Synthetic hydrogels for controlled stem cell differentiation
Shao Qiong
Liu†
a,
Richie
Tay†
b,
Majad
Khan
b,
Pui Lai
Rachel Ee
a,
James L.
Hedrick
c and
Yi Yan
Yang
*b
aDepartment of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore 117543, Singapore
bInstitute of Bioengineering and Nanotechnology, 31 Biopolis Way, Singapore, 138669, Singapore. E-mail: yyyang@ibn.a-star.edu.sg; Fax: +65-64789084; Tel: +65-68247106
cIBM Almaden Research Center, 650 Harry Road, San Jose, California 95120, USA
First published on 24th November 2009
Abstract
Stem cells offer great promise for regenerative medicine because of their pluripotency and their ability for self-renewal; however, their use in clinical treatments requires knowledge of the cues that control stem cell fate in vivo, and the ability to recapitulate those cues in tissue-engineered systems to direct differentiation into desired cell types and tissues. Hydrogels formed from poly(ethylene glycol) (PEG) are useful as scaffolds for promoting stem cell growth and differentiation towards the formation of tissues. The mechanical and biochemical microenvironment of these PEG hydrogels can be modified in a variety of ways to control cellular functions that are important in determining and maintaining stem cell phenotype. In this review, recent advances in the synthesis and modification of PEG hydrogels will be presented, along with important physicochemical considerations in the design of these hydrogels to better mimic the stem cell microenvironment and direct stem cell differentiation.
 Shao Qiong Liu | Shao Qiong Liu received her PhD in chemical and biomolecular engineering in 2006 from National University of Singapore (NUS). She has previously worked as a researcher at Nanyang Technological University (NTU) from 2005–2007. Currently, she is a research fellow at NUS, working with Dr Pui Lai Rachel Ee and Dr Yi Yan Yang on synthesis of injectable hydrogels as a scaffold for delivery of human mesenchymal stem cells to cartilage defect. She has co-authored 13 peer-reviewed journal publications. |
 Richie Tay | Richie Tay received his BS in Chemical and Biomolecular Engineering from Johns Hopkins University in 2008. He subsequently completed a one-year research stint in the Institute of Bioengineering and Nanotechnology in Singapore, during which he worked with Dr Yi Yan Yang on self-assembled and photopolymerized gene delivery platforms. He is now pursuing a PhD in tissue engineering in MIT under the Harvard-MIT Division of Health, Sciences and Technology. |
 Majad Khan | Majad Khan was born and bred in the northern English town of Halifax; he did his undergraduate studies in chemistry at Imperial College, UK, graduating in 2000 with an MSci. This was followed by PhD studies at Cambridge University in the area of organic polymer chemistry, focusing particularly on hyperbranched polymers for delivery purposes, under the supervision of Prof. Wilhelm Huck at the Melville Laboratory for Polymer Synthesis. After obtaining his PhD in 2004, Majad joined the institute of Bioengineering and Nanotechnology (IBN) in Singapore as a Postdoctoral Fellow, working primarily in the research group of Dr Yi Yan Yang. He currently works as a Research Scientist in Dr Yang's group specializing in synthesizing cationic based polymers/liposomes/hydrogels for gene and drug delivery purposes. |
 Pui Lai Rachel Ee | Rachel received her BSc. in Pharmacy with First Class Honors from the National University of Singapore in 1998 and has been a licensed pharmacist since 1999. She received her PhD in Pharmaceutics from the University of Illinois at Chicago in 2004. She was awarded a fellowship by the Singapore Economic Development Board and received overseas industrial training in drug discovery and development in ProSkelia, a spin-off company from the former Aventis Pharma (now Sanofi-aventis). In 2006, she joined the Department of Pharmacy at the National University of Singapore as Assistant Professor. Her research group is involved in the synthesis and evaluation of biofunctionalized scaffolds for stem cell/drug delivery for cartilage applications as well as cancer. She has published more than 15 peer-reviewed journal articles. |
 James L. Hedrick | James received his PhD from James McGrath at Virginia Tech in Material Science and Engineering. He joined IBM Research in 1985 in the Advance Organic Materials Group. James has focused on the synthesis and basic structure property relationships on synthetic polymers for advance microelectronic and biomedicinal related applications. Areas of emphasis include organocatalytic methods to biocompatible/degradable polymers, functional oligomers, copolymers and complex architectures. He is the recipient of the ACS, Division of Polymer Chemistry, Carl S. Marvel Award 2003, ACS, Division of Polymer Chemistry, Industrial Sponsors Award 2006, European Polymer Chemistry Award 200 and 2009 Cooperative Research Award in Polymer Science and Engineering with Robert Waymouth of Stanford (ACS PMSE Division). He has co-authored more than 300 papers and has more than 50 patents issued. |
 Yi Yan Yang | Dr Yi Yan Yang is a Group Leader at Institute of Bioengineering and Nanotechnology, Singapore. She received her PhD in Chemical Engineering from Tsinghua University, P.R. China in 1990. After working as an assistant and associated professor in Tsinghua University for 3 and 5 years respectively, she moved to Singapore in 1998 and worked in Institute of Materials Science and Engineering. In 2003, she moved to Institute of Bioengineering and Nanotechnology as a Group Leader. Since 2008, she has been an adjunct associate professor in the Department of Pharmacy, National University of Singapore. She is a co-inventor of 11 patents, and has authored/co-authored more than 70 peer-reviewed journal publications. Research of her laboratory mainly focuses on self-assembled nanostructures for drug/cell delivery and for applications in cancer and infectious disease therapies as well as cartilage repair. |
1. Introduction
The past decade has witnessed a surge of interest in using stem cells to regenerate diseased or damaged tissues. These cells are found in embryonic and many adult tissues, but can also be derived from adult somatic cells that have been reprogrammed via gene transfer or cell fusion.1 Regardless of their source, all stem cells have the capacity for self-renewal and differentiation down multiple lineages.2,3 Although this capacity is more restricted and decreases with age for adult stem cells, the latter is an autologous cell source (i.e. the cells can be obtained from patients) and their use thus avoids issues of immune rejection and ethical concerns currently plaguing the application of embryonic stem cells (ESCs).2,4The defining features of stem cells make them a potentially invaluable source for cell therapy in many pathologies, but clinical use is predicated on finding ways to reliably differentiate stem cell populations into desired phenotypes. Without such direction, implanted stem cells form bodies of heterogeneous cell types that could degenerate into tumors.4 Recent advances in stem cell biology have revealed many of the biochemical and biophysical cues that regulate stem cell proliferation and differentiation in vivo, and this knowledge has in turn spurred the design of artificial culture platforms capable of both nurturing and directing the fate of stem cells.
Hydrogels are the most extensively studied platforms because their 3D nature and high water content most resemble the native extracellular matrix (ECM) that surrounds many types of stem cells.5,6 ECM components like collagen, gelatin and hyaluronic acid (HA) have been used successfully as hydrogels for stem cell culture,7,8 but it is difficult to tailor the mechanical and degradation properties of these natural scaffolds according to tissue requirements. Synthetic hydrogels are superior in this respect, offering greater control over not just gel physical properties but also its chemical composition and overall architecture.9 In particular, hydrogels based on poly(ethylene glycol) (PEG) are especially popular because, on the one hand, the bioinertness of PEG prevents non-specific protein adhesion, thus reducing immune and inflammatory responses, while on the other, the remarkable versatility of PEG macromer chemistry facilitates the incorporation of chemical and physical cues for stem cell adhesion and controlled differentiation.10
This review describes the marriage of advanced PEG synthetic chemistry with our current understanding of stem cell biology to create 3D milieux that mimic the stem cell microenvironment. Although we focus on the differentiation of mesenchymal stem cells (MSCs), which currently constitute the bulk of hydrogel-based stem cell research, our discussion is broadly applicable to all types of stem cells. Part 2 introduces methods of synthesizing covalently crosslinked PEG hydrogels, while Parts 3 and 4 discuss methods to transform these passive scaffolds into dynamic niches for stem cell differentiation.
2. PEG hydrogel synthesis
PEG-based hydrogels have been made through either physically or chemically crosslinking, as illustrated in a number of excellent reviews.6,11 Physically crosslinked hydrogels are formed by molecular entanglement and/or weak secondary forces such as van der Waals interactions, ionic interactions, hydrogen bonding and hydrophobic interaction.12 For instance, aqueous amphiphilic block copolymers such as PEO-PPO-PEO and PEG-PLGA-PEG (ABA) undergo the reversible sol–gel transition in response to temperature.13,14 A series of PLGA-PEG-PLGA (BAB) triblock copolymers have also been developed, which could be tuned to form hydrogels under physiological temperature via aggregation of percolated micelles.15–17 These hydrogels have been shown to sustain the release of the entrapped drugs, and have potential applications in tissue engineering applications such as cartilage repair.18,19 Nonetheless, we focus on chemically crosslinked gels in this review (Fig. 1), since these are generally more robust than ones formed via ionic or physical interactions. Because chemical crosslinking often involves conditions (e.g. high temperatures, UV irradiation) or reagents (e.g. initiators and catalysts) that are potentially damaging to biomolecules and cells, we will highlight only those protocols with proven biocompatibility.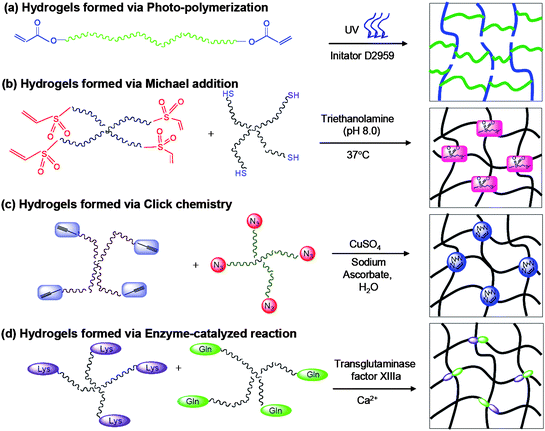 | ||
| Fig. 1 Reaction scheme for preparing PEG based hydrogels via (a) photo-polymerization; (b) Michael addition; (c) click chemistry; (d) enzyme-catalyzed reaction. | ||
2.1 Radical polymerization
Macromers containing two or more vinyl groups can be polymerized in a radical-mediated chain-growth mechanism in the presence of radical-generating initiators and conditions.6 By selecting appropriate initiators and initiator concentrations, stable gels can be produced within minutes under physiological conditions. In redox-initiated polymerization, radicals are formed by a pair of redox initiators, typically a persulfate with an ascorbate or N,N,N′,N′-tetramethyl ethylenediamine (TEMED).6 This type of polymerization was used to synthesize the first hydrogels in the 1960s,20 and it remains a common way of preparing PEG hydrogels for tissue engineering applications.21–23 More commonly, however, initiation is performed using light to activate aromatic compounds via photocleavage or hydrogen abstraction (Fig. 1a).24 Besides being faster than redox polymerization, photopolymerization also affords greater control over gel formation, since radical production can be restricted in time and space using focused beams of light.24 Both visible light and UV photoinitiators have been used successfully to encapsulate cells, but the latter is preferred due to their higher efficiency.25 Early studies established photoinitiation as being compatible for in situ polymerization both transdermally and onto the arterial walls of animals.26,27 In recent years, photopolymerized PEG hydrogels have also been extensively employed as scaffolds for the controlled differentiation of MSCs and ESCs by the Anseth and Elisseeff labs.28–32One drawback of chain-growth mechanisms like radical polymerization is that macromer conversion can be low, especially when cells and other biomolecules are present that can hinder radical propagation.11 Prolonging the polymerization process might increase conversion, but it also increases cell exposure to harmful free radicals and potentially damaging polymerization conditions (e.g. UV exposure, pH changes). Cytotoxicity is in fact a major concern with radical polymerization; studies show that, in addition to exposure time, initiator chemistry and concentration can also adversely affect cell viability, although the extent of damage is cell type-dependent.33 Recent work by Fedorovich and coworkers investigated the effects of photoexposure on mesenchymal stem cells.34 Conditions commonly used for photopolymerization—low-intensity, short-wavelength UV with Irgacure 2959, a mild photoinitiator—caused upwards of 20% cell death for MSCs in monolayers, while the remaining viable cells, though still capable of differentiation, were forced into cell cycle arrest probably as a result of DNA damage.34 Encapsulating MSCs in photopolymerizable hydrogels actually mitigated photodamage, as free radicals formed were rapidly consumed during polymerization before they could inflict cellular damage. In addition, a combined APS/TEMED system showed negative cooperative effect for the cytotoxicity at certain concentrations.35 These show that, with the right choice of conditions, radical polymerization can still be a safe way of encapsulating stem cells in situ.
2.2 Michael addition chemistry
Thiols react quickly and efficiently with α,β-unsaturated carbonyls in a Michael-type conjugate addition to form stable thioether linkages. This reaction proceeds at room temperature without catalysts or initiators, does not produce by-products, and is specific to thiols (amines, another class of reactive nucleophiles, are much slower to react).36 Hubbell and coworkers were the first to exploit conjugate addition to create PEG-based hydrogels (Fig. 1b),37–40 but this route has since become a common method of hydrogel synthesis.41 In a typical reaction scheme, di-functional or multi-arm PEG macromers end-functionalized with acrylate or vinyl sulfone groups are reacted with similar macromers containing terminal thiols. Because the reaction is fairly facile, thiols in proteins and other biomolecules could also participate, thus providing a convenient means of incorporating bioactive entities into the hydrogels (see section 3.4.1). The main disadvantages of this reaction are the (relatively) longer polymerization time, the risk of denaturing disulfide-bearing biomolecules, and the lack of spatial and temporal control over network structure, although the latter is partly compensated by greater control over the cross-inking reaction and thus the material properties of the gel.112.3 Click chemistry
Click chemistry comprises a class of reactions that are fast, high-yielding, require only mild reaction conditions, and produce neither by-products nor side reactions.42,43 The thiol-ene conjugate addition is an example, but the “cream of the crop” is probably the copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition between azide and terminal alkyne moieties (henceforth referred to simply as the click reaction).43 The cycloaddition is bio-orthogonal and highly specific, and has become a popular way of synthesizing hydrogels;44–47 for instance, Hedrick et al. created well-defined PEG hydrogels using the click reaction (Fig. 1c), with higher swelling capacities and significantly improved mechanical properties as compared to photochemically crosslinked hydrogels.48 As with thiol-ene conjugation, click reactions allow fine-tuning of gel network structure via small variations in azide : acetylene ratio.48 Gelation time is longer than with radical polymerization, but can be reduced with adjustments in temperature and catalyst concentration.44,45 The biggest concern with conventional click reactions is their reliance on cytotoxic copper ions as catalyst, which must be removed using chelators prior to cell culture. However, DeForest et al. recently demonstrated that copper-free click chemistry (with strained cyclic alkynes replacing linear ones) can be used to encapsulate cells in situ, which should pave the way for a wider application of this synthetic route.46,472.4 Mixed-mode polymerization
The advantages of chain-growth (high crosslinking density, fast gelation, spatial and temporal control over gelation) and step-growth mechanisms (no initiator, control over network structure) can be harnessed in mixed-mode polymerization reactions for hydrogel synthesis. One such system is mixed-mode thiol-ene photopolymerization developed by Anseth and coworkers to polymerize acrylated macromers and multifunctional thiols.49,50 The predominant mechanism (and thus the hydrogel properties) can be controlled by varying the thiol : acrylate ratio, as well as thiol functionality and chemistry.11,51 In thiol-yne photopolymerization, vinyl groups are replaced with alkynes, which can react consecutively with two thiols.52 This enables a higher crosslinking density than can be achieved with thiol-ene systems, without the network non-idealities commonly seen in photopolymerized hydrogels. Further, the introduction of alkyne functionalities facilitates bioconjugation via click chemistry.2.5 Enzyme-catalyzed cross-linking
The biological equivalent of synthetic click chemistry utilizes enzymes in place of chemical catalysts to crosslink hydrogels.53–56 A commonly employed reaction is the crosslinking of lysine (Lys) and glutamine (Gln) residues mediated by the transglutaminase Factor XIIIa (Fig. 1d). In vivo this reaction binds fibrin molecules together in the culminating step of the blood clotting cascade, and incorporating the transglutaminase substrate sequence into multi-arm PEG can induce similarly rapid gelation in the presence of the enzyme and its cofactor, Ca2+ ions.53–55 Enzymatic reactions have all the advantages of click reactions, but are unfortunately limited by diffusion limitations to the enzyme during gel formation, and by the prohibitive cost of enzymes.563. Approaches to biodegradable and biocompatible PEG hydrogels
PEG is inherently both biologically and chemically inert,1,57 so hydrogels based solely on PEG do not break down easily and cannot support cell culture. Fortunately, the versatility of PEG macromer chemistry has allowed a variety of functional moieties to be incorporated to enhance hydrogel bioactivity.51 In this section we describe several ways of imparting biodegradability and biocompatibility to PEG hydrogels, while in the next we offer more specific modifications necessary to direct stem cell fate.3.1 Hydrolytically degradable PEG hydrogels
Another biodegradable PEG-PPO-PEG (pluronic) containing hydrogel has also been investigated because of its temperature sensitive properties.61–64 The macromer was prepared with PEG-PPO-PEG and oligoCL or oligoLA terminated with acrylate group, which can be crosslinked in situ using a neutral redox initiation system to form hydrogels. The resultant hydrogels exhibited reversible temperature sensitivities and the degradation rate as well as gelation time were found to be well tuned.
A different group of ester-containing PEG hydrogels are those based on the unsaturated fumaric acid;65 these hydrogels contain double bonds in their backbone, which facilitate crosslinking as well as the incorporation of bioactive peptides.23 Various fumarate-containing macromers have been synthesized, including oligo[poly(ethylene glycol) fumarate] (OPF) and poly(propylene fumarate-co-ethylene glycol) [P(PF-co-EG)] by Mikos and colleagues,66 and poly(lactide-co-ethylene oxide-co-fumarate) (PLEOF) by He and Jabbari.65 PEG hydrogels were formed via aqueous-phase, redox-initiated polymerization of these macromers with PEGDA, and the degradation rate was controlled by varying the cross-linking density.
The main advantage of PEG-PPE hydrogels over other degradable hydrogels is that the phosphates from gel breakdown are able to bind calcium ions in solution to induce scaffold calcification, an event which occurs naturally during bone formation.69 Further, the autocalcified scaffolds are able to sequester cell-secreted proteins like osteopontin (which is involved in natural mineralization processes),31 making this class of hydrogels very attractive for bone tissue engineering. Table 1 summarizes the different hydrolytically degradable hydrogel systems used for stem cell culture.
| Chemical group added | Macromer used | Gelation mechanism | Application in cell culture | Ref. |
|---|---|---|---|---|
| a APS: ammonium persulfate; AA: ascorbic acid (AA); TEMED: N,N,N′,N′-tetramethylethylenediamine. | ||||
| Poly(α-hydroxy acid) | PLA-PEG-PLA diacrylate or dimethacrylate | Photo-polymerization with D2959 | Osteoblast differentiation | 59,71,72 |
| Chondrocyte culture | ||||
| Neural precursor expansion and differentiation | ||||
| PCL-PEG-PCL dimethacrylate | Cartilage regeneration | 73 | ||
| PCL-pluronic-PCL diacrylate | Redox polymerization with APS/TEMED | 61–64 | ||
| PLA-pluronic-PLA diacrylate | ||||
| Sebacic acid | PEG sebacate diacrylate | Photo-polymerization with D2959 | hMSC attachment and growth | 74 |
| Dithiothreitol (DTT) | PEG-DTT-PEG diacrylate | Photo-polymerization with D2959 | hMSC viability and spreading | 41 |
| Fumarate | Oligo(PEG-fumarate) | Redox polymerization with APS/AA | RMSC chondrogenesis | 75,76 |
| Poly(propylene fumarate-co-ethylene glycol) and PEGDA | Redox polymerization with APS/AA | 77 | ||
| Poly(lactide-co-ethylene oxide-co-fumarate) (PLEOF) and methylene bisacrylamide (BISAM) | Redox polymerization with APS/AA | BMSC osteogenesis | 78 | |
| Cyclic acetal | 5-Ethyl-5(hydroxymethyl)-β,β-dimethyl-1,3-dioxane-2-ethanol diacrylate (EHD) and PEGDA | Redox polymerization with APS/TEMED | Osteoprogenitor differentiation | 67 |
| PECA diacrylate | BMSC osteogenesis | 79 | ||
| Polyphosphoester (PPE) | PhosPEG | Photo-polymerization with D2959 | GMSC osteogenesis | 68–70 |
| (PPE-HA)-ACRYL | ||||
| PEG-PPE macromer |
3.2 Photodegradable PEG hydrogels
Recently, Anseth's group developed a novel photodegradable PEG hydrogel using a photolabile nitrobenzyl ether-derived moiety.22 Degradation rate was controlled using light intensity and wavelength, and varied according to the photolabile group used. The advantage of such gels over their hydrolytically degraded counterparts is the ability to remotely manipulate gel physical and chemical properties in a spatiotemporally defined manner. For instance, using laser rastering with a single- or two-photon laser scanning microscope (LSM), 3D channels were created rapidly (in the presence of cells) for cell migration. Upon tethering of biomolecules to the photolabile group, photolysis could also be used to temporally control the presentation of biological signals to gel-encapsulated cells to influence cell differentiation. While it remains to be seen if such systems are amenable to in vivo manipulation, they are undoubtedly useful as in vitro 3D culture platforms to elucidate the effects of microenvironmental physical and biochemical changes on stem cell behavior.3.3 Enzymatically degradable PEG hydrogels
Ideally, scaffold degradation should not rely on random hydrolysis or on externally administered triggers, but should instead be initiated by cells in synchrony with proliferation, migration and endogenous ECM production. In vivo, ECM remodeling is mediated by various secreted and membrane-bound proteases, including the cysteine and serine proteases and matrix metalloproteinases (MMPs), all of which target specific cleavage sites on ECM proteins and proteoglycans.80 Many of these enzyme-cleavable peptide sequences have been identified and incorporated as motifs in PEG hydrogels to enable cell-directed proteolysis (Table 2).Pioneering work by West and Hubbell involved photopolymerizing PEG macromers end-capped with acrylate-terminated oligopeptides,81 but this has largely been superseded by more versatile schemes that use separate peptide and PEG components.65,82,83 The most popular is that developed by Lutolf and co-workers, involving multifunctional PEG-vinyl sulfone (PEGVS) macromers and cysteine-terminated oligopeptides, which are cross-linked via a Michael-type addition reaction.82,83 The rate of degradation of these hydrogels could be altered by tuning the enzymatic sensitivity of the substrate peptides, or by varying the crosslinking density (which affects the extent of cell migration).82,84
3.4 Incorporating bioactive moieties
| Peptide sequence | ECM/Molecular source | Function | Ref. |
|---|---|---|---|
| RGD | Fibronectin, vitronectin, laminin, collagen, osteopontin and thrombospondin | Promotes hMSC adhesion, spreading, migration and proliferation | 32,49,50,78,85–87,89,93 |
| Regulates hMSC differentiation | |||
| IKVAV, YIGSR | Laminin | Enhances hMSC adhesion and viability | 74,94 |
| KIPKASSVPTELSAISTLYL | Bone morphogenetic protein-2 (BMP-2) | Promotes MSC differentiation and mineralization | 78 |
| GPQGIWGQ, LGPA | Collagen type I (MMP substrate) | Cell-mediated proteolytic degradation | 37,40,49,82–84,91,95,96 |
| QPQGLAK | Collagen type II (MMP-13 substrate) | Cell-mediated proteolytic degradation | 65 |
| VRN, YKNR | Fibrinogen (plasmin substrate) | Cell-mediated proteolytic degradation | 38,81,97 |
| Y(GPO)7 | Collagen type I | Collagen retention | 98 |
| Promotes hMSC differentiation | |||
| (GPO)4GFOGER(GPO)4GCG | Collagen type I | Collagen-integrin β1 interactions | unpublished data |
| Promotes hMSC differentiation | |||
| KLER | Decorin | Collagen type II retention and organization | 99 |
| Promotes hMSC differentiation | |||
| AKA3KA | Elastin | Enhances gel elasticity | 100 |
| K-βAla-FAKLAARLYRKA | Anti-thrombin III | Heparin binding | 38,97 |
| E6 | Osteonectin | Hydroxyapatite binding | 101 |
Oligopeptides can either be tethered to the hydrogel scaffold or incorporated as part of the hydrogel backbone. A common tethering approach is to conjugate the N-terminus of the peptides to PEG macromers terminated with an N-hydroxysuccinimide (NHS) group.32,85–87 This reaction, while simple and fast to perform, nevertheless gives relatively low yields and does not distinguish between terminal and side-chain amines.80 Greater chemoselectivity is provided by reacting thiol-containing peptides (the thiols provided by cysteine residues) and PEG-(meth)acrylates in a Michael-type conjugate addition.37,83 This approach has the advantage of being a “one-pot” process—peptide conjugation can occur concurrently with gel formation and cell encapsulation—but the peptide : macromer ratio needs to be carefully tuned to ensure adequate peptide concentration and gel robustness. Another highly selective reaction is the click reaction between azide and alkyne functionalities,78 which our group recently exploited to synthesize PEG-peptide hydrogels.45 In our scheme, tetra-acetylene PEG was crosslinked with peptide-diazides in which the azide groups were conjugated to a single terminal lysine. This allowed the peptides to be incorporated as tethers during polymerization (which enhances their presentation to cells, see section 4.2.2) without compromising the mechanical strength of the gel.
The same thiol and acrylate precursors used in Michael addition could be photopolymerized via mixed chain- and step-growth mechanisms, as has been described by Salinas et al.49,50,88,89 One advantage of photopolymerization is that it can be used to spatially pattern peptides in hydrogel networks (a process known as photolithography). For example, West et al. formed partially crosslinked hydrogels, then added acrylated peptides and photo-crosslinked them in defined regions using photomasks and LSM.90,91 In a different approach, Anseth and co-workers conjugated photoreactive allyl ester groups to backbone peptides, then induced thiol-ene coupling between those groups and additional oligopeptides using LSM photolithography.47,92
| Functional moieties | Function | Ref. |
|---|---|---|
| t-Butyl methacrylate | Promotes adipogenesis | 28 |
| Ethylene glycol methacrylate phosphate (EGMP) | Facilitates mineralization, enhances cell viability, promotes osteogenesis | 28,31 |
| 3,3,4,4-Tetrafluorobutyl methacrylate | Promotes adipogenesis | 28 |
| Methacrylic acid | Promotes chondrogenesis | 28 |
| Fluvastatin | Promotes BMP-2 production and initiates osteogenesis | 102 |
| Dexamethasone | Promotes osteogenesis | 103 |
4. PEG hydrogels for stem cell differentiation—constructing artificial stem cell niches
With the incorporation of degradable moieties and bioactive functionalities, PEG hydrogels are no longer mere 3D cages for stem cell culture, but tissue engineers now recognize a host of other physical and chemical cues that are also required to drive tissue formation. In vivo, adult stem cells reside in unique local microenvironments or niches, where a web of short- and long-range signals—arising from interactions with specialized niche cells, the surrounding ECM, and secreted factors both dissolved and ECM-sequestered—regulate their self-renewal and differentiation (Fig. 2).9,104–106 An improved understanding of niche biology in recent years has facilitated the reconstruction of this native environment within PEG hydrogels as a means of directing stem cell fate. Here we present several of the critical mechanical and biochemical features that have been explored.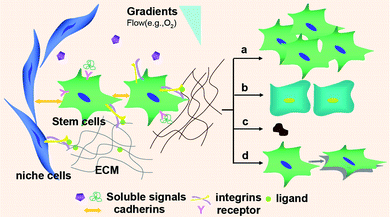 | ||
| Fig. 2 Aspects of the stem cell microenvironment that affect the behavior of stem cells. Soluble signals such as growth factors, cytokines and chemokines; interactions with niche cells via cadherins and selectins; and integrin-mediated cell-ECM interactions, as well as gradients, all affect stem cell self-renewal (a), differentiation (b), apoptosis (c), and migration (d). | ||
4.1 Growth factors, cytokines and chemical additives
During development, families of growth factors (GFs) and cytokines coordinate dozens of differentiation pathways open to embryonic and adult somatic stem cells. Despite this complexity, researchers tend to focus on a few members that show particular potency in guiding certain paths of differentiation. For instance, Hwang et al. showed that TGF-β1 induced chondrogenesis in embryoid bodies (EBs) encapsulated in PEG hydrogels,29 while others have used VEGF165 and BMP-2 successfully for vasculogenesis and osteogenesis respectively.65,107 (Ref. 108 provide a more comprehensive overview.)Although GFs and cytokines are commonly added to culture medium in vitro, diffusion limitations within hydrogels, coupled with the need to maintain appropriate concentrations following hydrogel implantation, demand an alternative mode of delivery. The pleiotropic nature of GF effects—a single growth factor can trigger multiple signaling pathways in many cell lines—also necessitates controlled release if differentiation is to be localized and guided.1 In tissues, ECM proteins and proteoglycans help to sequester and locally concentrate GFs;105 for tissue engineering, the addition of heparan sulfate, a charged glycosaminoglycan (GAG), to PEG hydrogels serves the same function.109 A variety of other ways to retain and release GFs in hydrogels, including covalent tethering using enzyme-sensitive linkers40 and encapsulation in biodegradable particulate carriers,76,107 have been outlined in recent excellent reviews.11,108 While most of these methods were designed for sustained release of GFs via diffusion or linker/scaffold degradation, certain GFs actually benefit from being permanently bound to the scaffold, and knowledge of the metabolism of GFs by stem cells is necessary to determine the appropriate mode of GF presentation.110
In addition to GFs and cytokines, several membrane-permeable small organic compounds can also direct differentiation. These include common medium supplements like dexamethasone, L-ascorbic acid-2-phosphate (AsAP) and β-glycerophosphate (β-GP) (for osteogenesis),111 and others like glucosamine (chondrogenesis),112 retinoic acid (neurogenesis),113 and 5-azacytidine (myogenesis, cardiomyogenesis)114,115 which are fast gaining favor due to their influence on both embryonic and adult stem cells (see also the review by Ding).116 These molecules are currently introduced in solution as a bolus, but advances in bioconjugation should eventually facilitate coupling to PEG hydrogels for co-delivery with GFs.
4.2 ECM components
The use of decellularized matrices raises issues of immunogenicity and compositional variations, which prompted Prestwich and coworkers to develop semi-synthetic ECMs (sECMs) based on chemically-modified ECM components cross-linked with PEG.126 The hydrogel components were designed to be modular to allow tissue-specific assembly and the addition of other biological cues to enhance cell adhesion and hydrogel degradation. Delivery of MSCs using these sECMs facilitated cartilage repair in a rabbit model within 12 weeks, and there was seamless integration with host tissue.126 A point of relevance regarding ECM-containing hydrogels is that they are susceptible to enzymatic degradation, which can alter bioactivity over time; for example, Shah et al. reported that HA degradation generated fragments that led to reduced collagen production.125 More work is required to characterize other effects of ECM degradation on stem cells.
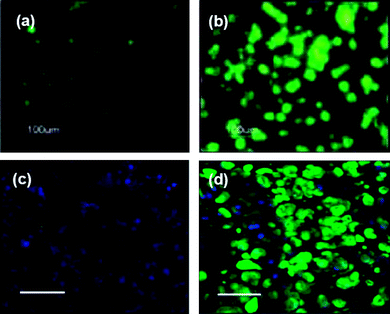 | ||
| Fig. 3 Enhanced chondrogenesis of human embryonic stem cell-derived cells (hESDCs) when exposed to RGD peptides, or chondrocyte-conditioned media. Pictures show hydrogel sections that were immunostained for collagen type II, an abundant cartilage ECM protein. hESDCs in unmodified PEGDA hydrogels showed little differentiation after 3 weeks in chondrogenic media (a), whereas neocartilage was formed by cells in RGD-conjugated hydrogels (b).73 Similarly, hESDCs cultured in normal growth medium for 12 weeks did not exhibit chondrogenesis (c), but those in conditioned medium clearly produced cartilage-like tissue in vivo (d).22 (Blue: nuclei; green: collagen II. All scale bars = 100 μm). (Reprinted with permission from ref. 73, Copyright (2006) by Mary Ann Liebert Inc. Publishers and ref. 22, Copyright (2008) by the National Academy of Sciences, USA). | ||
Other peptides with specific bioactivity include the collagen-mimetic Y(GPO)7 and the decorin-derived KLER peptides, both of which help to retain cell-secreted collagens and enhance MSC chondrogenesis. For instance, MSCs in Y(GPO)7/PEG hydrogel showed significantly more positive type II collagen as compared to that in PEG hydrogels (77% vs. 43%).98 There is also the KIPKASSVPTELSAISTLYL peptide from BMP-2, which acts synergistically with RGD to promote osteogenesis.78,98 Our group have recently synthesized a hybrid peptide containing a GFOGER sequence flanked by GPO repeats. The GFOGER sequence is derived from type I collagen and is known to maintain chondrocyte phenotype in suspension culture via collagen-integrin β1 interactions.133 By incorporating the peptide into PEG hydrogels using Michael addition, we are able to direct encapsulated hMSCs toward chondrogenesis, with enhanced secretion of GAG-rich ECM as compared to cells in hydrogels without the peptide (Fig. 4).
 | ||
| Fig. 4 Alcian blue staining of cartilage construct comprising a PEG hydrogel seeded with MSCs, after 4 weeks of culture (GAG stained blue, nuclei stained red). (left) Blank hydrogel containing TGF-β3; (right) hydrogel containing triple-helical (GPO)4GFOGER(GPO)4GCG sequence and TGF-β3. Construct containing (GPO)4GFOGER(GPO)4GCG produced an abundance of cartilage-specific ECM rich in GAG. Unpublished data. | ||
The use of oligopeptides comes with a few caveats, however, since short peptide sequences cannot recapitulate certain spatial effects seen with larger proteins.134 The RGD peptide presents an illustrative case. First, contextual presentation of the peptide is important: constrained RGD bound via two links to a hydrogel backbone promoted 60% cell viability, but survival increased to 88% with pendant RGD groups containing a polyglycine spacer, which reduced steric hindrance to integrin-peptide interactions.89 The spatial arrangement of the peptides is also important, as integrins require clustering prior to signal transduction, and ligands in ECM proteins are often arranged to engage multiple integrins and promote clustering.135 To reproduce this effect, polyvalent RGD-based molecules have been synthesized containing either linear RGD repeats, cyclic RGDfK, branched RGD arms, or multimeric self-assembled RGD peptides.136–140 These constructs have been proven useful for 2D cell adhesion, though their effect on stem cell behavior in 3D has not been assessed. In addition, nanopattern distribution of RGD has been shown to enhance cell adhesion on PEG hydrogel, while it is not the case with large distance.141 Further, Ding and Spatz et al. investigated this nanopatterned RGD quantitatively and found that integrin clustering and cell adhesion were dependent on the order of nanopattern RGD when the ligand spacing was larger than 70 nm.142
Second, subtle changes to peptide sequence and conformation can affect receptor binding: the residues flanking RGD (which vary in different ECM proteins) dictate both the affinity and selectivity of integrin binding, while the RGD loop (the native conformation in many proteins) binds more tightly and selectively than linear sequences.127,143 In fact, the use of different RGD sequences and cyclopeptides is one way of targeting specific integrins and avoiding the promiscuity of integrin binding seen with full-length proteins. For example, Hsiong et al. showed that the RGD cyclopeptide has greater affinity for the αv subunit, and binding leads to enhanced MSC osteogenesis due to the activation of relevant transcription factors.144
A third consideration is that two or more sequences in different protein domains sometimes act in concert during receptor binding, the best known being RGD and its neighboring epitope PHSRN in fibronectin (FN), both of which are required for effective binding to α5β1 integrin.127,145 Taking this into account, Benoit et al. incorporated both sequences into a single oligopeptide, separated by a polyglycine spacer to mimic their separation in FN.145 Osteoblasts cultured on RGD-PHSRN-immobilized PEG hydrogel discs displayed higher proliferation and metabolic activity compared to those on RGD-immobilized surfaces. Nonetheless, there are concerns that tandem peptides cannot fully replicate the precise orientation and interaction between protein domains, an issue that is better addressed through the use of recombinant protein fragments.93,146 Petrie et al. showed that a recombinant FN III7-10 fragment was more adhesive and bound α5β1 with greater specificity than RGD or RGD-PHSRN peptides, while Martino et al. demonstrated enhanced hMSC osteogenesis using recombinant domains as a result of more effective α5β1 binding.93,147
One final issue concerns the temporal regulation of ligand presentation. Studies indicate that the expression of ECM proteins and their cellular receptors changes with the stage of differentiation, for example, fibronectin and its α5β1 integrin receptor are required for mesenchymal condensation, but are subsequently downregulated during early chondrogenesis.148 With peptides however, signaling is constitutive, which might be disruptive to certain differentiation programs. Inspired by in vivo removal of redundant ECM components via cell-directed proteolysis, Anseth and coworkers tethered RGD moieties to a PEG hydrogel using MMP-cleavable linkers to allow temporal control of RGD-integrin signaling.49 They showed that hMSCs produced ten times more GAGs and close to twice the amount of collagen type II when cultured in RGD-cleavable hydrogels compared to uncleavable gels. This dependence of stem cell differentiation on temporal changes in ligand presentation is expected to receive further study in the coming years, especially with the recent development of photoresponsive hydrogel platforms that allow real-time manipulation of gel biochemical composition.22
Bioactive peptides streamline considerably the use of ECM proteins, but recent work has focused on even simpler functional groups that capture ECM features specific to certain tissue environments.28,31 In work by Benoit and Anseth, carboxylic acid functionalities that resembled anionic GAGs in cartilage ECM were found to improve chondrogenic differentiation of hMSCs when conjugated to PEG hydrogels. Similarly, phosphate groups (found in abundance in mineralized tissue) and t-butyl groups (mimicking the lipid-rich hydrophobic environment in fatty tissue) promoted osteogenesis and adipogenesis respectively, based on lineage-specific protein production and gene expression assays (Table 3).28 While it is not possible to distil the ECM environment of every tissue to a few functionalities, such studies could spearhead the development of simpler synthetic systems to replace the complicated chemical cocktails and conjugate systems currently in use.
4.3 Cell-cell interactions
Besides cell-ECM interactions, various homotypic and heterotypic cell-cell interactions in the niche also influence stem cell fate. Homotypic contacts between stem cells can be regulated by controlling cell density. For ESCs this means controlling the size of the embryoid bodies (EBs) that they form, which can impact differentiation outcomes.149 To avoid the broad size distributions commonly seen with suspension or hanging drop cultures, Karp et al. fabricated PEG hydrogel microwells with defined geometries, and demonstrated the production of homogeneous EBs with reduced variation in ESC differentiation markers.150,151 MSC clustering also affects differentiation: mesenchymal condensation is characterized by N-cadherin-mediated cell adhesion, and is the first stage towards chondrogenesis.152 However, N-cadherin expression eventually diminishes, and over-expression of this adhesion molecule can prevent further progression down the chondrogenic line.152 Such lineage-specific, temporally-varying expression patterns complicate the use of cell density alone to control differentiation, since other non-density-dependent regulatory factors must also be taken into account.Some of these factors likely arise from heterotypic interactions between stem cells and the many heterogeneous cell types that populate the niche. Niche cells anchor and align the stem cells, and regulate their self-renewal and differentiation via an array of adhesion molecules (e.g. cadherins and CAMs) as well as paracrine (e.g. Wnt and hedgehog proteins) and juxtacrine (e.g. Notch receptors and ligands) signaling pathways.153 These interactions are best captured by co-culture systems, and indeed, co-culture with chondrocytes stimulated chondrogenesis of both hMSCs and hESCs.154 However, such systems are difficult to design, and only autologous cell sources can be used. As an alternative, several groups demonstrated that bathing stem cells in cell-conditioned media could be an effective substitute for co-culture,155,156 which suggests that contact-mediated signals might not be as important as paracrine factors (Fig. 3c,d).30 Using conditioned media has its own limitations though; for instance, it is only suitable for in vitro differentiation, and batch variations in its contents might produce inconsistent results, as was seen in a few recent reports.1 A compromise was achieved in the co-culture system devised by Lee et al., which employed sequential photopolymerization to produce a bilayer of mES-derived cells atop mouse hepatic cells.157 In this way, morphogenetic factors secreted by the differentiated cells could directly influence stem cell differentiation even though the two cell lines are kept separate. The size and porosity of the hydrogel layers could be tuned to reduce diffusion limitations to short-distance paracrine signaling. Recently, even juxtacrine signals have made their way into the hydrogel environment, with conjugate chemistry and recombinant DNA technology allowing membrane-bound morphogens or their bioactive domains to be ligated to polymers, or expressed as part of ECM proteins.158 These innovations suggest that it might eventually be possible to engineer intercellular interactions in a more controlled fashion to direct differentiation.
4.4 Physical cues
This has implications for hydrogel design, as gel viscoelasticity needs to be tailored, along with the identity and density of additional ligands, to the desired cell lineage. But there are complications. For many PEG hydrogel systems, these parameters are intertwined, and 3D systems introduce steric effects that must also be taken into account.167 For instance, stiffening the matrix by increasing the cross-linking density can simultaneously affect ligand presentation and gel porosity, the latter in turn impacting cell clustering, migration and ECM deposition patterns.95,167,168 Hydrogel designs that uncouple these influences—such as the PEG-based interpenetrating networks of Saha et al. and the PEG-fibrinogen systems developed by Seliktar's group—would thus be useful as they allow mechanical and biochemical cues to be examined, then engineered, separately.169,170 Also, the requirements of 3D culture restrict the range of acceptable moduli for hydrogels, since excessively stiff gels hinder mass transport, while gels that are too soft are inadequate for cell support. Moduli that have proven effective on 2D substrates might not be suitable for 3D constructs, and more studies need to be done to elucidate the link between scaffold mechanics and cell behavior in 3D.
A third complication is the structural heterogeneity that most tissues exhibit (e.g. aligned myofibers in cardiac tissue, mechanically distinct zones in cartilage, vascular presence in most soft tissues), which contrasts with the isotropic structures produced by most synthesis schemes. To introduce mechanical anisotropy, Zawko et al. used a dual crosslinking method in which methacrylated HA was first crosslinked chemically at its –OH groups to produce a homogeneous gel, then a photomask was used to pattern photo-crosslinks to adjust gel swelling.171 Although HA was used in the reported system, this method could easily be adapted for use with PEG hydrogels. To create microvasculature within hydrogel networks—a feature essential for directing angio- and neurogenesis172,173—PEG could be photopolymerized around fibrillar proteins, and the proteins were subsequently removed using proteases. Namba et al. achieved this for PEG-PLA encapsulating a fibrin network.172 They showed that primary neurons in the hydrogel survived the polymerization-digestion procedure, and the highly interconnected network accelerated neurite outgrowth compared to non-porous gels. The rapid prototyping of even more complex structures within photopolymerized PEG hydrogels has been made possible in recent years by improvements in photolithography and computer-aided design (see ref. 174 for an excellent review). Using single- or multi-photon lasers, macromer solutions can be selectively cured layer by layer to build 3D constructs, or polymerized gels containing photolabile moieties can be selectively degraded to carve out 3D elements.22,175 Alternatively, the PEG macromer can be carefully extruded and gelled in a novel Bioplotting™ process.173,176 These new technologies will become indispensable for creating architecturally complex hydrogels that can better guide cell behavior and facilitate integration with host tissue.
One way to avoid potential mismatches is to let cells direct the degradation via proteolysis of incorporated peptide linkers (section 3.3).39,53 Because proteolysis is localized to the cell periphery, the bulk of the hydrogel retains its mechanical integrity and can sustain cell activity throughout the remodeling process.82,83 Such hydrogels have been used successfully to support the formation of cartilage, bone, and cardiac and smooth muscle.49,65,82,84,95,96 The catch is that in systems where the enzymatically sensitive group is part of the cross-linker, it is not possible to separate the influence of matrix stiffness and degradability. Nonetheless, recent work by Park et al. showed that cells increased protease secretion in response to higher network density, making it unnecessary to decouple the two influences.96 Other enzymatically sensitive systems rely on degradation of GAGs that make up the hydrogel network, but those are more useful for GAG-rich tissues in which cells secrete the necessary degradative enzymes.45,122,125
Scaffold degradation also affects cell-cell interactions that are important for stem cell differentiation. In a recent report, hMSCs in a hydrolytically degradable PEG hydrogel (cultured in osteogenic medium) started off with a rounded morphology and minimal intercellular contact (Fig. 5 left). Only with hydrogel degradation did cell migration and interaction occur to transform the cells into osteoblasts (Fig. 5 right).5 Kraehenbuehl et al. described a different response for encapsulated cardioprogenitor cells.95 These cells remained clustered for the first three weeks in culture, but degradation was essential for the clusters to grow and coalesce into critical-size masses that supported cardiomyogenesis. The multitude of cellular effects elicited by timely scaffold degradation makes it an important parameter to consider during hydrogel engineering.
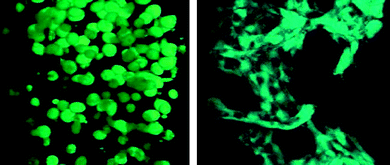 | ||
| Fig. 5 Osteogenesis of hMSCs encapsulated in degradable PEG hydrogels modified with signals. The cells were initially rounded with few intercellular interactions (left). With sufficient degradation over 2 weeks in vitro, there were enhanced cell-cell and cell-ECM interactions, leading to osteogenesis and tissue evolution (right). (Reprinted with permission from ref. 5, Copyright (2007) by The American Association for the Advancement of Science, USA). | ||
Also requiring investigation is the interaction between mechanical conditioning and other chemical and physical cues. Elisseeff's team recently reported that embryoid body-derived stem cells required pre-induction with TGF-β1 before they responded chondrogenically to mechanical compression, a dependence not observed for MSCs.182 For hESCs, strain actually inhibited differentiation and promoted self-renewal, but only when applied in concert with conditioned media.183 These findings suggest that biophysical effects might depend on cell biochemical profiles, which vary with cell line and stage of differentiation. Other groups demonstrated that mechanical loading could alter the degradation profile of scaffolds, and promote scaffold remodeling by cells, both of which affect the stability of pre-engineered constructs upon implantation into mechanically active tissues.184,185 These results again underscore the dynamism inherent in the stem cell microenvironment, and many interacting factors that must be taken into account when replicating this milieu in a synthetic hydrogel network.
5. Conclusion and future directions: engineering and exploring
An improved engineering toolbox, comprising new synthetic methods, new ways to pattern and provide controlled release of biochemical cues, and new ways of shaping hydrogel structure and mechanics, has allowed tissue engineers to transform previously biologically inert synthetic hydrogels into dynamic environments that can direct stem cell fate. The introduction of environmentally responsive materials revolutionized the field of therapeutics delivery two decades ago by localizing release to environments that provide appropriate stimuli. A parallel development has occurred in the field of tissue engineering with the creation of cell-responsive hydrogels. Salinas' recent work with PEG hydrogels that can temporally alter the presentation of RGD ligands in response to MSC chondrogenesis is very promising, as it heralds new designs that would allow the evolution of hydrogel physical and biochemical profiles in accordance with cellular feedback at various stages of differentiation.9,40,49 This would hopefully supersede traditional tissue engineering approaches in which cells and materials are treated as separate modalities to be used combinatorially rather than synergistically.While spatiotemporally controlled protein or ligand release is an important feature of many new hydrogel systems,11 the introduction of gene delivery strategies is poised to provide even greater guidance for differentiation. Potential problems with growth factor (GF) delivery—reduced bioactivity of recombinant or synthetic analogs, susceptibility to denaturation or degradation during hydrogel fabrication, the need to calibrate required concentrations—can be avoided by transfecting cells with genes for the relevant GFs.186 The genes can be physically or chemically incorporated into biodegradable hydrogels. The degradation rate of the hydrogel matrices controls the release rate of the genes.187 This approach holds great promise in cell differentiation.
The ability of hydrogels to promote host tissue responses is beneficial for tissue integration, but such influence is usually restricted to tissue-specific cells in the vicinity of the implant. MSCs are known to be mobilized to sites of injury in vivo,188 and recruiting these cells to the implant could potentially accelerate regeneration in a wider variety of tissues. It could also eliminate the need for cell delivery in regenerative medicine—and the associated cost and effort of procuring, seeding, culturing and differentiating the cells within hydrogels prior to implantation. Some of the chemical signals mediating stem cell homing have only recently been identified,188,189 and much more remains to be discovered about the regulatory networks behind this process, but the incorporation of such homing factors into PEG hydrogels (coupled with pro-angiogenic cues to facilitate their distribution) would open up new possibilities for hydrogel-based cell therapy.
Hitherto, our discussion has centered on ways of exploiting stem cell biology to engineer biomimetic PEG hydrogels, but these hydrogels could also form the basis of technological platforms to investigate stem cell differentiation. In fact, PEG synthetic chemistry lends itself well to scale-up operations for generating gel microarrays with controllable physical and biochemical properties. Such arrays then enable the multifactorial, high-throughput screening of niche factors that influence stem cell fate in 3D. Arrays formed from the photopolymerization of PEG macromers can be used to study the effects of network degradability and ligand presentation on hMSC viability;94 to probe how cell micro-organization affects stem cell behavior (when combined with dielectrophoresis to allow cell patterning);190–192 and to create embryoid bodies with controlled size and homogeneity for ESC differentiation studies.151 The incorporation of photolithographic techniques is expected to augment current testing options by enabling gradient patterning and temporal changes to gel properties.22 In other applications, Lutolf et al. developed gel microwells using Michael addition chemistry, and immobilized various intercellular adhesion molecules (e.g. N-cadherin) to the wells via micro-contact printing to study cell-cell interactions sans complicated co-culture systems.193 Such screening technologies using gel-based artificial niches should yield more information about the various aspects governing stem cell differentiation, which would in turn allow the design of more instructive hydrogel systems.
Another key consideration is how to maintain lineage commitment and differentiation of stem cells in vivo.3 It has been shown that in vitro predifferentiation may not guarantee stable commitment in an in vivo environment.194 Elisseeff et al. successfully induced a stable phenotype in PEG hydrogels for cartilage repair in vivo using either morphogenetic factors secreted from differentiated cells or co-culture techniques.30,154,156 However, it is important to understand how the differentiation of stem cells is determined and regulated in vivo. We believe that the symbiosis of biological research and technological development will eventually produce more efficient screening platforms to probe differentiation pathways under a simulated physiological environment, and the feedback thus produced as well as extensive in vivo evaluation would then propel the engineering of more advanced “designer PEG hydrogels” for controlled stem cell differentiation.
Acknowledgements
This work was funded by Institute of Bioengineering and Nanotechnology, Agency for Science, Technology and Research (A*STAR), Singapore and A*STAR-Singapore Stem Cell Consortium Grant No: SSCC-06-05 (to PLRE and YYY).References
- N. S. Hwang, S. Varghese and J. Elisseeff, Adv. Drug Delivery Rev., 2008, 60, 199–214 CrossRef CAS.
- B. C. Heng, T. Cao and E. H. Lee, Stem Cells, 2004, 22, 1152–1167 Search PubMed.
- M. Mimeault, R. Hauke and S. K. Batra, Clin. Pharmacol. Ther., 2007, 82, 252–264 CrossRef CAS.
- T. Ashan, A. M. Doyle and R. M. Nerem, in Principles of Regenerative Medicine, Academic Press, San Diego, 2008, pp. 28–47 Search PubMed.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS.
- G. D. Nicodemus and S. J. Bryant, Tissue Eng., Part B: Rev., 2008, 14, 149–165 Search PubMed.
- K. Brannvall, K. Bergman, U. Wallenquist, S. Svahn, T. Bowden, J. Hilborn and K. Forsberg-Nilsson, J. Neurosci. Res., 2007, 85, 2138–2146 CrossRef CAS.
- C. Chung and J. A. Burdick, Tissue Eng. A, 2009, 15, 243–254 Search PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS.
- D. L. Hern and J. A. Hubbell, J. Biomed. Mater. Res., 1998, 39, 266–276 CrossRef CAS.
- C. C. Lin and K. S. Anseth, Pharm. Res., 2009, 26, 631–643 CrossRef CAS.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2002, 54, 13–36 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37–51 CrossRef CAS.
- L. Yu, G. T. Chang, H. Zhang and J. D. Ding, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1122–1133 CrossRef CAS.
- L. Yu and J. D. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- L. Yu, H. Zhang and J. D. Ding, Angew. Chem., Int. Ed., 2006, 45, 2232–2235 CrossRef CAS.
- B. Jeong, K. M. Lee, A. Gutowska and Y. H. H. An, Biomacromolecules, 2002, 3, 865–868 CrossRef CAS.
- L. Yu, G. T. Chang, H. Zhang and J. D. Ding, Int. J. Pharm., 2008, 348, 95–106 CrossRef CAS.
- O. Wichterle and D. Lím, Nature, 1960, 185, 117–118 CrossRef.
- S. Kaihara, S. Matsumura and J. P. Fisher, Macromolecules, 2007, 40, 7625–7632 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS.
- H. Shin, P. Q. Ruhe, A. G. Mikos and J. A. Jansen, Biomaterials, 2003, 24, 3201–3211 CrossRef CAS.
- J. L. Ifkovits and J. A. Burdick, Tissue Eng., 2007, 13, 2369–2385 CrossRef CAS.
- P. X. Ma, J. H. Elisseeff, Scaffolding in Tissue Engineering, CRC Press, Florida, USA, 2005 Search PubMed.
- J. Elisseeff, K. Anseth, D. Sims, W. McIntosh, M. Randolph and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3104–3107 CrossRef CAS.
- J. L. Hill-West, S. M. Chowdhury, M. J. Slepian and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5967–5971 CrossRef CAS.
- D. S. W. Benoit, M. P. Schwartz, A. R. Durney and K. S. Anseth, Nat. Mater., 2008, 7, 816–823 CrossRef CAS.
- N. S. Hwang, M. S. Kim, S. Sampattavanich, J. H. Baek, Z. J. Zhang and J. Elisseeff, Stem Cells, 2006, 24, 284–291 Search PubMed.
- N. S. Hwang, S. Varghese, H. J. Lee, Z. J. Zhang, Z. H. Ye, J. Bae, L. Z. Cheng and J. Elisseeff, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20641–20646 CrossRef CAS.
- C. R. Nuttelman, D. S. W. Benoit, M. C. Tripodi and K. S. Anseth, Biomaterials, 2006, 27, 1377–1386 CrossRef CAS.
- F. Yang, C. G. Williams, D. Wang, H. Lee, P. N. Manson and J. Elisseeff, Biomaterials, 2005, 26, 5991–5998 CrossRef CAS.
- C. G. Williams, A. N. Malik, T. K. Kim, P. N. Manson and J. H. Elisseeff, Biomaterials, 2005, 26, 1211–1218 CrossRef CAS.
- N. E. Fedorovich, M. H. Oudshoorn, D. van Geemen, W. E. Hennink, J. Alblas and W. J. A. Dhert, Biomaterials, 2009, 30, 344–353 CrossRef CAS.
- S. F. Duan, W. Zhu, L. Yu and J. D. Ding, Chin. Sci. Bull., 2005, 50, 1093–1096 CrossRef CAS.
- D. L. Elbert, A. B. Pratt, M. P. Lutolf, S. Halstenberg and J. A. Hubbell, J. Controlled Release, 2001, 76, 11–25 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS.
- A. B. Pratt, F. E. Weber, H. G. Schmoekel, R. Muller and J. A. Hubbell, Biotechnol. Bioeng., 2004, 86, 27–36 CrossRef CAS.
- S. C. Rizzi, M. Ehrbar, S. Halstenberg, G. P. Raeber, H. G. Schmoekel, H. Hagenmuller, R. Muller, F. E. Weber and J. A. Hubbell, Biomacromolecules, 2006, 7, 3019–3029 CrossRef CAS.
- A. H. Zisch, M. P. Lutolf, M. Ehrbar, G. P. Raeber, S. C. Rizzi, N. Davies, H. Schmokel, D. Bezuidenhout, V. Djonov, P. Zilla and J. A. Hubbell, FASEB J., 2003, 17, 2260 CAS.
- G. A. Hudalla, T. S. Eng and W. L. Murphy, Biomacromolecules, 2008, 9, 842–849 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- T. P. Lodge, Macromolecules, 2009, 42, 3827–3829 CrossRef CAS.
- D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- S. Q. Liu, P. L. R. Ee, C. Y. Ke, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2009, 30, 1453–1461 CrossRef CAS.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- C. A. Deforest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- C. N. Salinas and K. S. Anseth, Biomaterials, 2008, 29, 2370–2377 CrossRef CAS.
- C. N. Salinas, B. B. Cole, A. M. Kasko and K. S. Anseth, Tissue Eng., 2007, 13, 1025–1034 CrossRef CAS.
- C. R. Nuttelman, M. A. Rice, A. E. Rydholm, C. N. Salinas, D. N. Shah and K. S. Anseth, Prog. Polym. Sci., 2008, 33, 167–179 CrossRef CAS.
- B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2009, 42, 211–217 CrossRef CAS.
- M. Ehrbar, S. C. Rizzi, R. G. Schoenmakers, B. S. Miguel, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomacromolecules, 2007, 8, 3000–3007 CrossRef CAS.
- T. J. Sanborn, P. B. Messersmith and A. E. Barron, Biomaterials, 2002, 23, 2703–2710 CrossRef CAS.
- J. J. Sperinde and L. G. Griffith, Macromolecules, 1997, 30, 5255–5264 CrossRef CAS.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- J. K. Kim, K. W. Lee, T. E. Hefferan, B. L. Currier, M. J. Yaszemski and L. C. Lu, Biomacromolecules, 2008, 9, 149–157 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- D. S. W. Benoit, A. R. Durney and K. S. Anseth, Tissue Eng., 2006, 12, 1663–1673 CrossRef CAS.
- M. A. Rice, J. Sanchez-Adams and K. S. Anseth, Biomacromolecules, 2006, 7, 1968–1975 CrossRef CAS.
- B. Wang, W. Zhu, Y. Zhang, Z. G. Yang and J. D. Ding, React. Funct. Polym., 2006, 66, 509–518 CrossRef CAS.
- Y. Zhang, W. Zhu, B. B. Wang and J. D. Ding, J. Controlled Release, 2005, 105, 260–268 CrossRef CAS.
- W. Zhu and J. D. Ding, J. Appl. Polym. Sci., 2006, 99, 2375–2383 CrossRef CAS.
- Y. Zhang, W. Zhu, B. B. Wang, L. Yu and J. D. Ding, J. Pharm. Sci., 2005, 94, 1676–1684 CrossRef CAS.
- X. He and E. Jabbari, Biomacromolecules, 2007, 8, 780–792 CrossRef CAS.
- E. Behravesh, K. Zygourakis and A. G. Mikos, J. Biomed. Mater. Res., 2003, 65a, 260–270 Search PubMed.
- E. E. Falco, M. Patel and J. P. Fisher, Pharm. Res., 2008, 25, 2348–2356 CrossRef CAS.
- Q. Li, J. Wang, S. Shahani, D. D. N. Sun, B. Sharma, J. H. Elisseeff and K. W. Leong, Biomaterials, 2006, 27, 1027–1034 CrossRef CAS.
- D. A. Wang, C. G. Williams, F. Yang, N. Cher, H. Lee and J. H. Elisseeff, Tissue Eng., 2005, 11, 201–213 CrossRef CAS.
- J. Z. Du, T. M. Sun, S. Q. Weng, X. S. Chen and J. Wang, Biomacromolecules, 2007, 8, 3375–3381 CrossRef CAS.
- J. A. Burdick, M. N. Mason, A. D. Hinman, K. Thorne and K. S. Anseth, J. Controlled Release, 2002, 83, 53–63 CrossRef CAS.
- M. J. Mahoney and K. S. Anseth, Biomaterials, 2006, 27, 2265–2274 CrossRef CAS.
- M. A. Rice and K. S. Anseth, Tissue Eng., 2007, 13, 683–691 CrossRef CAS.
- L. M. Weber, K. N. Hayda, K. Haskins and K. S. Anseth, Biomaterials, 2007, 28, 3004–3011 CrossRef CAS.
- H. Park, X. Guo, J. S. Temenoff, Y. Tabata, A. I. Caplan, F. K. Kasper and A. G. Mikos, Biomacromolecules, 2009, 10, 541–546 CrossRef CAS.
- H. Park, J. S. Temenoff, Y. Tabata, A. I. Caplan and A. G. Mikos, Biomaterials, 2007, 28, 3217–3227 CrossRef CAS.
- A. K. Shung, E. Behravesh, S. Jo and A. G. Mikos, Tissue Eng., 2003, 9, 243–254 CrossRef CAS.
- X. He, J. Ma and E. Jabbari, Langmuir, 2008, 24, 12508–12516 CrossRef CAS.
- M. W. Betz, J. F. Caccamese, D. P. Coletti, J. J. Sauk and J. P. Fisher, J. Biomed. Mater. Res. A, 2008, 90A, 819–829.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- J. L. West and J. A. Hubbell, Macromolecules, 1999, 32, 241–244 CrossRef CAS.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS.
- M. P. Lutolf, G. P. Raeber, A. H. Zisch, N. Tirelli and J. A. Hubbell, Adv. Mater., 2003, 15, 888–892 CrossRef CAS.
- M. R. Lutolf, F. E. Weber, H. G. Schmoekel, J. C. Schense, T. Kohler, R. Muller and J. A. Hubbell, Nat. Biotechnol., 2003, 21, 513–518 CrossRef CAS.
- J. A. Burdick and K. S. Anseth, Biomaterials, 2002, 23, 4315–4323 CrossRef CAS.
- N. S. Hwang, S. Varghese, Z. Zhang and J. Elisseeff, Tissue Eng., 2006, 12, 2695–2706 CrossRef CAS.
- C. R. Nuttelman, M. C. Tripodi and K. S. Anseth, Matrix Biol., 2005, 24, 208–218 CrossRef CAS.
- C. N. Salinas and K. S. Anseth, Macromolecules, 2008, 41, 6019–6026 CrossRef CAS.
- C. N. Salinas and K. S. Anseth, J. Tissue Eng. Regener. Med., 2008, 2, 296–304 Search PubMed.
- M. S. Hahn, L. J. Taite, J. J. Moon, M. C. Rowland, K. A. Ruffino and J. L. West, Biomaterials, 2006, 27, 2519–2524 CrossRef CAS.
- S. H. Lee, J. J. Moon and J. L. West, Biomaterials, 2008, 29, 2962–2968 CrossRef CAS.
- B. D. Polizzotti, B. D. Fairbanks and K. S. Anseth, Biomacromolecules, 2008, 9, 1084–1087 CrossRef CAS.
- M. M. Martino, M. Mochizuki, D. A. Rothenfluh, S. A. Rempel, J. A. Hubbell and T. H. Barker, Biomaterials, 2009, 30, 1089–1097 CrossRef CAS.
- L. Jongpaiboonkit, W. J. King and W. L. Murphy, Tissue Eng. A, 2009, 15, 343–353 Search PubMed.
- T. P. Kraehenbuehl, P. Zammaretti, A. J. Van der Vlies, R. G. Schoenmakers, M. P. Lutolf, M. E. Jaconi and J. A. Hubbell, Biomaterials, 2008, 29, 2757–2766 CrossRef CAS.
- Y. Park, M. P. Lutolf, J. A. Hubbell, E. B. Hunziker and M. Wong, Tissue Eng., 2004, 10, 515–522 CrossRef CAS.
- S. Halstenberg, A. Panitch, S. Rizzi, H. Hall and J. A. Hubbell, Biomacromolecules, 2002, 3, 710–723 CrossRef CAS.
- H. J. Lee, C. Yu, T. Chansakul, N. S. Hwang, S. Varghese, S. M. Yu and J. H. Elisseeff, Tissue Eng. A, 2008, 14, 1843–1851 Search PubMed.
- C. N. Salinas and K. S. Anseth, J. Biomed. Mater. Res. A, 2009, 90, 456–464 CrossRef.
- S. E. Grieshaber, A. J. E. Farran, S. Lin-Gibson, K. L. Kiick and X. Jia, Macromolecules, 2009, 42, 2532–2541 CrossRef CAS.
- A. S. Sarvestani, X. He and E. Jabbari, Biopolymers, 2007, 85, 370–378 CrossRef CAS.
- D. S. Benoit, C. R. Nuttelman, S. D. Collins and K. S. Anseth, Biomaterials, 2006, 27, 6102–6110 CrossRef CAS.
- C. R. Nuttelman, M. C. Tripodi and K. S. Anseth, J. Biomed. Mater. Res. A, 2006, 76, 183–195 CrossRef.
- E. Fuchs, T. Tumbar and G. Guasch, Cell, 2004, 116, 769–778 CrossRef CAS.
- F. M. Watt and B. L. Hogan, Science, 2000, 287, 1427–1430 CrossRef CAS.
- C. M. Kolf, E. Cho and R. S. Tuan, Arthritis Res. Ther., 2007, 9, 204 CrossRef.
- L. S. Ferreira, S. Gerecht, J. Fuller, H. F. Shieh, G. Vunjak-Novakovic and R. Langer, Biomaterials, 2007, 28, 2706–2717 CrossRef CAS.
- J. K. Tessmar and A. M. Gopferich, Adv. Drug Delivery Rev., 2007, 59, 274–291 CrossRef CAS.
- D. S. Benoit and K. S. Anseth, Acta Biomater., 2005, 1, 461–470 CrossRef.
- I. D. Dinbergs, L. Brown and E. R. Edelman, J. Biol. Chem., 1996, 271, 29822–29829 CrossRef CAS.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143–147 CrossRef CAS.
- N. S. Hwang, S. Varghese, P. Theprungsirikul, A. Canver and J. Elisseeff, Biomaterials, 2006, 27, 6015–6023 CrossRef CAS.
- H. Wichterle, I. Lieberam, J. A. Porter and T. M. Jessell, Cell, 2002, 110, 385–397 CrossRef CAS.
- A. Burlacu, A. M. Rosca, H. Maniu, I. Titorencu, E. Dragan, V. Jinga and M. Simionescu, Eur. J. Cell Biol., 2008, 87, 173–184 CrossRef CAS.
- S. C. Choi, J. Yoon, W. J. Shim, Y. M. Ro and D. S. Lim, Exp. Mol. Med., 2004, 36, 515–523 Search PubMed.
- S. Ding and P. G. Schultz, Nat. Biotechnol., 2004, 22, 833–840 CrossRef.
- S. S. Chen, W. Fitzgerald, J. Zimmerberg, H. K. Kleinman and L. Margolis, Stem Cells, 2007, 25, 553–561 Search PubMed.
- J. Kim, I. S. Kim, T. H. Cho, K. B. Lee, S. J. Hwang, G. Tae, I. Noh, S. H. Lee, Y. Park and K. Sun, Biomaterials, 2007, 28, 1830–1837 CrossRef CAS.
- S. Gerecht, J. A. Burdick, L. S. Ferreira, S. A. Townsend, R. Langer and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11298–11303 CrossRef CAS.
- L. Y. Lum, N. L. Cher, C. G. Williams and J. H. Elisseeff, IEEE Eng. Med. Biol. Mag., 2003, 22, 71–76 CrossRef.
- D. Philip, S. S. Chen, W. Fitzgerald, J. Orenstein, L. Margolis and H. K. Kleinman, Stem Cells, 2005, 23, 288–296 Search PubMed.
- S. Varghese, N. S. Hwang, A. C. Canver, P. Theprungsirikul, D. W. Lin and J. Elisseeff, Matrix Biol., 2008, 27, 12–21 CrossRef CAS.
- D. A. Wang, S. Varghese, B. Sharma, I. Strehin, S. Fermanian, J. Gorham, D. H. Fairbrother, B. Cascio and J. H. Elisseeff, Nat. Mater., 2007, 6, 385–392 CrossRef CAS.
- P. Buma, J. S. Pieper, T. van Tienen, J. L. van Susante, P. M. van der Kraan, J. H. Veerkamp, W. B. van den Berg, R. P. Veth and T. H. van Kuppevelt, Biomaterials, 2003, 24, 3255–3263 CrossRef CAS.
- D. N. Shah, S. M. Recktenwall-Work and K. S. Anseth, Biomaterials, 2008, 29, 2060–2072 CrossRef CAS.
- Y. Liu, X. Z. Shu and G. D. Prestwich, Tissue Eng., 2006, 12, 3405–3416 CrossRef CAS.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS.
- J. A. Hubbell, Bio/Technology, 1995, 13, 565–576 CrossRef CAS.
- C. R. Nuttelman, M. C. Tripodi and K. S. Anseth, Matrix Biol., 2005, 24, 208–218 CrossRef CAS.
- C. H. Streuli, J. Cell Sci., 2009, 122, 171–177 CrossRef CAS.
- M. P. Ruiz-Torres, G. Perez-Rivero, M. L. Diez-Marques, M. Griera, R. Ortega, M. Rodriguez-Puyol and D. Rodriguez-Puyol, Int. J. Biochem. Cell Biol., 2007, 39, 133–145 CrossRef CAS.
- R. Ortega-Velazquez, M. L. Diez-Marques, M. P. Ruiz-Torres, M. Gonzalez-Rubio, M. Rodriguez-Puyol and D. Rodriguez Puyol, FASEB J., 2003, 17, 1529–1531 CAS.
- A. Gigout, M. Jolicoeur, M. Nelea, N. Raynal, R. Farndale and M. D. Buschmann, J. Biol. Chem., 2008, 283, 31522–31530 CrossRef CAS.
- A. J. Garcia and C. D. Reyes, J. Dent. Res., 2005, 84, 407–413 CrossRef CAS.
- E. A. Cavalcanti-Adam, D. Aydin, V. C. Hirschfeld-Warneken and J. P. Spatz, HFSP J., 2008, 2, 276–285 CrossRef CAS.
- M. Dettin, M. T. Conconi, R. Gambaretto, A. Pasquato, M. Folin, C. Di Bello and P. P. Parnigotto, J. Biomed. Mater. Res., 2002, 60, 466–471 CrossRef CAS.
- M. Kreiner, Z. Li, J. Beattie, S. M. Kelly, H. J. Mardon and C. F. van der Walle, Protein Eng., Des. Sel., 2008, 21, 553–560 CrossRef CAS.
- S. Lucie, G. Elisabeth, F. Stephanie, S. Guy, H. Amandine, A. R. Corinne, B. Didier, S. Catherine, G. Alexei, D. Pascal and C. Jean-Luc, Mol. Ther., 2009, 17, 837–843 CrossRef CAS.
- G. Thumshirn, U. Hersel, S. L. Goodman and H. Kessler, Chem.–Eur. J., 2003, 9, 2717–2725 CrossRef.
- M. C. Porte-Durrieu, F. Guillemot, S. Pallu, C. Labrugere, B. Brouillaud, R. Bareille, J. Amedee, N. Barthe, M. Dard and C. Baquey, Biomaterials, 2004, 25, 4837–4846 CrossRef CAS.
- S. V. Graeter, J. H. Huang, N. Perschmann, M. Lopez-Garcia, H. Kessler, J. D. Ding and J. P. Spatz, Nano Lett., 2007, 7, 1413–1418 CrossRef CAS.
- J. H. Huang, S. V. Grater, F. Corbellinl, S. Rinck, E. Bock, R. Kemkemer, H. Kessler, J. D. Ding and J. P. Spatz, Nano Lett., 2009, 9, 1111–1116 CrossRef CAS.
- L. Perlin, S. MacNeil and S. Rimmer, Soft Matter, 2008, 4, 2331–2349 RSC.
- S. X. Hsiong, T. Boontheekul, N. Huebsch and D. J. Mooney, Tissue Eng., Part A, 2008, 15, 263–272 Search PubMed.
- D. S. Benoit and K. S. Anseth, Biomaterials, 2005, 26, 5209–5220 CrossRef CAS.
- S. M. Cutler and A. J. Garcia, Biomaterials, 2003, 24, 1759–1770 CrossRef CAS.
- T. A. Petrie, J. R. Capadona, C. D. Reyes and A. J. Garcia, Biomaterials, 2006, 27, 5459–5470 CrossRef CAS.
- S. Tavella, G. Bellese, P. Castagnola, I. Martin, D. Piccini, R. Doliana, A. Colombatti, R. Cancedda and C. Tacchetti, J. Cell Sci., 1997, 110(Pt 18), 2261–2270 CAS.
- J. M. Messana, N. S. Hwang, J. Coburn, J. H. Elisseeff and Z. Zhang, J. Tissue Eng. Regener. Med., 2008, 2, 499–506 Search PubMed.
- J. M. Karp, J. Yeh, G. Eng, J. Fukuda, J. Blumling, K. Y. Suh, J. Cheng, A. Mahdavi, J. Borenstein, R. Langer and A. Khademhosseini, Lab Chip, 2007, 7, 786–794 RSC.
- H. C. Moeller, M. K. Mian, S. Shrivastava, B. G. Chung and A. Khademhosseini, Biomaterials, 2008, 29, 752–763 CrossRef CAS.
- A. M. DeLise and R. S. Tuan, J. Cell. Biochem., 2002, 87, 342–359 CrossRef CAS.
- R. N. Moore, A. Dasgupta, N. Rajaei, M. L. Yarmush, M. Toner, L. Larue and P. V. Moghe, Biotechnol. Bioeng., 2008, 101, 1332–1343 CrossRef CAS.
- N. S. Hwang, S. Varghese and J. Elisseeff, PLoS One, 2008, 3, e2498 CrossRef.
- K. Bentz, M. Molcanyi, S. Hess, A. Schneider, J. Hescheler, E. Neugebauer and U. Schaefer, Cell. Physiol. Biochem., 2006, 18, 275–286 CrossRef CAS.
- N. S. Hwang, S. Varghese, C. Puleo, Z. Zhang and J. Elisseeff, J. Cell. Physiol., 2007, 212, 281–284 CrossRef CAS.
- H. J. Lee, C. Yu, T. Chansakul, S. Varghese, N. S. Hwang and J. H. Elisseeff, Stem Cells Dev., 2008, 17, 555–563 CrossRef CAS.
- S. Eshghi and D. V. Schaffer, Engineering microenvironments to control stem cell fate and function, in StemBook, ed. T.S.C.R. Community, 2008 Search PubMed.
- W. P. Daley, S. B. Peters and M. Larsen, J. Cell Sci., 2008, 121, 255–264 CrossRef CAS.
- D. M. Cohen and C. S. Chen, Mechanical control of stem cell differentiation, in StemBook, ed. T.S.C.R. Community, 2008 Search PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- A. S. Rowlands, P. A. George and J. J. Cooper-White, Am. J. Physiol.: Cell Physiol., 2008, 295, C1037–C1044 CrossRef CAS.
- Y. S. Pek, A. C. Wan, A. Shekaran, L. Zhuo and J. Y. Ying, Nat. Nanotechnol., 2008, 3, 671–675 CrossRef CAS.
- E. Cukierman, R. Pankov, D. R. Stevens and K. M. Yamada, Science, 2001, 294, 1708–1712 CrossRef CAS.
- M. H. Zaman, L. M. Trapani, A. L. Sieminski, D. Mackellar, H. Gong, R. D. Kamm, A. Wells, D. A. Lauffenburger and P. Matsudaira, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10889–10894 CrossRef CAS.
- S. J. Bryant and K. S. Anseth, J. Biomed. Mater. Res., 2002, 59, 63–72 CrossRef CAS.
- K. Saha, A. J. Keung, E. F. Irwin, Y. Li, L. Little, D. V. Schaffer and K. E. Healy, Biophys. J., 2008, 95, 4426–4438 CrossRef CAS.
- D. Dikovsky, H. Bianco-Peled and D. Seliktar, Biophys. J., 2008, 94, 2914–2925 CrossRef CAS.
- S. A. Zawko, S. Suri, Q. Truong and C. E. Schmidt, Acta Biomater., 2009, 5, 14–22 CrossRef CAS.
- R. M. Namba, A. A. Cole, K. B. Bjugstad and M. J. Mahoney, Acta Biomater, 2009 Search PubMed.
- M. S. Stosich, E. K. Moioli, J. K. Wu, C. H. Lee, C. Rohde, A. M. Yoursef, J. Ascherman, R. Diraddo, N. W. Marion and J. J. Mao, Methods, 2009, 47, 116–121 CrossRef CAS.
- J. Miller and J. West, in Bio-Materials and Prototyping Applications in Medicine, eds. P. Bartolo and B. Bidanda, Springer, US, 2007, pp. 49–65 Search PubMed.
- Y. Lu, G. Mapili, G. Suhali, S. Chen and K. Roy, J. Biomed. Mater. Res. A, 2006, 77, 396–405 CrossRef.
- P. S. Maher, R. P. Keatch, K. Donnelly, R. E. Mackay and J. Z. Paxton, Rapid Prototyping J., 2009, 15, 204–210 CrossRef.
- J. H. Wang and B. P. Thampatty, Int. Rev. Cell Mol. Biol., 2008, 271, 301–346 Search PubMed.
- C. K. Kuo and R. S. Tuan, Tissue Eng. A, 2008, 14, 1615–1627 Search PubMed.
- C. Y. Huang, K. L. Hagar, L. E. Frost, Y. Sun and H. S. Cheung, Stem Cells, 2004, 22, 313–323 Search PubMed.
- H. Huang, Y. Nakayama, K. Qin, K. Yamamoto, J. Ando, J. Yamashita, H. Itoh, K. Kanda, H. Yaku, Y. Okamoto and Y. Nemoto, J. Artif. Organs, 2005, 8, 110–118 Search PubMed.
- B. T. Estes, J. M. Gimble and F. Guilak, Curr. Top. Dev. Biol., 2004, 60, 91–126 CAS.
- V. Terraciano, N. Hwang, L. Moroni, H. B. Park, Z. Zhang, J. Mizrahi, D. Seliktar and J. Elisseeff, Stem Cells, 2007, 25, 2730–2738 Search PubMed.
- S. Saha, L. Ji, J. J. de Pablo and S. P. Palecek, J. Cell. Physiol., 2006, 206, 126–137 CrossRef CAS.
- E. J. Lee, J. W. Holmes and K. D. Costa, Ann. Biomed. Eng., 2008, 36, 1322–1334 CrossRef.
- G. D. Nicodemus, K. A. Shiplet, S. R. Kaltz and S. J. Bryant, Biotechnol. Bioeng., 2009, 102, 948–959 CrossRef CAS.
- H. Storrie and D. J. Mooney, Adv. Drug Delivery Rev., 2006, 58, 500–514 CrossRef CAS.
- Y. Lei and T. Segura, Biomaterials, 2009, 30, 254–265 CrossRef CAS.
- H. Kawada, J. Fujita, K. Kinjo, Y. Matsuzaki, M. Tsuma, H. Miyatake, Y. Muguruma, K. Tsuboi, Y. Itabashi, Y. Ikeda, S. Ogawa, H. Okano, T. Hotta, K. Ando and K. Fukuda, Blood, 2004, 104, 3581–3587 CrossRef CAS.
- H. S. Hong, J. Lee, E. Lee, Y. S. Kwon, W. Ahn, M. H. Jiang, J. C. Kim and Y. Son, Nat. Med., 2009, 15, 425–435 CrossRef CAS.
- D. R. Albrecht, V. L. Tsang, R. L. Sah and S. N. Bhatia, Lab Chip, 2005, 5, 111–118 RSC.
- D. R. Albrecht, G. H. Underhill, T. B. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–375 CrossRef CAS.
- G. H. Underhill, A. A. Chen, D. R. Albrecht and S. N. Bhatia, Biomaterials, 2007, 28, 256–270 CrossRef CAS.
- M. P. Lutolf, R. Doyonnas, K. Havenstrite, K. Koleckar and H. M. Blau, Integr. Biol., 2009, 1, 59–69 RSC.
- K. Pelttari, A. Winter, E. Steck, K. Goetzke, T. Hennig, B. G. Ochs, T. Aigner and W. Richter, Arthritis Rheum., 2006, 54, 3254–3266 CrossRef CAS.
Footnote |
| † These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2010 |