 Open Access Article
Open Access ArticleControlled two-photon photodegradation of PEG hydrogels to study and manipulate subcellular interactions on soft materials†
Mark W.
Tibbitt
a,
April M.
Kloxin
ab,
Kiran U.
Dyamenahalli
c and
Kristi S.
Anseth
*ab
aDepartment of Chemical and Biological Engineering, University of Colorado, Boulder, CO, USA. E-mail: kristi.anseth@colorado.edu; Fax: +1 303-735-0095; Tel: +1 303-492-7471
bHoward Hughes Medical Institute, University of Colorado, Boulder, CO, USA
cMedical Scientist Training Program, University of Colorado, Aurora, CO, USA
First published on 13th August 2010
Abstract
Cell adhesion and detachment to and from the extracellular matrix (ECM) are critical regulators of cell function and fate due to the exchange of mechanical signals between the cell and its microenvironment. To study this cell mechanobiology, researchers have developed several innovative methods to investigate cell adhesion in vitro; however, most of these culture platforms are unnaturally stiff or static. To better capture the soft, dynamic nature of the ECM, we present a PEG-based hydrogel in which the context and geometry of the extracellular space can be precisely controlled in situvia two-photon induced erosion. Here, we characterize the two-photon erosion process, demonstrate its efficacy in the presence of cells, and subsequently exploit it to induce subcellular detachment from soft hydrogels. A working space was established for a range of laser powers required to induce complete erosion of the gel, and these data are plotted with model predictions. From this working space, two-photon irradiation parameters were selected for complete erosion in the presence of cells. Micron-scale features were eroded on and within a gel to demonstrate the resolution of patterning with these irradiation conditions. Lastly, two-photon irradiation was used to erode the material at the cell–gel interface to remove cell adhesion sites selectively, and cell retraction was monitored to quantify the mesenchymal stem cell (MSC) response to subcellular detachment from soft materials.
Introduction
Approaches to expand and culture progenitor cells in vitro often focus on the role of soluble media additives to control cell function, such as proliferation and differentiation. For example, multipotent mesenchymal stem cells (MSCs) can be differentiated into chondrocytes with the exogenous addition of transforming growth factor-β3 (TGF-β3) or osteoblasts with the addition of dexamethasone, β-glycerol phosphate, and ascorbate.1 Less attention is given to the role of the cellular microenvironment in this process, but it may be equally important for regulating cell function; for MSCs, chondrogenesis is conducted in pellet culture while osteogenesis is conducted in high cell density plate culture.1 Similarly, the elasticity of the culture substrate has been shown to influence cell proliferation, adhesion, morphology, and migration and to direct MSC differentiation.2–6 Furthermore, patterning of substrates to control cell geometry has been shown to regulate relative cell growth and apoptotic rates.7,8 These results illustrate that a myriad of epigenetic factors, beyond soluble media additives, contribute to the control of cell function.9,10 In particular, the extracellular matrix (ECM) serves as a major regulator of cell phenotype11 by presenting mechanical cues and interacting with the cell through integrin binding.12,13 Mechanotransduction relays these physical signals via dynamic focal adhesion formation and cytoskeletal organization, which regulate cell morphology, proliferation, migration, differentiation, and apoptosis via changes in gene expression.12,14–16Seminal studies in cellular mechanobiology5,7 have effectively linked ECM mechanics and cell–material interactions to cell function, but this work has been conducted primarily on static substrates that fail to capture the active nature of the native ECM. In vivo, the ECM undergoes continual restructuring by ECM-cleaving molecules, such as matrix metalloproteinases, and cellular deposition of ECM components, creating a dynamic adhesive landscape.17 This landscape directs attachment and detachment to and from the ECM, controls the geometry of the cell niche, and regulates cytoskeletal organization, which all influence tissue formation, cell fate, wound repair, and cancer metastasis.18 In short, cells exert contractile forces as they actively engage with the ECM, introducing tensile stresses in the cytoskeleton that originate at focal adhesions and it is now evident that both dynamic adhesion and force generation play a major role in directing cell function.14
Researchers have developed several in vitro techniques to investigate the mechanisms of cell adhesion and cytoskeletal tension on the cellular level, including the cell-spreading assay,19,20 laser-induced ablation of actin stress fibers,21,22 pharmacological treatment to disrupt microtubules,23 and trypsin-induced detachment.24 In complementary approaches, investigators have developed responsive material substrates that can trigger changes in surface chemistry to temporally manipulate adhesive ligand presentation25,26 and cell attachment.27,28 For example, Wildt et al. presented a strategy to induce subcellular detachment by electrochemically releasing the fibronectin-derived adhesive ligand RGD from micropatterned gold features on glass and reported quantitative analysis of the detachment dynamics.28 This technique allows the user to control adhesion dynamically on stiff substrates in predefined geometries.
Building from these studies, we sought to engineer soft, yet tunable, photodegradable substrates that would allow user-defined manipulation of cell–material interactions at any location, over any area, and at any point in time. Photoactive polymeric biomaterials that can be manipulated exogenously with light have emerged in recent literature as a class of materials that provide unprecedented spatial and temporal control of both chemical and mechanical properties.29–32 Out of this work, photodegradable hydrogels have been developed that are fully compatible with cell culture.6,30,33,34 Studies with these materials have demonstrated control of the biochemical nature of the ECM to direct MSC differentiation,30 channel formation to direct cancer cell migration,30 and control of ECM elasticity to mediate the myofibroblast-to-fibroblast transition34 or to direct cell morphology in 2D6 and 3D.33
In this manuscript, our objective is to employ a poly(ethylene glycol) (PEG)-based photodegradable hydrogel to study the dynamics of subcellular detachment from soft materials by spatially defined substrate erosion. First, we demonstrate how the focal point of a mode-locked, femtosecond pulsed two-photon laser can be rastered directly below cells seeded on photodegradable hydrogels to erode the underlying substrate completely and disrupt cell–material interactions. Second, a simple model is presented to determine the irradiation conditions that afford complete erosion via two-photon excitation. Laser-induced degradation, and ultimately complete erosion, is demonstrated over a broad range of pulse energies, and features are generated within the gel on size scales relevant to cells. As a final proof of principle, surface erosion is conducted in the presence of cells to analyze quantitatively the retraction dynamics of erosion-induced subcellular detachment.
Results and discussion
Preparation of a two-photon degradable hydrogel
Photodegradable hydrogels were formed by redox initiated, free-radical chain polymerization of a photolabile PEG-based crosslinker (PEGdiPDA, Mn ≈ 4070 g mol−1, Fig. 1a) with a monoacrylated PEG macromer (PEGA, Mn ≈ 400 g mol−1) in phosphate buffered saline (PBS). Since PEG-based materials are bioinert and present a poorly adhesive substrate for cells,35 fibronectin (300 nM, Rg ≈ 14 nm36) was included in the macromer solution used for all cell experiments; by this method, the adhesive protein is entrapped within or covalently attached to the hydrogel (mesh size ξ ≈ 10 nm, as approximated using rubber elasticity and Flory–Rehner theories) and presents integrin-binding adhesive sites. By this approach, cells sense the hydrogel's adhesive context and geometry, spread on the surface, and form an organized cytoskeleton (Fig. 3b).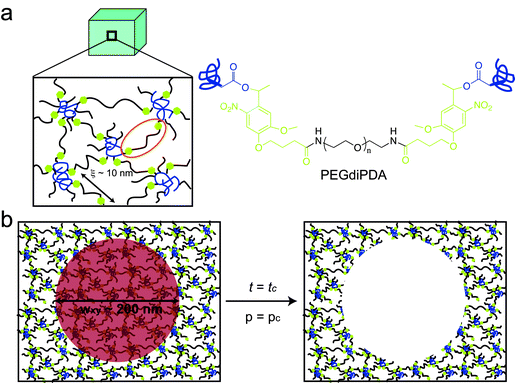 | ||
| Fig. 1 Two-photon induced degradation and complete erosion of photodegradable hydrogels. (a) Poly(ethylene glycol) (PEG)-based photodegradable hydrogels were formed (a, left) by redox-initiated free radical chain polymerization of PEGdiPDA (a, right) with PEGA. The crosslinking density of these gels corresponds to a mesh size (ξ) of approximately 10 nm (a, left), and the gels allow fibronectin to be physically entrapped within or covalently linked to the gel structure. (b) Focused, two-photon irradiation induces complete gel erosion when the focal point (red circle) dwells for enough time (t = tc) such that a sufficient fraction of PEGdiPDA crosslinks can be cleaved locally (p = pc) for a given average laser power. This photocleavage releases the polymer chains that comprise the hydrogel selectively from irradiated regions (b, right). | ||
The crosslinker, PEGdiPDA, was rendered photoactive by attaching an acrylated ortho-nitrobenzylether (NBE) moiety to both sides of a PEG-bis-amine, as previously described.30 This moiety was chosen due to its capability of undergoing two-photon photolysis37 (at 740 nm the two-photon cross-section δuΦu ≈ 0.01 to 0.03 GM38) and its previous use in biological systems.39 Upon two-photon excitation, PEGdiPDA cleaves between the NBE and acrylate functionalities, releasing modified PEG and polyacrylate chains with pendant PEG. Previous studies have shown that the degradation products of this hydrogel formulation are cytocompatible.30,33,34
While the ability to erode completely NBE modified PEG-based hydrogels with two-photon excitation has been demonstrated,30,31 these experiments were not performed in the presence of cells. Here, we sought to better understand how these materials might provide a unique substrate for probing cell–material interactions and to exploit this knowledge to direct cell function in vitro. Toward this end, there are critical aspects of the photodegradation process that need to be understood: (i) complete two-photon erosion should occur with micron-scale resolution; (ii) features should be generated on relevant time scales (ms to s); and (iii) the irradiation conditions should not disrupt normal cell function. For example, two-photon erosion can be used to define the geometry of the stem cell niche only if the features are formed rapidly on the cellular size scale and the cell is not affected solely by the irradiation. Therefore, we first characterized the resolution and time-scale of two-photon erosion within PEGdiPDA hydrogels, and subsequently employed previously reported cytocompatible irradiation conditions to induce subcellular detachment of MSCs.
Resolution of two-photon erosion of PEGdiPDA hydrogels
Mode-locked, pulsed laser directed two-photon excitation induces degradation within PEGdiPDA hydrogels over multiple length scales from µm to mm by cleaving the NBE moieties within the PEGdiPDA crosslinks (Fig. 1b). The spatial resolution of patterning within the gel is theoretically limited by the focal volume, which for a Gaussian laser beam, is governed by the lateral (wxy) and axial (wz) 1/e radii.40 The focal radii are dictated by the numerical aperture (NA), the wavelength of irradiation (λ), and the refractive index of the material (n) as given by40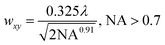 | (1) |
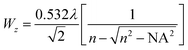 | (2) |
For the experiments in this study with NA = 1.0, λ = 740 nm, and n = 1.33, wxy is 170 nm, and wz is 610 nm. The theoretical diameter for the focal volume is an order of magnitude larger than the approximate mesh size of the hydrogels (Fig. 1b), confirming that the focal point is the limiting factor for spatial resolution of degradation and not the material itself. Further, this focal volume confirms that micron-scale patterning is attainable in both the x–y and z-dimensions, which is demonstrated in experimental results found in Fig. 2c and 3.
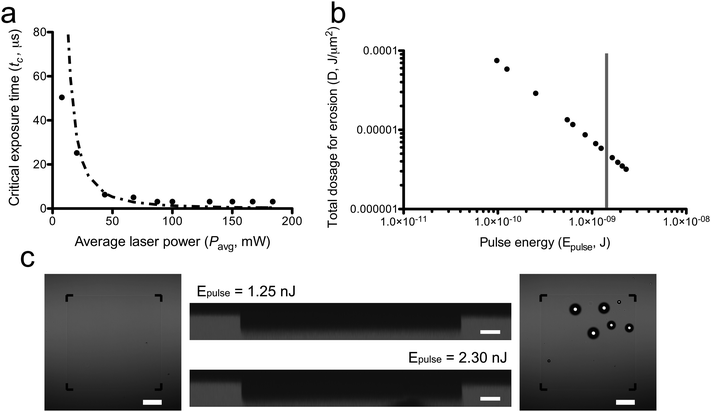 | ||
| Fig. 2 Parameter space for two-photon induced erosion of PEGdiPDA hydrogels. (a) Critical exposure times (tc) for irradiation to induce complete erosion of PEGdiPDA hydrogels were determined experimentally for a broad range of average laser powers (Pavg) (solid circles) and plotted with model predictions (dashed line). (b) The pulse energy (Epulse) for each average laser power used was calculated and combined with tc to determine the total dosage required to erode the material and these data were plotted (solid circles). Previous studies have concluded that pulse energies above 1.5 nJ thermally ablate intracellular components (gray vertical line), demonstrating that complete erosion can be performed below the cut-off for laser-induced ablation of intracellular components. (c) Locally confined erosion of PEGdiPDA hydrogels was imaged in brightfield (outside images, black corner marks indicate the region of erosion) and with cross-sections of fluorescent confocal stacks (central images) and was confirmed with profilometry (data not shown). For the pulse energy used in cells studies and feature formation (Epulse = 1.25 nJ, left outside image and top central image), selective erosion was achieved with an average depth of 8 ± 1 µm for 7 µm deep scans. At the highest pulse energies studied (Epulse = 2.3 nJ, right outside image and bottom central image), nonspecific erosion was induced with an average depth of 11 ± 1 µm for 7 µm deep scans. Furthermore, cavitation was observed within the hydrogels at such high pulse energies as evidenced by bubble formation in the material. Scale bars represent 20 µm in the fluorescent images and 50 µm in the brightfield images. | ||
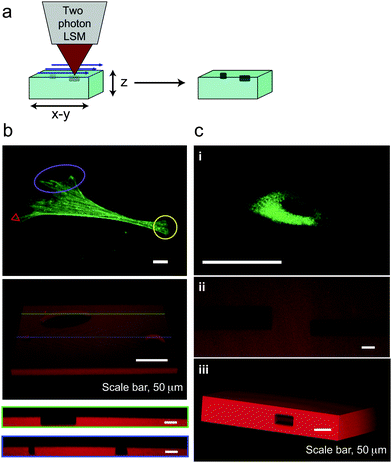 | ||
| Fig. 3 Feature formation to control ECM context and geometry. (a) Defined features can be patterned at the surface or within PEGdiPDA hydrogels by rastering the focal point of a two-photon laser scanning microscope (LSM, Zeiss LSM 710) through specific geometries using region of interest software. (b) Surface feature formation can be performed on size scales relevant to the cell (∼1 to 100 µm) and spatially confined to desired regions to disrupt adhesion at the anterior or posterior of adhered cells (purple oval and yellow circle) or to disrupt adhesion at individual filopodia (red triangle). To demonstrate this strategy, feature formation was performed in the absence of cells on the order of microns (red triangle) to 100 µm (purple oval) and was monitored with confocal microscopy (3D renderings of fluorescent confocal stacks and the corresponding cross-sections, green and blue lines). (c) Features were also patterned within the bulk of PEGdiPDA hydrogels to motivate the utility of this approach for directing encapsulated cells (c.i) to migrate down specific channels (c.ii) or for defining the geometry of the cell niche (c.iii). 20 µm and 30 µm wide channels were patterned into PEGdiPDA gels (c.ii) for representative channel formation, and a 45 µm wide square cylinder was patterned into a gel (c.iii) as a representative change to the geometry of the cell niche. Scale bars represent 20 µm, except as noted. | ||
Critical exposure time for two-photon erosion
A statistical-kinetic model of two-photon degradation and reverse gelation was developed to determine the critical exposure times needed to completely erode the focal volume within this material. In this manuscript, we use the terms degrade or degradation to refer to the cleavage of individual PEGdiPDA crosslinks due to two-photon excitation and the term reverse gelation to refer to the critical extent of degradation when the gel undergoes a solid-to-liquid transition, releasing soluble PEG crosslinks and polyacrylate chains with pendant PEG into solution. The release of these soluble fragments from the gel leads to mass loss, which we term erosion. Thus, reverse gelation refers to the point at which features are formed within the material through complete erosion and degradation is the process of cleaving bonds that ultimately leads to reverse gelation.To reach reverse gelation, such that the gel erodes completely and undergoes a solid-to-liquid transition, a critical fraction of the NBE molecules must be degraded within the focal point. Based on a statistical treatment of chain growth networks, the critical extent of cleavage (pc) is dictated by the weight average number of crosslinks per polyacrylate kinetic chain (N = 20 for this system33), such that 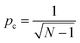 .41 The degradation reaction as a function of irradiation time (t) is modeled as a first-order process as previously described:38,42
.41 The degradation reaction as a function of irradiation time (t) is modeled as a first-order process as previously described:38,42
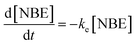 | (3) |
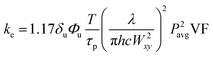 | (4) |
Here, δuΦu is the two-photon cross-section; T is the period of the laser pulses (12.5 ns); τp is the duration of the laser pulses (140 fs); h is Planck's constant; c is the speed of light; Pavg is the average laser power; and VF is a volume factor (0.63 for an axial cylinder).38,42 For these studies, a conservative estimate of 0.01 GM for the two-photon cross-section was used based on previous literature.38Eqn (3) was solved to give a form for the extent of degradation (p):
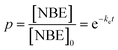 | (5) |
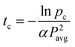 | (6) |
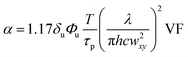 . This relationship combines photodegradation kinetics, two-photon physics, and statistical models of reverse gelation to predict the exposure time required for complete hydrogel erosion. Accordingly, this model provides a working parameter space for two-photon induced erosion of PEGdiPDA hydrogels (Fig. 2a) and a methodology for determining critical exposure times (tc) for two-photon labile materials based on Pavg. Experimentally determined critical exposure times required to erode the material completely were plotted with the statistical-kinetic model (Fig. 2a) over a broad range of average laser powers (7.8 to 183.8 mW). Experimentally determined critical exposure times agree well with the model over the range of Pavg from 20.2 to 87.5 mW (i.e., the absolute value of the deviation = (tcexperimental − tcpredicted)/tcexperimental < 0.5). Outside of this range, larger deviations are observed at high Pavg because of the limitation of the fastest scan speed on the LSM and at low Pavg because of excitation from other planes that can lead to out-of-plane cleavage at slower pixel dwell times. Using this approach, the critical exposure time for erosion of any photolabile network can be predicted for a broad range of irradiation conditions. In addition, this information can be used rationally to select irradiation parameters for modifying gel properties and thus the cell microenvironment.
. This relationship combines photodegradation kinetics, two-photon physics, and statistical models of reverse gelation to predict the exposure time required for complete hydrogel erosion. Accordingly, this model provides a working parameter space for two-photon induced erosion of PEGdiPDA hydrogels (Fig. 2a) and a methodology for determining critical exposure times (tc) for two-photon labile materials based on Pavg. Experimentally determined critical exposure times required to erode the material completely were plotted with the statistical-kinetic model (Fig. 2a) over a broad range of average laser powers (7.8 to 183.8 mW). Experimentally determined critical exposure times agree well with the model over the range of Pavg from 20.2 to 87.5 mW (i.e., the absolute value of the deviation = (tcexperimental − tcpredicted)/tcexperimental < 0.5). Outside of this range, larger deviations are observed at high Pavg because of the limitation of the fastest scan speed on the LSM and at low Pavg because of excitation from other planes that can lead to out-of-plane cleavage at slower pixel dwell times. Using this approach, the critical exposure time for erosion of any photolabile network can be predicted for a broad range of irradiation conditions. In addition, this information can be used rationally to select irradiation parameters for modifying gel properties and thus the cell microenvironment.
Irradiation conditions for in situ degradation
An array of laser powers results in erosion of PEGdiPDA hydrogels (Fig. 2a), but the end application of erosion in the presence of cells limits the user to cytocompatible irradiation conditions. Focused two-photon irradiation has been used previously to thermally ablate intracellular components, such as actin stress fibers,22,43,44 neurofilaments,45 and mitochondria.44,46 These investigations have demonstrated that femtosecond pulsed two-photon irradiation can be used in the presence of cells, but that there is a pulse energy (Epulse) threshold for photo-induced ablation and a threshold for maintaining cell viability. It was reported that pulse energies below 1.5 nJ fail to induce intracellular ablation44 and that cells remain viable when exposed to pulse energies at or below 4 nJ.46 In other words, to prevent non-specific ablation of intracellular components, such as the actin cytoskeleton, and to maintain high cell viability the user should operate below the pulse energy threshold of 1.5 nJ.The pulse energy for a specific laser system is the energy delivered by each femtosecond laser pulse and is calculated as Epulse = Ppeakτp, where 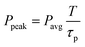 is the peak energy delivered by the laser (τp = 140 fs for the laser used in this study). To explore the relationship between pulse energy and gel erosion, the pulse energies were determined for the irradiation conditions used in this study (Fig. 2b). From these energies and the tc, the total dosage (D) required to erode the material was also calculated,
is the peak energy delivered by the laser (τp = 140 fs for the laser used in this study). To explore the relationship between pulse energy and gel erosion, the pulse energies were determined for the irradiation conditions used in this study (Fig. 2b). From these energies and the tc, the total dosage (D) required to erode the material was also calculated, 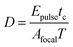 with
with 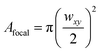 . This analysis demonstrates that higher pulse energies require less total dosage to erode the material (Fig. 2b). However, sufficiently high pulse energies (2.3 nJ) induce cavitation within the PEGdiPDA hydrogels causing non-specific, uncontrolled degradation (Fig. 2c). At lower pulse energies (Epulse < 2.0 nJ), no cavitation was observed (Fig. 2c). Based on previous findings for the cellular response to pulse energies44,46 and our characterization of photoerosion in PEGdiPDA hydrogels, a working space for achieving complete gel erosion while not affecting cell function is established. Correspondingly, gel erosion in this study was conducted with a laser pulse energy of 1.25 nJ to minimize the total dosage while avoiding ablation of cellular components and uncontrolled gel erosion due to cavitation. Further, exposure to this pulse energy of 1.25 nJ does not affect the normal function of cells seeded on non-degradable PEGDA hydrogels (ESI†, Fig. S1).
. This analysis demonstrates that higher pulse energies require less total dosage to erode the material (Fig. 2b). However, sufficiently high pulse energies (2.3 nJ) induce cavitation within the PEGdiPDA hydrogels causing non-specific, uncontrolled degradation (Fig. 2c). At lower pulse energies (Epulse < 2.0 nJ), no cavitation was observed (Fig. 2c). Based on previous findings for the cellular response to pulse energies44,46 and our characterization of photoerosion in PEGdiPDA hydrogels, a working space for achieving complete gel erosion while not affecting cell function is established. Correspondingly, gel erosion in this study was conducted with a laser pulse energy of 1.25 nJ to minimize the total dosage while avoiding ablation of cellular components and uncontrolled gel erosion due to cavitation. Further, exposure to this pulse energy of 1.25 nJ does not affect the normal function of cells seeded on non-degradable PEGDA hydrogels (ESI†, Fig. S1).
Two-photon control of ECM context and geometry
Studies of the cellular response to dynamic changes in the ECM necessitate modifying the adhesive landscape on size scales relevant to the cell (∼1 to 100 µm) in a spatially and temporally defined manner. Recently, this has been achieved in predefined geometries on the surface of glass with an array of gold features that electrochemically releases RGD.28 In addition, adhesive ligands have been photochemically exposed on glass via single and two-photon irradiation of photoactive SAMs.25,26,37 Here, two-photon induced photodegradation of PEGdiPDA hydrogels offers a soft substrate with tunable elasticity and user-defined geometrical and temporal control of the ECM landscape at both the surface of 2D hydrogel scaffolds and within 3D hydrogels. In each case, erosion is achieved by rastering the focal point for the critical exposure time (a pixel dwell time of 1.58 µs) given the pulse energy (1.25 nJ) at each focal volume within the desired geometries (Fig. 3a). Due to the short pixel dwell time, features can be formed rapidly within the gel: ∼10 ms for a 10 µm wide square and ∼4 s for a 250 µm diameter circle. As such, this platform is appropriate to study (i) erosion-induced motility and the dynamics of subcellular detachment on 2D substrates and (ii) migration through channels and the effect of the geometry of the cell niche on cell function within 3D scaffolds.Based on the degradation parameters outlined above, pulsed two-photon laser excitation was used to spatially direct feature formation at the surface of a PEGdiPDA hydrogel (Fig. 3a), demonstrating selective removal of specific cell–material interactions. This technique allows for spatiotemporal removal of the ECM associated with the anterior or posterior of polarized cells (Fig. 3b, purple oval and yellow circle), which has implications in understanding the mechanisms of cell motility, as well as individual filopodia (Fig. 3b, triangle) to study the dynamics of subcellular detachment. Further, this material allows for three-dimensional scaffold erosion within the gel, which could be used to direct cell migration and connectivity or to define the geometry of the cell niche. A cell's geometrical constraints have been shown to influence relative growth and apoptotic rates on 2D substrates,7,8 but due to material limitations, this has been difficult to study in 3D. Here, we demonstrate the ability to pattern channels on relevant size scales or geometric niches within 3D scaffolds using cytocompatible irradiation conditions (Fig. 3c). This method presents a unique approach that provides full spatial and temporal control of the ECM context for hypothesis testing of how the adhesive landscape on 2D and within 3D soft materials affects cell function.
Erosion of PEGdiPDA hydrogels to study subcellular detachment
Cell adhesion and morphology are important regulators of cell function and fate. To demonstrate the efficacy and utility of this material for cell studies, subcellular adhesion was controlled dynamically by selective two-photon irradiation. Specifically, MSCs were transfected with a GFP–actin construct for real time monitoring of cytoskeletal organization and morphology. These cells were seeded on 2D PEGdiPDA hydrogels and given time (∼48 h) to attach to the substrate, spread, and form an organized cytoskeleton. Subsequently, regions of the scaffold were eroded to disrupt cell–material interactions selectively (less than 20% of the initial cell area was eroded in all cases, Fig. 4a). Removal of the substrate material just at the cell interface induces subcellular detachment (Fig. 4, ESI†, Movies S1 and S2), which was initiated at the site of scaffold erosion and was followed by cellular retraction. The morphological response of each cell was tracked over an elapsed time between 10 and 60 minutes using confocal microscopy (Fig. 4a). Control MSCs seeded on non-degradable PEG diacrylate (PEGDA) gels exposed to the same irradiation conditions did not detach or undergo cytoskeletal reorganization (ESI†, Fig. S1, Movie S3).![Two-photon erosion of PEG substrates induces subcellular detachment. (a) Specific regions of cell–material interactions between GFP–actin transfected MSCs and PEGdiPDA hydrogels were disrupted by two-photon erosion (t = 0 s, region outlined in gray). This spatiotemporal erosion induced cell retraction, which was monitored with confocal microscopy (representative images of the first 400 s for an individual cell are shown). (b) Using confocal microscopy, cell area was monitored in response to subcellular detachment, and a fractional area decrease (ΔA(t)/A0) was calculated via image analysis. The solid line is a fit to the equation ΔA(t)/A0 = ΔAmax/A0[1 − exp(−(t − t0)/τc)], and good agreement is observed. (c) Average values for the induction time (t0), retraction time (τc), and the maximum fractional area change (ΔAmax/A0) were compiled for retracting cells (n = 6, mean ± s.e.m), where slowed induction and retraction times were observed on these soft hydrogels as compared to stiff culture substrates.28 Scale bars represent 20 µm.](/image/article/2010/SM/c0sm00174k/c0sm00174k-f4.gif) | ||
| Fig. 4 Two-photon erosion of PEG substrates induces subcellular detachment. (a) Specific regions of cell–material interactions between GFP–actin transfected MSCs and PEGdiPDA hydrogels were disrupted by two-photon erosion (t = 0 s, region outlined in gray). This spatiotemporal erosion induced cell retraction, which was monitored with confocal microscopy (representative images of the first 400 s for an individual cell are shown). (b) Using confocal microscopy, cell area was monitored in response to subcellular detachment, and a fractional area decrease (ΔA(t)/A0) was calculated via image analysis. The solid line is a fit to the equation ΔA(t)/A0 = ΔAmax/A0[1 − exp(−(t − t0)/τc)], and good agreement is observed. (c) Average values for the induction time (t0), retraction time (τc), and the maximum fractional area change (ΔAmax/A0) were compiled for retracting cells (n = 6, mean ± s.e.m), where slowed induction and retraction times were observed on these soft hydrogels as compared to stiff culture substrates.28 Scale bars represent 20 µm. | ||
To quantify the dynamics of subcellular detachment from PEGdiPDA hydrogels, the fractional decrease in area of each cell was followed as ΔA(t)/A0 (Fig. 4b), where ΔA(t) is the change in cell area at time t (A0 − A(t)) and A0 is the initial cell area, similar to the approach of Wildt et al.28 The following expression that describes the relationship between cell area and retraction time was fit to the data corresponding to each retracting cell:
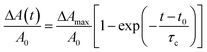 | (7) |
Conclusions
A strategy is presented that allows users to manipulate precisely the context and geometry of a cell's underlying substrate or surrounding microenviroment at any point in space and time via two-photon irradiation of a photodegradable PEG-based hydrogel. A statistical-kinetic model of gel erosion was combined with previous literature to determine a cytocompatible parameter space for spatiotemporal two-photon induced erosion of PEGdiPDA hydrogels. Based on this parameter space, irradiation conditions were selected that preclude uncontrolled erosion and nonspecific ablation of intracellular components while minimizing the total light dosage. These cytocompatible irradiation parameters were subsequently employed to generate features at the surface and within the bulk of PEGdiPDA gels. Subsequently, surface erosion was exploited to disrupt the cell–material interface and induce subcellular detachment. Using this approach we quantified subcellular detachment of MSCs from soft materials, which was observed to be ∼6 fold slower than detachment from stiffer substrates.28 PEGdiPDA hydrogels combined with two-photon induced erosion are useful tools for studying cell adhesion and detachment to and from soft substrates with tunable elasticity, and these materials should facilitate fundamental investigations of the dynamics of cell mechanobiology in both 2D and 3D culture.Experimental
Synthesis of a photodegradable crosslinker
A nitrobenzyl-based photodegradable monomer, photodegradable acrylate (PDA), was synthesized by acrylation of ethyl 4-(4-(1-hydroxyethyl)-2-methoxy-5-nitrophenoxy)butanoic acid and subsequent purification as previously described.30 Through a pendant carboxylic acid, this photodegradable monomer can easily be coupled to any primary amine containing molecule, such as amine functionalized PEG and peptides. A divinyl crosslinking macromer (PEGdiPDA) was synthesized by coupling the PDA to PEG-bis-amine (Mn ≈ 3400 g mol−1, Laysan Bio).30 Briefly, in a glass scintillation vial, PDA (6 mmol) was dissolved in n-methylpyrrolidone (NMP, 156 mmol, Applied Biosystems). 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 6.5 mmol, Anaspec) and 1-hydroxybenzotriazole (HOBt, 6.5 mmol, Anaspec) were added and the solution was heated intermittently with vortexing into complete dissolution of all components. Diisopropylethylamine (DIEA, 11.8 mmol, Anaspec) was subsequently added to the solution and stirred for 5 min. In a single-neck round bottom flask with argon purge, PEG-bis-amine (0.6 mmol) was dissolved in NMP (104 mmol) by intermittent heating and stirring. The activated acid solution was added to this PEG-bis-amine solution, and the flask was purged with argon and stirred overnight at room temperature. The reaction solution was precipitated in cold ethyl ether (0 °C on ice, Fisher Scientific). The precipitated product was recovered by centrifugation (5 min at 3000 rpm) and washed twice with cold ethyl ether. After the final wash, ether was decanted, and the pelleted product was stored under vacuum overnight to remove any remaining ether. The crude product (<10 wt%) was dissolved in deionized water (DI water, 18 MΩ cm, Barnstead NANOpure II). The resulting mixture was centrifuged (30 min at 3000 rpm) to remove any soluble byproducts that remained from the reaction. The dissolved product was subsequently dialyzed (SpectraPor 7, CO 1000 g mol−1) and lyophilized. The purity of the PEGdiPDA was verified with proton NMR (>85% modification of PEG-bis-amine with the PDA). 1H NMR ((CD3)2SO): δ = 8.0 (t, C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)NHCH2CH2O), δ = 7.6 (s, aromatic-H), δ = 7.2 (s, aromatic-H), δ = 6.4, 6.05 (d, d, OC(O)CHCH2), δ = 6.35 (m, aromatic-CH(CH3)OC(O)CHCH2), δ = 6.25 (m, OC(O)CHCH2), δ = 4.1 (t, aromatic-OCH2CH2CH2CO2H), δ = 3.9 (s, aromatic-OCH3), δ = 3.5 (m, [CH2CH2O]n, n ≈ 77), δ = 2.75 (t, NH2CH2CH2O), δ = 2.4 (t, aromatic-OCH2CH2CH2CO2H), δ = 2.0 (m, aromatic-OCH2CH2CH2CO2H), and δ = 1.4 (d, aromatic-CHCH3).
O)NHCH2CH2O), δ = 7.6 (s, aromatic-H), δ = 7.2 (s, aromatic-H), δ = 6.4, 6.05 (d, d, OC(O)CHCH2), δ = 6.35 (m, aromatic-CH(CH3)OC(O)CHCH2), δ = 6.25 (m, OC(O)CHCH2), δ = 4.1 (t, aromatic-OCH2CH2CH2CO2H), δ = 3.9 (s, aromatic-OCH3), δ = 3.5 (m, [CH2CH2O]n, n ≈ 77), δ = 2.75 (t, NH2CH2CH2O), δ = 2.4 (t, aromatic-OCH2CH2CH2CO2H), δ = 2.0 (m, aromatic-OCH2CH2CH2CO2H), and δ = 1.4 (d, aromatic-CHCH3).
Fabrication of PEGdiPDA hydrogels
Photodegradable hydrogels were synthesized using redox-initiated free radical chain polymerization as previously described.33 Briefly, stock solutions of each component were prepared, sterile filtered, and diluted to final concentrations of 8.2 wt% PEGdiPDA, 6.8 wt% PEG monoacrylate (PEGA, Mn ≈ 400 g mol−1, Monomer-Polymer and Dajac Labs), and 0.2 M ammonium persulfate (Acros) in phosphate buffered saline (PBS, pH ≈ 7.4, Invitrogen) to make the gel-forming monomer solution. This monomer solution was aliquoted into individual tubes for each gel sample. To initiate polymerization, tetraethylmethylenediamine (0.1 M in PBS, Sigma Aldrich) was added to an aliquot of the monomer solution while vortexing and quickly pipetted into a mold between glass coverslips (0.25 mm thick spacer, McMaster-Carr; 25 mm × 25 mm cover glass, No. 2, Fisher Scientific). To aid in handling, one of these coverslips was modified with an acrylate silane to covalently link the gel to the coverslip during polymerization. Coverslip modification was performed as previously described:34 the slip was cleaned with Piranha (30 min), rinsed with DI water, dried with an acetone rinse, and modified by chemical vapor deposition with (3-acryloxypropyl)-trimethoxysilane (120 µL, Gelest) in a sealed chamber at 60 °C for 3 h. For gels used in cell culture, fibronectin (FN, human, BD Biosciences) was added to the monomer solution (300 nM in PBS) for entrapment within the gel during polymerization. For gels used in demonstrations of feature formation, methacryloxyethyl thiocarbamoyl rhodamine B (Polysciences, Inc.) was added to the monomer solution (300 µM in PBS, sterile filtered) to covalently modify the polymer network with a fluorophore for subsequent pattern imaging.Culture and transfection of hMSCs
Adult human mesenchymal stem cells (MSCs) (Cambrex Bio Science) were plated on 15 cm diameter tissue culture polystyrene Petri dishes (BD Bioscience) at 5000 cells cm−2. The MSCs were cultured in low-glucose DMEM containing 10% fetal bovine serum, 1 µg mL−1 amphotericin B, 50 units mL−1 penicillin, and 50 µg mL−1 streptomycin (Invitrogen). The cells were grown at standard cell culture conditions (incubated at 37 °C with 5% CO2) and the media were refreshed every third day. Cells were passaged at ∼75 to 85% confluency and MSCs at passage 2 or 3 were used in this study. To monitor the cytoskeletal and morphological responses to subcellular detachment, MSCs were transfected with a GFP–actin plasmid (BD Biosciences), which is a mammalian expression vector that encodes a fusion protein of the green fluorescent protein (GFP) from Aequorea coerulescens to human cytoplasmic β-actin. Transfections were conducted with 1.25 to 1.5 × 106 cells and 1.8 µg of the GFP–actin plasmid in 100 µL of Human MSC Nucleofector® Solution (Lonza) with an Amaxa Nucleofector® II. Upon transfection, cells were seeded on PEGdiPDA hydrogels at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells cm−2, based on hemocytometer counts prior to transfection. Cells were given ∼48 h to adhere to the hydrogels, spread on the surface, and express the GFP–actin plasmid. Subsequently, the gels were transferred to a custom imaging chamber for two-photon irradiation and monitoring of the cell response.
000 cells cm−2, based on hemocytometer counts prior to transfection. Cells were given ∼48 h to adhere to the hydrogels, spread on the surface, and express the GFP–actin plasmid. Subsequently, the gels were transferred to a custom imaging chamber for two-photon irradiation and monitoring of the cell response.
Laser setup and power measurement
Two-photon induced photoerosion of PEGdiPDA hydrogels was carried out on an LSM 710 (Zeiss) with a mode-locked Ti:Sapphire, femtosecond pulsed, multiphoton laser (Chameleon Ultra II, Coherent, Inc.) at a wavelength of 740 nm. A 20× water immersion objective (NA = 1.0, Plan-Apochromat) was used for all eroding scans. ZEN region of interest (ROI) software (Zeiss) was used to raster the focal point through defined geometries of the in-focus x–y plane. Features were patterned into the surface of PEGdiPDA hydrogels by rastering the focal point through desired ROIs and scanning in the z-direction from the surface of the gel to 7 µm below the surface of the gel with a step size of 1 µm. Similarly, features were patterned within the bulk of PEGdiPDA hydrogels by scanning through selected z-dimensions with a step size of 1 µm. A pixel dwell time of 1.58 µs and an average laser power of 0.1 W was used for pattern formation and to ensure complete erosion each scan was performed twice. The average power of the laser through the objective was measured with a power meter (FieldMaxII-TO, Coherent, Inc.) for all two-photon irradiation conditions used. Imaging of pattern formation and cells was also performed on the LSM 710.
Gel characterization
Rhodamine-labeled PEGdiPDA hydrogels were imaged on the LSM 710 before and after feature formation. Both brightfield and fluorescent images were taken to monitor two-photon induced erosion and the height (z-dimension) of feature formation was monitored with fluorescent confocal z-stacks. Feature formation and height measurements were confirmed by profilometry (DekTak 6M Stylus Profiler).Quantification of subcellular detachment
Confocal microscopy (LSM 710, Zeiss) was used to monitor cell area as a function of time post-irradiation. Specifically, a time-series of images (Δt = 15 or 30 s) of each cell was captured for an elapsed time between 10 and 60 minutes. A MATLAB® script was written using the Image Processing Toolbox to quantify the cell area over the course of the time-series corresponding to each cell.Statistical analysis
All data are represented as mean ± standard error of the mean (s.e.m.). GraphPad© software was used for curve fitting, plotting, and data analysis.Acknowledgements
The authors would like to thank Prof. R. R. McLeod for helpful discussions on optics and two-photon physics and for careful reading of earlier versions of the manuscript and Prof. C. N. Bowman for use of the profilometer. The authors acknowledge the NIH (DE16523) and HHMI for funding this work. M. W. Tibbitt acknowledges the US Department of Education's Graduate Assistantships in Areas of National Need program and the National Institutes of Health (T32 GM-065103) for fellowship assistance. K. U. Dyamenahalli acknowledges the NIH Medical Scientist Training Program (GM008497) for funding assistance.References
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143–147 CrossRef CAS.
- R. J. Pelham and Y. L. Wang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13661–13665 CrossRef CAS.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS.
- P. C. Georges and P. A. Janmey, J. Appl. Physiol., 2005, 98, 1547–1553 CrossRef.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS.
- M. T. Frey and Y. L. Wang, Soft Matter, 2009, 5, 1918–1924 RSC.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425–1428 CrossRef CAS.
- R. Singhvi, A. Kumar, G. P. Lopez, G. N. Stephanopoulos, D. I. C. Wang, G. M. Whitesides and D. E. Ingber, Science, 1994, 264, 696–698 CrossRef CAS.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS.
- D. O. Freytes, L. Q. Wan and G. Vunjak-Novakovic, J. Cell. Biochem., 2009, 108, 1047–1058 CrossRef CAS.
- F. Guilak, D. M. Cohen, B. T. Estes, J. M. Gimble, W. Liedtke and C. S. Chen, Cell Stem Cell, 2009, 5, 17–26 CrossRef CAS.
- M. A. Schwartz, M. D. Schaller and M. H. Ginsberg, Annu. Rev. Cell Dev. Biol., 1995, 11, 549–599 CrossRef CAS.
- F. G. Giancotti and E. Ruoslahti, Science, 1999, 285, 1028–1032 CrossRef CAS.
- D. E. Ingber, Prog. Biophys. Mol. Biol., 2008, 97, 163–179 CrossRef.
- A. Mammoto, K. M. Connor, T. Mammoto, C. W. Yung, D. Huh, C. M. Aderman, G. Mostoslavsky, L. E. H. Smith and D. E. Ingber, Nature, 2009, 457, 1103–1108 CrossRef CAS.
- A. Birgersdotter, R. Sandberg and I. Ernberg, Semin. Cancer Biol., 2005, 15, 405–412 CrossRef.
- W. P. Daley, S. B. Peters and M. Larsen, J. Cell Sci., 2008, 121, 255–264 CrossRef CAS.
- D. A. Lauffenburger and A. F. Horwitz, Cell, 1996, 84, 359–369 CAS.
- D. Raucher and M. P. Sheetz, J. Cell Biol., 2000, 148, 127–136 CrossRef CAS.
- H. G. Dobereiner, B. Dubin-Thaler, G. Giannone, H. S. Xenias and M. P. Sheetz, Phys. Rev. Lett., 2004, 93, 038102 CrossRef.
- Z. Rajfur, P. Roy, C. Otey, L. Romer and K. Jacobson, Nat. Cell Biol., 2002, 4, 286–293 CrossRef CAS.
- S. Kumar, I. Z. Maxwell, A. Heisterkamp, T. R. Polte, T. P. Lele, M. Salanga, E. Mazur and D. E. Ingber, Biophys. J., 2006, 90, 3762–3773 CrossRef CAS.
- E. J. Ezratty, M. A. Partridge and G. G. Gundersen, Nat. Cell Biol., 2005, 7, 581–590 CrossRef CAS.
- S. Sen and S. Kumar, Cell. Mol. Bioeng., 2009, 2, 218–230 Search PubMed.
- S. Petersen, J. M. Alonso, A. Specht, P. Duodu, M. Goeldner and A. del Campo, Angew. Chem., Int. Ed., 2008, 47, 3192–3195 CrossRef CAS.
- Y. Ohmuro-Matsuyama and Y. Tatsu, Angew. Chem., Int. Ed., 2008, 47, 7527–7529 CrossRef CAS.
- W. S. Yeo and M. Mrksich, Langmuir, 2006, 22, 10816–10820 CrossRef CAS.
- B. Wildt, D. Wirtz and P. C. Searson, Nat. Methods, 2009, 6, 211–213 CrossRef CAS.
- J. A. Johnson, J. M. Baskin, C. R. Bertozzi, J. T. Koberstein and N. J. Turro, Chem. Commun., 2008, 3064–3066 RSC.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS.
- D. Y. Wong, D. R. Griffin, J. Reed and A. M. Kasko, Macromolecules, 2010, 43, 2824–2831 CrossRef CAS.
- S. Murayama and M. Kato, Anal. Chem., 2010, 82, 2186–2191 CrossRef CAS.
- A. M. Kloxin, M. W. Tibbitt, A. M. Kasko, J. F. Fairbairn and K. S. Anseth, Adv. Mater., 2010, 22, 61–66 CrossRef CAS.
- A. M. Kloxin, J. A. Benton and K. S. Anseth, Biomaterials, 2010, 31, 1–8 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- M. Rocco, E. Infusini, M. G. Daga, L. Gogioso and C. Cuniberti, EMBO J., 1987, 6, 2343–2349 CAS.
- M. Alvarez, A. Best, S. Pradhan-Kadam, K. Koynov, U. Jonas and M. Kreiter, Adv. Mater., 2008, 20, 4563–4567 CrossRef CAS.
- I. Aujard, C. Benbrahim, M. Gouget, O. Ruel, J. B. Baudin, P. Neveu and L. Jullien, Chem.–Eur. J., 2006, 12, 6865–6879 CrossRef CAS.
- Y. R. Zhao, Q. Zheng, K. Dakin, K. Xu, M. L. Martinez and W. H. Li, J. Am. Chem. Soc., 2004, 126, 4653–4663 CrossRef CAS.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1368–1376.
- S. K. Reddy, K. S. Anseth and C. N. Bowman, Polymer, 2005, 46, 4212–4222 CrossRef CAS.
- N. I. Kiskin, R. Chillingworth, J. A. McCray, D. Piston and D. Ogden, Eur Biophys. J. Biophys. Lett., 2002, 30, 588–604 Search PubMed.
- A. Heisterkamp, I. Z. Maxwell, E. Mazur, J. M. Underwood, J. A. Nickerson, S. Kumar and D. E. Ingber, Opt. Express, 2005, 13, 3690–3696 CrossRef CAS.
- N. Shen, D. Datta, C. B. Schaffer, P. LeDuc, D. E. Ingber and E. Mazur, Mech. Chem. Biosystems, 2005, 2, 17–26 Search PubMed.
- M. F. Yanik, H. Cinar, H. N. Cinar, A. D. Chisholm, Y. S. Jin and A. Ben-Yakar, Nature, 2004, 432, 822–822 CrossRef CAS.
- W. Watanabe, N. Arakawa, S. Matsunaga, T. Higashi, K. Fukui, K. Isobe and K. Itoh, Opt. Express, 2004, 12, 4203–4213 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and data for control cell experiments with non-degradable PEG hydrogels as well as movies S1-S3. See DOI: 10.1039/c0sm00174k |
| This journal is © The Royal Society of Chemistry 2010 |