Anti-inflammatory activity of natural dietary flavonoids
Min-Hsiung
Pan
*a,
Ching-Shu
Lai
a and
Chi-Tang
Ho
*b
aDepartment of Seafood Science, National Kaohsiung Marine University, No.142, Haijhuan Rd., Nanzih District, Kaohsiung, 81143, Taiwan. E-mail: mhpan@mail.nkmu.edu.tw; Fax: (+886)-7-361-1261; Tel: (+886)-7-361-7141 Ext 3623
bDepartment of Food Science, Rutgers University, 65 Dudley Road, New Brunswick, New Jersey 08901-8520, USA. E-mail: ho@aesop.rutgers.edu; Fax: +1-732-932-6776
First published on 13th September 2010
Abstract
Over the past few decades, inflammation has been recognized as a major risk factor for various human diseases. Acute inflammation is short-term, self-limiting and it's easy for host defenses to return the body to homeostasis. Chronic inflammatory responses are predispose to a pathological progression of chronic illnesses characterized by infiltration of inflammatory cells, excessive production of cytokines, dysregulation of cellular signaling and loss of barrier function. Targeting reduction of chronic inflammation is a beneficial strategy to combat several human diseases. Flavonoids are widely present in the average diet in such foods as fruits and vegetables, and have been demonstrated to exhibit a broad spectrum of biological activities for human health including an anti-inflammatory property. Numerous studies have proposed that flavonoids act through a variety mechanisms to prevent and attenuate inflammatory responses and serve as possible cardioprotective, neuroprotective and chemopreventive agents. In this review, we summarize current knowledge and underlying mechanisms on anti-inflammatory activities of flavonoids and their implicated effects in the development of various chronic inflammatory diseases.
Introduction
Inflammation is a normal biological process in response to tissue injury, microbial pathogen infection and chemical irritation. This biological process also involves the innate and adaptive immune systems. At a damaged site, inflammation is initiated by migration of immune cells from blood vessels and release of mediators, followed by recruitment of inflammatory cells and release of reactive oxygen species (ROS), reactive nitrogen species (RNS) and proinflammatory cytokines to eliminate foreign pathogens, resolving infection and repairing injured tissues.1,2 Thus, the main function of inflammation is beneficial for a host's defense. In general, normal inflammation is rapid and self-limiting, but aberrant resolution and prolonged inflammation causes various chronic disorders.3Chronic inflammation can inflict more serious damage to a host tissue than bacterial infection. Diverse ROS and RNS such as •O2− (superoxide anion), •OH (hydroxyl radical), H2O2 (hydrogen peroxide), nitric oxide (NO), and 1O2 (singlet oxygen) generated by inflammatory cells injure cellular biomolecules including nucleic acids, proteins and lipids, causing cellular and tissue damage, which in turn augments the state of inflammation.4 These also trigger a series of signaling molecules, inflammatory gene expression and activation of enzymes involved in chronic inflammation. Inflammatory chemicals produced by inflamed and immune cells also attack normal tissues surrounding the infected tissue, causing oxidative damage and extensive tissue inflammation.1,4
Studies show that chronic inflammation is linked to a wide range of progressive diseases, including cancer, neurological disease, metabolic disorder and cardiovascular disease.3,4 Numbers of studies suggest elimination of chronic inflammation as a major way to prevent various chronic diseases.1,3 Epidemiological studies provide convincing evidence that natural dietary compounds that humans consume as food possess many biological activities. Among these natural bioactive compounds, flavonoids are widely recognized for their biological and pharmacological effects, including antiviral, anti-carcinogenic, antioxidant, antimicrobial, anti-inflammatory, anti-angiogenic and anti-thrombogenic properties.1,5 Epidemiologic studies indicate that the incidence of chronic disease and cancer is inversely correlated with the consumption of fruits and vegetables rich in flavonoids,5,6 and this is attributed to their possible anti-inflammatory activities. This review forcuses on the molecular basis of the anti-inflammatory potential of flavonoids, with special emphasis on their effect on molecular mechanisms and signaling pathways involved in inflammation, as agents in reducing or eliminating different chronic inflammation-associated human diseases.
The role of inflammation in human disease
Inflammation is a complicated process, driven by preexisting conditions (infection or injury) or genetic changes, that results in triggering signaling cascades, activation of transcription factors, gene expression, increased of levels of inflammatory enzymes, and release of various oxidants and proinflammatory molecules in immune or inflammatory cells.2 In this condition, excessive oxidants and inflammatory mediators have a harmful effect on normal tissue, including toxicity, loss of barrier function, abnormal cell proliferation, inhibiting normal function of tissues and organs, and finally leading to systemic disorders.1,2 Over the past few decades, many studies reveal that chronic inflammation is a critical component in many human diseases and conditions, including obesity, cardiovascular diseases, neurodegenerative diseases, diabetes, aging, and cancers2,4 (Fig. 1).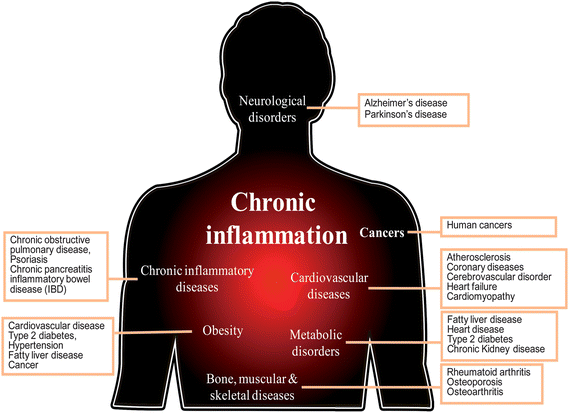 | ||
| Fig. 1 Chronic inflammation is linked to human diseases. | ||
Cardiovascular diseases
Cardiovascular disease (CVD) is becoming the leading cause of death in the world. Chronic inflammation, such as atherosclerosis, coronary diseases, cerebrovascular disorder, heart failure and cardiomyopathy is common in CVD.7 In the past, researchers suggested a number of traditional risk factors implicated in the pathogenesis of CVD including age, hypertension, dyslipidemia, hypercholesterolemia, glucose tolerance and metabolic symptoms. However, recent studies focus on the relationship between endothelial dysfunction and inflammatory condition.7,8 Vascular endothelium is very important in regulation of vascular homeostasis, and inhibition of leukocyte adhesion and platelet aggregation by release of mediators such as nitric oxide (NO).8 Increase of NO production, damage of endothelial cells, increase of oxidative stress and an enhanced proinflammatory state lead to the alteration of vascular integrity, the reduction of vasodilator capacity and the appearance of endothelial dysfunction.Atherosclerosis is a chronic inflammatory disease and a major cause of CVD. Recent studies demonstrate that vascular inflammation is the earliest event in the development of atherosclerosis.7,9 The process involves stimulation of cholesterol, oxidized low-density-lipoprotein (ox-LDL) and oxidative free radicals, which initiate activation of vascular endothelial cells and enhance their adhesive property with monocytes by expression of adhesion molecules selectins, vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule-1 (ICAM-1).10 Once monocytes firmly attach on the surface of endothelium, they transmigrate into the arterial intima and differentiate to macrophages. This transmigration is triggered by chemoattractant molecules such as monocyte chemoattractant protein-1 (MCP-1), proinflammatory cytokines (TNF-α and ILs) as well as growth factors (PDGF and TGF-β) produced by activated T cells and macrophage.11 Among these, studies indicate that MCP-1 is important for recruitment of monocytes into intima. Differentiated macrophages that expresse scavenger receptors become foam cells via uptake of ox-LDL generated in the intima resulting in formation of fatty streaks.12 The molecules secreted by monocytes, macrophages and arterial cells maintain an inflammatory response within the artery and promote proliferation and migration of vascular smooth muscle cells.9,11 Proliferative smooth muscle cells release fibrogenic mediators and build a dense extracellular matrix around foam cells and monocytes, finally causingfatty streaks to progress into fibrous plaque.13
In pathogenesis and progression of atherosclerosis, chronic inflammation is involved in every stage that is characterized by infiltration of monocytes/macrophages and production of proinflammatory cytokines9,11 (Fig. 2). Hence, the modulation or regulation of the interaction between endothelial and inflammatory cells and the transformation of macrophages to foam cells could be the basis for the beneficial effects that prevent or slow down the progression of this disease. This diversity of cytokine expression and function might also lead to the identification of selective therapeutic targets for the prevention and treatment of atherosclerosis.10,11
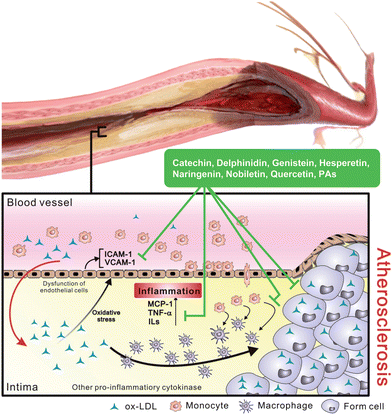 | ||
| Fig. 2 Induction of inflammatory responses by ox-LDL in development of atherosclerosis. Stimulated ox-LDL increases chemoattractant concentration and induces monocyte transmigrating into intima then transforming to macrophage. Differentiated macrophage that specific to uptake ox-LDL becomes foam cell and leads to the formation of fatty streak in intima. | ||
Moreover, clinical research shows that elevated levels of systemic inflammatory molecules including IL-6, ICAM-1, P-selectin, E-selectin and C-reactive protein (CRP), are classic acute-phase markers occurring in patients with coronary disease, and, therefore that might be a predictor of cardiovascular risk.14,15 Different treatments of atherosclerosis are associated with reduction of these inflammatory markers, providing a new target for blockage or therapy of atherosclerosis by inhibition of inflammation.9,16
Neurological disorders
Many neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, multiple sclerosis, amyotrophic lateral sclerosis and other nervous system pathologies are associated with inflammatory injury (Fig. 3). This complex inflammatory process involves different cellular components in the central nervous system, such as microglial cells, astrocytes, ependymal cells, macrophages and mast cells.17 Microglial cells are macrophages in the brain and the central nervous system that are the first response to a neuronal injury.18 In the case of Alzheimer's disease, a cleavage product of amyloid precursor protein (APP), an integral membrane protein, and the aggregation of amyloid β peptides (Aβ1-42 and Aβ1-40) trigger activation of microglials and astrocytes, following activation of transcription factors such as nuclear factor-kappa B (NF-κB) and activator protein (AP)-1 that induce production of ROS and various proinflammatory mediators including NO, PGE2, ILs and TNF-α.17,18 These proinflammatory cytokines and ROS produced by activated microglial and astrocytes may cause neuronal damage or neurotoxicity resulting in apoptosis and necrosis.17 Moreover, proinflammatory mediators released from microglials and astrocytes can activate each other to amplify inflammatory signals to neurons.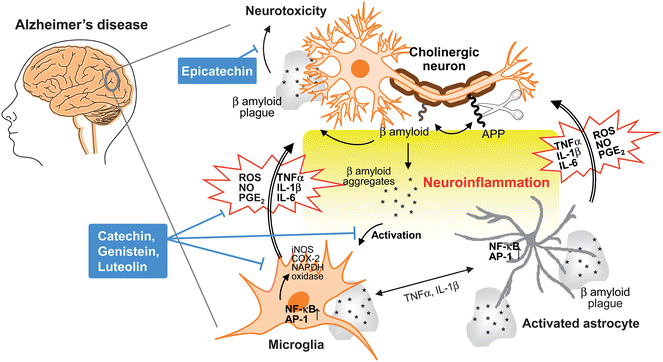 | ||
| Fig. 3 Neuroinflammation in Alzheimer's disease. During the development of Alzheimer's disease, β amyloid peptide is produced by cleavage of amyloid precursor protein (APP), aggregates and accumulates as β amyloid plaques. Both aggregates and plaques cause neurotoxicity or activation of microglia through up-regulating NF-κB and AP-1 transcription factors, which in turn release ROS and pro-inflammatory cytokines such as NO, PGE2, IL-1, IL-6, and TNF-α that damage cholinergic neuron. These pro-inflammatory cytokines also directly activate astrocytes that also produce cytokines to amplify inflammatory signals and result in neuroinflammation and neurodegeneration. Flavonoids act through avoiding β amyloid induced-neuron injury and death, modulation of pro-inflammatory cytokines and ROS production as well as inhibiting the activation of microglia and astrocyte as neuroprotective mechanisms. | ||
Parkinson's disease is a neurodegenerative disorder in the central nervous system with a pathology similar to Alzheimer's disease. Aggregation and accumulation of α-synuclein protein of Lewy bodies and loss of dopaminergic neurons are major neuropathological alterations in Parkinson's disease.19 Lewy bodies and intracellular and/or extracellular α-synuclein protein aggregates released from neurons lead to the activation of microglial cells,20,21 resulting in the activation of transcription factor NF-κB, gene expression of iNOS, COX-2 and NAPDH oxidase, and production of inflammatory mediators.22 Proinflammatory cytokines such as TNF-α and ILs derived from activated microglial cells also trigger activation of astrocytes. Studies show that released factors from activated microglials and astrocytes have a combinational effect in promoting neurotoxicity.17,22
In neurodegenerative diseases, the activation of immune cells such as microglials and astrocytes is a critical step in neuropathology. Oxidative stress is also important in neurodegenerative diseases, both in Alzheimer's disease and Parkinson's disease.17,18,22 Several therapeutic approaches that target inflammatory responses demonstrate the ability to interfere with activation of transcription factor and inhibit function of inflammatory enzymes and production of ROS.23,24
Obesity
Obesity, resulting from excessive fat sorted in adipose tissue is a highly prevalent condition that is related to many metabolic disorders throughout the world. Numerous studies indicate the high risk of obesity in the development of cardiovascular disease, type 2 diabetes, hypertension, fatty liver disease and cancer.25 It is now clear that the function of adipose tissue is not only as fat storage, but also as a major endocrine and secretary organ that produces adipokines such as leptin and adiponectin.26 Leptin is an important hormone in the regulation of energy expenditure and caloric intake for maintaining energy balance.27 Clinical and animal studies show that leptin deficiency results in body weight increase and a high risk for the development of type 2 diabetes.27,28In recent years, many studies document that obesity is significantly associated with a chronic low-grade inflammation29 (Fig. 4). The first connection between obesity and inflammation is evidenced by the release of TNF-α from adipocytes. As the lipid content increases in adipose tissue, adipocytes synthesize TNF-α and several cytokines (IL-1β and IL-6) that change the number and size of cells, influencing lipoprotein lipase and increasing the inflammatory state.30 TNF-α also induces insulin resistance by downregulation of insulin receptor phosphorylation, decrease of glucose uptake and expression of GLUT4 transporter.29,31 Another important inflammatory feature in adipose tissue is recruitment of immune and inflammatory cells such as neutrophils, eosinophils and macrophages.32 Studies in both mice and humans show that while in an obese state, macrophage infiltration is increased in adipose tissue.32 Large adipocytes secret chemotactic signals, such as monocyte chemoattractant protein-1 (MCP-1), to trigger infiltration of macrophages, that then leads to the creation of a chronic, low-grade inflammation in obesity.32,33 Increased levels of acute phase protein CRP are found in many obese individuals,34 and circulating CRP concentrations are related to the development of cardiovascular disease,14 indicating the association of obesity and cardiovascular disease. Some lines of evidence also suggest that obesity is linked to fat storage in the liver that can lead to the development of fatty liver diseases.25 It is suggested that IL-6 derived from adipocytes may drive the production of CRP in the liver.35 Overexpression of MCP-1 in adipose tissue leads to increase of hepatic triglyceride content.33 In addition, elevated levels of circulating TNF-α in an obese state is often associated with an increase in insulin resistance.31,36 These observations emphasize the correlation among obesity, inflammation and metabolic disorders.
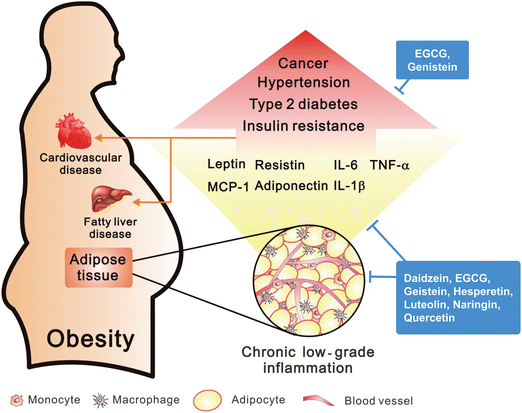 | ||
| Fig. 4 Obesity in the induction of inflammation. Adipose tissue of visceral obesity induced chronic low-grade inflammation through macrophage infiltration by MCP-1 and secretion of pro-inflammatory factors. However, obesity may cause the high risk in development of several diseases. | ||
Metabolic disorders
Extensive research reveals that inflammation is a characteristic feature of metabolic disorders, including fatty liver disease, type 2 diabetes, chronic kidney disease and heart disease (Fig. 5). Inflammatory responses are considered to be a critical stage of metabolic dysfunction characterized by abnormal proinflammatory cytokine production, increased acute phase protein and activation of inflammatory signaling pathways.37 In addition, obesity correlates with an increase in inflammatory conditions and metabolic syndromes.29 Type 2 diabetes is the most prevalent and serious metabolic disease caused by insulin resistance derived from pancreatic beta cell dysfunction. Both experimental and clinical studies demonstrate that several inflammatory cytokines are closely related to pathogenesis of insulin resistance.38 Increased levels of IL-1β in plasma are shown to have detrimental effects on the function of IL-1 receptor antagonist proteins (IL-1ra) that promote beta cell destruction and alter insulin sensitivity.39 Moreover, IL-6 acts on activation of tyrosine phosphatase and interferes with the interaction between insulin receptor and suppressor of cytokine signaling (SOCS) proteins that result in insulin resistance.40 In patients with chronic kidney disease, elevated levels of serum acute phase proteins such as C reactive protein (CRP), TNF-α and ILs are associated with an increase in chronic inflammatory states and insulin resistance.41 In addition to insulin resistance and Type 2 diabetes, an elevated concentration of CRP and various cytokines also occur in chronic heart failure with fluid overload and cardiovascular disease.34,41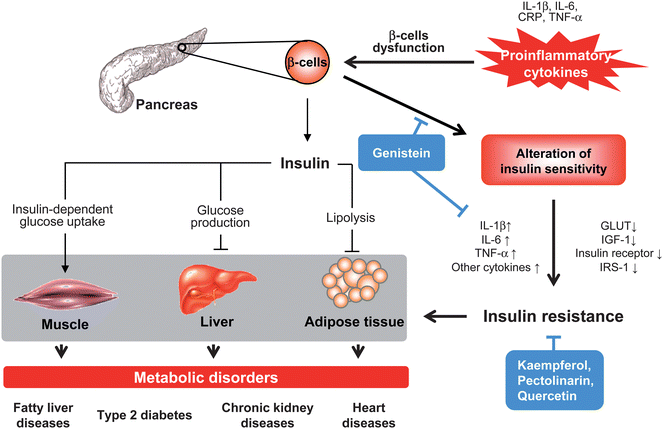 | ||
| Fig. 5 Proinflammatory cytokines in insulin resistance and metabolic disorders. Insulin synthesized and secreted from β cells in pancreas acts as normal function in different organs and tissues, includes reducing glucose production and output in liver, facilitating glucose uptake in skeletal muscle, and decreasing lipolysis in adipose tissue. Excessive pro-inflammatory cytokines (CRP, IL-1, IL-6, and TNF-α) cause dysfunction of β cell or recruit of inflammatory cells (monocytes and macrophages) that affect both insulin secretion and insulin action, promote pathogenesis of insulin resistance and subsequently reducing insulin-dependent signalling. This local insulin resistance also contributes to its target tissues such as increase concentration of glucose and fatty acids in skeletal muscle, liver and adipose tissue that lead to various metabolic disorders. Flavonoids act through interfering with pro-inflammatory cytokines-induced β cells dysfunction and cell death, decreasing cytokines production, up-regulation of insulin-dependent signaling and improving glucose uptake in different cell types. | ||
Non-alcoholic fatty liver disease (NAFLD) is a common liver disorder associated with obesity and insulin resistance that results from abnormal adipokine and cytokine production.42 In liver tissue, these mediators, such as leptin, TNF-α and ILs decrease insulin signaling to hepatocytes by the activation of several signaling molecules and kinases. This results in hepatic insulin resistance, hyperglycemia and fatty liver development caused by increased fatty acid uptake and VLDL production.37,42 Hepatic insulin resistance also stimulates the production of CRP and cytokines that promote atherosclerosis by the inhibition of NO production, an increase in the adhesion property in endothelial cells, and increasing macrophage uptake ox-LDL.40 This information indicates that hepatic insulin resistance is related to the induction of metabolic syndromes and the acceleration of cardiovascular disease progression.
Bone, muscular and skeletal diseases
Rheumatoid arthritis is an autoimmune disease that causes joint destruction and functional disability, often characterized by chronic inflammatory responses primarily affecting the synovium of diarthrodial joints (Fig. 6). Besides infections and genetic factors, rheumatoid arthritis is indicated in the interaction between immune cells, such as T cells, B cells, macrophages, dendritic cells and fibroblasts and as such plays an important role in the pathogenesis of this disease.43 In patients with rheumatoid arthritis, large numbers of activated T cells are present in inflamed joints. Recruited T cells develop a lymphoid structure in synovium with B cells, macrophages and fibroblast-like synoviocytes that create a complex network between cells through secreting various cytokines, such as TNF-α, IL-1 and IL-6.44 Among these cell types in synovium, fibroblast-like synoviocytes is known to produce prostaglandins and proteases that destroy bone and cartilage.45 Moreover, activated B cells and macrophage continuously secrete IL-1 and TNF-α that maintain the synovial fibroblast in an inflamed state.44,45 Enrichment of these immune cells and derived-proinflammatory cytokines in synovium causes varying degrees of joint destruction and also extra-articular organ involvement.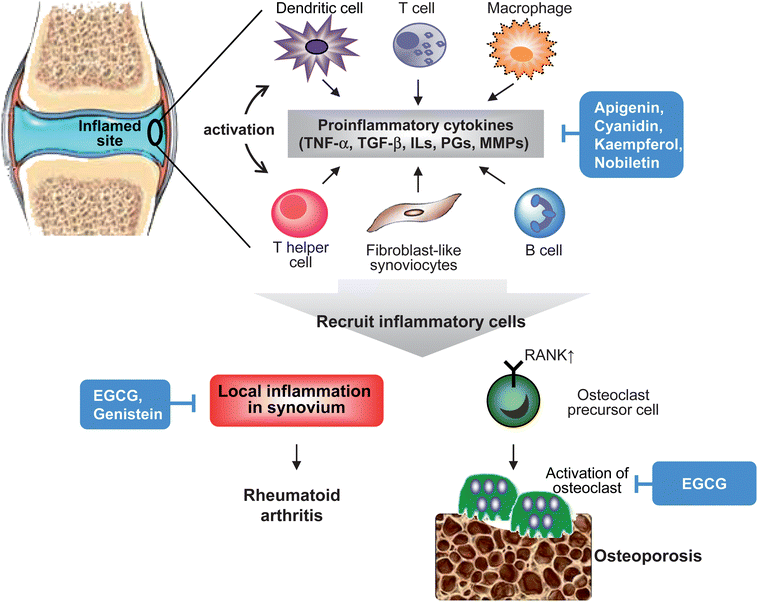 | ||
| Fig. 6 Mechanisms of inflammation-associated pathogenesis in rheumatoid arthritis and osteoporosis. In inflamed rheumatoid synovium and bone tissue, pro-inflammatory cytokines produced by recruited inflammatory cells (macrophages, T cells and B cells), endothelial cells and synovial fibroblasts are central to the inflammatory process in rheumatoid arthritis and osteoporosis. This pathological process also involves in innate and adaptive immunity responses. These pro-inflammatory cytokines result in activation of synovial fibroblasts and produce proteases that lead to tissue destruction. In addition, cytokines-trigger activation and differentiation of osteoclasts are important in bone loss. Flavonoids act through reducing recruitment of inflammatory cells, cytokines production, MMPs expression, and activation or differentiation of osteoclasts. | ||
Osteoporosis is also a chronic inflammation condition that is characterized by the loss of bone density (Fig. 6). Studies identify proinflammatory cytokines TNF-α, IL-1 and IL-6 as important mediators of bone resorption through increased expression of receptor activator of NF-κB (RANK), activation and differentiation of osteoclast, and decreased osteoblast survival.46 Despite several in vitro and in vivo studies indicating that proinflammatory cytokines contribute to osteoporosis, and increased levels of IL-1β, IL-6 and TNF-α in whole blood culture from patients with osteoporosis, the mechanism involved in bone loss is still unclear. In addition, recent studies also reveal that elevated systemic CRP is associated with poor bone health.46,47
Chronic inflammatory diseases
A continued chronic inflammatory state in different organs and tissues leads to a various chronic inflammatory diseases such as chronic obstructive pulmonary disease (COPD), psoriasis, rheumatoid arthritis, chronic pancreatitis and inflammatory bowel disease (IBD) all of which are frequently associated with infiltration of immune and inflammatory cells. For example, inflammatory bowel disease leads to ulcerative colitis (UC) and Crohn's disease (CD), based on clinical features and histopathology, which result from abnormal regulation of the immune system in the intestine.48 In IBD, damage of epithelium in the colon increases recruitment of immune cells such as activated T lymphocytes and leucocytes that are mediated by the expression of adhesion molecules.49 This is thought to be a key step of inflammatory process involved in IBD (Fig. 7).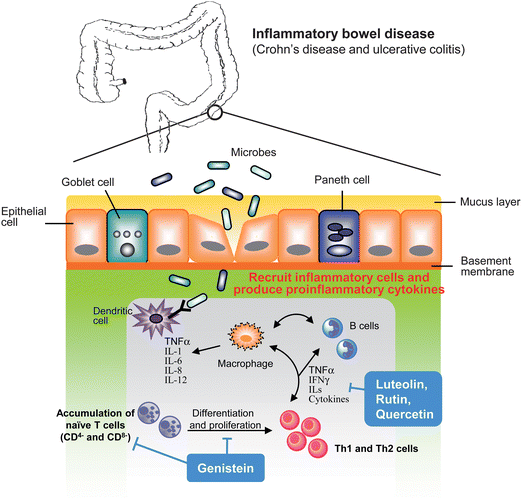 | ||
| Fig. 7 Underlying mechanisms in inflammatory bowel disease. As bacteria infection or environmental factors that cause colonic endothelium damage result in recruitment of inflammatory and immune cells from bloodstream. Accumulated inflammatory cells produce pro-inflammatory mediators that trigger proliferation and activation of T cells, lead to differentiate to Th1 and Th2 cells that result in amplification of inflammatory cascade and cause tissue injury. Flavonoids act through decreasing inflammatory cytokines production, reducing recruitment of inflammatory cells and modulation of differentiation and proliferation of T cells. | ||
A number of studies indicate that an increased level of proinflammatory cytokines is important for pathogenesis of IBD.48 In general, epithelial cells, paneth cells, enterocytes and immune cells of colon form a complex barrier by the expression of cytokines, chemokines and metabolite from microbes or hosts in order to fight pathogens and to maintain intestinal homeostasis. Also, an alteration of the immune system toward luminal antigens is thought to play a crucial role in pathogenesis in IBD.48 In an immune response, T helper (Th) cells recognize self-antigens (from food consumption or intestinal bacteria) that present from lymphocytes and phagocytes, and then start to produce cytokines. As the specific-antigen occurs, it enhances production of various cytokines in colonic epithelium that promote CD4 and CD8 cells which then differentiate to Th1 and Th2 cells. These two Th cells produce a dramatic amount of proinflammatory cytokines that affect colon mucosa and intestinal cells.50 Studies show that Crohn's disease is mediated by Th1 cells that are characterized by the production of IL-1, IL-6, interferon-γ (IFN-γ) and TNF-α, whereas ulcerative colitis is thought to be a Th2 cell mediated response by the secretion of IL-4, IL-5 and IL-10.48,50 Overexpression of these proinflammatory cytokines is found in the intestinal mucosa from IBD patients and is related to the alteration of intestinal homeostasis and results in an abnormal inflammatory response in the intestinal mucosa. Indeed, increased proinflammatory cytokines in colon mucosa are also linked to the enhanced expression of anti-apoptotic molecules, leading to apoptotic resistance and promotion of accumulation of T cells.48,50
Cancers
Several lines of evidence indicate that cancer development in humans is a multistep and long-term process which requires six properties, including limitless replication potential, evasion of apoptosis, self-sufficiency in growth signals, insensitivity to anti-growth signals, sustained angiogenesis, and tissue invasion and metastasis.51 Since Virchow observed in 18th century, that cancers frequently occur at sites of chronic irritation, much research confirms the concept that chronic inflammation is a critical component of tumor promotion and progression, including colorectal, gastric, pancreatic, pulmonary, cystic, hepatocellular, ovarian, skin and esophageal cancers.4,52 In view of inflammation involved in different cancers, increasing evidence suggests that inflammation should be the seventh hallmarker in cancer development.53The pathological mechanism of inflammation involved in tumorigenesis is very complicated.54 In tissue injury or inflammatory stimulation, inflammatory cells are recruited and production of pro-inflammatory cytokines and diverse ROS and RNS that induce genetic change which enhances malignant transformation and proliferation of initiated cells. Subsequently, as tumor tissue forms, inflammation continues to promote development of cancer by creating an inflammatory microenvironment, which consists of stromal cells, inflammatory cells and the extracellular matrix from surrounding tissues. The inflammatory and immuosuppressive cytokines and chemokines secreted from these cells not only promotes proliferation, angiogenesis, invasion and metastasis but also suppresses the host's immune system and facilitates tumor growth and development54,55 (Fig. 8).
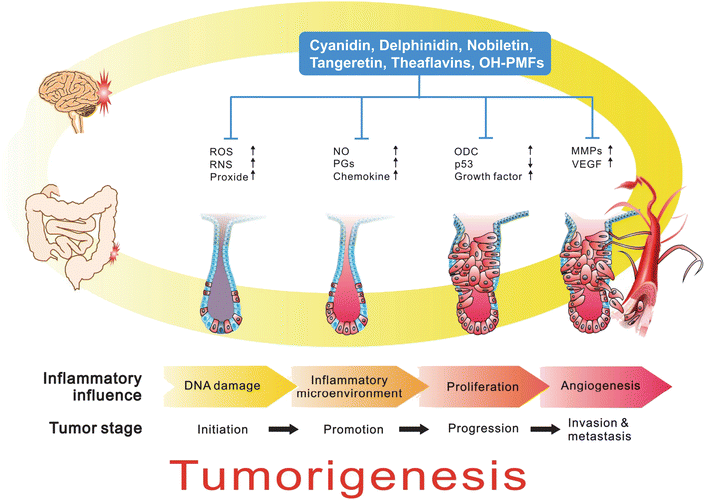 | ||
| Fig. 8 Role of inflammation in cancer development. Chronic inflammation is a critical component of tumor promotion and progression, including colorectal, gastric, pancreas, lung, bladder, hepatocellular, ovary, skin and esophageal cancers. In colonic tumorigenesis, inflammatory stimulation, inflammatory cells are recruited and production of pro-inflammatory cytokines and diverse ROS and RNS that induction of genetic change, enhanced malignant transformation and proliferation of initiated cells. Subsequently, as tumor tissue formation, inflammation also promotes development of cancer by creating an inflammatory microenvironment. The inflammatory and immuosuppressive cytokines and chemokines secreted from these cells not only promote proliferation, angiogenesis, invasion and metastasis but also suppress the host's immune system and facilitates tumor growth and development. | ||
There are many key molecules that link inflammation and cancer, including transcription factors, signal transducers and activators of transcription 3 (STAT3), nuclear factor-κB (NF-κB), nuclear factor of activated T-cells (NFAT), activator protein-1 (AP-1), CCAAT enhancer binding protein (C/EBP), cAMP response element binding protein/p300 (CBP/p300), activator transcription factor (ATF), downstream genes iNOS, COX-2, interleukin-6 (IL-6), IL-1β, tumor necrosis factor-R (TNF-R), 5-lipoxygenase (5-LOX), hypoxia inducible factor-1α (HIF-1α), and vascular endothelial growth factor (VEGF), resulting in inflammation and tumorigenesis.54,56 Besides the above, inflammatory signaling is regulated by upstream kinases, such as NFκB-inducing kinase (NIK), IκB kinase (PKC), mitogen-activated protein kinase (MAPK), phosphoinositide-3 kinase (PI3K)/Akt and protein kinase C (PKC), also important in inflammation linked tumorigenesis. These critical molecules can be considered important in the modulation of an inflammatory response, and thus could block or prevent inflammation-associated carcinogenesis.54,56
Chemoprevention: inflammation as potential target
The term “chemoprevention” was coined by Sporn in the mid-1970s and is defined as the use of a chemical substance of either natural or synthetic origin to prevent, hamper, arrest, or reverse a disease.57 It is suggested that inflammation is a multifaceted and complicated process implicated in infiltration and activation of various immune and inflammatory cells, cytokine production, signal transduction and molecular mechanism that results in a widespread physiological state in organ and tissue. There are many clinical drugs and dietary natural compounds that are demonstrated to be able to exert protective action by different mechanisms, on a number of pathological aspects through targeting interference with chronic inflammatory response. For example, anti-inflammatory drugs, including nonsteroidal anti-inflammatory drugs (NSAID), are found to have some neuroprotective effects.58 Resveratrol and curcumin are found to prevent cell damage and death through their antioxidative activities that reveal potential neuroprotective activities.59,60 Anti-α4 integrin monoclonal antibody acts by disrupting adhesion and migration of immune cells that attenuate cytokine release in Crohn's disease individuals.61 Using monoclonal antibodies against B cells function, TNF-α and IL-6 signaling are beneficial for autoimmune diseases, including rheumatoid arthritis and inflammatory bowel disease.62 In patients with type 2 diabetes, treatment with an IL-1 receptor antagonist may restore the function of pancreatic β cells and improve insulin-resistance.39 Kinase Inhibitors such as SB203580, a p38 MAPK inhibitor, are found to decrease IL-1β production and reduce rheumatoid arthritis in mice.63 Oral intake of tyrosine kinase inhibitor showed a reduction of inflammatory markers and improved quality in rheumatoid arthritis and psoriasis patients.62 In an in vivo study, PPARγ agonist reduced the expression of proinflammatory cytokines that then decreased immune complex deposition and renal inflammation, and lowered atherosclerotic lesions.64 PPARγ agonist also lowered plasma glucose and serum CRP and TNF-α, and increased the production of adiponectin in adipose tissue, thus improving insulin resistance in humans with type 2 diabetes.62The relationship between inflammation and cancer is established.54,55 Numerous evidence demonstrates that inflammatory pathways are critical targets in cancer treatment and prevention.65 Many natural bioactive compounds are reported to interfere with the initiation, promotion/progression, and invasion/metastasis of cancer through the control of intracellular signaling cascades as the process of inflammation progresses. These bioactive compounds include flavonoids, flavonolignans, isothiocyanates, proanthocyanidins, terpenoids, and other polyphenolic compounds.1,56 These bioactive compounds act by avoiding the causes of tissue damage, inhibiting signaling pathways and the activation of transcription factors, inhibiting oxidant-generating enzymes and mediators of inflammation, scavenging reactive oxygen and nitrogen species generated by inflammatory cells, and modulating angiogenesis and metastasis.
Flavonoids: new approach in chronic inflammatory diseases
Flavonoids are plant secondary metabolites that are ubiquitous in fruits, vegetables, nuts, seeds and plants. These polyphenolic compounds are a subgroup of chemically related polyphenols that possess a basic 15-carbon skeleton and can be represented as C6-C3-C6, consisting of two benzene rings (C6) joined by a linear three carbon chain (C3).1 Based on the differences in the structure of the aglycones C ring, flavonoids can be classified into seven groups: flavones, flavanones, flavonols, flavanonols, isoflavones, flavanols (catechins) and anthocyanidins (Table 1). The structural variation of flavonoids may come from various patterns of substitution through enzymatic reactions including hydroxylation, methoxylation, sulfonation, acylation, prenylation, or glycosylation. Flavonoids are most frequently present as conjugates in glycosides and polymers that are water soluble and degraded to variable extents in the digestive system.66| Groups | Structure | Examples |
|---|---|---|
| Flavones |

|
Apigenin, luteolin, tangeretin, nobiletin, 5-hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone |
| Flavonols |
|
Kaempferol, myricetin, quercetin, isorhamnetin |
| Flavanols |

|
Catechin, gallocatechin, epicatechin, epigallocatechin-3-gallate |
| Flavanones |

|
Naringenin, hesperetin, eriodictyol |
| Isoflavones |
|
Daidzein, genistein, glycitein |
| Anthocyanidins |
|
Cyanidin, delphinidin, pelargonidin |
There are also a wide variety of types of naturally occurring flavonoids—at least 2000. Some of them exhibit a broad spectrum of pharmacological properties such as antioxidant, free radical-scavenging, anti-inflammatory, anti-carcinogenic, antiviral, anti-bacterial, anti-thrombogenic and anti-atherogenic activities. It is reported that human intake of all flavonoids is a few hundred milligrams to 650 mg per day in our diet.66 Significant scientific evidence shows that flavonoids have many beneficial health effects for human beings. Many studies show that flavonoid intake improves health and fights off chronic diseases.67 Among the biological properties of flavonoids, anti-inflammatory activity is attracting growing interest in managing chronic inflammatory diseases. The biochemical and molecular mechanisms, as well as the signaling pathways, of flavonoids implicated in chronic inflammatory diseases are described below.
Flavones
Apigenin, a flavonoid present in parsley and celery (Fig. 9), is found to inhibit HIF-1α and VEGF expression by blocking PI3K/Akt signaling or LPS-induced pro-inflammatory cytokines expression by inactivating NF-κB through the suppression of p65 phosphorylation.1 Lupus is autoimmune disease, characterized by production of autoantibodies to attack nuclear antigens and immune complex deposition in organs.68 In an in vivo study, apigenin decreased response of Th1 and Th17 cells to major lupus autoantigen, and subsequently suppressed the ability of lupus B cells to produce pathogenic autoantibodies that limit the inflammatory state in SFN-1 mice. Apigenin also downregulated the expression of COX-2 and cellular FLICE-like inhibitory protein (c-FLIP) in immune cells as well as causing activated immune cells to undergo apoptosis, thus suppressing inflammation in lupus.69 In the pathogenesis of rheumatoid arthritis, it is reported that inflammatory cytokines produced by fibroblast-like synoviocytes are involved in joint destruction. Apigenin is known to induce ROS production and cause apoptosis through oxidative stress-activated ERK1/2 pathway in fibroblast-like synoviocytes.70 Moreover, intake of apigenin also showed immunomodulating effects triggered by TNF-α in a mouse model of rheumatoid arthritis.71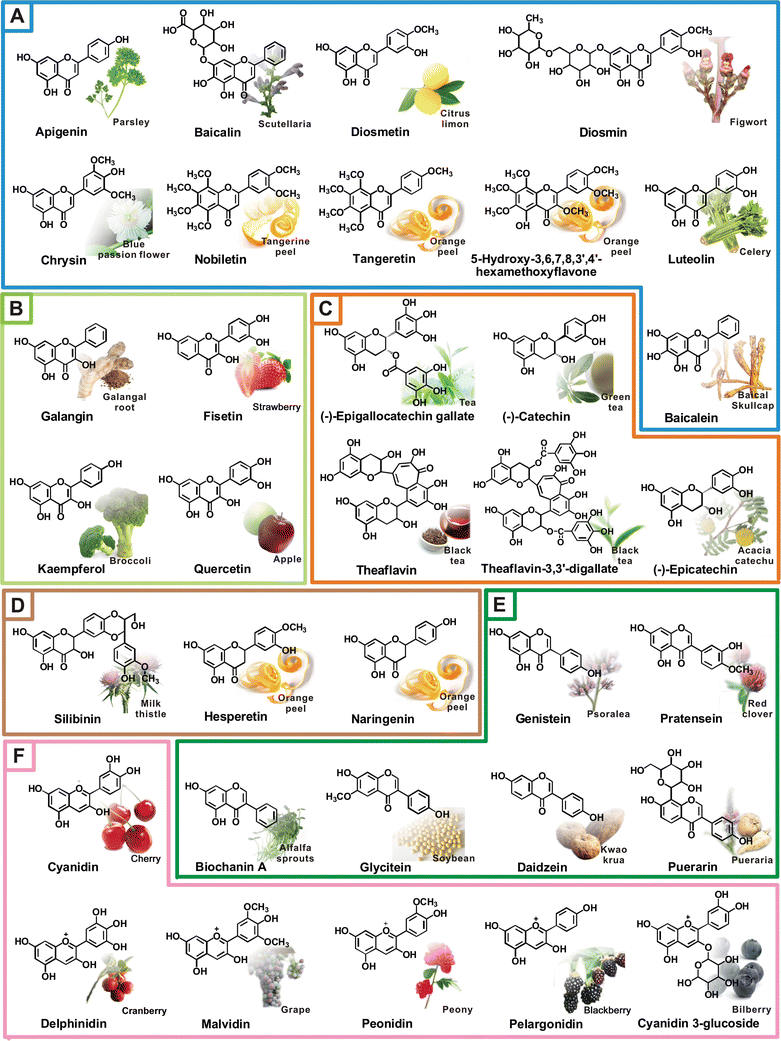 | ||
| Fig. 9 Representative natural flavonoids and their dietary sources. (A) flavones, (B) flavonols, (C) flavanols, (D) flavanones, (F) isoflavones, (G) anthocyanidins. | ||
Luteolin is prevalent in thyme and also exists in beets, brussels sprouts, cabbage and cauliflower, and is shown to possess great antioxidative activity. It is known that the activation of microglia and cytokine production is important in neurodegenerative diseases. Treatment with luteolin strongly suppressed IFNγ-induced CD40 expression and the production of TNF-α and IL-6 through downregulated phosphorylation of STAT-1 in microglia.72 Also, luteolin significantly inhibited LPS-induced activation of microglia by inhibiting JNK phosphorylation and activation of AP-1, increasing dopamine uptake and decreasing excessive TNF-α, NO and superoxide production in microglia,73,74 suggesting the neuroprotective effect of luteolin against brain injury. In addition, luteolin was found to regulate MAPK and NF-κB signaling that inhibits TNF-α induced IL-8 production, which is an important inflammatory cytokine involved in maintaining the inflammatory state in inflammatory bowel diseases.75 Luteolin also decreases TNF-α, IL-1 and MCP-1 gene expression and increases adiponectin and leptin levels through the enhancement of transcriptional activity of PPARγ in 3T3-L1 adipocytes that might improve obesity-driven insulin resistance.76
Citrus peel is a rich source of polymethoxyflavones, such as tangeretin and nobiletin, and exhibits a broad spectrum of biological activities, including modulation of inflammatory-derived cancer development.77 It is shown that tangeretin suppresses IL-1β-induced COX-2 expression through inhibiting activation of MAPK and Akt.1 In an in vivo study, nobiletin was found to lower levels of eotaxin, a potent eosinophil chemoattractant cytokine that relieves the infiltration of eosinophils and airway inflammation in asthmatic rats.78 It is suggested that the inhibition of foam cells forming macrophage and ox-LDL uptake is one of the targets for atherosclerosis. Nobiletin inhibited macrophage foam-cell formation through reducing metabolism of β-VLDL, is primarily taken up by macrophages via the LDL receptor in cultured murine J774A.1 macrophages.79 Several studies demonstrate that nobiletin can improve arthritic diseases as evidenced by decreasing proinflammatory cytokine production in human synovial cells and downregulating gene expression of MMPs in human synovial fibroblasts,80 as well as in collagen-induced arthritic (CIA) mice.81 Nobiletin can also inhibit leukocyte infiltration, protein expression of iNOS and COX-2 as well as tumorigenesis in mouse skin.1 In our previous studies, we reported that a metabolite of nobiletin (3′,4′-didemethylnobiletin) and 5-hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone, a hydroxylated PMF in citrus peel, inhibited 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-induced skin inflammation and tumor promotion by suppressing MAPK and PI3K/Akt signaling pathways.82,83
Recently, two new flavones isolated from Cirsium japonicum DC, pectolinarin and 5,7-dihydroxy-6,4′-dimethoxyflavone were found to reduce high-carbohydrate/high-fat diet-induced diabetes in rat through decreasing plasma glucose and increasing adiponectin levels that may improve glucose and lipid homeostasis.84
Flavonols
Quercetin, an ubiquitous plant secondary metabolite, is found abundant in onions, broccoli, apples, grapes, wine, tea, and leafy green vegetables, is well known as a potent antioxidant and anti-inflammatory agent. Recently, it was shown to possess good anti-atherosclerotic activity. In human umbilical vein endothelial cells (HUVECs), quercetin treatment strongly attenuated the inflammation-induced upregulated expression of VCAM-1, ICAM-1 and monocyte chemoattractant protein-1 (MCP-1), which may contribute to its interference with the interaction between monocytes and vascular endothelial cells during the early stages of atherosclerosis.85 Oral feeding of quercetin (64-mg/kg body weight daily) significantly inhibited atherosclerotic lesion size in the aortic sinus and thoracic aorta through reducing superoxide production, improving endothelial NO synthase (eNOS) function and decreasing plasma-sP-selectin levels in the apolipoprotein E (ApoE)(-/-) gene-knockout mouse.86 Quercetin also decreased circulating inflammatory markers, including IFNγ, IL-1α and IL-4 in high fat diet animal models and therefore may improve inflammation or obesity-associated disorder.87 In addition, consumption of quercetin is found to decrease systolic blood pressure and plasma oxidised LDL in obese subjects (aged 25–65 years) without affecting liver and kidney functions.88 When rats were fed a diet of rutin, a quercetin glycoside, there was markedly attenuated dextran sulfate sodium (DSS) induced gene expression of IL-1β and IL-6 in colonic mucosa and decreased intestinal colitis.89 Quercetin was found to inhibit 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis of rats possibly through downregulation of TNF-α-induced NF-κB activation.90Kaempferol, another flavonol-type flavonoid present in broccoli, tea and various vegetables, is considered to improve osteoporosis. For example, treatment with kaempferol stimulated differentiation and mineralization of murine pre-osteoblastic cell lines that may contribute to the prevention of bone loss.91 In the pathology of osteoporosis, proinflammatory cytokines TNF-α is important for bone disruption and osteoclastogenic cytokine production. Kaempferol was reported to antagonize TNF-α-induced p65 translocation and production of IL-6 and MCP-1as well as RANKL-triggered osteoclast precursor cell differentiation. These data indicate that kaempferol exerted a profound anti-osteoclastogenic effect.92 Advanced glycation end products (AGE) are oxidative products formed from nonenzymatic reaction of reducing sugars with free amino groups of proteins. AGE is reported to be involved in diabetic complications and various age/inflammation-related chronic diseases through generation of ROS and activation of inflammatory signaling cascades.93 Studies show that supplementation of kaempferol in mice reduced AGE-induced activation of NADPH oxidase and proinflammatory gene expression through modulating the NF-κB signaling cascade.94 When aged Sprague-Dawley rats were fed with a diet containing kaempferol, the activation of T cell was inhibited and COX-2, iNOS and MCP-1 gene expressions of kidney through modulation of NIK/IKK and MAPK signalings were reduced possibly reducing kidney disease.95 In addition, both kaempferol and quercetin could significantly improve insulin-stimulated glucose uptake in mature 3T3-L1 adipocytes by acting as agonists of PPARγ that may exert a beneficial effect on hyperglycemia and insulin resistance.96
Flavanols
Tea is a popular beverage worldwide. It is produced from the leaves of Camellia sinensis. There are more than 300 different kinds of tea made by different manufacturing processes. Among these, green tea has attracted attention for its health benefits contributed by catechin compounds including epigallocatechin-3-gallate (EGCG), epigallocatechin (EGC), epicatechin-3-gallate (ECG), and epicatechin (EC). Numerous studies demonstrate the potential of green tea in iron-chelating, radical-scavenging, anti-inflammatory and brain-permeable activities thus preventing cardiovascular, chronic and neurodegenerative diseases.97,98In vivo study shows that pretreatment with (±)-catechin protected dopaminergic neurons in the substantia nigra against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced toxicity through modulation of the phosphorylation of c-Jun N-terminal kinase (JNK) and GSK-3β.99 Pretreatment with epicatechin also attenuated ox-LDL induced neurotoxicity in mouse-derived striatal neurons.100EGCG is the most abundant polyphenolic compound in green tea. EGCG is found to inhibit LPS-induced microglial activation, NO and TNF-α production as well as subsequent neuronal injury both in the human dopaminergic cell line SH-SY5Y and in primary rat mesencephalic culture, suggesting the neuroprotective activity of EGCG may be inhibition of microglial activation.101 Neuronal damage and death caused by excessive NO is one of the pathological mechanisms in neurodegeneration. EGCG was found to inhibit NO-induced PC12 cell death by scavenging ROS.102
Obesity is a low-grade inflammatory state and is predisposed to an increased incidence of diabetes and CVD. A number line of evidence shows that EGCG possesses excellent activity against obesity-associated pathogenesis and metabolic disorders. In high fat diet-induced obesity animal models, supplementation with dietary EGCG reduced body weight gain and body fat, plasma cholesterol and MCP-1 levels, and decreased lipid accumulation in hepatocytes as well as attenuated insulin resistance.103 In addition, feeding EGCG improved high fat diet-induced non-alcoholic steatohepatitis in mice by decreasing triglyceride and cholesterol levels, lipid peroxidation, and expression of α-smooth muscle actin (α-SMA) in liver tissue. The liver protective effect of EGCG in mice on obesitywas further evidenced by expressions of insulin receptor substrate-1 (IRS-1) and phosphorylated IRS-1 (pIRS-1), and decreasing TNF-α and NF-κB signaling in liver tissues thus improving nonalcoholic steatohepatitis and insulin resistance.104 Treatment with EGCG on regulatory T cells from obese individuals significantly enhanced the function of regulatory T cells by the production of anti-inflammatory cytokine IL-10.105 Moreover, EGCG is reported to inhibit TNF-α stimulated activation of activator protein-1 (AP-1) and secretion of MCP-1 porcine aortic endothelial cells that work against vascular endothelial inflammation and atherosclerosis.106
EGCG also protects against bone, muscular and skeletal diseases. In synoviocytes and chondrocytes, upregulation of MAPK is critical for proinflammatory cytokine-induced signaling that causes production of several mediators of cartilage damage in an arthritic joint. EGCG was reported to modulate IL-1β-induced activation of MAPKs and DNA binding activity of AP-1 in osteoarthritis chondrocytes.107 Synovial fibroblasts produce metalloproteinases (MMPs) induced by proinflammatory cytokine TNF-α, which are involved in destroying cartilage and bone in Rheumatoid arthritis. Treatment with EGCG suppresses TNF-α-induced production of MMP-1 and MMP-3 through downregulating phosphorylation of MAPKs, such as ERK1/2, p38, JNK and the activation of AP-1 transcription factor in RA synovial fibroblasts.108 Furthermore, EGCG suppresses osteoclast differentiation through down-regulating expression of nuclear factor activated T cells c1 (NF-ATc1) and reduces histologic scores in experimental arthritis in mice that may improve osteoporosis and rheumatoid arthritis.109
Flavanones
Much of the activity of flavanones from citrus peels appears to have an impact on vascular endothelial cells that reveal protective effect against atherosclerosis and cardiovascular diseases. In high cholesterol-fed New Zealand white rabbits, diet supplemented with naringenin were shown to reduce gene expression of vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1) and MCP-1 in endothelial cells. Plasma lipoprotein levels, total cholesterol, triglyceride, and high-density lipoprotein (HDL) were also decreased by naringenin feeding.110,111 These findings suggest that naringenin can prevent hypercholesterolemia caused fatty liver and acts on interfering immune cells and vascular endothelial cells as well as macrophage infiltration against atherosclerosis. Naringin is shown to inhibit TNF-α-induced expression of MMP-9 in vascular smooth muscle cells (VSMC) through modulation of the PI3K/AKT/mTOR/p70S6K pathway and suppression of the transcriptional activity of AP-1 and NF-κB.112 Emerging studies demonstrate that obesity is a major risk factor for atherosclerosis and cardiovascular disease. Adiponectin secreted from adipocytes was found to suppress atherogenesis by inhibiting monocyte adhesion, reducing macrophage uptake ox-LDL and suppressing the accumulation of ox-LDL in vascular wall.113 Studies show that naringin and hesperetin, other flavanones found in large quantity in citrus peel, enhanced adiponectin transcription in differentiated 3T3-L1 adipocytes through activation of PPARγ.114 Naringin is also reported to inhibit high fat diet-induced atherosclerosis and normalize hyperinsulinemia and obesity in low-density lipoprotein receptor-null mice.115Cytokines promote proliferation and migration of vascular smooth muscle cells and play an important role in atherosclerosis. Hesperetin is found to inhibit platelet-derived growth factor (PDGF)-BB induced proliferation of primary cultured rat aortic vascular smooth muscle cells through regulation of Akt and MAPKs signaling that results in cell cycle arrest.116 Moreover, hesperetin also increased NO releases from human umbilical vein endothelial cells through up-regulation eNOS expression that may improve function of vascular endothelial cells.117 In addition to the anti-atherosclerosis effect, studies also demonstrate the protective activity of naringin and hesperetin against neuroinflammatory injury. Both naringin and hesperetin attenuated LPS/IFNγ-induced TNF-α production through glial cells, while naringin showed a greater effect as evidenced by inhibition of iNOS expression by modulation of p38 MAPK and STAT-1 signaling.118
Isoflavonoids
The health benefits of isoflavonoids from soybeans was recognized in recent years, especially about its anti-atherosclerotic functions. Consumption of soy-based diets is associated with a lower risk of cardiovascular disease in humans and reduced atherosclerosis in several experimental animals.119 These effects are related to their antioxidant, anti-inflammatory and antithrombogenic properties through inhibition of endothelial and inflammatory cell activation and reduction in recruitment of leukocytes, as well as foam cell formation. Genistein, the major isoflavone abundantly present in soybean, is known for its role in the regulation of vascular function and protection against atherosclerosis.120 Treatment with genistein markedly inhibited TNF-α-induced cell adhesion molecule CD62E and CD106 expression and subsequent monocyte adhesion in HUVECs and human brain microvascular endothelial cells.121 Genistein also decreased the interaction between monocyte and endothelial cells through the activation of PPARγ that inhibits of monocyte adhesion in culture cells and animals.122,123 In an in vitro study, genistein inhibited LPS-induced MCP-1 secretion from macrophages that contribute to reduced monocyte migration.124In vivo administration of genistein inhibits LPS-induced expression of iNOS and nitrotyrosine protein in vascular tissue that prevents hypotension and vascular alterations.125Genistein is also known to have a potential effect on rheumatoid arthritis, diabetes, metabolic disorders, neurodegenerative diseases and chronic colitis by modulation of inflammatory response. For instance, genistein inhibits production of proinflammatory molecules NO, IL-1β, and HC gp-39, known as markers of cartilage catabolism in LPS-stimulated human chondrocytes.126 In a collagen-induced rheumatoid arthritis rat, genistein modulated Th1-predominant immune response by suppressing the secretion of IFNγ and increasing IL-4 production that balances the inflammatory state.127 NAFLD is an obesity-related fatty liver disease caused by proinflammatory cytokine TNF-α and ILs and leads to the dysfunction of hepatocytes and increase fatty acid uptake. Genistein is reported to reduce high fat diet-induced steatohepatitis through decreasing plasma TNF-α levels and improved liver function in rat.128 Supplementation with genistein and daidzein also decreases mRNA levels of TNF-α, IL-1β and MCP-1 in plasma and liver tissue in obese Zucker rats, suggesting a preventive effect of dietary genistein on steatohepatitis through its anti-inflammatory activity.129 Furthermore, intake of high-dose isoflavones (genistein, daidzein, and glycitein) significantly reduced serum TNF-α and increased adiponectin levels in women, indicating that isoflavonids could regulate inflammatory conditions and improve metabolic parameters.130
In addition, genistein attenuated LPS-induced loss of dopamine uptake in rat mesencephalic neuron-glia cultures through reducing microglia activation and production of NO and TNF-α131 may protect dopaminergic neuron injury-caused pathogenesis of Parkinson's disease. In Alzheimer's disease, accumulation of astrocytes at sites of Aβ deposition is the earliest neuropathological changes that initiate the inflammatory response. Treatment with genistein reduced Aβ-induced production of inflammatory mediator iNOS, COX-2, TNF-α and IL-1β in astrocytes, possibly through activation of PPARs.132 Also, oral treatment with genistein reduced TNBS-induced chronic colitis by inhibiting expression of COX-2 mRNA and protein as well as the myeloperoxidase (MPO) activity in rat colon that exerts beneficial anti-inflammatory effects against inflammatory bowel disease.133 Genistein is a well-known tyrosine kinase inhibitor. In an in vitro study, genistein was found to prevent IL-1β/IFNγ-induced expression of COX-2 and iNOS as well as produce PGE2 and NO in human islets that may improve insulin resistance and prevent pathogenesis of diabetes.134
Anthocyanidins
Anthocyanidins are common plant pigments that give the red and blue colors in some fruits and vegetables such as blueberries and grapes. Epidemiological investigations and animal experiments indicate that anthocyanins may contribute to chemopreventive activities of various chronic inflammatory diseases.135,136 Anthocyanins-rich berries were demonstrated to possess a broad spectrum of biological properties, including antioxidant, cadioprotective, neuroprotective, anti-inflammatory and anticancer.137 In an animal study, cyanidin was reported to reduce PGE2 levels in paw tissues and TNF-α levels in serum in adjuvant-induced arthritis.138 Damage and apoptosis of vascular endothelial cells is frequently observed in atheromatous plaques and contributes to pathology of atherosclerosis. It has been shown that cyanidin inhibited TNF-α-induced endothelial cell apoptosis, elevated expression of eNOS and thioredoxin may improve vascular endothelial cell function and vasculopathy.139 VEGF is known as a major pro-angiogenic and pro-atherosclerotic factor. Both cyanidin and delphinidin, other major anthocyanidins present in pigmented fruits and vegetables, inhibit PDGF-induced VEGF expression through down-regulation of p38 MAPK and JNK signalings in vascular smooth muscle cells.140Delphinidin also shows protective effects against cardiovascular disease. It is suggested that proliferation of vascular endothelial cells is important in the pathogenesis of atherosclerosis.141 Delphinidin treatment inhibits serum and VEGF-induced bovine aortic endothelial cell proliferation through modulation of ERK and also results in cell cycle arrest.142 Also, delphinidin increased eNOS expression by mediating the MAP kinase pathway, thus preventing bovine aortic endothelial cell apoptosis.143 In addition, delphinidin was found to fight against ox-LDL-induced damage in HUVECs and regulate apoptotic molecule expression.144 These studies suggest delphinidin may be important in preventing both plaque development and atherosclerosis.
Proanthocyanidines and theaflavins
Proanthocyanidins (PAs), also called condensed tannins, are ubiquitous and present as the second most abundant group of natural phenolics after lignin. Oligomers and polymers of proanthocyanidins are widely found in the plant kingdom in fruits, cereals, berries, beans, nuts, cocoa and wine. The abundance of proanthocyanidins in plants makes them an important part of the human diet and are reported to exhibit a wide range of biological effects.145 A recent study demonstrated that dietary grape seed proanthocyanidins (GSPs) is effective against ultraviolet (UV) radiation-induced skin tumor in mice. Dietary GSPs inhibited UVB-induced infiltration of proinflammatory leukocytes and the levels of myeloperoxidase, cyclooxygenase-2 (COX-2), prostaglandin (PG) E(2), cyclin D1 and proliferating cell nuclear antigen (PCNA) in the skin and skin tumors.146 PAs are shown to mediate several anti-inflammatory mechanisms involved in the development of cardiovascular disease by modulation of monocytes adhesion in the inflammatory process of atherosclerosis.145 PAs also exhibit in vivo hepatoprotective and anti-fibrogenic effects against dimethylnitrosamine-induced liver injury.147Theaflavins, a mixture of theaflavin (TF-1), theaflavin-3-gallate (TF-2a), theaflavin-3′-gallate (TF-2b), and theaflavin-3,3′-digallate (TF-3) are the major components of black tea. We previously reported that TF-3 exerts its anti-inflammatory and cancer chemopreventive actions by suppressing the activation of NFκB through inhibition of IKK activity.148 We also found that epicatechins in green tea and theaflavins in black tea are able to reduce the concentration of methylglyoxal in physiological phosphate buffer conditions.149 Among these black tea components, TF-3 is generally considered to be the more effective component for a protective effect against inflammatory processes.150
Conclusion
Chronic inflammation is linked to numerous human diseases. Increasingly epidemiological and experimental studies demonstrate that modulation of inflammatory response by natural phytochemicals plays an important role in the prevention, mitigation, and treatment of many chronic inflammatory diseases. Flavonoids are a group of natural compounds widely present in vegetables, fruits and edible plants that possess potent biological activities. Dietary intake of flavonoids is suggested to prevent and lower the risk of chronic diseases. In this review, we discussed the possible mechanisms by which flavonoids play a role in the regulation of the inflammatory processes associated with atherosclerosis (cardiovascular disease), neurodegenerative diseases, obesity, metabolic disorders, bone, muscular and skeletal diseases, and chronic inflammatory diseases, as well as cancers. The anti-inflammatory activity of flavonoids is seen through several mechanisms involving the modulation of inflammatory signaling, reduction of inflammatory molecule production, diminishing recruitment and activation of inflammatory cells, regulation of cellular function and their antioxidative property. Regarding the safety, ability and the anti-inflammatory effects of flavonoids, they are likely to have a potential role in preventive and therapeutic roles in chronic inflammatory conditions. However, additional, extensive research of flavonoids in strengthening the network of inflammatory response needs to be studied in the future.Abbreviations
| Aβ | amyloid β peptide |
| AGE | advanced glycation end products |
| AP-1 | activator protein-1 |
| ApoE | apolipoprotein E |
| APP | amyloid precursor protein |
| α-SMA | α-smooth muscle actin |
| ATF | activator transcription factor |
| CBP/p300 | cAMP response element binding protein/p300 |
| CD | Crohn's disease |
| C/EBP | CCAAT enhancer binding protein |
| c-FLIP | cellular FLICE-like inhibitory protein |
| CIA | collagen-induced arthritic |
| COPD | chronic obstructive pulmonary disease |
| COX-2 | cyclooxygenase-2 |
| CRP | c-active protein |
| CVD | cardiovascular disease |
| DSS | dextran sulfate sodium |
| EC | epicatechin |
| ECG | epicatechin-3-gallate |
| EGC | epigallocatechin |
| EGCG | epigallocatechin-3-gallate |
| ERK1/2 | external signal regulated kinase 1/2 |
| eNOS | endothelial NO synthase |
| GLUT4 | glucose transporter type 4 |
| GSK-3β | glycogen synthase kinase 3β |
| HC gp-39 | human cartilage glycoprotein-39 |
| HDL | high-density lipoprotein |
| HIF-1α | hypoxia inducible factor-1α |
| H2O2 | hydrogen peroxide |
| HUVEC | human umbilical vein endothelial cell |
| ICAM-1 | intercellular adhesion molecule-1 |
| IL | Interleukin |
| IL-1ra | IL-1 receptor antagonist protein |
| IBD | inflammatory bowel disease |
| IFN-γ | interferon-γ |
| IKK | IκB kinase |
| iNOS | inducible nitric oxide synthase |
| IRS-1 | insulin receptor substrate-1 |
| JNK | c-Jun N-terminal kinase |
| 5-LOX | 5-lipoxygenase |
| LPS | lipopolysaccharides |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| MMP | matrix metalloproteinase |
| MPO | myeloperoxidase |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mTOR | mammalian target of rapamycin |
| NADPH oxidase | nicotinamide adenine dinucleotide phosphate-oxidase |
| NAFLD | non-alcoholic fatty liver disease |
| NF-ATc1 | nuclear factor of activated T cells c1 |
| NF-κB | nuclear factor-kappa B |
| NFAT | nuclear factor of activated T-cells |
| NIK | NF-κB-inducing kinase |
| NO | nitric oxide |
| NSAID | nonsteroidal anti-inflammatory drugs |
| 1O2 | singlet oxygen |
| •O2− | superoxide anion |
| •OH | hydroxyl radical |
| ox-LDL | oxidized low-density-lipoprotein |
| p70S6K | p70 ribosomal S6 kinase |
| PDGF | platelet-derived growth factor |
| PGE2 | prostaglandin E2 |
| PI3K | phosphoinositide-3 kinase |
| pIRS-1 | phosphorylated IRS-1 |
| PKC | protein kinase C |
| PPARγ | peroxisome proliferator-activated receptor γ |
| RA | rheumatoid arthritis |
| RANKL | receptor activator for nuclear factor κB ligand |
| ROS | reactive oxygen species |
| SOCS | suppressor of cytokine signaling |
| STAT | signal transducers and activators of transcription |
| TGF-β | transforming growth factor-β |
| Th cell | T helper cell |
| TNBS | 2,4,6-trinitrobenzene sulfonic acid |
| TNF-α | tumor necrosis factor-α |
| TPA | 12-O-tetradecanoyl-phorbol-13-acetate |
| UC | ulcerative colitis |
| VCAM-1 | vascular cell adhension molecule-1 |
| VEGF | vascular endothelial growth factor |
| VLDL | very-low-density lipoprotein |
| VSMC | vascular smooth muscle cells |
References
- M. H. Pan, C. S. Lai, S. Dushenkov and C. T. Ho, J. Agric. Food Chem., 2009, 57, 4467 CrossRef CAS.
- R. Medzhitov, Nature, 2008, 454, 428 CrossRef CAS.
- P. C. Calder, R. Albers, J. M. Antoine, S. Blum, R. Bourdet-Sicard, G. A. Ferns, G. Folkerts, P. S. Friedmann, G. S. Frost, F. Guarner, M. Lovik, S. Macfarlane, P. D. Meyer, L. M'Rabet, M. Serafini, E. W. van, L. J. van, D. W. Vas, S. Vidry, B. M. Winklhofer-Roob and J. Zhao, Br.J.Nutr, 2009, 101(Suppl. 1), S1.
- P. Libby, Nutr. Rev., 2007, 65, S140 Search PubMed.
- A. Garcia-Lafuente, E. Guillamon, A. Villares, M. A. Rostagno and J. A. Martinez, Inflammation Res., 2009, 58, 537 CrossRef CAS.
- L. I. Mennen, D. Sapinho, B. A. de, N. Arnault, S. Bertrais, P. Galan and S. Hercberg, J. Nutr., 2004, 134, 923 CAS.
- P. M. Ridker and J. D. Silvertown, J. Periodontol., 2008, 79, 1544 CrossRef CAS.
- Y. Higashi, K. Noma, M. Yoshizumi and Y. Kihara, Circ. J., 2009, 73, 411 CrossRef CAS.
- P. Libby, Y. Okamoto, V. Z. Rocha and E. Folco, Circ. J., 2010, 74, 213 CrossRef CAS.
- J. Mestas and K. Ley, Trends Cardiovasc. Med., 2008, 18, 228 CrossRef CAS.
- E. A. Liehn, A. Zernecke, O. Postea and C. Weber, Arch. Physiol. Biochem., 2006, 112, 229 CrossRef CAS.
- K. J. Moore and M. W. Freeman, Arterioscler., Thromb., Vasc. Biol., 2006, 26, 1702 Search PubMed.
- R. L. Wilensky and D. Hamamdzic, Curr. Opin. Cardiol., 2007, 22, 545 CrossRef.
- E. S. Nakou, E. N. Liberopoulos, H. J. Milionis and M. S. Elisaf, Curr. Vasc. Pharmacol., 2008, 6, 258 Search PubMed.
- G. J. Blake and P. M. Ridker, J. Intern. Med., 2002, 252, 283 CrossRef CAS.
- R. Klingenberg and G. K. Hansson, Eur. Heart J., 2009, 30, 2838 CrossRef CAS.
- C. K. Glass, K. Saijo, B. Winner, M. C. Marchetto and F. H. Gage, Cell, 2010, 140, 918 CrossRef CAS.
- S. T. Dheen, C. Kaur and E. A. Ling, Curr. Med. Chem., 2007, 14, 1189 CrossRef CAS.
- M. G. Tansey and M. S. Goldberg, Neurobiol. Dis., 2010, 37, 510 CrossRef CAS.
- R. E. Mrak and W. S. Griffin, J. Neuropathol. Exp. Neurol., 2007, 66, 683 CrossRef CAS.
- H. Braak, M. Sastre and T. K. Del, Acta Neuropathol., 2007, 114, 231 CrossRef CAS.
- R. L. Miller, M. James-Kracke, G. Y. Sun and A. Y. Sun, Neurochem. Res., 2009, 34, 55 CrossRef CAS.
- A. P. Kulkarni, L. A. Kellaway, D. K. Lahiri and G. J. Kotwal, Ann. N. Y. Acad. Sci., 2004, 1035, 147 CrossRef CAS.
- C. Infante-Duarte, S. Waiczies, J. Wuerfel and F. Zipp, J. Mol. Med., 2008, 86, 975 CrossRef CAS.
- G. Reaven, F. Abbasi and T. McLaughlin, Recent Prog.Horm.Res., 2004, 59, 207 CrossRef CAS.
- A. Fortuno, A. Rodriguez, J. Gomez-Ambrosi, G. Fruhbeck and J. Diez, J. Physiol Biochem., 2003, 59, 51 Search PubMed.
- J. M. Friedman and J. L. Halaas, Nature, 1998, 395, 763 CrossRef CAS.
- V. R. Drel, N. Mashtalir, O. Ilnytska, J. Shin, F. Li, V. V. Lyzogubov and I. G. Obrosova, Diabetes, 2006, 55, 3335 CrossRef CAS.
- S. Mehta and J. A. Farmer, Curr. Atheroscler. Rep., 2007, 9, 134 Search PubMed.
- G. S. Hotamisligil, N. S. Shargill and B. M. Spiegelman, Science, 1993, 259, 87 CrossRef CAS.
- T. Tzanavari, P. Giannogonas and K. P. Karalis, Current Directions in Autoimmunity, 2010, 11, 145 Search PubMed.
- J. G. Neels and J. M. Olefsky, J. Clin. Invest., 2005, 116, 33 CrossRef.
- H. Kanda, S. Tateya, Y. Tamori, K. Kotani, K. Hiasa, R. Kitazawa, S. Kitazawa, H. Miyachi, S. Maeda, K. Egashira and M. Kasuga, J. Clin. Invest., 2006, 116, 1494 CrossRef CAS.
- H. Florez, S. Castillo-Florez, A. Mendez, P. Casanova-Romero, C. Larreal-Urdaneta, D. Lee and R. Goldberg, Diabetes Res. Clin. Pract., 2006, 71, 92 CrossRef CAS.
- M. Bluher, Exp. Clin. Endocrinol. Diabetes, 2009, 117, 241 Search PubMed.
- K. T. Uysal, S. M. Wiesbrock and G. S. Hotamisligil, Endocrinology, 1998, 139, 4832 CrossRef CAS.
- G. S. Hotamisligil, Nature, 2006, 444, 860 CrossRef CAS.
- S. E. Kahn, Diabetologia, 2003, 46, 3 CAS.
- S. Malozowski and J. T. Sahlroot, N. Engl. J. Med., 2007, 357, 302 CrossRef CAS.
- R. Meshkani and K. Adeli, Clin. Biochem., 2009, 42, 1331 CrossRef CAS.
- W. W. Cheung, K. H. Paik and R. H. Mak, Pediatr. Nephrol., 2010, 25, 711 CrossRef.
- R. Anty, P. Gual, P. M. Huet, Y. L. Marchand-Brustel and A. Tran, Gastroenterol.Clin.Biol., 2007, 31, 1127 Search PubMed.
- R. Scrivo, F. M. Di, A. Spadaro and G. Valesini, Ann. N. Y. Acad. Sci., 2007, 1108, 312 CrossRef CAS.
- A. Manzo, M. Bombardieri, F. Humby and C. Pitzalis, Immunol. Rev., 2010, 233, 267 CrossRef CAS.
- B. Bartok and G. S. Firestein, Immunol. Rev., 2010, 233, 233 CrossRef CAS.
- G. R. Mundy, Nutr. Rev., 2007, 65, S147 Search PubMed.
- R. R. McLean, Curr.Osteoporos.Rep., 2009, 7, 134 Search PubMed.
- M. G. Neuman, Translational Research, 2007, 149, 173 Search PubMed.
- G. Monteleone and F. Caprioli, Clin. Sci., 2010, 118, 707 Search PubMed.
- B. Singh, S. Read, C. Asseman, V. Malmstrom, C. Mottet, L. A. Stephens, R. Stepankova, H. Tlaskalova and F. Powrie, Immunol. Rev., 2001, 182, 190 CrossRef CAS.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57 CAS.
- F. Balkwill and A. Mantovani, Lancet, 2001, 357, 539 CrossRef CAS.
- F. Colotta, P. Allavena, A. Sica, C. Garlanda and A. Mantovani, Carcinogenesis, 2009, 30, 1073 CrossRef CAS.
- A. Mantovani, P. Allavena, A. Sica and F. Balkwill, Nature, 2008, 454, 436 CrossRef CAS.
- L. M. Coussens and Z. Werb, Nature, 2002, 420, 860 CrossRef CAS.
- M. H. Pan and C. T. Ho, Chem. Soc. Rev., 2008, 37, 2558 RSC.
- M. B. Sporn, N. M. Dunlop, D. L. Newton and J. M. Smith, Fed.Proc., 1976, 35, 1332 Search PubMed.
- M. Hirohata, K. Ono and M. Yamada, Curr. Pharm. Des., 2008, 14, 3280 CrossRef CAS.
- V. Vingtdeux, U. Dreses-Werringloer, H. Zhao, P. Davies and P. Marambaud, BMC Neurosci., 2008, 9(suppl 2), S6 CrossRef.
- B. Ray and D. K. Lahiri, Curr. Opin. Pharmacol., 2009, 9, 434 CrossRef CAS.
- A. G. van, S. Vermeire and P. Rutgeerts, Minerva Gastroenterol.Dietol., 2007, 53, 249 Search PubMed.
- C. A. Dinarello, Cell, 2010, 140, 935 CrossRef CAS.
- K. K. Brown, S. A. Heitmeyer, E. B. Hookfin, L. Hsieh, M. Buchalova, Y. O. Taiwo and M. J. Janusz, Journal of Inflammation, 2008, 5, 22 Search PubMed.
- W. Zhao, S. G. Thacker, J. B. Hodgin, H. Zhang, J. H. Wang, J. L. Park, A. Randolph, E. C. Somers, S. Pennathur, M. Kretzler, F. C. Brosius, III and M. J. Kaplan, J. Immunol., 2009, 183, 2729 CrossRef CAS.
- B. B. Aggarwal, R. V. Vijayalekshmi and B. Sung, Clin. Cancer Res., 2009, 15, 425 CrossRef CAS.
- R. H. Liu, J.Nutr., 2004, 134, 3479S CAS.
- P. Knekt, J. Kumpulainen, R. Jarvinen, H. Rissanen, M. Heliovaara, A. Reunanen, T. Hakulinen and A. Aromaa, Am.J.Clin.Nutr., 2002, 76, 560 CAS.
- J. C. Oates, Autoimmunity, 2010, 43, 56 CrossRef CAS.
- H. K. Kang, D. Ecklund, M. Liu and S. K. Datta, Arthritis Res. Ther., 2009, 11, R59 CrossRef.
- G. C. Shin, C. Kim, J. M. Lee, W. S. Cho, S. G. Lee, M. Jeong, J. Cho and K. Lee, Chem.-Biol. Interact., 2009, 182, 29 CrossRef CAS.
- Y. Kumazawa, K. Kawaguchi and H. Takimoto, Curr. Pharm. Des., 2006, 12, 4271 CrossRef CAS.
- K. Rezai-Zadeh, J. Ehrhart, Y. Bai, P. R. Sanberg, P. Bickford, J. Tan and R. D. Shytle, J.Neuroinflammation., 2008, 5, 41 Search PubMed.
- S. Jang, K. W. Kelley and R. W. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7534 CrossRef CAS.
- H. Q. Chen, Z. Y. Jin, X. J. Wang, X. M. Xu, L. Deng and J. W. Zhao, Neurosci. Lett., 2008, 448, 175 CrossRef CAS.
- J. A. Kim, D. K. Kim, O. H. Kang, Y. A. Choi, H. J. Park, S. C. Choi, T. H. Kim, K. J. Yun, Y. H. Nah and Y. M. Lee, Int. Immunopharmacol., 2005, 5, 209 CrossRef CAS.
- L. Ding, D. Jin and X. Chen, J.Nutr.Biochem., 2009 Search PubMed.
- S. Li, M. H. Pan, C. Y. Lo, D. Tan, Y. Wang, F. Shahidi and C. T. Ho, Journal of Functional Foods, 2009, 1, 2 Search PubMed.
- Y. Q. Wu, C. H. Zhou, J. Tao and S. N. Li, Life Sci., 2006, 78, 2689 CrossRef CAS.
- S. C. Whitman, E. M. Kurowska, J. A. Manthey and A. Daugherty, Atherosclerosis, 2005, 178, 25 CrossRef CAS.
- N. Lin, T. Sato, Y. Takayama, Y. Mimaki, Y. Sashida, M. Yano and A. Ito, Biochem. Pharmacol., 2003, 65, 2065 CrossRef CAS.
- K. Imada, N. Lin, C. Liu, A. Lu, W. Chen, M. Yano, T. Sato and A. Ito, Biochem. Biophys. Res. Commun., 2008, 373, 181 CrossRef CAS.
- C. S. Lai, S. Li, C. Y. Chai, C. Y. Lo, S. Dushenkov, C. T. Ho, M. H. Pan and Y. J. Wang, Carcinogenesis, 2008, 29, 2415 CrossRef CAS.
- C. S. Lai, S. Li, C. Y. Chai, C. Y. Lo, C. T. Ho, Y. J. Wang and M. H. Pan, Carcinogenesis, 2007, 28, 2581 CrossRef CAS.
- Z. Liao, X. Chen and M. Wu, Arch. Pharmacal Res., 2010, 33, 353 Search PubMed.
- S. Tribolo, F. Lodi, C. Connor, S. Suri, V. G. Wilson, M. A. Taylor, P. W. Needs, P. A. Kroon and D. A. Hughes, Atherosclerosis, 2008, 197, 50 CrossRef CAS.
- W. M. Loke, J. M. Proudfoot, J. M. Hodgson, A. J. McKinley, N. Hime, M. Magat, R. Stocker and K. D. Croft, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 749 Search PubMed.
- L. K. Stewart, J. L. Soileau, D. Ribnicky, Z. Q. Wang, I. Raskin, A. Poulev, M. Majewski, W. T. Cefalu and T. W. Gettys, Metab., Clin. Exp., 2008, 57, S39 Search PubMed.
- S. Egert, A. Bosy-Westphal, J. Seiberl, C. Kurbitz, U. Settler, S. Plachta-Danielzik, A. E. Wagner, J. Frank, J. Schrezenmeir, G. Rimbach, S. Wolffram and M. J. Muller, Br. J. Nutr., 2009, 102, 1065 CrossRef CAS.
- K. H. Kwon, A. Murakami, T. Tanaka and H. Ohigashi, Biochem. Pharmacol., 2005, 69, 395 CrossRef CAS.
- H. Kim, H. Kong, B. Choi, Y. Yang, Y. Kim, M. J. Lim, L. Neckers and Y. Jung, Pharm. Res., 2005, 22, 1499 CrossRef CAS.
- M. Miyake, N. Arai, S. Ushio, K. Iwaki, M. Ikeda and M. Kurimoto, Biosci., Biotechnol., Biochem., 2003, 67, 1199 CrossRef CAS.
- J. L. Pang, D. A. Ricupero, S. Huang, N. Fatma, D. P. Singh, J. R. Romero and N. Chattopadhyay, Biochem. Pharmacol., 2006, 71, 818 CrossRef CAS.
- S. F. Yan, R. Ramasamy and A. M. Schmidt, Nat.Clin.Pract.Endocrinol.Metab, 2008, 4, 285 Search PubMed.
- J. M. Kim, E. K. Lee, D. H. Kim, B. P. Yu and H. Y. Chung, Age, 2010, 32, 197 CrossRef CAS.
- M. J. Park, E. K. Lee, H. S. Heo, M. S. Kim, B. Sung, M. K. Kim, J. Lee, N. D. Kim, S. Anton, J. S. Choi, B. P. Yu and H. Y. Chung, J. Med. Food, 2009, 12, 351 CrossRef CAS.
- X. K. Fang, J. Gao and D. N. Zhu, Life Sci., 2008, 82, 615 CrossRef CAS.
- O. Weinreb, S. Mandel, T. Amit and M. B. Youdim, J. Nutr. Biochem., 2004, 15, 506 CrossRef CAS.
- P. V. Babu and D. Liu, Curr. Med. Chem., 2008, 15, 1840 CrossRef CAS.
- H. Ruan, Y. Yang, X. Zhu, X. Wang and R. Chen, Neurosci. Lett., 2009, 450, 152 CrossRef CAS.
- H. Schroeter, J. P. Spencer, C. Rice-Evans and R. J. Williams, Biochem. J., 2001, 358, 547 CrossRef CAS.
- R. Li, Y. G. Huang, D. Fang and W. D. Le, J. Neurosci. Res., 2004, 78, 723 CrossRef CAS.
- J. Y. Jung, C. R. Han, Y. J. Jeong, H. J. Kim, H. S. Lim, K. H. Lee, H. O. Park, W. M. Oh, S. H. Kim and W. J. Kim, Neurosci. Lett., 2007, 411, 222 CrossRef CAS.
- M. Bose, J. D. Lambert, J. Ju, K. R. Reuhl, S. A. Shapses and C. S. Yang, J.Nutr., 2008, 138, 1677 CAS.
- T. Ueno, T. Torimura, T. Nakamura, R. Sivakumar, H. Nakayama, S. Otabe, X. Yuan, K. Yamada, O. Hashimoto, K. Inoue, H. Koga and M. Sata, Int.J.Mol.Med., 2009, 24, 17 Search PubMed.
- J. M. Yun, I. Jialal and S. Devaraj, Br. J. Nutr., 2010, 103, 1771 CrossRef CAS.
- Y. Zheng, M. Toborek and B. Hennig, Metabolism, 2010 Search PubMed.
- R. Singh, S. Ahmed, C. J. Malemud, V. M. Goldberg and T. M. Haqqi, J. Orthop. Res., 2003, 21, 102 CrossRef CAS.
- H. J. Yun, W. H. Yoo, M. K. Han, Y. R. Lee, J. S. Kim and S. I. Lee, Rheumatol. Int., 2008, 29, 23 CrossRef CAS.
- A. Morinobu, W. Biao, S. Tanaka, M. Horiuchi, L. Jun, G. Tsuji, Y. Sakai, M. Kurosaka and S. Kumagai, Arthritis Rheum., 2008, 58, 2012 CrossRef CAS.
- C. H. Lee, T. S. Jeong, Y. K. Choi, B. H. Hyun, G. T. Oh, E. H. Kim, J. R. Kim, J. I. Han and S. H. Bok, Biochem. Biophys. Res. Commun., 2001, 284, 681 CrossRef CAS.
- S. C. Choe, H. S. Kim, T. S. Jeong, S. H. Bok and Y. B. Park, J. Cardiovasc. Pharmacol., 2001, 38, 947 CrossRef CAS.
- E. J. Lee, D. I. Kim, W. J. Kim and S. K. Moon, Mol. Nutr. Food Res., 2009, 53, 1582 CrossRef CAS.
- H. Ekmekci and O. B. Ekmekci, Clin.Appl.Thromb.Hemost., 2006, 12, 163 Search PubMed.
- L. Liu, S. Shan, K. Zhang, Z. Q. Ning, X. P. Lu and Y. Y. Cheng, Phytother. Res., 2008, 22, 1400 CrossRef CAS.
- E. E. Mulvihill, J. M. Assini, B. G. Sutherland, A. S. DiMattia, M. Khami, J. B. Koppes, C. G. Sawyez, S. C. Whitman and M. W. Huff, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 742 Search PubMed.
- Y. R. Jin, X. H. Han, Y. H. Zhang, J. J. Lee, Y. Lim, T. J. Kim, H. S. Yoo and Y. P. Yun, J. Cell. Biochem., 2008, 104, 1 CrossRef CAS.
- L. Liu, D. M. Xu and Y. Y. Cheng, J. Agric. Food Chem., 2008, 56, 824 CrossRef CAS.
- K. Vafeiadou, D. Vauzour, H. Y. Lee, A. Rodriguez-Mateos, R. J. Williams and J. P. Spencer, Arch. Biochem. Biophys., 2009, 484, 100 CrossRef CAS.
- S. Nagarajan, J. Nutr. Biochem., 2010, 21, 255 CrossRef CAS.
- H. Si and D. Liu, Curr. Med. Chem., 2007, 14, 2581 CrossRef CAS.
- Y. W. Lee and W. H. Lee, J. Nutr. Biochem., 2008, 19, 819 CrossRef CAS.
- B. K. Chacko, R. T. Chandler, T. L. D'Alessandro, A. Mundhekar, N. K. Khoo, N. Botting, S. Barnes and R. P. Patel, J.Nutr., 2007, 137, 351 CAS.
- S. Kim, H. J. Shin, S. Y. Kim, J. H. Kim, Y. S. Lee, D. H. Kim and M. O. Lee, Mol. Cell. Endocrinol., 2004, 220, 51 CrossRef CAS.
- S. Nagarajan, R. L. Burris, B. W. Stewart, J. E. Wilkerson and T. M. Badger, J.Nutr., 2008, 138, 332 CAS.
- A. Bermejo, A. Zarzuelo and J. Duarte, J. Cardiovasc. Pharmacol., 2003, 42, 329 CrossRef CAS.
- S. Hooshmand, d. Y. Soung, E. A. Lucas, S. V. Madihally, C. W. Levenson and B. H. Arjmandi, J. Nutr. Biochem., 2007, 18, 609 CrossRef CAS.
- J. Wang, Q. Zhang, S. Jin, D. He, S. Zhao and S. Liu, Maturitas, 2008, 59, 405 CrossRef CAS.
- M. Yalniz, I. H. Bahcecioglu, N. Kuzu, O. K. Poyrazoglu, O. Bulmus, S. Celebi, B. Ustundag, I. H. Ozercan and K. Sahin, J. Gastroenterol. Hepatol., 2007, 22, 2009 CrossRef CAS.
- O. A. Gudbrandsen, H. Wergedahl and R. K. Berge, Nutrition, 2009, 25, 574 CrossRef CAS.
- C. Charles, J. Yuskavage, O. Carlson, M. John, A. S. Tagalicud, M. Maggio, D. C. Muller, J. Egan and S. Basaria, Menopause, 2009, 16, 395 CrossRef.
- X. Wang, S. Chen, G. Ma, M. Ye and G. Lu, NeuroReport, 2005, 16, 267 CrossRef CAS.
- S. L. Valles, P. Dolz-Gaiton, J. Gambini, C. Borras, A. Lloret, F. V. Pallardo and J. Vina, Brain Res., 2010, 1312, 138 CrossRef CAS.
- J. Seibel, A. F. Molzberger, T. Hertrampf, U. Laudenbach-Leschowski and P. Diel, Eur. J. Nutr., 2009, 48, 213 CrossRef CAS.
- J. A. Corbett, G. Kwon, M. H. Marino, C. P. Rodi, P. M. Sullivan, J. Turk and M. L. McDaniel, Am.J.Physiol, 1996, 270, C1581 CAS.
- D. X. Hou, M. Fujii, N. Terahara and M. Yoshimoto, J. Biomed. Biotechnol., 2004, 2004, 321 Search PubMed.
- W. R. Leifert and M. Y. Abeywardena, Nutr. Res., 2008, 28, 729 CrossRef CAS.
- S. Zafra-Stone, T. Yasmin, M. Bagchi, A. Chatterjee, J. A. Vinson and D. Bagchi, Mol. Nutr. Food Res., 2007, 51, 675 CrossRef CAS.
- Y. H. He, C. Xiao, Y. S. Wang, L. H. Zhao, H. Y. Zhao, Y. Tong, J. Zhou, H. W. Jia, C. Lu, X. M. Li and A. P. Lu, Zhongguo Zhong.Yao Za Zhi., 2005, 30, 1602 Search PubMed.
- J. W. Xu, K. Ikeda and Y. Yamori, Atherosclerosis, 2007, 193, 299 CrossRef CAS.
- M. H. Oak, J. E. Bedoui, S. V. Madeira, K. Chalupsky and V. B. Schini-Kerth, Br. J. Pharmacol., 2006, 149, 283 CrossRef CAS.
- J. C. Choy, D. J. Granville, D. W. Hunt and B. M. McManus, J. Mol. Cell. Cardiol., 2001, 33, 1673 CrossRef CAS.
- S. Martin, L. Favot, R. Matz, C. Lugnier and R. Andriantsitohaina, Biochem. Pharmacol., 2003, 65, 669 CrossRef CAS.
- S. Martin, G. Giannone, R. Andriantsitohaina and M. C. Martinez, Br. J. Pharmacol., 2003, 139, 1095 CrossRef CAS.
- C. Y. Chen, L. Yi, X. Jin, M. T. Mi, T. Zhang, W. H. Ling and B. Yu, Chem.-Biol. Interact., 2010, 183, 105 CrossRef CAS.
- S. E. Rasmussen, H. Frederiksen, K. K. Struntze and L. Poulsen, Mol. Nutr. Food Res., 2005, 49, 159 CrossRef CAS.
- S. D. Sharma and S. K. Katiyar, Pharm. Res., 2010, 27, 1092 CrossRef CAS.
- M. O. Shin, S. Yoon and J. O. Moon, Arch. Pharmacal Res., 2010, 33, 167 Search PubMed.
- M. H. Pan, S. Y. Lin-Shiau, C. T. Ho, J. H. Lin and J. K. Lin, Biochem. Pharmacol., 2000, 59, 357 CrossRef CAS.
- C. Y. Lo, S. Li, D. Tan, M. H. Pan, S. Sang and C. T. Ho, Mol. Nutr. Food Res., 2006, 50, 1118 CrossRef CAS.
- Y. C. Liang, D. C. Tsai, S. Y. Lin-Shiau, C. F. Chen, C. T. Ho and J. K. Lin, Nutr. Cancer, 2002, 42, 217 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |