Hydrogen storage in water-stable metal–organic frameworks incorporating 1,3- and 1,4-benzenedipyrazolate†
Hye Jin
Choi
a,
Mircea
Dincă
a,
Anne
Dailly
b and
Jeffrey R.
Long
*a
aUniversity of California, Berkeley, Department of Chemistry, Berkeley, CA 94720-1460, USA. E-mail: jrlong@berkeley.edu
bGeneral Motors Corporation, Chemical & Environmental Sciences Laboratory, Warren, MI 48090-9055, USA
First published on 4th November 2009
Abstract
Pyrazolate-bridged metal–organic frameworks incorporating tetrahedral Zn2+ ions are shown to exhibit a high chemical stability in boiling water, organic solvents, and acidic media, and are assessed for their hydrogen storage properties.
Broader contextThe development of a safe and effective means of storing hydrogen is critical to its utilization as a clean fuel in automobiles. Owing to their high surface areas, microporous metal–organic frameworks have emerged as promising candidates for storage materials, particularly at cryogenic temperatures. Many of the best frameworks, however, are constructed from organocarboxylate-bridging ligands and decompose with prolonged exposure to water vapor. The present work reports two new frameworks, built up of tetrahedral Zn2+ ions connected through 1,3- or 1,4-benzenedipyrazolate, which exhibit excellent stability to water, extreme temperatures, and even dilute acid. One of these materials displays a high hydrogen storage capacity, adsorbing 4.7 excess wt% of H2 at 77 K and 40 bar. Such materials are of importance for their ability to tolerate adventitious water that may be present in the storage tank or the hydrogen fuel. |
Introduction
Microporous metal–organic frameworks have attracted much recent attention, particularly for their potential applications in gas storage, molecular separations, and heterogeneous catalysis.1 Certain of these materials have even shown considerable promise for the high-density storage of hydrogen, which poses an extraordinarily difficult challenge owing to its high fugacity.1e,2,3 While the H2 uptake in such compounds at cryogenic temperatures can approach capacities compatible with the targets set forth by the US Department of Energy for mobile applications,4 capacities near ambient temperature are very much diminished. This has prompted exploration of several approaches for increasing the H2 binding affinity of the framework surfaces, including via introduction of open metal coordination sites.2e,3,5Despite the promise offered by high-surface area metal–organic frameworks, many that incorporate carboxylate-based bridging ligands are prone to decomposition upon exposure to atmospheric water. For example, Zn4O(BDC)3 (BDC2− = 1,4-benzenedicarboxylate), an iconic metal–organic framework exhibiting the highest known density for cryogenic hydrogen storage (66 g L−1 at 77 K and 100 bar), decomposes upon exposure to air.2l Consequently, the preparation, handling, and utilization of such materials require moisture-free conditions that limit or impair their applicability.
The discovery of new high-surface area metal–organic frameworks that are stable to air, water, other chemical agents, and high temperatures is an important step in extending the versatility of these metal–organic frameworks. To this end, it has been shown that microporous imidazolate-bridged frameworks can exhibit high thermal stability and remarkable chemical resistance in boiling alkaline water and other solvents.6 This behavior is likely due to the greater basicity of imidazolate, as reflected in the significantly higher pKa of imidazoles relative to carboxylic acids, which results in stronger metal–ligand bonds. Given that pyrazoles have similar high pKa values, pyrazolate-based metal–organic frameworks should also exhibit good thermal and chemical stability. Accordingly, we are working to extend our previous metal–tetrazolate framework chemistry3,7 to triazolate-8 and pyrazolate-based ligands, and recently reported the synthesis of Co(1,4-BDP) (1,4-H2BDP = 1,4-benzenedi(4-pyrazolyl)), which displays remarkable flexibility and a large Langmuir surface area of 2670 m2 g−1.9 Here, we demonstrate the use of 1,4-H2BDP and its meta isomer 1,3-benzenedi(4-pyrazolyl) (1,3-H2BDP) in the synthesis of rigid zinc-containing frameworks exhibiting good cryogenic H2 adsorption properties and excellent stability to boiling solvents, including water.
Experimental
Water was distilled and deionized prior to use. All other reagents were obtained from commercial vendors, and were used without further purification.Synthesis of 1,3-benzenedi(4′-pyrazolyl) (1,3-H2BDP)
Under a nitrogen atmosphere, 7.0 mL of phosphorous oxychloride was added to 6.0 mL of anhydrous DMF in a three-neck round-bottomed flask, and the mixture was chilled in an ice bath for 10 min. A solution of 1,3-phenylenedicarboxylic acid (2.0 g, 0.010 mol) in 10 mL of DMF was added, and the reaction mixture was heated at 70 °C for 18 h. While still hot, the resultant dark solution was poured into ca. 200 mL of ice water, and a solution of NaClO4 (4.4 g, 0.036 mol) in 100 mL of water was added to afford an off-white precipitate. The solid was collected by filtration, washed with successive aliquots of water (5 × 20 mL), methanol (3 × 20 mL), and diethylether (3 × 20 mL), and dried under reduced pressure to yield 4.5 g (86%) of 1,3-benzenebis(2′-(1′,3′-dimethylamino)-trimethinium)diperchlorate (A). A 50 mL aliquot of 0.01 M NaOH was added to a suspension of A (4.5 g, 8.5 mmol) in 150 mL of water, and the solution was stirred and heated at 70 °C for 45 min. The resulting colorless solution was titrated with a 1.2 M aqueous HCl solution until the pH of the solution was 3. Hydrazine monohydrate (4.0 mL, 79 mmol) was added and the mixture was heated at 70 °C for 1 h to give an off-white precipitate. The solid was collected by filtration, washed with successive aliquots of water (3 × 50 mL), and dried in air to afford 1.29 g (61%) of product. IR (neat, ATR): νN–H 3141, 3114, 3069, νC–H 2932, 2857, 2674, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N 1610, 1593, 1522 cm−1. 1H NMR (DMSO-d6): δ 12.92 (s, br, 2H), 8.031 (d, br, 4H), 7.86 (s, 1 H), 7.426 (d, 2H), 7.310 (tr, 1H) ppm. Anal. calcd for C12H10N4: C, 68.56; H, 4.79; N, 26.65. Found: C, 68.79; H, 4.98; N, 26.49.
N 1610, 1593, 1522 cm−1. 1H NMR (DMSO-d6): δ 12.92 (s, br, 2H), 8.031 (d, br, 4H), 7.86 (s, 1 H), 7.426 (d, 2H), 7.310 (tr, 1H) ppm. Anal. calcd for C12H10N4: C, 68.56; H, 4.79; N, 26.65. Found: C, 68.79; H, 4.98; N, 26.49.
Synthesis of Zn(1,4-BDP)·2DEF·H2O (1)
A borosilicate tube (1.2 cm o.d. × 15 cm length) was charged with Zn(CF3SO3)2 (120 mg, 0.33 mmol), 1,4-H2BDP (60 mg, 0.29 mmol), and 2.0 mL of N,N′-diethylformamide (DEF). The reaction mixture was degassed by the freeze–pump–thaw method (5 cycles), and the tube was sealed under reduced pressure. The tube was heated in an oil bath at 150 °C for 5 days to afford an off-white solid. Upon cooling to room temperature, the tube was broken open in air and the solid was immediately collected by filtration. The solid was then washed with successive aliquots of DEF (5 × 10 mL) and dried under reduced pressure for 30 min to afford 0.13 g (95%) of product. IR(neat, ATR): νCC 1610,1559, νCN 1436, νCO 1668 cm−1. Anal. calcd for C22H30N6O2Zn (Zn(1,4-BDP)·2DEF): C, 55.52; H, 6.35; N, 17.66. Found: C, 55.43; H, 6.29; N, 17.49. The solid quickly loses water in air.
Synthesis of Co(1,3-BDP)·DEF·0.5H2O (2)
A borosilicate tube (0.7 cm o.d. × 8 cm length) was charged with Co(CF3SO3)2 (30 mg, 0.084 mmol), 1,3-H2BDP (16 mg, 0.076 mmol), and 1.5 mL of N,N′-diethylformamide (DEF). The reaction mixture was degassed by the freeze–pump–thaw method (5 cycles), and the tube was sealed under reduced pressure. The tube was heated in an oil bath at 140 °C for 3 days to afford a purple microcrystalline solid. Upon cooling to room temperature, the tube was broken open in air and the solid was immediately collected by filtration. The solid was then washed with successive aliquots of DEF (5 × 10 mL) and dried under reduced pressure for 30 min to afford 0.015 g (54%) of product. IR(neat, ATR): νCC 1610,1559, νCN 1436, νCO 1668 cm−1. Anal. calcd for C17H20CoN5O1.5: C, 53.97; H, 4.98; N, 18.33. Found: C, 54.12; H, 5.34; N, 18.56. Single crystals suitable for X-ray analysis† were obtained from a reaction controlled by programmable heat ramping. A borosilicate tube (0.7 cm o.d. × 8 cm length) was charged with Co(CF3SO3)2 (30 mg, 0.084 mmol), and H2BDP (15 mg, 0.071 mmol), and DEF (0.4 mL), and after degassing and sealing, it was heated in a programmable furnace. The temperature was increased at a rate of 0.1 °C min−1 to 130 °C, which was maintained for 4 days prior to cooling at a rate of 0.1 °C min−1 to room temperature.
Synthesis of Zn(1,3-BDP)·0.7DMF·0.5H2O (3)
Solid 1,3-H2BDP (0.262 g, 1.25 mol) was added to a solution of Zn(NO3)2·6H2O (0.402 g, 1.35 mmol) in 12 mL of DMF, affording a yellow solution. The reaction mixture was heated at 110 °C for 60 h, resulting in formation of a white precipitate. Upon cooling to room temperature, the solid was filtered, washed with successive aliquots of DMF (5 × 10 mL) and dried under reduced pressure to afford 0.285 g (63%) of product. IR (neat, ATR): νCC 1611, 1592, νCN 1384, νCO 1676 cm−1. Anal. calcd for C14.1H13.9N4.7O1.2Zn: C, 50.74; H, 4.20; N, 19.72. Found: C, 50.65; H, 3.96; N, 19.55.
Low-pressure gas adsorption measurements
Gas adsorption isotherms for pressures in the range 0–1.2 bar were measured by the volumetric method using a Micromeritics ASAP2020 instrument. Solid samples of 1 and 3 were transferred to preweighed analysis tubes, which were capped with Transeals and evacuated by heating at 160 °C under dynamic vacuum until an outgas rate of less than 2 mTorr min−1 (0.27 Pa min−1) was achieved (ca. two days). The evacuated analysis tubes containing the degassed samples of Zn(1,4-BDP) (1d) and Zn(1,3-BDP) (3d) were then carefully transferred to an electronic balance and weighed again to determine the mass of sample (215 mg and 123 mg, respectively). The tube was then placed back on the analysis port of the gas adsorption instrument. The outgas rate was again confirmed to be less than 2 mTorr min−1 (0.27 Pa min−1). For all isotherms, warm and cold free-space correction measurements were performed using ultra-high-purity He gas (UHP grade 5.0, 99.999% purity); N2, and H2 isotherms at 77 and 87 K were measured in liquid nitrogen and liquid argon baths, respectively, using UHP-grade gas sources. Oil-free vacuum pumps and oil-free pressure regulators were used for all measurements to prevent contamination of the samples during the evacuation process or of the feed gases during the isotherm measurements.Determination of isosteric heat of H2 adsorption
To probe how the heat of adsorption varies for the two different frameworks, the adsorbed amounts of H2 at 77 K and 87 K were fit using a virial-type expression, composed of ai and bi parameters,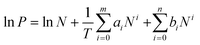 | (1) |
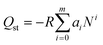 | (2) |
High-pressure H2 adsorption measurements and data analysis
Solids 1 and 3 were degassed by the method described in the previous section. For each measurement, the sample was then loaded into the sample holder under a nitrogen atmosphere. Excess adsorption of H2 was measured using an automated Sieverts' apparatus (PCT-Pro 2000 from Hy-Energy LLC) over a pressure range of 0–90 bar. At least 200 mg of adsorbent was used in each experiment, and UHP-grade hydrogen and helium (99.999% purity) were used for all measurements. Measurements at 77 K were performed by submerging the sample in liquid nitrogen. The volume of the sample holder and the connecting gas manifold were determined prior to sample measurement using He at 298 K and 77 K. Helium was used to determine the dead space volume correction for a non-porous sample of known volume; this correction accounted for the change in effective sample volume observed when cooling the sample holder from room temperature to low temperature. The adsorbent was then introduced into the sample holder and He gas was used to determine the volume of the sample at room temperature. Since He gas penetrates the pores of the sample without being significantly adsorbed onto the surface, the volume measured with He corresponds to the volume of the framework walls, also referred to as the framework skeleton. Consequently, the skeletal density of the material, dsk, can be obtained from the following expression.| dsk = m/Vsk | (3) |
Gravimetric water adsorption measurement
A sample of 1 was degassed and weighed by the method detailed above. The evacuated analysis tube containing the degassed solid of Zn(1,4-BDP) (1d) was weighed, and then attached to a Micromeritics ASAP2020 instrument, which was exposed to saturated water vapor at room temperature. After 24 h, the tube was transferred to an electronic balance and weighed to determine the adsorbed amount of water.X-ray structure determination†
A single crystal of Co(1,3-BDP)·DEF·0.5H2O (2) was coated in Paratone-N oil, attached to a Kapton loop, quickly transferred to a Bruker Platinum 200 Instrument at the Advanced Light Source at the Lawrence Berkeley National Laboratory, and cooled in a stream of nitrogen. Preliminary cell data were collected to give a unit cell consistent with the tetragonal Laue group, and the unit cell parameters were later refined against all data. A full hemisphere of data was collected, and the crystal didn't show significant decay during data collection. Data were integrated and corrected for Lorentz and polarization effects using SAINT v7.3411a and were corrected for absorption effects using SADABS 2.10.11b Crystal and refinement parameters are listed in Table S1.† Space group assignments were based upon systematic absences, E statistics, and successful refinement of the structures. The structure was solved by direct methods and expanded through successive difference Fourier maps. It was refined against all data using the SHELXTL 5.0 software package.11c Thermal parameters for all non-hydrogen atoms pertaining to the framework skeleton were refined anisotropically. Due to severe disorder of the solvent molecules in the pores, the residual electron density within the pores was squeezed out using PLATON,11d and the final refinement was performed based on the resulting data.Other physical measurements
Infrared spectra were collected on a Nicolet Avatar 360 FTIR spectrometer equipped with an attenuated total reflectance (ATR) accessory. Carbon, hydrogen, and nitrogen analyses were obtained from the Microanalytical Laboratory of the University of California, Berkeley. Powder X-ray diffraction data were collected using Cu Kα (λ = 1.5406 Å) radiation on a Bruker D8 Advance diffractometer. Thermogravimetric analyses were carried out at a ramp rate of 1 °C min−1 in a nitrogen flow with a TA Instruments TGA Q5000 V3.1 Build 246.Results and discussion
Employing synthetic conditions analogous to those used in the synthesis of Co(1,4-BDP),9 Zn(CF3SO3)2 was found to react with 1,4-H2BDP in N,N-diethylformamide (DEF) to afford colorless microcrystals of Zn(1,4-BDP)·2DEF·H2O (1) in 95% yield. Powder X-ray diffraction data for 1 revealed a pattern matching that observed for Co(1,4-BDP)·2DEF·H2O, indicating the two phases to be isostructural.12 The crystal structure of 1 therefore consists of chains of tetrahedrally coordinated Zn2+ ions bridged by pyrazolate groups from BDP2− ligands to form square channels running along the c axis of the crystal. As shown in Fig. S1 of the ESI,† these channels have a width of approximately 10 × 10 Å2, with much narrower openings connecting channels along the a and b axes.Reaction of the bent linker 1,3-H2BDP with Co(CF3SO3)2 in DEF at 140 °C in a sealed borosilicate tube produced purple microcrystals of Co(1,3-BDP)·DEF·0.5H2O (2) in 54% yield. X-ray analysis13 of a single crystal of 2 revealed a structure (see Fig. 1) wherein Co2+ ions are again tetrahedrally coordinated by N atoms from four independent pyrazolate rings. In contrast to Co(1,4-BDP), however, the meta-oriented pyrazolate substituents give rise to zig-zag chains that orient in an alternating fashion to form cylindrical 3.9-Å diameter channels running along the a and b crystal axes. The benzene rings from pairs of neighboring 1,3-BDP2− linkers display weak π–π stacking interactions (with a centroid–centroid distance of 3.419(2) Å) to give a double-walled framework. Some disorder occurs in the arrangement of these aromatic rings in the structure, suggesting that the 3.9 × 3.9 Å2 square channels running along the c axis may be disrupted by occasional blockages.
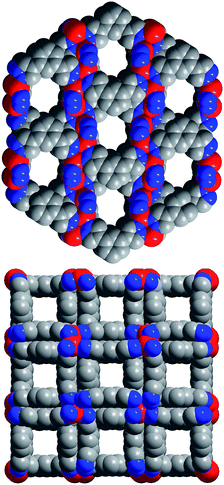 | ||
| Fig. 1 A portion of the crystal structure of 2, as viewed along the a (upper) and c (lower) crystal axes. Red, blue, and grey spheres represent Co, N, and C atoms, respectively; H atoms are omitted for clarity. Each Co atom resides on a two-fold rotation axis and the aromatic rings lie across a mirror plane. Selected interatomic distances (Å) and angles (°): Co–N, 1.985(5); N–N, 1.357(1); C–N, 1.337(1); Co⋯Co, 6.66(8); N–Co–N, 107.5(2); Co–N–C, 129.2(8); Co–N–N, 123.3(7); C–N–N, 107.3(6). | ||
A similar reaction between Zn(NO3)2·6H2O and 1,3-H2BDP in N,N-dimethylformamide (DMF) at 110 °C yielded Zn(1,3-BDP)·0.7DMF·0.5H2O (3). As evidenced by its powder X-ray diffraction pattern, this compound is isostructural to 2. Preliminary studies have also shown that other first row transition metal ions, such as Mn2+, Fe2+, Ni2+, and Cu2+, do not share the preference of Co2+ and Zn2+ for tetrahedral coordination with pyrazolate ligands. Indeed, reactions of these ions with 1,3-H2BDP and 1,4-H2BDP produced different crystalline phases whose structures have thus far eluded us.
Thermogravimetric analyses performed on compounds 1 and 3 demonstrated the high thermal stability of their pyrazolate-bridged frameworks. As shown in Fig. S5,† solvent loss steps at 120 °C for 1 and 180 °C for 3 are followed by extended plateaus of compositional stability up to 400 °C for 1 and 500 °C for 3. Significantly, the powder X-ray diffraction pattern for a sample of 3 that had been heated to 450 °C was identical to that of the as-synthesized phase (see Fig. S9†). Such high thermal stability is extremely rare among transition metal-carboxylate frameworks,14 which commonly decompose in the range 200–350 °C, and has mostly been observed previously in imidazolate-, triazolate-, or other pyrazolate-bridged frameworks.6,8,15
To probe the water sensitivity of 1, as-synthesized samples were immersed in water at elevated temperature for various periods of time and the resulting solids were characterized by powder X-ray diffraction (see Fig. 2, upper). Interestingly, a phase change occurs within minutes of immersing 1 in water heated at 40 °C, and the resulting pattern is retained during all subsequent manipulations, even after immersion in boiling water for 3 days. The original phase is rapidly regenerated, however, upon re-immersion of this modified phase in DEF for only 3 min.16 The results suggest that no bonds are broken in the framework upon heating in water, and that only a slight, reversible change in its conformation occurs during the treatment. Accordingly, the (100) reflection at 2θ = 6.6° does not shift or disappear during the experiment. Complete regeneration of the porous structure was also proven by a low-pressure H2 adsorption measurement at 77 K, which, as discussed below, showed no change in uptake capacity subsequent to treatment with hot water. Gravimetric analysis further showed that the desolvated form of 1 (1d, as discussed below) adsorbs 28 wt% water at room temperature, which is comparable or superior to the water uptake capacity of zeolites-AX (A = Li+, Na+, K+, Rb+, Cs+),17 and points to its potential use as a water scavenger or in water adsorption-based heating/cooling applications.18 The behavior of 1 in boiling methanol is similar to its behavior in water: an initial phase change is completely reversed upon treatment with DEF for 3 min (see Fig. S8†).
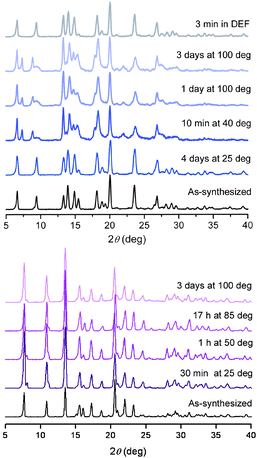 | ||
| Fig. 2 Powder X-ray diffraction patterns for 1 (upper) and 3 (lower) after treatment in water for various durations at various temperatures. | ||
Compound 3 exhibits an even more marked chemical stability. As shown at the bottom of Fig. 2, the structure is fully retained even after immersion in boiling water for 3 days. Boiling methanol or benzene also leave the framework intact, and no changes in peak position or intensity are visible in the associated diffraction patterns (see Fig. S10†). The unusual robustness of 3 is likely due to its double-walled framework structure, which is reminiscent of interpenetrated frameworks that are known to exhibit increased stability relative to their non-interpenetrated counterparts.5b Intriguingly, 3 is also stable in an acidic aqueous medium with pH = 3 for up to 30 min at 90 °C. Although some imidazolate-, triazolate-, and pyrazolate-based frameworks show extended stability in neutral or alkaline solutions,6,8,15 stability at low pH has not been reported thus far for metal–organic frameworks, and could open new opportunities for the use of these materials in acid–base catalysis.
In order to investigate their gas sorption properties, desolvated phases 1d and 3d were prepared by heating 1 and 3 at 160 °C under dynamic vacuum. The permanent porosity of the resulting solids was evaluated by N2 adsorption measurements performed at 77 K (see Fig. 3). Both compounds displayed Type I adsorption isotherms characteristic of microporous solids,19 and adsorbed significant amounts of N2, with 1d taking up 23 mmol g−1 and 3d taking up 13 mmol g−1. Fits to the data gave Langmuir surface areas of 2320(4) and 1161(3) and BET surface areas of 1710(20) and 820(10) m2 g−1 for 1d and 3d, respectively. Note that the reduction of the surface area of 3d to roughly half of that observed for 1d is consistent with its double-walled framework structure.
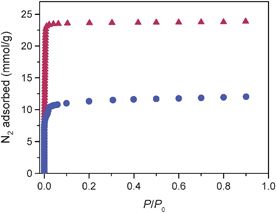 | ||
| Fig. 3 Nitrogen adsorption isotherms at 77 K for the solid 1d (red triangles), and 3d (blue circles) that were activated at 160 °C. | ||
The hydrogen adsorption capacity of each desolvated phase was also measured at 77 and 87 K. As depicted at the top of Fig. 4, at pressures of up to 900 mm Hg, both 1d and 3d exhibit a fully reversible H2 uptake of 1.6 wt% at 77 K. Importantly, these isotherms are unchanged after treatment of the samples with boiling water followed by dehydration of the pores. At 87 K, the H2 capacities for 1d and 3d in this pressure range dropped to 1.0 and 1.2 wt%, respectively. The H2 adsorption isotherms at 77 and 87 K were fit using a virial-type expression to obtain the isosteric heat of H2 adsorption (Qst) for each material.2e As shown at the bottom of Fig. 4, Qst ranges from −6.9 to −5.4 kJ mol−1 for 1d and from −6.6 to −5.9 kJ mol−1 for 3d. The greater average magnitude of Qst for 3d is consistent with the steeper rise of its adsorption isotherm at low pressures, and is likely a consequence of the smaller pore size of the framework. In addition, the lesser degree of variation in the values observed for 3d is consistent with its relatively isotropic pore shape, which should result in less variation of the binding affinity. Furthermore, the relatively low zero-coverage binding energy for both materials agrees with the lack of open metal-H2 binding sites, which are responsible for the high zero-coverage binding energy and more pronounced enthalpy changes observed for metal–organic frameworks with unsaturated metal sites.3,5
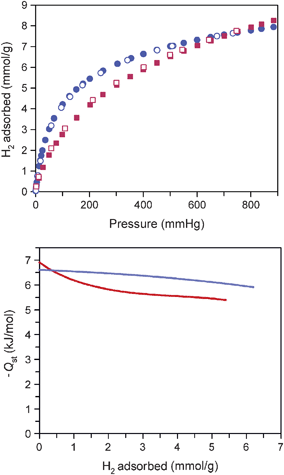 | ||
| Fig. 4 Low-pressure H2 adsorption isotherms measured at 77 K (top) and plot of the isosteric heat of H2 adsorption (bottom), as determined from adsorption data collected at 77 and 87 K for 1d (red) and 3d (blue). Filled and empty symbols represent adsorption and desorption, respectively. | ||
Interestingly, the hydrogen uptake at 900 mm Hg and 77 K is nearly the same for both compounds. This seems counterintuitive, given that the surface area of 1d is roughly double that of 3d. However, the expected difference in capacity becomes evident in the high-pressure H2 adsorption isotherms collected at 77 K (see Fig. 5). Although the excess H2 uptake in 1d initially lags below that of 3d, it exhibits a second, relatively broad step with notable hysteresis upon desorption. On the other hand, the H2 adsorption for 3d shows no inflection as it rapidly climbs below 10 bar to near its maximum. Overall, the two compounds exhibit excess H2 uptake capacities of 4.7 wt% for 1d and 2.1 wt% for 3d. To our knowledge, the former corresponds to the highest loading yet reported for a metal–organic framework with a well-demonstrated water stability.
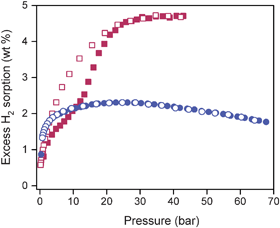 | ||
| Fig. 5 Excess surface adsorption of H2 on 1d (red) and 3d (blue) at high pressure and 77 K. Filled and empty symbols represent adsorption and desorption, respectively. | ||
The hysteretic H2 adsorption observed for 1d is reminiscent of the data observed for Co(1,4-BDP) at 77 K, which exhibits a much broader hysteresis with a more prominent adsorption step occurring at ca. 25 bar.9 This effect was assigned to an accordion type flexibility of the framework, which is associated a change in the coordination geometry around the Co2+ ions. Although the intense peak corresponding to the (100) reflection in the powder X-ray diffraction pattern of 1 is retained in 1d, their diffraction patterns are not quite identical (see Fig. S3†). This together with the slight hysteresis suggests that there may be a minor relaxation in the framework structure of 1 upon removal of the guest solvent, with the initial structure being reinstated upon pressurization with more than 10 bar of H2.
Conclusions
The foregoing results demonstrate a high degree of chemical and thermal stability for two new zinc(II)–pyrazolate metal–organic frameworks. Most significantly, with a BET surface area of 1710 m2 g−1, Zn(1,4-BDP) exhibits an excess H2 uptake of 4.7 wt% at 40 bar and 77 K. Future research will focus on generating related, air- and water-stable frameworks with higher surface areas and greater capacities for cryogenic hydrogen storage.Acknowledgements
This research was funded by the General Motors Company. A portion of the work was conducted at the Advanced Light Source facility at Lawrence Berkeley National Laboratory, which is operated by the DoE under contract DE-AC03-76SF00098.References
- (a) O. M. Yaghi, M. O'Keefe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS; (b) S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; (c) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC; (d) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC; (e) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC; (f) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (g) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- (a) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS; (b) G. Férey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guégan, Chem. Commun., 2003, 2976 RSC; (c) J. L. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666 CrossRef CAS; (d) E. Y. Lee and M. P. Suh, Angew. Chem., Int. Ed., 2004, 43, 2798 CrossRef CAS; (e) D. Sun, S. Ma, Y. Ke, D. J. Collins and H.-C. Zhou, J. Am. Chem. Soc., 2006, 128, 3896 CrossRef CAS; (f) J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304 CrossRef CAS; (g) A. G. Wong-Foy, A. J. Matzger and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 3494 CrossRef CAS; (h) X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358 CrossRef; (i) M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J.-H. Lee, J.-S. Chang, S. H. Jhung and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 8227 CrossRef CAS; (j) B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203 CrossRef CAS; (k) H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197 RSC; (l) S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176 CrossRef CAS; (m) K. Sumida, M. R. Hill, S. Horike, A. Dailly and J. R. Long, J. Am. Chem. Soc., 2009, 131, 15120 CrossRef CAS.
- (a) M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef CAS; (b) M. Dincă, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419 CrossRef CAS; (c) M. Dincă and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172 CrossRef CAS.
- DoE Office of Energy Efficiency and Renewable Energy Hydrogen, Fuel Cells & Infrastructure Technologies Program Multi-Year Research, Development and Demonstration Plan, 2005, available at: http://www1.eere.energy.gov/hydrogenandfuelcells/mypp.
- (a) S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506 CrossRef; (b) Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 726 CrossRef CAS; (c) Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 8136 CrossRef CAS; (d) K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604 CrossRef CAS; (e) M. Dincă and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766 CrossRef CAS; (f) K. L. Mulfort, O. K. Farha, C. L. Stern, A. A. Sarjeant and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 3866 CrossRef CAS; (g) D. Himsl, D. Wallacher and M. Hartmann, Angew. Chem., Int. Ed., 2009, 48, 4639 CrossRef CAS.
- (a) X.-C. Huang, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557 CrossRef CAS; (b) Y. Liu, V. Ch. Kravtsov, R. Larsen and M. Eddaoudi, Chem. Commun., 2006, 1488 RSC; (c) K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keefe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS; (d) R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS.
- (a) M. Dincă, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904 CrossRef CAS; (b) M. Dincă, A. Dailly, C. Tsay and J. R. Long, Inorg. Chem., 2008, 47, 11 CrossRef CAS; (c) M. Dincă, A. Dailly and J. R. Long, Chem.–Eur. J., 2008, 14, 10280 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 CrossRef CAS.
- H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848 CrossRef CAS.
- R. Gentleman and R. Ihaka, The R Project for Statistical Computing, the Statistics Department of the University of Auckland, New Zealand, 1997; download, instructions, and further details on the use and capabilities of this software package area available online at http://www.r-project.org Search PubMed.
- (a) SAINT v7.34 Software for the Integration of CCD Detector System, Bruker Analytical X-ray Systems, Madison, WI, 2001 Search PubMed; (b) G. M. Sheldrick, SADABS v2.10, Program for adsorption corrections, Institute for Inorganic Chemistry, University of Göttingen, Germany, 1996 Search PubMed; R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 Search PubMed; (c) G. M. Sheldrick, SHELXTL 5.0, Program for solution and refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed; (d) P. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- The orthorhombic unit cell of 1 was refined using the program Powdercell to give a = 13.212 Å, b = 13.275 Å, and c = 14.419 Å.
- Crystal data: C17H20CoN5O1.5, MW = 377.31, Tetragonal, space group I41/amd, a = 22.847(15) Å, c =
12.458(16) Å;, V = 6503(10) Å3, Z = 14, Dc = 1.317 g cm−3, F(000) = 2674, λ(Mo Kα) = 0.77490 Å, μ(Mo Kα) = 0.936 mm−1, T = 150(2) K, total reflections = 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 681, unique reflections = 559, (Rint = 0.1155), observed data (I > 2σ(I)) = 414, R1 = 0.0763 (I > 2σ(I)), wR2 = 0.2192, and GoF = 1.704. Complete modeling of all the solvent molecules was not possible due to severe disorder, and the SQUEEZE subroutine in the PLATON software package was applied to mask the electron density in the cavities. This treatment gave R1 = 0.0635 (I > 2σ(I)), wR2 = 0.1642, and GoF = 1.341.
681, unique reflections = 559, (Rint = 0.1155), observed data (I > 2σ(I)) = 414, R1 = 0.0763 (I > 2σ(I)), wR2 = 0.2192, and GoF = 1.704. Complete modeling of all the solvent molecules was not possible due to severe disorder, and the SQUEEZE subroutine in the PLATON software package was applied to mask the electron density in the cavities. This treatment gave R1 = 0.0635 (I > 2σ(I)), wR2 = 0.1642, and GoF = 1.341. - (a) E. Y. Lee, S. Y. Jang and M. P. Suh, J. Am. Chem. Soc., 2005, 127, 6374 CrossRef CAS; (b) J. Luo, H. Xu, Y. Liu, Y. Zhao, L. L. Daemen, C. Brown, T. V. Timofeeva, S. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 9626 CrossRef CAS; (c) Y. E. Cheon, J. Park and M. P. Suh, Chem. Commun., 2009, 5436 RSC.
- (a) J. He, Y.-G. Yin, T. Wu, D. Li and X.-C. Huang, Chem. Commun., 2006, 2845 RSC; (b) L. Hou, Y.-Y. Lin and X.-M. Chen, Inorg. Chem., 2008, 47, 1346 CrossRef CAS.
- It seems that guest solvent molecules can diffuse rapidly through the large tetragonal pores of 10 × 10 Å2 in 1d. Given that DEF diffused into micron size crystals within 3 min, the diffusion rate can approximated as at least 6 × 10−11 m2 s−1 which falls into the range of 10−5-10−14 m2 s−1 observed for other porous materials, such as zeolites. See: H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844 Search PubMed , and references therein.
- O. M. Dzhigit, A. V. Kiselev, K. N. Mikos, G. G. Muttik and T. A. Rahmanova, Trans. Faraday Soc., 1971, 67, 458 RSC.
- S. K. Henninger, H. A. Habib and C. Janiak, J. Am. Chem. Soc., 2009, 131, 2776 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional crystal data, powder X-ray diffraction patterns, thermogravimetric analysis data and adsorption isotherms. CCDC reference number 745475. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b917512a |
| This journal is © The Royal Society of Chemistry 2010 |