2 catalyst in the ongoing research, especially more precise control over the alignment of energy levels, will afford a viable way to develop TiO2-based visible-light photocatalysts suitable for practical applications, as well as leading to improved catalyst design.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 50872056 and No. 51072082). T. H. thanks National Research Fund for Fundamental Key Projects No. 973 (2011CB933200) and the Hundred-Talent Program of Chinese Academy of Sciences.Notes and references
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735 CrossRef CAS
.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemannt, Chem. Rev., 1995, 95, 69 CrossRef CAS
- X. B. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515 CrossRef CAS
- M. Zhang, C. C. Chen, W. H. Ma and J. C. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730 CrossRef CAS
- G. Granados-Oliveros, E. A. Páez-Mozo, F. M. Ortega, C. Ferronato and J. Chovelon, Appl. Catal., B, 2009, 89, 448 CrossRef CAS
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS
- O. Lorret, D. Francová, G. Waldner and N. Stelzer, Appl. Catal., B, 2009, 91, 39 CrossRef CAS
- A. Zaleska, E. Grabowska, J. W. Sobczak, M. Gazda and J. Hupka, Appl. Catal., B, 2009, 89, 469 CrossRef CAS
- C. K. Xu, R. Killmeyer, M. L. Gray and S. U. M. Khan, Appl. Catal., B, 2006, 64, 312 CrossRef CAS
- C. Adán, A. Bahamonde, M. F. García and A. M. Arias, Appl. Catal., B, 2007, 72, 11 CrossRef CAS
- A. Kubacka, M. F. García and G. Colón, J. Catal., 2008, 254, 272 CrossRef CAS
- Z. F. Bian, J. Zhu, S. Wang, Y. Cao, X. Qian and H. X. Li, J. Phys. Chem. C, 2008, 112, 6258 CrossRef CAS
- W. Zhao, W. H. Ma, C. C. Chen, J. C. Zhao and Z. G. Shuai, J. Am. Chem. Soc., 2004, 126, 4782 CrossRef CAS
- B. F. Gao, Y. J. Kim, A. K. Chakraborty and W. I. Lee, Appl. Catal., B, 2008, 83, 202 CrossRef CAS
- X. F. Chen, X. C. Wang, Y. D. Hou, J. H. Huang, L. Wu and X. Z. Fu, J. Catal., 2008, 255, 59 CrossRef CAS
- G. S. Shao, F. Y. Wang, T. Z. Ren, Y. P. Liu and Z. Y. Yuan, Appl. Catal., B, 2009, 92, 61 CrossRef CAS
- M. Sathish, B. Viswanathan, R. P. Viswanath and C. S. Gopinath, Chem. Mater., 2005, 17, 6349 CrossRef CAS
- M. Y. Xing, J. L. Zhang and F. Chen, Appl. Catal., B, 2009, 89, 563 CrossRef CAS
- S. Sakthivel, M. Janczarek and H. Kisch, J. Phys. Chem. B, 2004, 108, 19384 CrossRef CAS
- Y. Cong, J. L. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976 CrossRef CAS
- T. Ihara, M. Miyoshi, Y. Iriyama, O. Matsumoto and S. Sugihara, Appl. Catal., B, 2003, 42, 403 CrossRef CAS
- X. B. Chen and C. Burda, J. Phys. Chem. B, 2004, 108, 15446 CrossRef CAS
- X. F. Qiu, Y. X. Zhao and C. Burda, Adv. Mater., 2007, 19, 3995 CrossRef CAS
- Y. Cong, J. L. Zhang, F. Chen, M. Anpo and D. N. He, J. Phys. Chem. C, 2007, 111, 10618 CrossRef CAS
- T. C. Jagadale, S. P. Takale, R. S. Sonawane, H. M. Joshi, S. I. Patil, B. B. Kale and S. B. Ogale, J. Phys. Chem. C, 2008, 112, 14595 CrossRef CAS
- B. F. Gao, Y. Ma, Y. A. Cao, W. S. Yang and J. N. Yao, J. Phys. Chem. B, 2006, 110, 14391 CrossRef CAS
- H. B. Yu, S. Chen, X. Quan, H. M. Zhao and Y. B. Zhang, Environ. Sci. Technol., 2008, 42, 3791 CrossRef CAS
- K. Naeem and F. Ouyang, E-J. Chem., 2009, 6(S1), S422 Search PubMed
- M. Anpo, K. Chiba, M. Tomonari, S. Coluccia, M. Che and M. A. Fox, Bull. Chem. Soc. Jpn., 1991, 64, 543 CAS
- Y. A. Cao, W. S. Yang, W. F. Zhang, G. Z. Liu and P. Yue, New J. Chem., 2004, 28, 218 RSC
- Y. Q. Cao, T. He, L. S. Zhao, E. J. Wang, W. S. Yang and Y. A. Cao, J. Phys. Chem. C, 2009, 113, 18121 CrossRef CAS
- E. Arpac, F. Sayılkan, M. Asiltürk, P. Tatar, N. Kiraz and H. Sayılkan, J. Hazard. Mater., 2007, 140, 69 CrossRef CAS
- G. Liu, Y. N Zhao, C. H. Sun, F. Li, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4516 CrossRef CAS
- D. Li, H. Haneda, S. Hishita and N. Ohashi, Chem. Mater., 2005, 17, 2588 CrossRef CAS
- D. Li, H. Haneda, S. Hishita and N. Ohashi, Chem. Mater., 2005, 17, 2596 CrossRef CAS
- Q. C. Xu, D. V. Wellia, M. A. Sk, K. H. Lim, J. S. C. Loo, D. W. Liao, R. Amal and T. T. Y. Tan, J. Photochem. Photobiol., A, 2010, 210, 181 CrossRef CAS
- E. J. Wang, W. S. Yang and Y. A. Cao, J. Phys. Chem. C, 2009, 113, 20912 CrossRef CAS
- V. Subramanian, W. W. Burke, H. W. Zhu and B. Q. Wei, J. Phys. Chem. C, 2008, 112, 4550 CrossRef CAS
-
Y. L. Xu, The Basics of Semiconducting Oxides and Compounds, Press of Xi'an Electronic Science and Technology University, Xi'an, 1991, p. 48 Search PubMed
- N. C. Saha and H. C. Tomkins, J. Appl. Phys., 1992, 72, 3072 CrossRef CAS
- C. S. Gopinath, J. Phys. Chem. B, 2006, 110, 7079 CrossRef CAS
- S. Sato, R. Nakamura and S. Abe, Appl. Catal., A, 2005, 284, 131 CrossRef CAS
- H. Q. Sun, Y. Bai, H. J. Liu, W. Q. Jin, N. P. Xu, G. J. Chen and B. Q. Xu, J. Phys. Chem. C, 2008, 112, 13304 CrossRef CAS
- X. B. Chen, Y. B. Lou, A. C. S. Samia, C. Burda and J. L. Gole, Adv. Funct. Mater., 2005, 15, 41 CrossRef CAS
- Q. C. Xu, D. V. Wellia, R. Amal, D. W. Liao, J. S. C. Loo and T. T. Y. Tan, Nanoscale, 2010, 2, 1122 RSC
- J. A. Taylor, G. M. Lancaster and J. W. Rabalais, J. Electron Spectrosc. Relat. Phenom., 1978, 13, 435 CrossRef CAS
- T. Lindgren, J. M. Mwabora, E. Avendano, J. Jonsson, A. Hoel, C. G. Granqvist and S. E. Lindquist, J. Phys. Chem. B, 2003, 107, 5709 CrossRef CAS
- H. Q. Sun, Y. Bai, W. Q. Jin and N. P. Xu, Sol. Energy Mater. Sol. Cells, 2008, 92, 76 CrossRef CAS
- N. Serpone, D. Lawless and R. Khairutdinov, J. Phys. Chem., 1995, 99, 16655 CrossRef CAS
- J. C. Yu, W. Ho, J. Yu, S. K. Hark and K. Iu, Langmuir, 2003, 19, 3889 CrossRef CAS
- L. V. Saraf, S. I. Patil, S. B. Ogale, S. R. Sainkar and S. T. Kshirsager, Int. J. Mod. Phys. B, 1998, 12, 2635 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2011 |