DNA fluorocode: A single molecule, optical map of DNA with nanometre resolution†
Robert K.
Neely
*a,
Peter
Dedecker
a,
Jun-ichi
Hotta
a,
Giedrė
Urbanavičiūtė
b,
Saulius
Klimašauskas
b and
Johan
Hofkens
a
aDepartement Chemie, Katholieke Universiteit Leuven, Celestijnenlaan 200F, B-3001, Heverlee, Belgium. E-mail: Robert.neely@chem.kuleuven.be; Fax: +0032 (0)16 327990; Tel: +0032 (0)16 327399
bInstitute of Biotechnology, V.A. Graiciuno 8, LT-02241, Vilnius, Lithuania
First published on 11th August 2010
Abstract
We present a new method for single-molecule optical DNA mapping using an exceptionally dense, yet sequence-specific coverage of DNA with a fluorescent probe. The method employs a DNA methyltransferase enzyme to direct the DNA labelling, followed by molecular combing of the DNA onto a polymer-coated surface and subsequent sub-diffraction limit localization of the fluorophores. The result is a ‘DNA fluorocode’; a simple description of the DNA sequence, with a maximum achievable resolution of less than 20 bases, which can be read and analyzed like a barcode. We demonstrate the generation of a fluorocode for genomic DNA from the lambda bacteriophage using a DNA methyltransferase, M.HhaI, to direct fluorescent labels to four-base sequences reading 5′-GCGC-3′. A consensus fluorocode that allows the study of the DNA sequence at the level of an individual labelling site can be generated from a handful of molecules.
Introduction
The DNA sequencing of individual genomes is rapidly becoming a reality. Recent developments in single molecule sequencing allow the analysis of an individual genome in a timeframe of around one week.1 Such methods employ massively parallel DNA sequencing strategies, which sequence short regions of the genome, from 302 up to 15003 bases in length and follow this with the assembly of the genome from these fragments. In principle, the approach is a simple and incredibly effective one, yet it has one significant flaw and this occurs where the DNA sequence repeats with a length that is greater than the size of the sequenced fragments. In such a case the linear assembly of the genome becomes ambiguous.Such repeats of the DNA sequence are surprisingly common. Known as copy number variations (CNVs), they are measured relative to a reference genome4 and are of greater than 1 kilobase in length5 and can reach lengths of several megabases. On a study of the genomes of 270 individuals, copy number variable regions were found to cover a total of 360 megabases, or approximately 12% of the human genome.5 They have been implicated in a variety of genetic disorders including schizophrenia6 and congenital heart defects.7 Repeats can be detected using third-generation sequencing methods1 but these techniques represent a rather labour and material-intensive route to studying CNVs. Further, given the variable number of copies that may be present and the hugely variable length of these repeats, the suitability of parallel sequencing methods for studying copy number variations is debatable.
Optical mapping of DNA is a complementary technique to DNA sequencing and in principle it provides a simple and intuitive route to visualize the sequence of a DNA molecule, typically on the scale of kilo- to mega-bases.8 Such mapping is critical to validate the assembly of short DNA sequence reads, particularly in complex and repetitive genomes.9 Optical mapping utilizes molecular combing10 in order to linearly align large DNA molecules on a surface, allowing for their subsequent imaging and the linear positioning of, for example, restriction enzyme sites along the DNA. Indeed, optical mapping using restriction enzymes has been pioneered by the Schwartz lab11,12 and the technique has been critical in validating the final versions of many genomes.13,14 Typically, it utilizes restriction enzymes that recognize 6- or 8-base sequences, giving a cleavage site on average every ∼4 kilobases or ∼65 kilobases, respectively (though these figures vary significantly depending on the genome).
‘DNA barcodes’ offer an alternative strategy to optical restriction mapping that also yields a genomic-scale map of the DNA sequence. These methods use sequence-specific fluorescent labelling of DNA and have the potential to be combined with sub-diffraction limit imaging techniques to significantly improve on the resolution that results from restriction mapping. Yet no study to date has been able to successfully combine both the sequence-specificity of restriction mapping and sub-diffraction limit positioning of fluorescent probes. Gad et al.15 have reported DNA barcodes for the BRCA1 and BRCA2 genes, variations in which are known to increase susceptibility to breast cancer. Using fluorescent antibodies the detection of a large deletion (∼24 kb) in the BRCA1 gene at the single molecule level is readily achieved. The sub-diffraction-limit positioning of fluorophores on DNA has previously been achieved by Qu et al.16 who used 7-base-long bis-PNA molecules that bind to DNA. However, the binding of the bis-PNA molecules was found to be rather non-specific. Sequence specific fluorescent labelling of DNA is achievable using ‘nick-translation’.17 In combination with molecular combing, nick-translation has been used to produce DNA barcodes using standard optical microscopy.18 DNA nicking enzymes produce single-strand breaks in DNA with high sequence specificity (typically at sequences 6 bases in length). The breaks can be labelled using a DNA polymerase enzyme and a fluorescently labelled nucleotide. Furthermore, using this approach DNA molecules have been mapped as they are driven through ‘nanoslits’ by an electric potential.19 In such a high-throughput format, fluorophore positions were determined with a standard deviation of around 3.5 kb.
We report a significant advance on the current state-of-the-art in optical DNA mapping by using a DNA methyltransferase to label the DNA at sequences reading 5′-GCGC-3′. The unique and reproducible pattern produced by this labelling, in combination with the high labelling density and sub-diffraction-limit localization of the fluorophores, enables identification of elements of the DNA at the level of single genes.
Results and discussion
We present a method to produce what we term a DNA fluorocode (since we find the use of ‘DNA barcode’ rather conflicts with the more common, taxonomic use of this term); an optical map of DNA derived from the observation of one or more DNA molecules that are sequence-specifically labelled using a DNA methyltransferase enzyme, and stretched onto a polymer-coated surface.Sequence-specific fluorescent labelling of DNA
In order to generate sequence-specifically labelled DNA with an exceptionally high labelling density, we employed the previously reported ‘methyltransferase-directed transfer of activated groups’ (mTAG) method.22 The reaction results in a covalent modification of DNA at target locations determined by the specificity of the DNA methyltransferase enzyme. The density of labelling is tuneable, depending on the methyltransferase enzyme used to carry out the mTAG reaction, but can far exceed that achievable using either nick-translation, PCR-based methods or non-covalent methods of sequence-specific labelling, such as triple helix formation.Fluorescent labelling using mTAG is a simple two-step procedure. The first step is a DNA methyltransferase-catalyzed covalent attachment of a linear side chain with a terminal amino group to the DNA. This reaction occurs upon incubation of the DNA along with a DNA methyltransferase and a modified methyltransferase cofactor (see Supplementary Fig. 1†), which is synthetically prepared.22 We employed an engineered version of the HhaI DNA methyltransferase enzyme (M.HhaI)23, which recognizes the four-base sequence 5’-GCGC-3′ and targets the italicised cytosine for modification at the C5-position to direct the fluorescent labelling of genomic DNA from the lambda bacteriophage. DNA methyltransferases, which typically work with these modified cofactors as wild-type enzymes or sterically engineered variants,20,21 offer a broad range of recognition site specificities.24 Hence, sequence coverage can be tailored to suit the DNA molecule and problem of interest. The resulting ‘derivatized DNA’ can be fluorescently labelled by incubation with a standard, commercially available amine-reactive fluorophore (succinimidyl ester). For this, we used the highly photostable dye, Atto647N.
There are a total of 215 target sites for HhaI on the 48.5 kilobases of the lambda phage genome, which have a distinctive distribution along the molecule, as indicated in Fig. 1. 149 HhaI sites lie between base 1 and 22500, a ∼5000 base gap defines the central region of the lambda DNA molecule and a less densely labelled region, from 27500 bases to the end of the molecule contains the remaining 66 HhaI sites. Fig. 1 simulates the appearance of an ideal lambda DNA molecule, that is uniformly stretched and labelled at every HhaI site on the molecule, under the microscope.
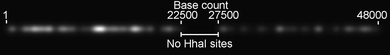 | ||
| Fig. 1 Generated image for a simulated lambda phage DNA. Each fluorophore position is displayed with a (Gaussian) point spread function that has a full-width half maximum (FWHM) of 305 nm, the expected size of a diffraction-limited spot for a single molecule emitting at 700 nm. The molecule is shown with a step between base pairs of 3.4 Å and has a length of 16.5 μm. | ||
Combing the labelled DNA
Lambda DNA molecules labelled at HhaI sites with Atto647N, were stretched onto a PMMA-coated surface using an evaporating droplet.25–27 This method gives reproducible stretching using small sample volumes. To form the droplet, we use 1 μL of solution containing ∼10 pM Atto647N-labelled lambda DNA and deposit this onto a PMMA-coated coverslip. The droplet is left uncovered and allowed to evaporate gradually. The stretching of single DNA molecules was readily visualized on the microscope, as shown in Fig. 2 and in Supplementary Video 1.† We favoured the use of the PMMA-coated surface for these experiments, since the great majority of the DNA molecules are deposited as single and linearly stretched molecules on this surface. Similar experiments on a silanized surface, for example, resulted in the deposition of DNA aggregates and molecules with complex topologies (data not shown), relative to those deposited on PMMA.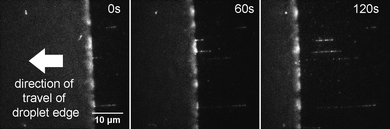 | ||
| Fig. 2 DNA combing using an evaporating droplet. Stills taken from a movie of DNA combing (see Supplementary Video 1†). Exposure time is 1 s and each frame is 41.5 μm by 41.5 μm in size. DNA molecules that are adsorbed to the surface in the early frames of the movie are swept away by the receding edge of the droplet. Deposition occurs at the air–water interface, which is clearly seen in the movie because of the bright but blurred fluorescence intensity from several DNA molecules that are rapidly diffusing there. DNA molecules are combed and stretched to around 1.6× their crystallographic length. | ||
Visualization and localization of fluorophores
The DNA molecules were visualized using a standard wide-field fluorescence microscope. In order to determine the position of each of the fluorophores along the DNA molecule we must first isolate individual chromophores and then fit a 2-dimensional Gaussian profile to the observed diffraction-limited spots in the experimental data.28,29 Doing so enables us to localize any given fluorophore with sub-diffraction-limit precision. Indeed, we found that, by manually fitting the position of a single fluorophore over 20 subsequent frames of a movie, the distribution of localized positions has a standard deviation of just 9.1 nm (this equates to 17 base pairs, where the step between pairs is 5.51 Å due to the overstretching of the DNA). Hence, a measurement between two localized fluorophores is possible, in principle, with a standard deviation of just 12.9 nm (simply derived from the square root of the sum of the squares of the error in fitting an individual fluorophore).In the context of the densely labelled DNA molecule, sub-diffraction-limit localization of a fluorophore necessitates the isolation and identification of the emission from individual fluorophores on the DNA. One established approach to enable this is the dSTORM30–32 technique, which utilizes on/off switching in organic fluorophores to ensure that single emitters can be readily isolated and their positions accurately determined. Whilst our labelling approach allows the use of this technique in principle, in practice we found that the DNA immediately dissociated from the surface upon addition of a solution (used to enable the on/off switching in dSTORM experiments) to the sample. Hence, we used an approach which utilizes the single-step photobleaching of individual fluorophores as a means to identify and localize them16,33 (Supplementary Figure S2†). This approach enables the use of a wide range of fluorophores for these experiments and does not require the use of an imaging buffer. Movies of the photobleaching of the labels on single DNA molecules were recorded, typically using a relatively long exposure time (i.e. 0.3 s) and low excitation power in order to minimize the effect of fluorophore blinking on our analysis. Fig. 3 shows the result of one such analysis. Following localization of each of the fluorophores on a DNA molecule, a line is projected along the molecule and the distance of each fluorophore along this line is determined, as shown in Fig. 3C. 20 individual DNA molecules were analyzed in this way. Molecules were selected for analysis where the labelling was sufficient that it was clear that the DNA molecule was approximately full length and where the DNA-strand was not obviously composed of more than one molecule. The number of localized fluorophores on a single DNA molecule was found to vary between 64 and 109 with a mean of 87 fluorophores.
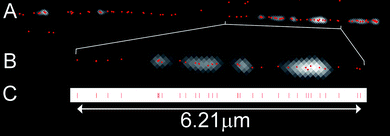 | ||
| Fig. 3 Positioning of the fluorophores. A) Image showing a single lambda DNA molecule. The average intensity image of the DNA taken from the movie is overlaid with the calculated fluorophore positions (red spots). B) Enlarged region of the DNA molecule in A. One pixel is 81 nm (∼150 bases). C) Positions from the DNA molecule in B projected onto a line. | ||
By convoluting each of the fitted points with a (Gaussian) point-spread function (PSF) with a full-width half maximum of 305 nm we can directly compare the fit to the raw experimental data. Fig. 4 shows the generated fit for one such molecule, along with the first frame from the movie and an image based on the average intensity of the emission over the entire movie.
 | ||
| Fig. 4 A comparison of the fluorocode to the raw data. A) Image taken from the first frame from the recorded photobleaching movie. B) An average image from all of the frames of the movie and (C) The DNA fluorocode, where each localized fluorophore is shown with a PSF with a FWHM of 305 nm. | ||
The high experimental resolution and sequence-specific labelling reveal heterogeneity in the stretching of the DNA molecules and deviations in the path described by the DNA molecules on the PMMA surface (for example, as shown in Fig. 3). This has important consequences for our measurements, since we ultimately want to know to which base a given fluorophore is attached. In fact, the error in determining the labelling site on the DNA can be significantly greater than the error in fitting its absolute position in the field of view. In order to estimate the error in our measurements along the DNA molecule we measured the observed gap between the fluorophores at the centre of the 20 DNA molecules we measured. Here, we find a standard deviation in the measurement of this ∼5000 base gap of 190 bases; approximately a 4% standard deviation in the distance measurement. This level of precision is unprecedented in any optical mapping study and, as we will show, allows the unambiguous alignment of single DNA molecules to a reference sequence.
DNA mapping using the fluorocode
Although we derive a discrete position (point) for each localized fluorophore, we choose to convolute this data with a point spread function in order to indicate the experimental uncertainty. The use of a PSF gives a defined probability distribution for the fluorophore position, regardless of the scale upon which the DNA molecule is displayed. Furthermore, fluorophore positions are also mapped with a brightness that indicates the number of fluorophores present at a given position. We term this display a fluorocode, and generate them for each of the DNA molecules by convoluting the localized fluorophores with a PSF of 42 nm (76 base pairs). We derive this error from the error in the fitting (10 nm) combined with the error resulting from inhomogeneous stretching of the DNA (based on an average distance of approximately 700 bases between fluorophores and a 4% error in the measurement of this distance). The image is stretched 5-fold perpendicular to the axis of the DNA to improve the clarity of the fluorocode, which is somewhat difficult to inspect and intuitively align as a series of small dots.In order to translate localized positions into labelling sites on the DNA molecule the experimental data is compared to a reference map of the known HhaI sites on lambda DNA34 (referred to as the “HhaI map”, henceforth). This is achieved by comparison of the intensity profiles of the two fluorocodes (experiment and HhaI map) and uses a simple convolution of the two profiles, stretching and shifting them relative to one another in order to maximize their overlap. We aligned 20 lambda DNA molecules in this way, with the result shown in Fig. 5A. The determined stretching factors (detailed in Supplementary Table 1†) vary between 1.50 and 1.67 with an average value of 1.62 implying an average step between base pairs of 0.55 nm for the combed DNA. Direct comparison between the experimental data and the HhaI map allows some quantification of the quality of the fluorocode as an optical map. For each DNA molecule, experimentally determined labelling sites were matched with the closest site (within 200 bases) on the HhaI map. On average 74 of the 87 (85%) fluorophores on a single DNA molecule were matched with a mean standard deviation of 71 bases (39 nm) between the fitted positions and their closest match on the HhaI map. By comparison, optical restriction mapping typically results in one cut to the DNA every 20 kilobases12 (though fragments as small as 700 bases can be characterized) and so one might expect to observe just three or four cut-sites on the lambda DNA molecule.11
 | ||
| Fig. 5 A) Automatically generated alignments of fluorocodes recorded for twenty lambda DNA molecules. Positions have been determined and all localized fluorophores are displayed with a 42 nm PSF. Each molecule is stretched 5-fold perpendicular to the DNA axis in order to enable simple inspection and intuitive alignment of the fluorocode. B) Top: The consensus fluorocode derived from the experimental data where more than three counts are required in a given 33-base bin before that bin is added to the consensus. Middle: The consensus fluorocode derived from the experimental data where more than two counts are required in a given 33-base bin before that bin is added to the consensus. Bottom: The fluorocode derived from the reference ‘HhaI map’ to which all of the experimental data is aligned. | ||
Notably, however, 15% of the localized fluorophores cannot be matched to the HhaI map. This is likely due in part to non-specific association of free dye molecules or short fragments of labelled DNA with the longer DNA molecules that were the focus of our experiments. Inhomogeneities on the PMMA surface and variable stretching (including breaks) of the DNA are also likely to contribute to the count of unassigned fluorophores.
Approximately one third of the available sites on the DNA are labelled and matched to a known HhaI target site. Previous work has shown the efficiency of DNA modification by the DNA methyltransferase/mTAG cofactor to be near complete.21 Hence, the efficiency of the coupling of the fluorophore to the modified DNA is relatively low. We cannot reliably count all of the emitters on a molecule, since some are bleached before imaging begins but we estimate a labelling efficiency of 50–60%, which is in line with the manufacturer's expectations for the amine-succinimidyl ester coupling. We see no dependence of the labelling efficiency on HhaI site density (Supplementary Figure S3†) though we do note a surprising lack of labelled sites below the 5000th base pair of the DNA. We attribute this is to breakage of the DNA molecules during the labelling and combing processes. The apparent bias toward molecules only missing a small fragment at one end of the DNA likely results from our selection of only the longest DNA molecules (missing short fragments from their ends) for analysis. We found that alignments of short fragments of DNA, containing relatively few fluorophores, to the HhaI map were not reliable.
Consensus fluorocode
Whilst an average density of localized sites of around one every 650 bases represents a significant advance compared to other DNA mapping technologies, this density is achieved by assigning only 34% of the 215 available HhaI labelling sites on lambda DNA. To further increase the number of fluorophores in the experimentally derived map we generated a ‘consensus’ fluorocode. All of the determined labelling sites (from 1732 localized emitters) from the 20 individual DNA molecules were gathered together and a histogram of their locations along the DNA molecule was created, as shown in Fig. 6. The histogram was used to determine the number of fluorophores falling into bins of 33 bases in width (approximately one standard deviation) along the axis of the DNA molecule. For example, bins covering bases 11616–11649 (which maps to the HhaI site at 11639) and 21054–21087 (which maps to the HhaI site at 21079) contain the highest count of localized emitters, with nine each. Whenever the number of fluorophores in a given bin was found to exceed a threshold value, the mid-point of that bin was added to a list of positions that we used to create a consensus fluorocode. We show two consensus fluorocodes in Fig. 5B, one requiring greater than three counts in a bin before it is added to the consensus and the other, more than two counts.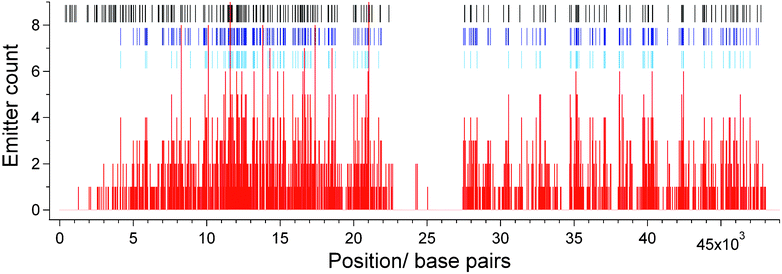 | ||
| Fig. 6 Histogram (red) showing number of localized fluorophores falling into bins of 33 bases in width along the DNA molecule. The positions of the HhaI sites on the DNA are shown (black tick marks) as are the sites where the counts in a bin exceed two (dark blue tick marks) and three (light blue tick marks). These positions are used to produce the consensus fluorocode, shown in Fig. 5B. | ||
The consensus fluorocode using a >2 count threshold contains 248 localized fluorophores. We can associate 163 (66%) of these positions with HhaI sites on the lambda molecule with a standard deviation between the experimentally derived and reference positions of 59 bases (32 nm) (see Supplementary Table 2 and Figure S4†). Raising the consensus threshold to >3 counts gives a fluorocode containing 120 fluorophore positions, 109 (91%) of which can be associated to known HhaI sites on the DNA with a standard deviation of 62 bases between the experimentally derived and expected positions of the fluorophores (see Supplementary Figure S5†). Take into consideration the fact that the sites below 5000 base pairs along the DNA are significantly underrepresented in the data and 86% of the 189 sites between 4158 base pairs and the end of the lambda molecule are assigned in the fluorocode with the threshold of two counts. This is an average of approximately one label every 270 bases for this section of the DNA. Hence, by combining the data from just twenty single molecules, sub-genetic resolution of the optical map can be achieved. The close match of the consensus to the HhaI map demonstrates the validity of the experimental approach and confirms that the DNA can be mapped with a precision of less than 50 nm, at a density of greater than one label every 300 bases. Inhomogeneous stretching of the DNA, non-specific association of free dye molecules and surface effects, which can be significant at the single molecule level, are negated by the consensus fluorocode.
A reference-free optical map
A potentially significant application of the fluorocode is in the study of copy number variations. Using a reference sequence, such as the HhaI map, allows for the detection of short variations in the sequence, depending on the labelling density. Significantly, however, optical mapping allows for the independent detection of CNVs, i.e. detection of sequence repeats in the absence of a reference map of the DNA. We demonstrate that such an approach is valid by constructing a consensus fluorocode that is ‘internally referenced’, i.e. we selected the DNA molecule with the most localized fluorophores from the experimental data and aligned the fluorocodes of the other molecules to it. In this instance, a consensus fluorocode was generated from nineteen aligned molecules (1668 individual emitters) using the same methodology as for the consensus produced using the HhaI map as a reference. This internally referenced fluorocode contains 190 localized emitters and can be aligned by eye to the HhaI map, as shown in Fig. 7. Notably, however, the inhomogeneous stretching of the ‘reference’ molecule in this case means that the left and right halves of the consensus cannot be simultaneously matched to the HhaI map. Whilst repeating DNA sequences can be identified by inspection of the fluorocode, this approach will be greatly improved by two-colour labelling of the DNA, which we plan to investigate in the near future. | ||
| Fig. 7 Internally referenced consensus fluorocode aligned at the right-hand end (top) and the left-hand end (bottom) to the fluorocode of the HhaI map (middle). All emitters are shown with a 50 nm PSF. | ||
Experimental
DNA labelling using methyltransferase-directed transfer of activated groups (mTAG)
20 μg of lambda DNA (Fermentas) was incubated with M.HhaI (variant Q82A/Y254S/N304A) (equimolar amount to the target sites) and 20 μM synthetic cofactor Ado-11-amino23 (see Supplementary Fig. 1 for details†) in 400 μl of M.HhaI buffer (50 mM Tris·HCl pH 7.4, 15 mM NaCl, 0.01% 2-mercaptoethanol, 0.5 mM EDTA, 0.2 mg ml−1 BSA) for 30 min at 37 °C. The completion of the modification reaction was verified by treating a 10 μl aliquot with R.Hin6I (Fermentas) and agarose gel electrophoresis. The modified DNA was then incubated with 187 μg of Proteinase K (Fermentas) in the M.HhaI buffer supplemented with 0.025% SDS for 1 h at 55 °C. DNA was purified by passing through a 1.6 ml Sephacryl™ S-400 column in PBS buffer followed by isopropanol precipitation. The pellet was dissolved in 0.15 M NaHCO3 (pH 8.3) and incubated with a 75-fold molar access of ATTO-647N NHS ester (ATTO-TEC) for 6 h at room temperature. Fluorophore-labelled DNA was purified and redissolved in water as described above.Coverslip preparation
Coverslips were mounted in a Teflon rack and then washed by sonication in acetone, then 1 M NaOH, followed by MilliQ-water (×2). Each sonication was carried out for 15 min. Polymethylmethacrylate (PMMA) (0.1% wt/vol) in chloroform was spin-coated (2000 rpm) onto the cleaned coverslips. The PMMA was subsequently annealed to the coverslips by baking at 120 °C for 1 h.DNA combing
Droplets of 1 μL volume, containing approximately 0.2 μg/ml of the labelled lambda DNA in 50 mM MES buffer at pH 5.7 were deposited onto the PMMA-coated coverslips. The coverslips were placed on a heat block at 60 °C and droplets allowed to evaporate for 30 min.Fluorescence microscopy
Movies of photobleaching, labelled DNA molecules were recorded using an Olympus IX71 microscope coupled to a Hammamatsu Image-EM C9100-13 CCD camera. The microscope setup has been described in detail previously.33 A Spectra Physics 635C-60 diode laser (635 nm) was used as an excitation source and fluorescence emission from the sample was detected via a Chroma Q660LP Dichroic filter and an HQ700/75m emission bandpass filter. Exposure time and laser intensity varied from sample to sample but were set such that the photobleaching of all of the fluorophores on a single DNA molecule required around 1000 frames of movie (typically 2–3 min).Sub-diffraction-limit positioning of fluorophores
We developed a program to fit the position of each of the fluorophores along a DNA molecule with sub-diffraction-limit precision making use of the fact that the emission for different fluorophores is additive. Whilst it is very difficult to localize several emitters when their emission profiles lie within an area whose dimensions are sub-diffraction-limit (∼250 nm), the stochastic nature of photobleaching means that any such group of emitters inevitably photobleaches until only one remains. The emission that we observe (a diffraction-limited spot) from this last fluorophore can be modelled and fitted using a two-dimensional Gaussian profile. By subtracting this emission from all previous frames in the movie, the emission of the penultimate emitter can be resolved. By applying this strategy recursively, in principle, the contribution of every emitter in the movie can be extracted. However, this strategy is prone to failure if more than one emitter within a diffraction-limited spot bleaches simultaneously or if the emitters display complex fluorescence dynamics, such as ‘photoblinking’. In the system measured here the linear distribution of the fluorophores means that we can predict a maximum of eight emitters can lay within a diffraction-limited region. Hence, simultaneous bleaching of more than one fluorophore in such a region is rare. While some blinking was indeed observed, we minimized its effect through longer integration times (200–500 milliseconds) and by binning adjacent frames of the movie before running the bleaching analysis.Visualization and alignment of the DNA fluorocodes
Fluorophore positions were visualized, creating the fluorocodes, for individual DNA molecules using a Matlab routine which convolves a Gaussian point spread function with the projected position of each of the fluorophores on a line. In order to align a fluorocode from an individual molecule (data) to another fluorocode (reference) an intensity profile along each fluorocode is generated using a chosen PSF for each fluorophore. The two intensity profiles are aligned by laterally shifting and stretching the reference profile to fit the profile of the data. The stretching factor applied to the reference map is allowed to vary between 1.4 and 1.8 and this and the lateral shift parameter are optimized by maximizing the output from the convolution of the two. The Matlab code is available on request.Conclusions
DNA fluorocoding potentially enables true single-molecule DNA profiling thanks to a combination of sequence-specificity, fluorophore coverage of the DNA and diffraction-unlimited resolution in the determination of fluorophore positions that restriction mapping and other previously reported methods for creating DNA bar codes cannot approach. For an individual DNA molecule, on average, we are able to position 34% (74 of 215 fluorophores) of the target sites for HhaI with a standard deviation of just 71 bases. In other words, on average, we are able to localize one fluorophore every 660 bases. The maximum resolution of our experiment is determined only by our optical resolution, which is as low as 10 nm, or just 18 bases. Hence, we expect the fluorocode to enable the first single-molecule studies of copy number variations, where the sequence repeats are of the order of kilobases in size.We have shown that we can significantly improve sequence coverage by combining data from several DNA molecules to generate a consensus fluorocode. Indeed, 76% of the target sites for HhaI are described in our consensus fluorocode (Fig. 5B), constructed from 20 DNA molecules. If we consider the lack of experimental data describing the ends of the DNA molecules then, in fact, we see 86% of the sites (163 of 189) between the HhaI site at 4158 bps and the end of the lambda molecule assigned in the consensus fluorocode. On average this equates to one fluorophore every 272 bases. The standard deviation in the position of the fluorophores assigned to each of these sites is just 59 bases. Such labelling density and experimental precision enables the construction of an optical map of genomic material with unrivalled detail and the unambiguous study of DNA motifs on the scale of the single gene.
A fundamental advantage of both optical restriction mapping and the fluorocode over other methods of optical mapping is their lack of necessity for a priori targeting of specific DNA sequences (as in PCR- or antibody-based labelling approaches). This enables an holistic approach to genome analysis and, in theory, makes mapping the genome possible in a single experiment and without any prior knowledge of the DNA sequence. Indeed, as we show in Fig. 7, the fluorocode enables the study of the DNA sequence in the complete absence of a reference map permitting entirely independent detection of repeat sequences of DNA, such as copy number variations.
Using a fluorescent labelling approach to map genomic DNA has distinct advantages over optical mapping using restriction enzymes. We have shown that these include the use of a far higher density of targeted (labelled) sites on the DNA and improved precision in determining the location of these sites. Yet there are significant advances still to be made using the fluorocoding approach. For example, multi-colour labelling of the DNA using two or more methyltransferases to direct the labelling will create a colour fluorocode that allows a high degree of confidence in the analysis and interpretation of the fluorocode. Mapping of DNA methylation status would also be possible using a two-colour approach and one enzyme, such as HhaI, whose activity is blocked by CpG methylation. Multi-colour labelling also enables an optical readout of DNA sequence by flowing a DNA molecule through a nanoslit, such as those designed by Jo et al.19 In all, the fluorocode offers a novel and versatile route to optically map genomic DNA in unprecedented detail.
Acknowledgements
The authors thank Gražvydas Lukinavičius for the kind gift of the Ado-11-amino cofactor and Audronė Lapinaitė for preparing the engineered version of M.HhaI. We gratefully acknowledge financial support from the European Research Council (RKN, Marie Curie IEF), the “Fonds voor Wetenschappelijk Onderzoek FWO” (Grant G.0366.06), the K.U.Leuven Research Fund (GOA 2006/2, Center of Excellence CECAT, CREA2007), the Flemish government (Long term structural funding- Methusalem funding and Tournesol 2009 project T2009.05) and the Federal Science Policy of Belgium (IAP-VI/27).Notes and references
- D. Pushkarev, N. F. Neff and S. R. Quake, Nat. Biotechnol., 2009, 27, 847–852 CrossRef CAS.
- T. D. Harris, P. R. Buzby, H. Babcock, E. Beer, J. Bowers, I. Braslavsky, M. Causey, J. Colonell, J. DiMeo, J. W. Efcavitch, E. Giladi, J. Gill, J. Healy, M. Jarosz, D. Lapen, K. Moulton, S. R. Quake, K. Steinmann, E. Thayer, A. Tyurina, R. Ward, H. Weiss and Z. Xie, Science, 2008, 320, 106–109 CrossRef.
- J. Eid, A. Fehr, J. Gray, K. Luong, J. Lyle, G. Otto, P. Peluso, D. Rank, P. Baybayan, B. Bettman, A. Bibillo, K. Bjornson, B. Chaudhuri, F. Christians, R. Cicero, S. Clark, R. Dalal, A. deWinter, J. Dixon, M. Foquet, A. Gaertner, P. Hardenbol, C. Heiner, K. Hester, D. Holden, G. Kearns, X. Kong, R. Kuse, Y. Lacroix, S. Lin, P. Lundquist, C. Ma, P. Marks, M. Maxham, D. Murphy, I. Park, T. Pham, M. Phillips, J. Roy, R. Sebra, G. Shen, J. Sorenson, A. Tomaney, K. Travers, M. Trulson, J. Vieceli, J. Wegener, D. Wu, A. Yang, D. Zaccarin, P. Zhao, F. Zhong, J. Korlach and S. Turner, Science, 2009, 323, 133–138 CrossRef CAS.
- L. Feuk, A. R. Carson and S. W. Scherer, Nat. Rev. Genet., 2006, 7, 85–97 CrossRef CAS.
- R. Redon, S. Ishikawa, K. R. Fitch, L. Feuk, G. H. Perry, T. D. Andrews, H. Fiegler, M. H. Shapero, A. R. Carson, W. Chen, E. K. Cho, S. Dallaire, J. L. Freeman, J. R. Gonzalez, M. Gratacos, J. Huang, D. Kalaitzopoulos, D. Komura, J. R. MacDonald, C. R. Marshall, R. Mei, L. Montgomery, K. Nishimura, K. Okamura, F. Shen, M. J. Somerville, J. Tchinda, A. Valsesia, C. Woodwark, F. Yang, J. Zhang, T. Zerjal, J. Zhang, L. Armengol, D. F. Conrad, X. Estivill, C. Tyler-Smith, N. P. Carter, H. Aburatani, C. Lee, K. W. Jones, S. W. Scherer and M. E. Hurles, Nature, 2006, 444, 444–454 CrossRef CAS.
- T. Walsh, J. M. McClellan, S. E. McCarthy, A. M. Addington, S. B. Pierce, G. M. Cooper, A. S. Nord, M. Kusenda, D. Malhotra, A. Bhandari, S. M. Stray, C. F. Rippey, P. Roccanova, V. Makarov, B. Lakshmi, R. L. Findling, L. Sikich, T. Stromberg, B. Merriman, N. Gogtay, P. Butler, K. Eckstrand, L. Noory, P. Gochman, R. Long, Z. Chen, S. Davis, C. Baker, E. E. Eichler, P. S. Meltzer, S. F. Nelson, A. B. Singleton, M. K. Lee, J. L. Rapoport, M. King and J. Sebat, Science, 2008, 320, 539–543 CrossRef CAS.
- F. Erdogan, L. A. Larsen, L. Zhang, Z. Tümer, N. Tommerup, W. Chen, J. R. Jacobsen, M. Schubert, J. Jurkatis, A. Tzschach, H. Ropers and R. Ullmann, J. Med. Genet., 2008, 45, 704–709 CrossRef CAS.
- C. Aston, Trends Biotechnol., 1999, 17, 297–302 CrossRef CAS.
- X. Michalet, R. Ekong, F. Fougerousse, S. Rousseaux, C. Schurra, N. Hornigold, M. V. Slegtenhorst, J. Wolfe, S. Povey, J. S. Beckmann and A. Bensimon, Science, 1997, 277, 1518–1523 CrossRef CAS.
- A. H. Samad, W. W. Cai, X. Hu, B. Irvin, J. Jing, J. Reed, X. Meng, J. Huang, E. Huff, B. Porter, A. Shenkar, T. Anantharaman, B. Mishra, V. Clarke, E. Dimalanta, J. Edington, C. Hiort, R. Rabbah, J. Skiada and D. C. Schwartz, Nature, 1995, 378, 516–517 CrossRef CAS.
- X. Meng, K. Benson, K. Chada, E. J. Huff and D. C. Schwartz, Nat. Genet., 1995, 9, 432–438 CrossRef CAS.
- S. Zhou, F. Wei, J. Nguyen, M. Bechner, K. Potamousis, S. Goldstein, L. Pape, M. R. Mehan, C. Churas, S. Pasternak, D. K. Forrest, R. Wise, D. Ware, R. A. Wing, M. S. Waterman, M. Livny and D. C. Schwartz, PLoS Genet., 2009, 5, e1000711 Search PubMed.
- S. Zhou, A. Kile, E. Kvikstad, M. Bechner, J. Severin, D. Forrest, R. Runnheim, C. Churas, T. S. Anantharaman, P. Myler, C. Vogt, A. Ivens, K. Stuart and D. C. Schwartz, Mol. Biochem. Parasitol., 2004, 138, 97–106 CrossRef CAS.
- S. Zhou, M. C. Bechner, M. Place, C. P. Churas, L. Pape, S. A. Leong, R. Runnheim, D. K. Forrest, S. Goldstein, M. Livny and D. C. Schwartz, BMC Genomics, 2007, 8, 278 CrossRef.
- S. Gad, M. Klinger, V. Caux-Moncoutier, S. Pages-Berhouet, M. Gauthier-Villars, I. Coupier, A. Bensimon, A. Aurias and D. Stoppa-Lyonnet, J. Med. Genet., 2002, 39, 817–821 CrossRef CAS.
- X. Qu, D. Wu, L. Mets and N. F. Scherer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11298–11303 CrossRef CAS.
- R. B. Kelly, N. R. Cozzarelli, M. P. Deutscher, I. R. Lehman and A. Kornberg, J. Bio. Chem., 1970, 245, 39–45 CAS.
- M. Xiao, A. Phong, C. Ha, T. Chan, D. Cai, L. Leung, E. Wan, A. L. Kistler, J. L. DeRisi, P. R. Selvin and P. Kwok, Nucleic Acids Res., 2007, 35, e16 CrossRef.
- K. Jo, D. M. Dhingra, T. Odijk, J. J. de Pablo, M. D. Graham, R. Runnheim, D. Forrest and D. C. Schwartz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2673–2678 CrossRef CAS.
- S. Klimasauskas and E. Weinhold, Trends Biotechnol., 2007, 25, 99–104 CrossRef CAS.
- C. Dalhoff, G. Lukinavicius, S. Klimasauskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS.
- G. Lukinavičius, V. Lapienė, Z. Stasevskij, C. Dalhoff, E. Weinhold and S. Klimasauskas, J. Am. Chem. Soc., 2007, 129, 2758–2759 CrossRef CAS.
- A. Lapinaitė, G. Lukinavičius, S. Klimašauskas, manuscript in preparation .
- R. J. Roberts, T. Vincze, J. Posfai and D. Macelis, Nucleic Acids Res., 2010, 38, D234–236 CrossRef CAS.
- W. Wang, J. Lin and D. Schwartz, Biophys. J., 1998, 75, 513–520 CAS.
- J. H. Kim, W. Shi and R. G. Larson, Langmuir, 2007, 23, 755–764 CrossRef CAS.
- Y. Liu, P. Wang, S. Dou, W. Wang, P. Xie, H. Yin, X. Zhang and X. G. Xi, J. Chem. Phys., 2004, 121, 4302–4309 CrossRef CAS.
- R. E. Thompson, D. R. Larson and W. W. Webb, Biophys. J., 2002, 82, 2775–2783 CrossRef CAS.
- A. Yildiz, J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman and P. R. Selvin, Science, 2003, 300, 2061–2065 CrossRef CAS.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS.
- M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS.
- M. Heilemann, P. Dedecker, J. Hofkens and M. Sauer, Laser Photonics Rev., 2009, 3, 180–202 Search PubMed.
- P. Dedecker, B. Muls, A. Deres, H. Uji-i, J. Hotta, M. Sliwa, J. Soumillion, K. Müllen, J. Enderlein and J. Hofkens, Adv. Mater., 2009, 21, 1079–1090 CrossRef CAS.
- T. Vincze, J. Posfai and R. J. Roberts, Nucleic Acids Res., 2003, 31, 3688–3691 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical structure of the mTAG cofactor, a movie showing DNA combing from an evaporating droplet and detailed results from the single molecule and consensus alignments. See DOI: 10.1039/c0sc00277a |
| This journal is © The Royal Society of Chemistry 2010 |