Amphiphilic peptides and their cross-disciplinary role as building blocks for nanoscience
Silvia
Cavalli
*ab,
Fernando
Albericio
*abc and
Alexander
Kros
*d
aCIBER-BBN, Networking Centre on Bioengineering, Biomaterials, and Nanomedicine, Barcelona Science Park, 08028 Barcelona, Spain. E-mail: silvia.cavalli@irbbarcelona.org; albericio@irbbarcelona.org; Fax: (+34) 93 403 71 26; Tel: (+34) 93 403 71 27
bInstitute for Research in Biomedicine, 08028 Barcelona, Spain
cDepartment of Organic Chemistry, University of Barcelona, Martí i Franqués 1-11, 08028 Barcelona, Spain
dInstitute of Chemistry, University Leiden, P.O. Box 9502, 2300 RA, Leiden, The Netherlands. E-mail: a.kros@chem.leidenuniv.nl; Fax: (+31) 715 274 397
First published on 13th October 2009
Abstract
Peptides are particularly attractive as molecular building blocks in the bottom-up fabrication of supramolecular structures based on self-assembly and have potential in many important applications in the fields of biotechnology and bioengineering. In the first part of this critical review the main categories of peptide-based amphiphiles will be discussed by showing some relevant examples, which demonstrate the importance of amphiphilic peptides as molecular building blocks for nanostructures. In the second part of this review we will review the cross-disciplinary role of peptide-based supramolecular nanoarchitectures ranging from chemistry to biology, medicine, materials science, and engineering through discussing several examples of applied nanomaterials (216 references).
 Silvia Cavalli | Silvia Cavalli obtained her Master’s degree in Chemistry (2002) at the University of Milan in Italy and her PhD (2006) under the supervision of Prof. Fraaije, Dr Kros and Dr Overhand, at Leiden University, the Netherlands. After working as a postdoctoral researcher in the group of Dr Ovaa at the Netherlands Cancer Institute NKI-AVL, she recently joined the laboratory of Prof. Dr Fernando Albericio at The Institute for Research in Biomedicine (Barcelona, Spain), were she is currently working as a research assistant. Her research interest is mainly related to the synthesis of self-assembling amphiphilic peptides and their biological applications. |
 Fernando Albericio | Professor Fernando Albericio received his PhD in Chemistry at the University of Barcelona, in 1981. Following postdoctoral work at Tufts University (Boston), at the Université d’Aix-Marseille (France) and at the University of Minnesota (1981–1984), he returned to Barcelona as an Associate Professor. During the 1992–1994 period, he was the Director of Peptide Research with Milligen/Biosearch at Boston. He rejoined the University of Barcelona, where he was promoted to a Professor in 1995. Nowadays, he holds various appointments: General Director of the Barcelona Science Park, Professor at the University of Barcelona, and Group Leader at the Institute for Research in Biomedicine. |
 Alexander Kros | Alexander Kros completed his PhD in physical organic chemistry in 2000 at Nijmegen University, the Netherlands with Prof. Nolte. After a period of postdoctoral research at Caltech, USA, with Prof. Tirrell he returned to Europe and became an Assistant Professor at Leiden University, the Netherlands. His scientific interests are in the design and assembly of lipidated peptides, peptide-based polymers, hydrogel-based drug delivery systems and model systems for membrane fusion. |
1. Introduction
The process of self-assembly is based on the spontaneous diffusion and specific interaction among molecules governed by non-covalent bonds, including electrostatic, hydrophobic, van der Waals, metal–ligand and hydrogen bonds as well as aromatic π-stacking.1–3 Although these interactions are individually weak, if sufficient in number, they can generate highly stable assemblies. Richard Feynman presented in 1959 a lecture entitled “There’s plenty of room at the bottom”, proposing the idea of a “bottom-up” approach for the fabrication of higher ordered structures via self-assembly using individual atoms and molecules as building blocks.4 One of the main challenges in supramolecular chemistry involves the issue of forming homogeneous and structurally well-defined architectures with tuneable properties to cover a wide range of possible applications. Therefore, an accurate design and good understanding of the rules governing the molecular assembly of specific monomeric building blocks are key features for the successful engineering of “smart” supramolecular architectures with predictable properties and functions.1Complexity in nature stems from a hierarchical organization of biomolecular components and levels of interactions between them. At the base of the hierarchy is a set of basic building blocks (i.e. amino acids, nucleic acids, sugars and lipids). One level of complexity above these are tectons,5 programmed nanoscale building blocks (i.e. an amino acid-based tecton would be a polypeptide designed to form α-helix or β-strands). Tectons can interact to give self-assembled units, which can combine and organize further to produce functional assemblies and systems. One challenge is to increase the number of building blocks and the repertoire of chemical tools. A rapidly growing field, synthetic biology,6 has emerged in a multidisciplinary effort among biologists, chemists, physicists, mathematicians, and engineers with the aim to improve understanding of biological systems through mimicry and to produce bio-orthogonal systems, non-native and non-perturbing chemical structures with new functions compared to biological systems. Among other peptides and proteins there appear to be useful building blocks (or tectons) for generating programmed biomolecules able to self-organize into higher hierarchical biomolecular systems, due to their relatively easy preparation and predictable structural folding.5
Amphiphilic peptides are particularly attractive as molecular building blocks7 in the bottom-up fabrication of supramolecular structures based on self-assembly and have potential in many important applications in the fields of biotechnology and bioengineering. During the past decade, many examples of supramolecular assemblies based on amphiphilic peptides as monomeric building blocks have been published and a selection of representative examples is discussed in detail in the following sections of this review.8,9 Peptides are a particularly attractive class of molecules, which can be used as molecular building blocks since their structural folding and stability have already been studied in detail.10–13 Amino acids and peptides can be seen as information carriers, which introduce structural “smartness” in nanostructures, particularly due to their ability to respond to external parameters (i.e. changes in solvent, pH and temperature or sensitivity to electronic or photonic energy and to the presence of chelating metals),14–16 which is of particular interest when the responsiveness is reversible. The availability of a variety of peptide-based building blocks has been mainly fuelled by the advent of straightforward and fast synthetic methodologies, mainly based on solid phase protocols that offer easy access to a wide variety of (oligo)peptides with virtually any amino acid sequence of about 5–50 residues.17–19 Moreover, the possibility to incorporate non-natural amino acids or functional moieties in the peptide sequence is particularly valuable for the introduction of an increased level of functionality in the assemblies.8 In addition, the intrinsic chiral nature of amino acids can lead to the expression of handedness to a higher hierarchical level.20 Finally, the use of biologically relevant peptide sequences can generate new materials, at the nanometre-scale with possible applications in the field of biotechnology and bioengineering.21,22
Although the current research efforts have already led to an enhanced understanding of the criteria that govern the assembly processes in amphiphilic peptides, due to their complexity, more investigation is required to gain a better insight into the way aggregation and peptide secondary structure influence each other. The construction of tailor made self-assembling peptides, with high levels of structural and functional control has a high potential, especially in the biomedical and materials science fields. Therefore the activities within this area have been intensified considerably with the aim to design functional types of amphiphilic peptide architectures. Finding specific methodology to build “smartness” into peptide-based responsive nanomaterials is a particularly fascinating emerging area and the extensive potential of sequence manipulations enables the specific fabrication of a vast number of different structures that can be fine tuned for many important applications, ranging from chemistry to materials science and engineering (i.e. nanomaterials have been successfully employed in catalysis, tissue repair, patterning and for the preparation of optical and electronic devices).
In the first part of this critical review the main categories of peptide-based amphiphiles are discussed by highlighting some relevant examples, which demonstrate the importance of amphiphilic peptides as molecular building blocks for nanostructures. In the next part of this review an overview of the synthetic strategies exploited for the preparation of the amphiphilic peptides is given. While in the last section several examples of recent innovations which incorporate “smart” peptides into tuneable hybrid materials are shown, demonstrating the multifunctional and cross-disciplinary role of amphiphilic peptide-based nanoarchitectures.
2. Amphiphilic peptides as building blocks for the bottom-up construction of nanometre-scale assembled structures
The first part of this critical review gives an overview of four main categories of peptide-based amphiphiles23 as molecular building blocks and will discuss some relevant examples that demonstrate the importance of amphiphilic peptides as molecular construction moieties for nanostructures. First, amphiphilic peptides are examined, followed by an overview on long chains alkylated/acylated peptides, and peptide-phospholipid conjugates. A final important category comprises peptide-based block copolymers.2.1 Amphiphilic peptides (amino acids only)
Amphiphilic peptides constituted of only amino acids are organized in amphipathic sequences comprised of both hydrophobic and hydrophilic domains.Zhang7 introduced the concept of “Peptide Lego”, based on ionic self-complementary peptides. These peptide building blocks contain two distinct surfaces, one being hydrophilic and the other hydrophobic, similar to the “Lego bricks” that have both pegs and holes positioned in such a well-ordered fashion, allowing precise assembly into a predetermined organization. In aqueous solutions, the hydrophobic side shields itself from water driving the self-assembly of the peptides, comparable to spontaneous protein folding as observed in nature. The structural feature of these “Lego bricks” is based on complementary ionic bonds with regular repeats on the hydrophilic surface due to the alternation of positively- and negatively-charged amino acid residues at specific intervals. Lysine (Lys) and arginine (Arg) are typically used as the positively-charged residues, while glutamic (Glu) and aspartic (Asp) acids are employed to generate the negative charge.
The complementary ionic sides have been classified by Zhang into several moduli (i.e. modulus I, II, III, IV, etc.), depending on the alternation of the charges (Fig. 1). For example, in the case of modulus I, the (+) positively- and (−) negatively-charged amino acid residues are alternated by one (+ − + − + − + − and − + − + − + in the complementary peptide). Consecutively, for modulus II, III, IV etc., charges are alternated by two, three, four etc. (+ + − − + + − −,+ + + − − − and + + + + − − − − etc.). The charge orientation can also be designed with a mixed order to yield entirely different molecules (mixed moduli). The first member of the “Peptide Lego” was serendipitously discovered from a segment in a left-handed Z-DNA binding protein in yeast, named Zuotin (Zuo means left in Chinese while tin means protein in biology. Zuotin is a yeast protein that was initially identified for its ability to bind preferentially to left-handed Z-DNA).25 The self-complementary sixteen-residue [(Ala-Glu-Ala-Glu-Ala-Lys-Ala-Lys)2] peptides, originally found in a region of alternating hydrophobic and hydrophilic residues in Zuotin, interacted strongly with each other to form a stable structure promoted by the hydrated salt ions.25 These molecules represent a class of self-assembling β-sheet peptides that spontaneously undergo association, in aqueous solutions, into a macroscopic membrane composed of well-ordered nanofibers. The architecture of the membrane resembled a high-density felt. Scanning electron microscopy (SEM) investigations at low magnification revealed that the structure looked like a flat membrane, which consisted of interwoven individual filaments, as seen at high magnification.
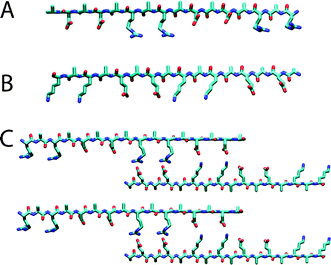 | ||
| Fig. 1 Examples of modulus II. Molecular models of the extended β-strand structures of individual molecules are shown for (A) ARARADADARARADAD (RAD16-II, R, arginine, A, alanine and D, aspartate) and (B) EAEAKAKAEAEAKAKA (EAK16-II, A, alanine, E, glutamate and K, lysine). The distance between the charged side chains along the backbone is approximately 6.8 Å; the methyl groups of Ala are found on one side of the sheet and the charged residues are on the other side. Conventional β-sheet hydrogen bond formation between the oxygens and hydrogens on the nitrogens of the peptide backbones are perpendicular to the page. (C) A proposed staggered assembly of molecular models for EAK16. The complementary ionic bonds and hydrophobic alanines are shown. Although an antiparallel β-sheet is illustrated, a parallel β-sheet model is also possible. Reproduced with permission from ref. 24. | ||
Amphiphilic peptides have been used as stimuli-responsive elements for the preparation of hydrogels14
As an example, Rajagopal et al.26 prepared a class of peptides whose ability to self-assemble into hydrogel was dependent on their folded state. Under unfolding conditions soluble peptides were freely flowing in aqueous solutions.
As shown in Fig. 2, when the folded peptides were triggered by external stimuli (i.e. changing the pH), they adopted a β-hairpin conformation and self-assembled into a highly crosslinked network of fibrils affording mechanically rigid hydrogels.26,27
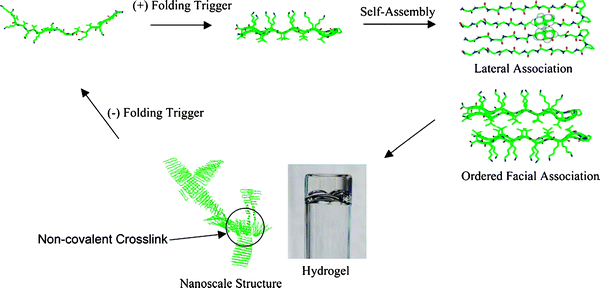 | ||
| Fig. 2 Environmentally triggered folding, self-assembly and non-covalent fibril crosslinking leading to hydrogel formation. Crosslinks are formed by the irregular facial self-assembly of hairpins. Reproduced from ref. 26 With kind permission from Springer Science + Business Media. | ||
Whitehouse and Boden have shown the absorption and self-assembly behaviour of a model peptide CH3CO-QQRFQWQFEQQ-CONH2 on mica substrates.28 At concentrations well below the critical concentration at which self-assembly into tapes occurs in bulk solution, peptide was found to self-assemble at solution–mica interfaces into planar tapes, a single molecule in thickness, and having a cross-beta structure. When the solvent was allowed to evaporate, quite different aggregate morphologies were obtained depending on the method used to prepare the initial film. In particular, a close-packed monolayer of parallel-aligned tapes, may be of practical utility as a functionalized protein-like surface.
Aggeli and co-workers thoroughly investigated the spontaneous self-assembly of β-sheet peptides into tapes, ribbons, fibrils and fibers.29,30 In one example, they have demonstrated that polyelectrolyte β-sheet complexes (PECs) could be formed on mixing aqueous solutions of cationic and anionic peptides, resulting in the spontaneous self-assembly of fibrillar networks and the production of hydrogels in appropriate pH windows.31 In general, the two peptides must have a propensity to form antiparallel β-sheets, appropriate complementarity in the disposition of their charged amino acid side chains, and at least one additional charged amino acid per peptide pair to stabilize the peptide PEC fibrillar network against flocculation. Furthermore, as the presence of salt attenuate the electrostatic forces, this must be accounted for in the peptide design. Such biocompatible and biodegradable hydrogels could be use for encapsulation, immobilization, and separation of cells, proteins, antibodies, or enzymes.
Well-defined assemblies of β-sheet structures have been achieved also at the air–water interface. Rapaport et al.32 investigated a family of amphiphilic peptides comprised of alternating hydrophobic and hydrophilic moieties with the generic sequence X-Y-(Z-Y)n-X, where the N- and C-terminal residues (X) bear charged ammonium and carboxylate groups, respectively, and Y and Z are alternating hydrophilic and hydrophobic amino acids. Variations in amino acid sequence and in the number of dyads (n) participating in hydrogen-bond formation were expected to tune the intermolecular interactions. Peptides Pro-Glu-(Phe-Glu)n-Pro (n = 4, 5 or 7) formed secondary structures composed of two-dimensional self-assembled β-sheet monolayers at the air–water interface confirmed by in situ grazing-incidence X-ray diffraction (GIXD) investigations. The alternating hydrophobic phenylalanine (Phe) and the hydrophilic glutamic acid (Glu) residues caused the peptide chains to orient as β-pleated sheets parallel to the water surface.
The flexibility of the peptide backbone and the repetitive nature of the amino acid sequence may induce dislocation defects (Fig. 3a) that limit long-range order to one-dimension (1D), in the direction normal to the peptide backbone (a direction). Noteworthy, the two-dimensional (2D) registry of the self-assembled architectures was induced by placement of proline (Pro) residues at the peptide termini. Without the Pro only 1D order was achieved, demonstrating the importance of a careful design of the peptide sequence for the successful self-assembling process. Pro was chosen to prevent the formation of disordered β-sheets along the a direction (Fig. 3a), on the base of three characteristic features: the tertiary amide, which cannot participate as a donor in the hydrogen bond array; the restricted dihedral angle (Φ) of Pro (ca. −60°) that is significantly different from that of β-sheet peptides (Φ ca. −120 to −150°), therefore making inclusion of Pro in the interior of a β-sheet ribbon sterically unfavourable, and the cyclic Pro side chain, which determines geometric constraints, minimizing disorder at the ribbon edge. In addition, attractive electrostatic interactions between the chain termini were expected to juxtapose the β-sheet ribbons along the b direction (Fig. 3b), facilitating the formation of the two-dimensional order. A schematic representation of the peptide Pro-Glu-(Phe-Glu)4-Pro in the β-pleated conformation and the targeted β-sheet crystalline assembly at the air–water interface is shown in Fig. 3c.
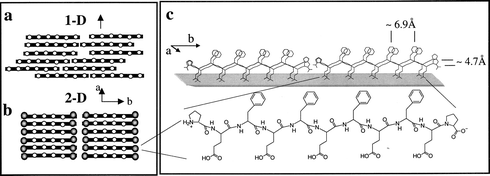 | ||
| Fig. 3 Schematic diagrams of β-strand assemblies at the air–water interface (rods and open dots represent peptide backbones and hydrophobic amino acids, respectively). View down the normal to the β-sheet of (a) one-dimensional order and (b) two-dimensional order induced by distinct chain termini. (c) Schematic representation of the peptide Pro-Glu-(Phe-Glu)4-Pro in the β-pleated conformation and the targeted β-sheet crystalline assembly at the air–water interface. Reprinted with permission from ref. 32. Copyright 2000 American Chemical Society. | ||
Well-defined β-sheet arrays at the interface were also observed for larger peptides able to form triple-stranded β-sheets.33 It was shown by Rapaport and co-workers34 that varying the amino acid sequence, parallel β-sheet assemblies may form as well.
The group of Ghadiri studied the design principles and the preparation strategies to achieve synthetic organic nanotubes, with special emphasis on noncovalent processes such as self-assembly and self-organization.35,36 As an example, hollow β-sheet tubular structures could be formed by the stacking of cyclic peptide rings. The cyclic peptides, containing an even number of alternating D and L-amino acids, in which the amide bonds are perpendicular to the plane of the ring, are shown to aggregate into microcrystalline tubes via a pH-controlled assembly strategy.37 The sequence of octapeptide cyclo[-(L-Gln-D-Ala-L-Glu-D-Ala)2-] was chosen to impart solubility in basic aqueous solution and thereby to prevent subunit association through coulombic repulsion. Controlled acidification promoted hydrogen bond interactions, thus allowing the self-assembly into hollow tubes composed of ring-shaped subunits stacked through antiparallel β-sheet hydrogen bonding. These structures exhibited a hydrophobic exterior and a hydrophilic interior and could insert into bilayer membranes introducing pores. Varying the number of amino acid residues and, hence, the ring size, modulated the porosity, as the smaller 8-residue rings only transport small ions while the larger 10-membered rings could also transport compounds like glucose and glutamate.
The coiled-coil motif38–40 is α left-handed superhelix composed of two or more right-handed α-helices and it has been identified in proteins ranging from muscle proteins to DNA transcription factors.
It has a characteristic amino-acid heptad repeating units designated as a to g in one helix and a′ to g′ in the other. The hydrophobic residues occupy positions at the interface of the two helices (a, d and a′, d′), whereas e, g and e′, g′, which are solvent exposed, are generally polar residues that give specificity between the two helices through electrostatic interactions (Fig. 4).
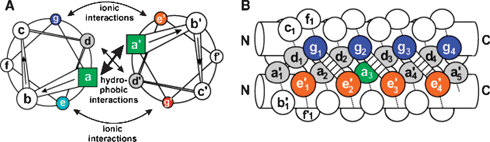 | ||
| Fig. 4 A parallel dimeric coiled-coil in a schematic representation. The helical wheel diagram in (A) top view down the axis of the α-helices from N-terminus to C-terminus. Panel (B) provides a side view. The residues are labeled a–g in one helix and a′–g′ in the other. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission from ref. 39. | ||
Papapostolou et al.41 engineered a second-generation self-assembling fiber (SAF) system based on two complementary leucine zippers (LZ). Specifically, the SAF peptides were based on the heptad repeat associated with coiled-coil protein sequences (abcdefg), with hydrophobic residues at the a and d sites, which formed the hydrophobic core of the coiled-coil helical bundle. The combination of isoleucine at a and leucine at d best specifies dimeric leucine zippers (LZs). The helical interface was connected further with interhelical charge–charge interactions between successive g and e sites. Complementary charge clusters were introduced to the surfaces of the interacting coiled-coils, a pair of negatively charged glutamates on one peptide, to complement an identically spaced pair of positively charged arginine side chains on the other was used to achieve the sticky-ended heterodimer designed to propagate the SAFs. To make the resulting coiled-coils as soluble as possible and introduce controllable aggregation, charged residues were placed at the b, c and f positions. Surfaces were therefore supercharged and highly hydrophilic with random charged patches. As a result fibers could be thinned down to ∼10 nm and made much more flexible such that they wrapped around one another to form loose networks.
The same group also described the design of a single-peptide, based on an α-helical coiled-coil motif, named MagicWand, which self-assembled into extended and thickened nano-to-mesoscale fibers of high stability and order.42 The peptide had a heptad sequence repeat, abcdefg, with isoleucine and leucine residues at the a and d sites to ensure dimerization. In addition, to direct staggered assembly of peptides and to foster fibrillogenesis the terminal quarters of the peptide were cationic and the central half anionic with lysine and glutamate, respectively, at core-flanking e and g positions. This +, −, −, + arrangement gave the peptide its name. Mutagenesis of the outer surface of the peptides i.e., at the b and f positions combined with stability and microscopy measurements, highlights the role of electrostatic and cation–π interactions in driving fiber formation, stability and thickening.
2.2 Long chains alkylated/acylated peptides
The second category of peptide amphiphiles discussed in this section is constituted by hydrophilic amino acid sequences coupled to hydrophobic alkyl chains at either the N- or the C-terminus.The laboratory of Stupp has given an important contribution in understanding the self-assembly of β-sheet peptide amphiphiles into cylindrical or spherical micelles and fiber structures.43–46 The same group has demonstrated the use of peptide amphiphile fibres in a broad range of applications. Many examples of their work will be discussed in more details in the following sections of this critical review.
In the past, the groups of Fields and Tirrell have investigated several amphiphilic peptides, which have been modified at the N-terminal with mono-alkyl hydrocarbon chains as minimal lipidation moieties for the stabilization of protein-like molecular architectures.47,48 In these studies mono-alkyl hydrocarbon chains were used for inducing protein-like structures such as α-helices and collagen-like triple helices. A potentially α-helical 16-residue peptide sequence, H-Lys-Ala-Glu-Ile-Glu-Ala-Leu-Lys-Ala-Glu-Ile-Glu-Ala-Leu-Lys-Ala-NH2, was designed based on a repeating heptad sequence, (Glu-Ile-Glu-Ala-Leu-Lys-Ala)n, known to associate to form a highly α-helical coiled-coil structure at a chain length of at least 23 residues.49 Although the peptide alone did not form a distinct secondary structure in solution, it adopted predominantly an α-helical structure upon acylation at the N-terminus with hexanoic acid (C6) or palmitic acid (C16) mono-alkyl chain, as demonstrated by circular dichroism (CD) spectroscopy. 1H NMR spectra showed that the thermal stability of the peptide structural conformation was already enhanced by N-acylation with a C6 alkyl chain. Increasing the alkyl chain length resulted in even higher thermal stability. This enhancement may partly be correlated to the extent of peptide–amphiphile aggregation. These studies demonstrated that hydrocarbon chains may be useful as general tools for induction and stabilization of protein-like secondary and tertiary structures in short peptide sequences and might be used for studying the mechanisms of peptide folding as well as for the generation of new biomaterials.
Löwik et al.50 demonstrated that certain peptide sequences conjugated by single C18 alkyl chains to both the N- and C-termini resulted in control over the secondary structure upon incorporation into a liposome membrane. A sequence derived from the circumsporozoite (CS) protein of the malaria parasite Plasmodium falciparum was chosen, as within the natural protein the Asn-Pro-Asn-Ala repeat is known to adopt a β-turn. Both the unmodified peptide in solution and the analogous peptide with only one alkyl chain showed random coil folding characteristics. However, when the double alkylated peptide was inserted into 1,2-dimyristoyl-sn-glycero-3-phosphoethanol-amine (DSPC) liposomes the peptide folded into a β-hairpin. This simple approach is a convenient way for stabilizing a variety of peptides into their preferred secondary structure on a lipid bilayer and might be employed in the presentation of multiple (hairpin) epitopes.
Lipid-modified proteins play important roles in numerous biological processes like signal transduction and vesicular trafficking, it is particularly interesting therefore to prepare molecules that can allow the study of these roles in precise molecular detail. The availability of a flexible solid-phase technique for the fast synthesis of lipidated peptide conjugates is of fundamental importance. Ideally such a technique would give access to peptides carrying different combinations of acid and base-labile lipid groups and allow for the introduction of additional reporter and/or linking groups required for application of the target peptides in further biological experiments. In this respect, Waldmann and co-workers51 developed a method for the efficient solid-phase synthesis of S-farnesylated and S-palmitoylated peptides, which are based on an oxidation-sensitive hydrazide linker that can be cleaved by oxidation with Cu(OAc)2 in the presence of pyridine and acetic acid. Furthermore, biologically fully functional Ras proteins were also prepared by the same group.52,53
Giralt and co-workers54 incorporated fatty acyl moieties into a proline rich (Pro-rich) sequence, a family of cell penetrating peptides. Confocal microscopy and flow cytometry showed that when a myristyl tail was added to the peptide, its internalization into HeLa cells was strongly improved. On the contrary, this improvement was not observed in the case of a caproyl (C6)-peptide conjugate. Monolayer studies with model dioleoylphosphatidylcholine (DOPC) membrane and transmission electron microscopy (TEM) further demonstrate the importance of the length of the fatty acyl moiety in the cell membrane penetration.
Tirrell and co-workers55 prepared biomimetic membranes from vesicle solutions functionalized with RGD lipopeptides. In their work two peptide–lipid conjugates containing the RGD amino acid sequence, (C16)2-Glu-C2-GRGDSP and (C16)2-Glu-PEO-GRGDSP, were synthesized and assembled into vesicles. Mouse fibroblast cell adhesion and growth were observed for RGD bilayers containing the polyethylene oxide spacer (PEO) lipopeptides but not for bilayers containing the shorter carbon spacer (C2) lipopeptides or for pure lipid bilayers. Furthermore, lithographic patterning and a microfluidic device were used to prepare RGD surface composition gradients that enabled cell response to varying surface properties.
For a better understanding of membrane insertion, assembly dynamics and membrane penetration, You and Gokel56 studied the aggregation behavior of a family of synthetic anion transporters (SATs) in liposomal bilayers. Pyrene, indole, dansyl and NBD fluorescent derivatives of the general type (n-C18H37)2N-COCH2OCH2CO-(Gly)3-Pro-(Gly)3-O-n-C7H15 were prepared. These amphiphilic peptides formed anion-conducting pores in bilayer membranes. Fluorescence resonance energy transfer (FRET) between pyrenyl- and indol-derivatives demonstrated aggregation of SAT monomers within the bilayer. This self-assembly led to pore formation, which was detected as C![[l with combining macron]](https://www.rsc.org/images/entities/char_006c_0304.gif) release from the liposomes. Furthermore, acrylamide quenching of fluorescent SATs supported membrane insertion. Finally, studies with NBD-labeled SATs showed that peptide partition into the bilayer is relatively slow. All together, this study functions as a synthetic model to investigate ion transport over membrane thought mimicry.
release from the liposomes. Furthermore, acrylamide quenching of fluorescent SATs supported membrane insertion. Finally, studies with NBD-labeled SATs showed that peptide partition into the bilayer is relatively slow. All together, this study functions as a synthetic model to investigate ion transport over membrane thought mimicry.
Peptide amphiphiles (PA) with hydrophobic alkyl tails have also been used for the noncovalent functionalization of carbon nanotubes. Arnold et al.57 showed that while carbon nanotubes without functionalization are insoluble in water, these nonpolar, one-dimensional nanostructures could be dispersed by ionic peptide amphiphiles in aqueous solutions. Furthermore, by adjustment of the pH, the solubility of functionalized carbon nanotubes can be controlled. Peptide amphiphiles self-assembled on the nonpolar and hydrophobic surface of carbon nanotubes, minimizing the interfacial energy of the nanotube–water interface. Inspection of the PA-functionalized multiwalled carbon nanotubes (MWNTs) by transmission electron microscopy (TEM) confirmed the existence of an organic coating on the exterior sidewall of the outermost nanotube shell. This approach offers the benefit that the nanotube sidewalls did not need to be chemically modified, thus maintaining the electrical, mechanical, and optical properties of unmodified carbon nanotubes.
2.3 Peptide–phospholipid conjugates
Peptide sequences have also been covalently coupled to phospholipids in order to facilitate the incorporation of lipopeptides into the cell membrane. In general, cell-membrane proteins are anchored to the lipid bilayer by lipidation in co- and post-translational enzymatic processes, including acylation with fatty acids, prenylation, and rather commonly C-terminal amidation with glycosylphosphatidylinositols (GPI).58–60Musiol et al.61 have shown the preparation of semi-synthetic lipoproteins by C-terminal lipidation exploiting the copper(I)-catalyzed Huisgen’s 1,3-dipolar cycloaddition.62 A fragment 214-231 of the human prion protein with a C-terminal propargylglycine was reacted with an azido-modified 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE), mimicking the GPI anchor, to form a stable 1,4-disubstituted 1,2,3-triazole product. The “click” reaction was used by the ligation of the resulting N-cysteinyl-lipopeptides and N-terminally fluorescently labeled peptide thioesters.63,64 Incubation of HeLa cells with the micellar solution of the lipopeptide confirmed its fast uptake of the semi-synthetic lipoprotein, as visualized by confocal fluorescence microscopy.
Membrane fusion is a key process in all living cells, as it facilitates the transport of molecules between and within cells. The process is triggered by the specific interaction of fusion proteins. This interaction brings two membranes into close proximity and is followed by local disruption of the lipids and merging of the membranes. The required protein recognition for the fusion of transport vesicles with the neuronal membrane involves the coiled-coil interaction between three complementary SNARE proteins.65 To induce intracellular transport, a four-helix coiled-coil bundle forms between two membrane-bound SNARE proteins and a cytoplasmic SNARE protein and forces the two membranes within a distance of 2–3 nm from one another. Recently, Robson Marsden et al.66 designed reduced systems based on lipidated peptides anchored to liposome membranes to gain insight into the most important aspects of membrane fusion.
In this work, two lipidated oligopeptide hybrids (LPE and LPK) were synthesized as a minimum coiled-coil pair designed to assemble specifically into a stable heterodimer that possessed all functional aspects of membrane-bound SNARE proteins (Fig. 5a). In this model system, the recognition domain was mimicked by two three-heptad repeat coiled-coil-forming peptides (E and K). The formation of the LPE/LPK complex was the driving force to bring two different liposomes close together. The role of the flexible spacer was fulfilled by a short poly(ethylene glycol) chain, which enabled extension of the oligopeptide component from the surface of the liposomes. The lipidated oligopeptides were anchored in the membrane by means of a phospholipid tail, 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (DOPE) mimicking the function of the transmembrane domain of SNARE proteins. Furthermore, lipid and content mixing were proven using a fluorescence resonance energy transfer (FRET) assay. Broadly, this minimal fusion system extends the area of synthetic biology, enabling the understanding of aspect of nature-liposome fusion in eukaryotic cells through mimicry (Fig. 5).
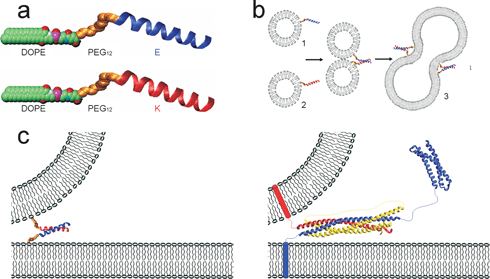 | ||
| Fig. 5 (a) Space-filling model of the lipidated oligopeptides LPE and LPK, consisting of a DOPE tail linked through a PEG12 spacer to the coiled-coil-forming oligopeptides E and K. The amino acid sequence of E is G(EIAALEK)3-NH2, and that of K is (KIAALKE)3GW-NH2. (b) The spontaneous incorporation of the DOPE tail in lipid bilayers results in liposomes decorated with either E or K peptides at the surface. When a liposome population carrying LPE (1) is mixed with a liposome population carrying LPK (2), coiled-coil formation (E/K) initiates liposome fusion (3). (c) Comparison of the minimal model (left) with the SNARE-protein-based model (right). Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission from ref. 66. | ||
Our group has designed a new series of amphiphilic lipopeptides (ALPs) consisting of an alternating hydrophobic (Leu) and hydrophilic (Glu) amino acid residue sequence coupled to a natural phospholipid tail. Three ALPs, each exhibiting a different length of the peptide part, (Leu-Glu)2-, (Leu-Glu)3- and (Leu-Glu)4, were synthesized and studied for their ability to form supramolecular assemblies at the air–water interface. In situ grazing-incidence X-ray diffraction (GIXD) revealed that lipopeptides, containing six to eight amino acid residues formed a new type of 2D self-organized monolayers that exhibit β-sheet ribbons segregated by lipid tails.67 These assemblies were able to act as well-defined two-dimensional templates for the mineralization of CaCO3. The conjugation of the phospholipid moiety to the octapeptide (Leu-Glu)4 not only enhanced the amphiphilic behavior of the molecule but also increased the flexibility of the monolayer, without compromising the β-sheet structure. A new morphological form of oriented calcite crystals nucleated underneath the flexible amphiphilic lipopeptide monolayer.68
2.4 Peptide-based block copolymers
The fourth class of peptide amphiphiles comprises peptide-based block copolymers. These polymers were recently reviewed in depth, so here we will show a limited number of highlights.69,70 Klok and co-workers71 synthesized a series of hybrid di- and triblock copolymers, which contained amphiphilic β-strand peptide sequences and poly(ethylene glycol) (PEG) segments. The block copolymers have been prepared via solid-phase synthesis, affording monodisperse peptide segments with a precisely defined α-amino acid sequence. The self-assembly properties of the peptide sequences were retained upon conjugation to PEG and the formation of an ordered superstructure was observed, consisting of alternating layers of PEG domains and peptide domains with a highly organized antiparallel β-sheet structure. This work suggests that the combination of biological structural motifs with synthetic polymers may be a versatile strategy for the development of novel self-assembled materials with complex internal structures and the potential to interface with biology. Smeenk et al.72 described the preparation and assembly of an ABA-type triblock copolymer consisting of a central β-sheet peptide block composed of the repetitive [(AG)3EG]n sequence conjugated to PEG end blocks. The [(AG)3EG]n β-sheet polypeptides, outfitted with N- and C-terminal cysteine (Cys) residues, were constructed by protein engineering. The thiol groups of the Cys were subsequently selectively alkylated with maleimide-functionalized PEG. Crystallization of the triblock copolymer resulted in well-defined fibrils, which were formed in the β-sheet stacking direction. The authors envisioned that control over the amino acid sequence would offer the possibility of introducing specific amino acid residues at the turns of the β-sheets, thereby creating a regular array of functional moieties at the fibril surface.The polymerization of α-amino acid-N-carboxyanhydride (NCA) monomers has been key for the chemical syntheses polypeptide in solution.73 The use of transition metal initiators in NCA polymerizations has allowed the preparation of very well-defined homopolypeptides and led to facile routes into peptide block copolymer materials. Kros and Cornelissen74 described the synthesis and self-assembly of hybrid block copolymers composed of a poly(γ-benzyl L-glutamate) block (PBLG) and two different polyisocyanide blocks, namely, poly((S)-(−)-α-methylbenzyl isocyanide) (PMBI) and poly(L-isocyanoalanyl-L-alanine methyl ester) (L,L-PIAA). The diblock copolymers were synthesized by the Ni-catalyzed living polymerization of γ-benzyl L-glutamate N-carboxyanhydride (Bn-Glu, NCA, vide infra) according to the method developed by Deming and co-workers75,76 followed by the polymerization of (S)-(−)-α-methylbenzyl isocyanide (MBI) or L-isocyanoalanyl-L-alanine methyl ester (L,L-IAA) to the reaction mixture. This new class of rod-rod block copolymers exhibited multiple structural motifs (for example, an α-helical peptide segment combined with a β-helical polyisocyanopeptide segment) and consequently unique properties. Remarkably, The PBLG-block-L,L-PIAA has three secondary structural motifs within one macromolecule, that is, an α-helical polypeptide segment and a polyisocyanide helix with side arms organized in a parallel β-sheet. Preliminary self-assembly studies were performed in organic solutions and the formation of polymersomes (with a uniform diameter of 7.5 mm) was observed upon fast drying of a solution of PBLG-block-L,L-PIAA revealed by confocal laser scanning microscopy. However, slow evaporation resulted in the formation of closed films, which suggested that the polymersomes were a kinetically trapped architecture.
Deming and co-workers77 synthesized diblock copolypeptide amphiphiles using the transition metal-mediated-amino-acid NCA polymerizations described above, which allowed control over polypeptide chain length and composition. Many of these diblock amphiphiles were found to form rigid hydrogels in water. The gelation process of the copolypeptides was found to be dependent not only on the amphiphilic nature of the polypeptides, but also on the type of secondary structures present in the chain, as α-helices or β-strands favoured the hydrogel formation, while random coil domains inhibited gelation. The poly(L-Lys·HBr) and poly(L-Glu sodium salt) domains, being highly charged polyelectrolytes at neutral pH, dissolved readily in water. The hydrophobic domains, when sufficiently large, could adopt regular conformations that aggregate and were insoluble in water, namely, rod-like α-helices for poly(L-Leu) and crystalline β-sheets for poly(L-Val). Copolypeptides of identical compositions, but of random sequences, were never found to form hydrogels at all.
Coiled-coil-based hydrogels represent an interesting area that continuously attracts the attention of many research groups especially for potential biomedical applications. Selected examples of hydrogels prepared exploiting the coiled-coil motif are given below.
For example the group of Kopeček78 exploited the coiled-coil motif for the preparation of hybrid hydrogels. Based on the observation that upon minor alteration of the primary structure many native and de novo designed coiled-coils can undergo conformational transition induced by changes in temperature, pH, ionic strength and solvent, Kopeček’s group studied engineered coiled-coil sequences as crosslinkers in synthetic polymer chains. For the primary chains of their hybrid hydrogels, a linear hydrophilic copolymer of N-(2-hydroxypropyl)methacrylamide (HPMA) and a metal-chelating monomer N-(N′,N′-dicarboxymethylaminopropyl)methacrylamide (DAMA) were prepared by radical copolymerization. A metal complex was formed by the pendant metal-chelating ligand-iminodiacetate (IDA)-Ni2+ and the terminal histidine residues (His tag) of the coiled-coils. Using this approach, genetically engineered His-tagged coiled-coils were connected to the polymethacrylamide polymer in a convenient manner resulting in the formation of a hydrogel. This hybrid system preserved the benefit of using synthetic polymer backbones that are well characterized, easy to manufacture and biocompatible. The temperature-responsiveness of the hybrid hydrogels was investigated and the gel structural transition was found to be related to the temperature-induced conformational change in the coiled-coil motif.
Among others Harden’s and Tirrell’s groups also extensively explored the area of coiled-coil-based hydrogels, as reported in recent works. As an example, in the former group the self-assembly of hydrogels composed of acidic and basic leucine zipper (LZ) associating domains and a soluble disordered coil block containing three copies of the Arg-Gly-Asp (RGD) integrin binding sequence were studied.79 The RGD sequences embedded in the disordered coil region supported the adhesion, spreading and polarization of human fibroblast cells on protein coated surfaces. Such hydrogel-forming bioactive proteins have potential for cell and tissue culture applications. In the latter group recombinant DNA methods were used to create artificial proteins that undergo reversible gelation in response to changes in pH or temperature.80 The proteins consisted of terminal LZ domains next to a flexible water soluble polyelectrolyte segment. Proteins bearing dissimilar helical coiled-coil end domains were found to degrade much more slowly than hydrogels formed from those bearing the same end domains.81 The mild conditions under which gel formation could be achieved (near-neutral pH and mild temperature) and the control over the erosion rate suggested that these materials have potential in bioengineering applications for encapsulation or controlled release of drugs.
Robson Marsden et al.82 demonstrated the formation of a noncovalent triblock copolymer based on a coiled-coil peptide motif. In their strategy the authors used two complementary, hydrophilic and hydrophobic, polymer-peptides. A Lys-functionalized poly(ethylene glycol) (K-PEG) and a Glu-functionalized polystyrene (PS-E).
These constructs were based on the coiled-coil-forming propensities of the specific peptide pair (E and K) to assemble into hetero coiled-coils (Fig. 6). The hierarchical self-assembly in solution resulted in the formation of coiled-coil complexes between the peptides and subsequently in the formation of the amphiphilic triblock copolymer PS-E/K-PEG. It was found by cryogenic transmission electron microscopy (cryo-TEM) that the noncovalent PS-E/K-PEG copolymer assembled into rodlike micelles, while the release of K-PEG led to a transformation to spherical micelles. Temperature-dependent studies revealed the reversible nature of the coiled-coil complex and the influence of this on the morphology of the aggregate. A possible mechanism for these transitions based on the interfacial free energy and the free energy of the hydrophobic blocks was also discussed by the authors.
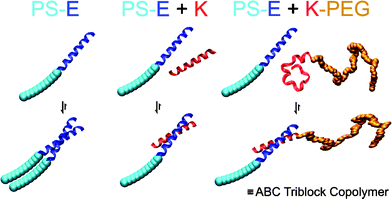 | ||
| Fig. 6 Schematic representation of the hierarchical self-assembly of the hybrids PS-E and K-PEG containing complementary peptide blocks. PS is polystyrene, PEG is poly(ethylene glycol), and E and K are peptides. Reprinted with permission from ref. 82. Copyright 2008 American Chemical Society. | ||
3. General synthetic and protein engineering strategies
From a chemical perspective, recent advances in synthetic strategies have allowed the preparation of (poly)peptides and (poly)peptide hybrids, which are able to assemble in a controlled fashion into supramolecular architectures and materials that mimic the structure and function of proteins in a controlled manner. These synthetic methods can be divided in three main classes: chemical synthesis based mostly on solid-phase strategies, protein engineering and ring-opening polymerization techniques.83 Next, an overview of other synthetic methodologies used to prepare peptide-based hybrids is given. Followed by a section dedicated to self-replicating peptides.3.1 Solid-phase peptide synthesis
Solid-phase peptide synthesis (SPPS) is a powerful method for the preparation of small to medium-sized peptides.17 In contrast to the ring-opening polymerization, SPPS allows the preparation of monodisperse peptides with precise control of the primary structure. As the yield of the majority of the reactions are very high (>98%), small and medium-sized peptides are easily obtained in high yields and purities. However, yields rapidly drop and purification becomes more difficult with increasing chain length. Synthesis of medium sized peptides can be faced by the “Hybrid” approach, where the protected peptides synthesized in solid-phase are combined in solution.84A further refinement that allows to overcome the limitations of SPPS with respect to the size and solubility of the peptides is the thioester-mediated native chemical ligation (NCL)63,64,85,86 of unprotected peptide segments. This method relies on the chemoselectivity of the reaction between a peptide-α-thioester and another segment containing an N-terminal cysteine residue. The different segments are synthesized using a solid-phase approach. NCL has been successfully used for the total chemical synthesis of a variety of proteins. This combination of solid-phase (synthesis of peptide segments) and solution (ligation) provides a convenient route for the synthesis of proteins composed of a larger number of peptide segments.
SPPS strategies are also useful for the synthesis of hybrid molecules containing, for example, hydrophobic alkyl or polymeric chains conjugated to peptide sequences. In some cases, the solid-phase supported synthesis, purification and analysis of these amphiphilic (poly)peptides is highly challenging. Strategies were developed to facilitate the synthesis of “difficult peptide sequences” (i.e. amyloid peptides), by integrating defined structure defects into peptides.87–89 Thus, the introduction of a reversible ester bond in the peptide sequence disrupts the amidic backbone suppressing the aggregation tendency, which benefits the synthesis, and improving the solubility, facilitating the purification. The “switch” ester was obtained by modification of the standard Fmoc protocols using a Boc-protected Thr/Ser derivative with an unprotected hydroxyl side chain functionality, followed by the coupling of the next Fmoc-amino acid to the β-hydroxyl group. At the end of the synthetic process after purification, the peptide backbone was subsequently re-established via a selective pH dependent rearrangement (O → N acyl switch). Hentschel et al.90 used this methodology to trigger the peptide guided assembly of poly(ethylene oxide)-peptide conjugates into tape structures.
3.2 Protein engineering
The synthesis of polypeptides by bacterial expression of artificial genes, also referred to as protein engineering, is a very attractive strategy, since it allows the preparation of high molecular weight and perfectly monodisperse polypeptides with a precisely defined primary structure.91 Protein engineering has been successfully used for the synthesis of natural structural proteins such as silk, collagen and elastin as well as for the preparation of de novo designed proteins. Protein engineering is not restricted to proteogenic α-amino acids and various methods are available that allow the incorporation of unnatural analogues.92,93 In this way, it has become possible to prepare artificial proteins that carry a variety of non proteogenic functional groups. The growing improvements in expression of recombinant protein polymers allowed to expand the use of protein-based biomaterials both in the investigation of basic cellular processes and in therapeutic applications.943.3 Ring-opening polymerization
Polypeptides can also be prepared by ring-opening polymerization of α- and β-amino acid N-carboxyanhydrides (NCA)95,96 and β-lactams97 affording poly(amino acid)s. Generally, the ring-opening polymerizations can be performed following well-established protocols, thus allowing a straightforward and high yielding synthesis of polypeptides with a broad molecular weight range at large scale. The down side of this method is that it is not possible to control the exact primary peptide sequence, in contrast to the solid phase-based protocols (SPPS). Polydispersities have been achieved as low as 1.03.98 However, for some applications high purity monomers could be required. In addition, side reactions may occur, which complicate the preparation of optically pure polypeptides with predictable molecular weights and narrow molecular weight distributions, also hampering the formation of well-defined block copolymers. A number of these drawbacks have been overcome by using transition metal initiators including for example Ni39,40,76,77 and Co99 complexes.3.4 Other synthetic approaches to peptide-based hybrids
Several other chemical strategies have been used in order to synthesize conjugates of non-peptidic segments, such as lipid or polymer tails, to peptides. Connectivity has also been achieved for example using amide100 or thiol-maleimide coupling101,102 as well as by imine,103 hydrazone linkage104 and chemoselective peptide ligation.105 A recent approach exploits the use of the 3 + 2 cycloaddition 62,106 between azides and alkynes as a highly useful chemical handle for conjugation either in a non Cu-mediated107–111 or Cu(I)-catalyzed112,113 manner. The so called “click” chemistry has recently emerged as a powerful tool for the synthesis of bioconjugates mainly due to the fact that the Cu(I)-catalyzed [2 + 3] cycloaddition compared to the non-catalyzed reaction, occurs with a dramatic rate increase and exclusive regioselectivity in aqueous media and at room temperature.112,114–117A versatile methodology to prepare peptide-based hybrid polymeric biomaterials exploits the atom transfer radical polymerization (ATRP), which allows the synthesis of polymers with well-controlled molecular weight and molecular weight distributions without the strict requirements on a water- and oxygen-free environment necessary for other types of living polymerizations.118 ATRP has been shown to be able to polymerize a wide range of bioinspired monomers such as peptide-based monomers that have been used to synthesize block copolymers either in solution119–121 or on solid-support.122 In a recent example, Ayres et al.123 used ATRP to polymerise a monomer based on the cyclic decapeptide gramicidin S, bearing a methacrylate moiety at the side chain of an hydroxy-proline residue (which was introduced at the place of one of the proline residues in the β-turn). Gramicidin S is a large cyclic peptide that is well known for its antibiotic properties124,125 and its ability to form inter- and intramolecular β-sheets.126,127 The ability to achieve controlled polymerization of such a bulky, biologically relevant, peptide-based monomer represents a new approach towards the preparation of well-defined, antimicrobial polymeric biomaterials.
3.5 Self-replication
Another interesting synthetic strategy is related to self-replication. Self-assembly of peptides has been used as a driving force for chemical catalysis in amide bond formation, resulting in self-replication.128The discovery that peptides have replicative properties has led to the design of a wide range of autocatalytic systems based on the coiled-coil motif and exploiting the native chemical ligation.63,64,85
In self-replication, the product (i.e. peptide) acts as a template to preorganize precursors (i.e. peptide fragments) based on its own sequence in order to catalyze the formation of the product itself (Scheme 1). As a result, a new product is formed that is identical to the template. The template’s ability to form a ternary complex with the two peptide fragments is mediated by interhelical hydrophobic interactions which, in turn, may promote the chemical ligation process. After dissociation, the newly synthesized peptide could then act as a template for a new set of fragments, resulting in exponential product growth. Improvements in the design of self-replicating peptides have been directed towards the enhancement of the autocatalytic efficiency.129–131 Ideas have also been discussed to promote the use of self-replicating peptides in novel biomaterial and biomedical applications.132
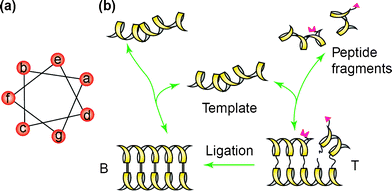 | ||
| Scheme 1 Self-replication cycle based on the coiled-coil motif. (a) Helical wheel representation of a coiled-coil peptide showing the heptad repeat. (b) The reaction cycle for a self-replicating peptide with its fragments. Reprinted from ref. 129, with permission from Elsevier. | ||
Ghadiri and co-workers133 highlighted a synthetic chemical approach, exploiting the native chemical ligation, towards the rational de novo design of complex self-organized molecular systems. The work showed the design and implementation of a network of nine α-helical peptides directing each other’s synthesis through a complex network of various autocatalytic and cross-catalytic cycles of complex topology, including two-, three-, and four-member pathways.
4. Applications of peptide-based nanostructures
The advances in synthetic tools allow unprecedented control over composition, structure and organization of artificial proteins and peptide hybrid materials. Undoubtedly, these developments and future advances will allow the integration of biological design concepts in materials science and lead to protein-inspired new materials.134 Applications of self-assembling peptide systems as simple and versatile molecular building blocks can provide new opportunities in biotechnology and engineering.135,136 An overview of the possible applications is given in the following sections of this review.4.1 Nanoreactors and catalysts
Guler and Stupp137 explored the ability of cylindrical supramolecular nanostructures to act in the hydrolysis of 2,4-dinitrophenyl acetate (DNPA), a common model compound to study the catalytic activity of enzymes and synthetic systems, selected as the substrate for mimicking esterase activity. A palmitoylated peptide sequence (KLLLAAA) with histidine residues attached to the lysine residue was shown to self-assemble into cylindrical β-sheet nanofibers and hydrolysis of DNPA by imidazolyl-functionalized molecules was monitored by UV-vis spectroscopy. A considerably higher hydrolysis rate was observed in the presence of internally high ordered supramolecular nanofibers compared to catalysts in solution or in spherical aggregates, used as control, validating the hypothesis that hydrolysis efficiency benefits by a high density of reactive sites displayed on the surface of a supramolecular catalytic particle with significant internal order. In future, these nanostructures could be further developed by co-assembling different molecules in a nanofiber so that various chemical events may be integrated into a single catalytic system.Amphiphilic peptides belong to the class of low-molecular weight amphiphiles (in nature, typical examples of this class are phospholipids with an average molecular volume of ∼0.5 nm3 and a molecular weight of ∼1 kDa). A more recently introduced class of super-amphiphiles138consists of hydrophilic–hydrophobic block-co-copolymers (i.e. diblock polymer of polystyrene and a polyisocyanopeptide with a molecular volume of ∼6.5 nm3 and a molecular weight of ∼6 kDa). Combining synthetic polymers with enzymes as head groups could lead to the development of a new class of giant amphiphiles with catalytic properties. In terms of both molecular volume and molecular weight the next generation of amphiphiles, the giant amphiphiles, are considerably larger than their low-molecular weight and polymeric counterparts (i.e. the n = 40 polystyrene-lipase biohybrid with a molecular volume of ∼25 nm3 and a molecular weight of ∼40 kDa).
Velonia et al.139 coupled the enzyme lipase B from Candida antarctica to an end maleimido-modified polystyrene. These giant amphiphiles self-assembled into fibres in a comparable manner to their low-molecular weight counterparts upon dispersion in aqueous solutions. It was proposed that the fibres were composed of micellar rods with a hydrophobic polystyrene core with the protein, acting as the hydrophilic head group, exposed to the aqueous environment. However, it was found that the catalytic activity of the enzyme was reduced 15-fold, which was ascribed to a destabilizing effect of the hydrophobic polystyrene tail on the active conformation and a possible lower accessibility of the catalytic site. A better understanding of the rules governing the assembly of these giant amphiphiles could lead to improvement in their activities.
The interest in applying polymeric microcapsules as small reactors is steadily increasing.140,141 Stimulus-responsive polymersomes based on polybutadiene-b-poly(γ-L-Glu) were investigated as possible nanoreactors.142,143 The size of the polymersome molecules could be reversibly altered by changing both the pH and the ion strength. Vriezema et al.144 described the encapsulation of Candida antarctica lipase B (CAL B) enzymes inside polymersomes of polystyrene-b-poly(isocyano-L-Ala (2-thiophen-3-yl-ethyl)amide) (PS-PIAT). It was demonstrated that the enclosed CAL B enzymes were still active and that the polymersome membrane was permeable to low molecular weight substrates, for example, 6,8-difluoro-4-methylumbelliferyl octanoate (DiFMU octanoate). Upon hydrolysis of the ester bond of this substrate, a fluorescent coumarin-type of product was formed, allowing the monitoring of the enzyme activity. It is worth noting that the membrane of the PS-PIAT vesicles was chiral and therefore potentially selective toward chiral substrates or chiral products.
Ryandov145 exploited two coiled-coils in order to build polynanoreactors. A noncovalent supradendrimer (SD) framework was built. This SD was obtained from the self-assembly of a single peptide sequence, SD-1 (Fig. 7A), which was based on a dimeric coiled-coil also known as the leucine zipper. SD-2 presented a leucine zipper sequence that was complementary to SD-1. Mixing SD-1 and SD-2 led to the formation of allowed polynanoreactors (SD-1,2) as shown in Fig. 7B. The cavities of SD-1,2 mimicked the constrained environments of natural protein vessels and was used to template the conversion of ionic metals into colloidals. Silver nanoparticles of 5.2 ± 0.5 nm were prepared within SD-1,2 cavities by citrate reduction of silver nitrate.
 | ||
| Fig. 7 Synthesis of silver nanoparticles within SD-1,2. (A) A growing silver nanoparticle (Ag) hosted by an SD-1,2 cavity is depicted as a gray circle. (B) A network of cavities filled with silver nanoparticles (gray circles). Arrows indicate the confined space of the cavity within which the particle is to grow. Cysteine residues forming encapsulating thiol (SH) clusters are shown as circles marked with crosses. (C) An electron micrograph of a spherical 1.2 μm wide spread of silver nanoparticles. (D) High magnification of an edge portion of the image shown in part (C). Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission from ref. 145. | ||
4.2 RGD functionalized materials
The tripeptide motif Arg-Gly-Asp (RGD) sequence, so-called “universal recognition site”, has been identified as a minimal essential cell adhesion sequence.146 Since its discovery, numerous materials have been RGD functionalized for a broad range of purposes, including (academic or) medical applications.147,148 RGD-related peptides are known to contribute to various cellular functions such as adhesion, invasion and to inhibit tumor metastasis.149 However, peptide-based drugs are generally rapidly hydrolyzed and eliminated from the bloodstream. In contrast, RGD-modified liposomes were shown to enable the half-lives and affinity of the unmodified peptide, resulting in enhancement of antimetastatic activity.150 Liposomal RGD was prepared using lipophilic derivatives of the peptide, which could easily be synthesized and incorporated into the liposomal bilayer.In another example, Yagi et al.151 have prepared liposomes whose surface is modified with peptides containing a five-time repeat of the GRGDS sequence, as found in the cell adhesion sequence of fibronectin. The peptide was lipidated by incorporation at the N-terminus of an aspartic acid residue modified with two C16 alkyl chains. The availability of the peptides on the surface was confirmed with immuno-electron microscopy studies, employing a specific antibody to the peptide that could be visualized with gold colloids. The liposomes were shown to bind mouse fibroblast cells and the association took place through interaction of the exposed peptide and the corresponding cell surface receptor.
Most approaches to display RGD-containing peptides immobilize the peptide by covalently linking it through the N-terminus, leaving the carboxy-terminus free. It is a well-known fact, however, that the RGD sequence exists in a conformational constrained loop in proteins such as fibronectin. It has been demonstrated that cyclic peptides which contain the RGD sequence can display higher affinities than their unconstrained linear counterparts. Pakalns et al.152 have shown that the same conformational constraint could be obtained by attaching doubly alkylated Glu derivatives to both the N- and C-termini. These peptide amphiphiles were utilized to prepare self-assembled monolayers which could be deposited as Langmuir–Blodgett films on a surface. On these surfaces functionalized with looped RGD amphiphiles, melanoma cells were able to spread in a concentration dependent manner.
A similar approach has been followed to enhance the activity of the RGD sequence towards integrin receptors. Marchi-Artzner et al.153,154 have attached a cyclized RGD containing pentapeptide to lipid alkyl chains connected through a short ethylene glycol spacer. A supported membrane containing these amphiphiles selectively adhered to endothelial cells of the human umbilical cord. Moreover, giant vesicles functionalized with cyclic RGD peptides adhered to the same endothelial cells, a process that could be inhibited by adding the corresponding soluble peptide. This suggests a specific interaction between the bilayer anchored peptide and the integrin receptors of the cells.
Hydrogels produced from self-assembling peptides and peptide derivatives are being investigated as synthetic extracellular matrices (ECM), cell culture substrates and scaffolds for regenerative medicine. In many cases, however, they are less stiff than the tissues or ECM. Jung et al.155 employed native chemical ligation to include unprotected RGD-functionalized peptides. In this respect, the authors designed and investigated a gel-forming system of peptide α-thioesters with N-terminal Cys residues based on an amino acid sequence that forms β-sheet fibrillar networks, Q11 (Ac-QQKFQFQFEQQ-NH2). Native chemical ligation led to significant matrix stiffening and improvements in primary human umbilical vein endothelial cell (HUVEC) proliferation. Furthermore, CD31 expression on the surface of the gels was observed.
By following a minimalist approach, Zhou et al.156 reported the design of a biomimetic nanofibrous hydrogel self-assembled bioactive hydrogels composed of simple fluorenylmethoxycarbonyl-diphenylalanine (Fmoc-FF)157 and fluorenylmethoxycarbonyl-arginine-glycine-aspartate (Fmoc-RGD) that assembled into β-sheets interlocked by π–π stacking of the Fmoc groups. Cylindrical nanofibers interwoven within the hydrogel with the presence of RGDs in tunable densities on the fibre surfaces were observed. This rapid gelling material was observed to promote adhesion of human dermal fibroblasts three-dimensionally in vitro.
Mata et al.158 were able to micro-pattern nanofiber gels of peptide amphiphiles (PAs) by incorporating polymerizable acetylene groups in the hydrophobic segments. PAs containing the cell adhesive epitope Arg-Gly-Asp-Ser (RGDS) were allowed to self-assemble within microfabricated molds to create networks of either randomly oriented or aligned nanofiber bundles (∼30 nm diameter) that were shaped into topographical patterns containing holes, posts, or channels up to 8 mm in height and down to 5 mm in lateral dimensions (Fig. 8). Human mesenchymal stem cell (hMSC) differentiation was investigated and was shown to be significantly affected by the type of substrate. For instance, when topographical patterns contained nanofibers aligned through flow prior to gelation, the majority of hMSCs oriented in the direction of the nanofibers. In topographical patterns with randomly oriented nanofibers, osteoblastic differentiation was enhanced on hole microtextures compared to all other surfaces. The hierarchical structures accessed by the simultaneous use of lithography and self-assembly may function for the construction of biomedical devices.
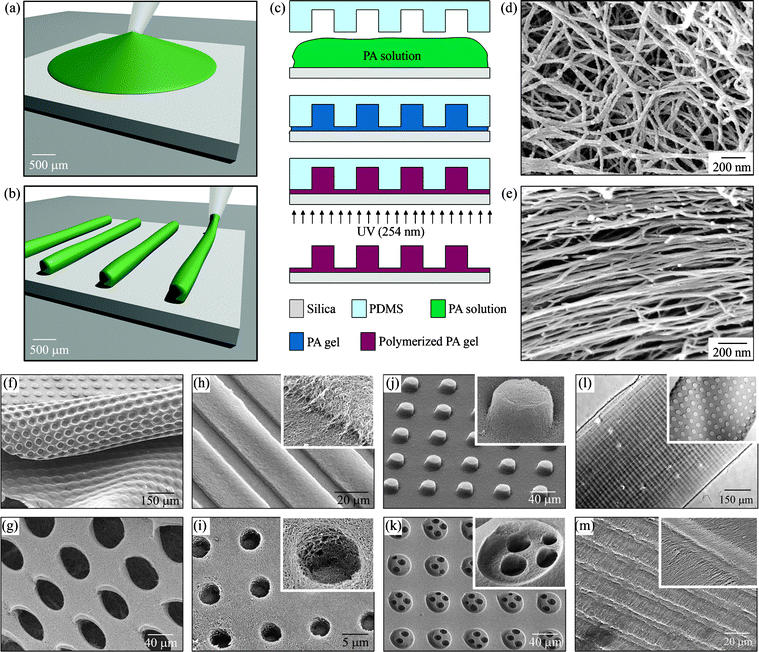 | ||
| Fig. 8 Fabrication techniques and resulting PA structures. (a–e) The fabrication process starts by either (a) dropping freshly dissolved PA for microtextures with randomly oriented nanofibers, or (b) dragging an aged PA solution for microtextures with aligned nanofibers on a silica substrate. (c) Then, a PDMS mold was used to cover the PA solution while allowing it to conform to the mold, self-assemble into nanofibers, and gel upon exposure to ammonium hydroxide (NH4OH). The PA gel was then polymerized under UV irradiation and released from the mold to realize the PA microtextures. The process in (a, c) was used to achieve well-defined three-dimensional (3D) PA structures with (d) randomly oriented nanofibers including (f) removable layers with microtextures or (g) pores and surface microtextures such as (h) channels, (i) holes, (j) posts, and (k) two-level topographies with features down to 5 μm in size. On the other hand, following the process in (b, c), microtextures with (l, m) channels and holes were also achieved but with aligned nanofibers (inset in m). Reproduced by permission of the Royal Society of Chemistry from ref. 158. | ||
4.3 Tissue regeneration materials
Culturing cells in three-dimensions has received a growing interest in the past few years.159 To grow in 3D culture, cells need to be embedded in a structure that mimics the ECM of structural proteins and other biological molecules found in real, living tissues. Many researchers use a commercially available material called Matrigel,160,161 which consists of structural proteins such as laminin and collagen, plus growth factors and enzymes, all taken from mouse tumours. Matrigel displays a lower critical solution temperature (LCST) and it is a liquid below 4 °C. Cells are mixed with the Matrigel solution and upon raising the temperature a 3D-gel is formed in which the cells can grow.Zhang and co-workers7,24,25,162 have developed a series of amphiphilic peptides that form stable hydrogels at low peptide concentrations (0.1–1%). They were characterized by an alternating sequence of hydrophobic and hydrophilic residues, in which the hydrophilic residues, in turn, alternate between being positively and negatively charged, such as in (KLDL)n, (EAKA)n and (RADA)n. The alternation between polar and non-polar residues promoted the formation of a β-strand building block with hydrophobic and hydrophilic faces. The solution to gel transition could be triggered rapidly when the ionic strength exceeded a specific threshold or the pH was adjusted to provide a zero net charge on the peptide. These types of peptides have been shown to be non-cytotoxic and of potential use in the repair of cartilage tissue. Chondrocytes were encapsulated within the hydrogel scaffold produced by the peptide Ac-(KLDL)3-CONH2.163 The scaffold was shown to maintain differentiated chondrocytes and to stimulate the synthesis and accumulation of extracellular matrix.
In another example, self-assembling peptides are being developed as scaffolds for tissue regeneration purposes, including cartilage repair and promotion of nerve cell growth.164 A major benefit of synthetic materials is that they minimize the risk of biological contamination. Self-assembling peptides also frequently show favorable properties concerning biocompatibility, immunogenicity and biodegradability, producing non-toxic waste products. Laminin is an extracellular matrix protein that influences neurite outgrowth. A peptide amphiphile shown to promote the regrowth of nerve cells in rats was made by including a neurite-promoting laminin epitope tag, IKVAV. Another construct, containing a heparin-binding site, showed preliminary results in being able to promote angiogenesis, the growth of blood vessels. These types of peptide amphiphiles have been further modified with biotin and a Gd3+ metal-chelating moiety suitable for detection by magnetic resonance imaging (MRI).164
Lee and co-workers165 designed self-assembling peptide nanofibers for prolonged delivery of insulin-like growth factor 1 (IGF-1), a cardiomyocyte growth and differentiation factor, using a ‘‘biotin sandwich’’ approach. Biotinylated IGF-1 was bound to tetravalent streptavidin and subsequently bound to biotinylated self-assembling peptides. This biotin sandwich strategy allowed binding of IGF-1 but did not prevent self-assembly of the peptides into nanofibers within the myocardium. When combined with transplanted cardiomyocytes, IGF-1 delivery by biotinylated nanofibers improved cell therapy compared to cells embedded within nanofibers alone or with loose IGF-1, demonstrating how engineering the local cellular microenvironment can improve cell therapy.
Recently, Stupp and co-workers166 reported the controlled assembly of polymers with peptide amphiphiles. High molecular weight polysaccharide hyaluronic acid (HA) and C-16 alkylated V3A3K3 were shown to self-assemble into hierarchically ordered sacs and membranes. In vitro studies using hMSCs were performed to investigate if these PA-HA structures can support cell viability and differentiation. Expanded hMSCs were incorporated within gel-filled sacs and cultured in growth media. The hMSCs remained viable within the sacs for up to 4 weeks in culture. Furthermore, hMSCs were able to differentiate toward a chondrogenic phenotype within the sacs. The self-assembling sacs could therefore provide sufficient nutrient diffusion necessary for cell survival and differentiation.
4.4 Drug-delivery vehicles
Molecular and supramolecular drug-delivery systems have attracted great attention as pharmaceutical formulations for the administration of otherwise poorly soluble, rapidly degradable, or even toxic molecules. Macromolecular self-assembly has been exploited recently to engineer materials for the encapsulation and controlled delivery of therapeutics.167 Synthetic carriers, including peptide-based delivery systems, have been developed in several laboratories as promising alternatives to inactivated viruses for gene delivery. Peptide-based vectors are particularly amenable to rational design and development in this field due to their exceptional adaptability.Zhang et al.12,168 developed a series of surfactant peptides comprising a hydrophobic tail attached to a polar head group consisting of one to two positively charged residues at the C- or N-terminus, one example being LLLLLLKK. These peptides self-assembled in water to produce nanovesicles and nanotubes. Also in the presence of DNA, the positively charged peptides self-assembled into a tube, thereby encapsulating the negatively charged DNA and were delivered to growing cells. Keller et al.169 introduced a dipalmitoylated cell penetrating peptide as a potent lead for the intracellular delivery of hydrophobic drugs using bilayer-mimetic nanocarriers. These supramolecular vehicles combined the advantages of both liposomes and micelles offering a hydrophobic environment, given by the palmitoyl tails, in spite of their small size. In vitro experiments demonstrated the rapid cellular internalization of a carboxyfluorescein-labeled derivative of the amphiphilic peptide into immortalized mouse brain capillary endothelial cells as visualized by confocal laser scanning microscopy.
Recently, Tirrell and co-workers170 reported on the internalization of a palmitoylated, pro-apoptotic peptide derived from p53 tumor suppressor protein with a human cancer cell line (SJSA-1 human osteosarcoma cell line, which overexpressed murine double minute, MDM2, and possesses wild-type p53). It was shown that these alkylated peptides formed elongated rod-like micelles above the critical micelle concentration. However, monomers instead of micelles were internalized, a finding that correlated with the dynamic nature of the assemblies and the noncovalent interactions holding them together. Internalization was shown to occur via adsorption mediated, energy-dependent pathways, resulting in accumulation of the material in endocytic vesicles. It was speculated that an increased micelle stability would be required for intact micelle internalization (i.e. using polymerizable peptide amphiphiles) to act as an effective way to transport peptide therapeutics inside the cell.
Bellomo et al.171 published pH-sensitive polymersomes composed of a diblock-polypeptides. Stimuli responsive release studies were demonstrated by fluorescence spectroscopy. A calcium sensitive Fura-2 dye was entrapped in the vesicles in the presence of external calcium and a wavelength shift for maximum emission intensity was observed when the pH dropped from 10.6 to 3.0, demonstrating the calcium binding of the dye upon bilayer disruption.
Holowka et al.172 reported the development of polypeptide vesicles composed of arginine-leucine (poly(L-arginine)60-block-poly(L-leucine)20, R60L20) diblock copolypeptide amphiphiles (Fig. 9). R60L20 vesicles, containing the model cargo Texas-Red-labelled, were internalized in both epithelial (T84) and endothelial (HULEC-5A) cell lines and found to be minimally cytotoxic (Fig. 9).
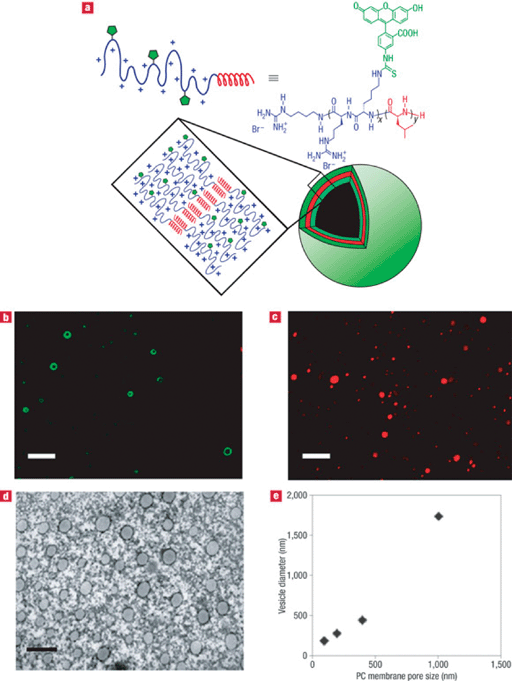 | ||
| Fig. 9 Formation and properties of R60L20 vesicles. (a) Schematic diagram of proposed self-assembly of R60L20 vesicles. (b) LSCM image of 1.0 μm extruded vesicles (scale bar = 5 μm). (c) LSCM image of vesicles containing Texas-Red-labelled dextran (total solution concentration = 1 μM). Scale bar = 5 μm. (d) Transmission electron micrograph of negatively stained vesicles that had been extruded through a 100 nm nucleopore polycarbonate (PC) membrane filter (scale bar = 200 nm). (e) Vesicle diameters determined using dynamic light scattering after extrusion through different PC membrane filters (100, 200, 400 and 1000 nm). Reprinted by permission from Macmillan Publishers Ltd: Nat. Mater., ref. 172, copyright 2007. | ||
4.5 Antimicrobial biomaterials
Fatty acid conjugation may improve the usefulness of peptides as antimicrobial agents by enhancing their ability to form secondary structures upon interacting with the bacterial membranes. Antimicrobial activity seams to be mediated by a complex, sensitive balance between various peptide parameters, including the length, net charge, hydrophobicity, secondary structure propensity, amphipathicity, size and depth of the helical polar/apolar sectors, flexibility, and resistance to degradation, and that the role of each parameter may change from one peptide to another.173In a work reported by Tirrell and co-workers,174 three peptides, YGAA[KKAAKAA]2 (AKK), KLFKRHLKWKII (SC4), and YG[AKAKAAKA]2 (KAK), were conjugated with lauric acid and tested for the effect on their structure, antibacterial activity, and eukaryotic cell toxicity. The conjugated AKK and SC4 peptides showed increased antimicrobial activity relative to unconjugated peptides, but the conjugated KAK peptide did not. The circular dichroism spectrum of AKK showed a significantly larger increase in its α-helical content in the conjugated form compared to KAK in a vesicle solution composed of phosphatidylethanolamine/phosphatidylglycerol, mimicking bacterial membranes. The KAK and AKK peptides and their corresponding fatty acid conjugates showed little change in their structure in the presence of phosphatidylcholine vesicles, which mimic the cell membrane of eukaryotic cells. The hemolytic activity of the KAK and AKK peptides and conjugates was low. However, the SC4 fatty acid conjugate showed strongly enhanced hemolytic activity and a corresponding increase in helical content in the presence of phosphatidylcholine vesicles.
Recently, Mas-Moruno et al.175 described the synthesis of several lipopolysaccharide (LPS)-neutralizing N-acylated peptides, which were obtained by acylation with distinct aliphatic acids of minimal LPS binding domain of the Limulus anti-LPS factor (LALF), the bactericidal permeability-increasing protein (BPI), and serum amyloid P (SAP). An antiparallel β-sheet folded 14-amino acid cyclic peptide, named LALF-14c, comprising residues 36–47, was used. Structural analysis of these peptides revealed that N-acylation with long acyl chains promoted the formation of micellar or fibril-like nanostructures. In all cases, peptides with long acyl chains showed greater LPS-neutralizing activities than the original acetylated peptide. On the basis of transmission electron microscopy (TEM) images, these enhanced activities could be associated with the capacity of the peptides to form nanostructures. TEM studies revealed that long fatty acyl chains promoted the formation of micellar and fibrilar superstructures. These results demonstrated that nanostructure formation can lead to anti-LPS peptides with higher LPS-neutralizing activities at cell-tolerable concentrations. These lipopeptides may provide the basis for the development of new antisepsis agents with improved cell-tolerated concentrations.
4.6 Templates for mineralization
Amphiphilic peptide-based systems can be used to create new class of materials at the molecular scale using self-assembly with a high impact in several fields of research. One example is the template-induced mineralization of inorganic crystals. Controlling the structure and organization of inorganic materials on the nanoscale remains a challenge for materials chemists. Spatially controlled assemblies of inorganic nanoparticles (i.e. Au or CdS) are of great interest from a technological perspective because of the unique electronic and optical properties of these materials.In this respect, Stupp and co-workers176,177 have extensively investigated the use of peptide-based nanofibers as scaffolds for the 1D assembly of lipophilic inorganic gold nanoparticles in apolar solvents as well as templates for CdS nanocrystals (Fig. 10).
 | ||
| Fig. 10 Bright field TEM micrographs of CdS mineralized suspensions of PA fibers at various Cd2+ : PA molar ratios: (a) Cd2+ : PA = 2.4 : 1; (b) Cd2+ : PA = 24 : 1. Left inset shows an electron diffraction pattern corresponding to the CdS zinc blende structure. Right inset shows an enlargement of a portion of an encapsulated fiber highlighting the strip of lower electron density through the middle corresponding to the hydrophobic core; (c) Cd2+ : PA = 240 : 1. Reprinted with permission from ref. 176. Copyright 2004 American Chemical Society. | ||
The group of Kelly178 investigated the use of peptidomimetics that assembled into 2D β-hairpin monolayers at the air-water interface for the nucleation of CdS nanocrystals via the {01.0} face. From this study it appeared that the degree of lattice matching played a major role in controlling the face nucleated and the crystallite size.
In CaCO3, biomineralization nucleation and growth of the crystals are related to the presence of carboxylate-rich proteins within a macromolecular matrix, often with organized β-sheet domains.
Volkmer et al.179 employed amphiphilic peptides, comprising alternating hydrophilic (Asp) and hydrophobic (Phe) amino acid residues, as acidic peptides designed to imitate the epitopes of acidic proteins from calcified tissues. CaCO3 crystallization experiments have shown that the amphiphilic peptides H-(Phe-Asp)2-OH and H-(Phe-Asp)4-OH specifically interact with distinct crystal faces of calcite. The morphological features of calcite crystals grown in their presence were very similar to calcite crystals grown from solutions containing natural acidic proteins.
Originally, researchers in the field of CaCO3 mineralization have searched for an epitaxial relation between template and mineral phase to explain observations of oriented nucleation. More recently, additional mechanisms such as orientational complementarity of functional groups and the role of electrostatic interactions have been promoted. A relatively soft macromolecular assembly, whose lattice parameters are rather immutable, would remain rigid enough to drive nucleation of a preferred crystal face. However, it is known that proteins, fatty acid films and possibly also collagenous tissue matrices do rearrange their structures upon exposure to mineral ions. To understand the interplay between the organic template and the mineral crystal it is important to explicitly address the issue of structural adaptation of the template during mineralization.180
In this respect, in a joint collaboration with the Sommerdijk group, we were able to show that 2D β-sheet self-organized monolayer, constituted by a phospholipid tail coupled to octapeptide (Leu-Glu)4, acted as a unique flexible template for the mineralization of CaCO3. Nucleation of different crystals’ faces was achieved, depending on the ability of the template to adapt to the structure of the inorganic phase. A new type of indented {10.0} oriented calcite crystals nucleated underneath the flexible amphiphilic lipopeptide monolayer. On the contrary, the formation of this new morphological form was significantly suppressed when a less adaptable monolayer, constituted by only an octapeptide (Leu-Glu)4, was used.68
More recently, Popescu et al.181 developed a series of self-organizing surfactants consisting of a dodecyl chain connected via a bisureido-heptylene unit to an amino acid head group (Gly, Ala, Val, and Leu were selected) as shown in Fig. 11. Structural arrangement in two-dimensional (2D) Langmuir monolayers showed that the spacing of these molecules in one direction was predetermined by the hydrogen-bonding distances between the bis-urea units. In the other direction, the intermolecular distance was determined by steric interactions introduced by the side groups of the amino acid moiety.
 | ||
| Fig. 11 (A) Molecular structure of self-organizing surfactants consisting of a dodecyl chain connected via a bisureido-heptylene unit to an amino acid head group (Gly, Ala, Val, and Leu for 1, 2, 3 and 4, respectively). (B) SEM images of (top left) crystals grown underneath monolayer of 1. No particular nucleation plane was favored; (top right) (01.2) oriented crystals grown under the monolayer of 2; (bottom left) and (bottom right) modified crystals with a concave indentation isolated from beneath a monolayer of 3 and 4, respectively ({10.0} calcite). Crystals viewed from the side that was exposed to the monolayer; the roughened (nucleating) faces have been attached to the monolayer. Reprinted with permission from ref. 181. Copyright 2007 American Chemical Society. | ||
Thus, the density of the surfactant molecules in a monolayer and the ability of these templates to respond to the presence of calcium ions could be alter by the choice of the amino acid (Fig. 11). The Gly and Ala based surfactants formed very rigid monolayers that were active in promoting the nucleation of calcium carbonate (predominately {10.0} calcite); however no particular nucleation plane was favored in the case of the glycine derived surfactant and some modified crystals, belonging to the {01.l} family with l = 1–2, were found in the case of the slightly more flexible alanine based surfactant. In contrast, the valine and leucine based surfactants formed more flexible monolayers that reorganized their structure upon complexation of calcium ions. Although in the latter two cases a relatively low nucleation density was observed, a high selectivity for the nucleation of {10.0} calcite was found. These results demonstrated that although the Langmuir monolayers of all compounds were active in the nucleation of calcium carbonate, habit modification was only observed when the size of the side group allowed the molecules to rearrange and adapt their organization in response to the mineral phase.
Another interesting application of peptide-based nanofibers is the preparation of tailor-made nanostructured composite materials suited for controlled biomineralization towards bone-like materials.
Hartgerink et al.182 exploited the self-assembly of single alkyl chain amphiphilic peptides for the controlled mineralization of hydroxyapatitite. The peptide was carefully designed bearing five different regions in order to introduce important features such as the hydrophobic character for promoting self-assembly imparted by the alkyl chain. Cysteines (Cys) were introduced to stabilize the aggregates, while the glycine (Gly) inferred some flexibility between the crosslinked area and the hydrophilic head group. The phosphorylated serine (Ser) could interact strongly with calcium ions to direct mineralization and finally an RGD sequence was attached as a cell adhesion ligand. This design allowed the fibers to be susceptible to pH-dependent crosslinking. At a pH of 8 and with all Cys reduced, the peptide amphiphiles were highly soluble. Upon acidification the compound became insoluble forming a fiber network stabilized by formation of disulfide bonds through oxidation and this process was found to be reversible. Mineralization of hydroxyapatite was directed by the obtained structure, forming a composite material with an alignment that is also observed between collagen fibrils and hydroxyapatite in bone. This highly dynamic amphiphilic peptide system appears to be an ideal scaffold for biomineralization and tissue engineering applications.
Lately, Huang et al.183 exploited the ability of branched peptide amphiphile molecules, containing the peptide motif Arg-Gly-Asp, BRGD-PA, to self-assemble in physiological environments into nanofibers, which displayed on their surfaces high densities of this biological signal. These nanofibers were used to study the effect of artificial bioactive nanostructures on ameloblasts with the long-term goal of developing cell-based strategies for tooth regeneration, using an in vitro cell and organ culture system. Ameloblast-like cells (line LS8) and primary enamel organ epithelial (EOE) cells were cultured within BRGD-PA hydrogels, ECM, and the mixture was injected into the EOE of mouse embryonic incisors, showing that the enamel organ epithelial cells participate in repair and regeneration with the BRGD-PA scaffold.
4.7 Probes and contrast agents for imaging
Molecular imaging has attracted more and more attention in the past decades. It has become clear that in vitro studies can not reliably predict the behaviour of cells in an in vivo environment. Therefore, there is a strong interest in developing and validating imaging technologies such as positron-emission tomography (PET), single photon emission computed tomography (SPECT), fluorescence imaging and magnetic resonance (MR) to image biological targets in vivo. Peptides play a crucial role in this field as they have several advantages such as small size and relatively easy synthesis, good tissue permeation and target accessibility but also no antigenicity. For diagnostic use, several Sandostatin peptide derivatives modified on the N-terminal moiety with bifunctional chelators suitable for labeling radiometals such as 99mTc, 111In,67,68 and Ga have been designed, synthesized, and used as in vivo contrast agents in nuclear medicine techniques such as PET and SPECT.184In a recent example, Kimura and co-workers185 demonstrated the importance of amphiphilic copolypeptide vesicles (peptosomes) labeled with a near-infrared fluorescence (NIRF) dye as a novel nanocarrier for in vivo molecular imaging of tumors in mice. Amphiphilic copolypeptides vesicles were composed of hydrophilic poly(sarcosine) and hydrophobic poly(γ-methyl-L-glutamate) blocks and were synthesized with varying chain lengths of the blocks via a sequential N-carboxyanhydride (NCA) polymerization method. The polypeptides, having a suitable hydrophilic/hydrophobic balance, were found to form vesicles of 100 nm size in Tris-buffered saline (TBS). These polymersomes showed to have the appropriate size for the passive targeting into the solid tumor by the enhanced permeability and retention (EPR) effect, allowing in vivo imaging of a small cancer on mouse.
Morisco et al.186 developed amphiphilic peptide-conjugates containing chelating agents able to coordinate a gadolinium ion. A hydrophobic moiety with two 18-carbon atom alkyl chains was connected to a cyclic eight-amino acid peptide, octreotide Sandostatin, able to target somatostatin receptors, which are widely overexpressed in tumor tissues such as gliomas, meningiomas, gastroenteropancreatic neuroendocrine, and breast tumors. These amphiphilic peptides aggregated in water solution giving, at very low concentrations, stable micelles. Fluorescence studies and circular dichroism experiments indicated the complete exposure of octreotide on the micelle surface and the predominant presence of β-turns. Furthermore, micelles showed high relaxivity values, suggesting that these aggregates could be used as tumor target-selective magnetic resonance imaging (MRI) contrast agents.
In another example, Stupp and co-workers187 chelated cylindrical nanofibers and gels, formed by self-assembling peptide amphiphiles, to a derivative of 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid (DOTA), which was selected for Gd(III) chelation. These amphiphilic peptide based-Gd(III) hybrids were shown to self-assemble into both spherical and fiber-like nanostructures, enhancing the T1 relaxation time and therefore suitable to use as magnetic resonance imaging (MRI) contrast agents (Fig. 12).
 | ||
| Fig. 12 AFM and TEM images of amphiphilic peptide based-Gd(III) hybrids (a) and without Gd(III) (b). Panel b is negatively stained with PTA to provide contrast, without staining no distinguishable structures are visible due to lack of electron density contrast. Reprinted with permission from ref. 187. Copyright 2005 American Chemical Society. | ||
In this journey from chemistry to biology, medicine, and materials science, molecular self-assembling peptides pave the way to the next frontier: new technologically relevant engineered nanostructures. Recent advancements in patterning technologies have significantly enhanced the ability to spatially control surfaces at the micrometre level. Common patterning methods include photolithography188,189 and soft lithographic approaches such as microcontact printing,190 microfluidic patterning,191,192 and micromolding.193,194 These techniques have been widely used for the high fidelity patterning of rigid substrates, such as modified silicon or glass195,196 and recently of deformable, solvated, biocompatible platforms such as hydrogels. In this way well-defined surfaces have been prepared, which can be useful for the construction of photosensitive switches, nanowires or light-harvesting devices. Finally, the interesting emerging of peptide-based well-defined structures for the fabrication of molecular machines or robots is highlighted.
4.8 Photosensitive switches and nanowires
Chemical strategies to build photosensitive switches into peptides as modulators of structure have been pioneered by Bredenbeck et al.197 They have shown that the inclusion of various azobenzene derivatives as crosslinkers of amino acid residues at well-defined positions can modulate helix formation upon irradiation with light of specific frequencies. This concept has been applied to the regulation of tertiary structures, including coiled-coils and also provides a means for studying the kinetics of protein folding. Inclusion of such a switch in biomaterials designs would be useful in creating photo-responsive materials.A significant amount of research has been dedicated to the synthesis of conducting polymers in confined environments.198,199 Polyaniline (PAni),200,201 polypyrrole (PPy)202 and polythiophene (PT)203,204 have all been polymerized from their respective organic monomers in aqueous media within the hydrophobic cores of self-assembled surfactant micelles, exploiting their ability to sequester soluble aromatic monomers within their hydrophobic regions.205 Recently, Tovar et al.206 used a peptide amphiphile (HOOC-VAVKIEGGGAAAA-NHCOC15H31), which self-assembled into cylindrical nanostructures with hydrophobic cores, to sequester 3,4-ethylene dioxythiophene (EDOT) monomer from the aqueous solution. The addition of ammonium persulfate resulted in the oxidative polymerization of EDOT yielding a encapsulated conductive polymer, as shown by electron microscopy and electrochemical measurements. The polymer exhibited good conductivity at physiologically compatible applied potentials. These materials may find application as both biologically and electronically active matrices for the transduction of biological events, as assembly in water buried the lipid segments within the conductive nanostructure, while the laminin-binding sequences (IKVAV) were located at the external surface and accessible for specific cellular interactions with neuronal cells.
The possibility of fabricating conducting nanowires by molecular means using peptide scaffolds is of particular interest to the electronics industry. Nanotubes made from self-assembling peptides are used as templates for mineralization of metals interesting for conductive purposes (i.e. copper). Once the organic scaffold has been removed, a pure conducting wire, immobilized on a surface, is obtained. Matsui and co-workers207 have been able to prepare nanowires of copper and nickel from peptide nanotubes. Reches and Gazit208 have demonstrated that the short dipeptide (Phe)2 was able to form nanotubes. After diffusion of silver ions into the tubes, the silver ions were reduced with citric acid. And finally the peptides were removed either enzymatically, chemically or through heat treatment yielding pure silver wires.
Scheibel et al.209 described the use of self-assembling amyloid protein fibers to construct nanowire elements. Self-assembly of a prion determinant from Saccharomyces cerevisiae, the N-terminal and middle region (NM) of Sup35p, produced 10 nm-wide protein fibers that were stable under a wide variety of conditions. The length could be varied in the range of 60 nm to several hundred micrometres by controlling the assembly conditions. A genetically modified NM variant that presented reactive, surface accessible cysteine residues was used to covalently link NM fibers to colloidal gold particles. These fibers were placed across gold electrodes and additional metal was deposited by highly specific chemical enhancement of the colloidal gold by reductive deposition of metallic silver and gold from the corresponding salts. These biotemplated metal wires, with a diameter of ∼100 nm, demonstrated the conductive properties of a solid metal wire, such as low resistance and ohmic behavior. With these materials it should be possible to harness the extraordinary diversity and specificity of protein functions to nanoscale electrical circuitry.
Ryu et al.210–212 were able to grow a micro-pattern of vertically well-aligned peptide nanowires on a silicon substrate by combining an aging “amorphous” diphenylalanine film at temperatures above 100 °C by aniline vapour to a soft-lithography technique using a microchanneled polydimethylsiloxane (PMDS) mold (Fig. 13). Films remained in the amorphous form when no high-temperature or other solvent vapours were used (i.e. benzene or toluene).
 | ||
| Fig. 13 (A) A schematic illustration for the micro-patterned growth of vertically well-aligned peptide nanowires. Microchanneled PDMS mold was used to prepare a micro-pattern of amorphous peptide thin film. The film was then aged at 150 °C under aniline vapor. (B) Electron micrograph of the resultant micro-pattern of peptide nanowires on a silicon substrate. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission from ref. 210. | ||
Raines and co-workers213 exploited self-assembly of collagen-related synthetic peptides into fiber templates to create new metallic nanowires. Such functional organic–inorganic hybrid nanomaterials have potential applications as nanoelectronic devices. Micrometre-long fibers were assembled from collagen-related peptides through π–π stacking interactions between C- and N-terminal aromatic residues. These peptides feature a Lys residue within their amino acid sequence which allowed the attachment of gold nanoparticles to both the side-chain Lys residue and the N-terminal amino functionality. After subsequent electroless silver plating under mild conditions, the peptidic fibers were grown into metallic nanowires. Electroless silver plating, also termed ‘‘silver enhancement’’, has been already used in the fabrication of nanowires from DNA214 and recombinant amyloid proteins.209 In the latter case, gold nanoparticles attached to the protein surface served as nucleation sites to promote the electroless reduction of silver and gold enabling nanowire growth. The conductivity of such metallic nanowires were measured using electron-beam (e-beam) lithography to define two terminal devices of single nanowires.
4.9 Molecular machines and motors
Finally, an exciting emerging field is the development of molecular machines or robots that can be turned on and off in response to a signal. Most of this work has, thus far, been carried out using large proteins (i.e. motors proteins such as myosin, kinesins and dyneins) or small interlocked catenane- and rotaxane-based organic molecules as described in a review by Kinbara et al.215To investigate the influence of supramolecular architecture on biomotor cooperativity, the group of Tirrell216 engineered a model multimotor system that allows the precise regulation of intermotor coupling. The model was composed of a rigid block, comprised of strongly associated acidic and basic leucine zipper domains that anchored motor proteins at specific distances along the polymer backbone (Fig. 14). The artificial protein scaffolds incorporated a basic zipper into the polymer backbone, whereas the complementary acidic zipper was fused to the C-terminus of a truncated kinesin-1 motor. The flexible polymer block was derived from the elastomeric poly(VPGVαG) structural motif of the protein elastin and conferred well-characterized mechanical compliance on the assembly. Every fifth α-valine (Vα) residue was replaced by a phenylalanine (F9 residue in the elastin sequence). This substitution provided a means to control the thermoresponsive behavior of the polymers, as discussed in more detail below. Variation in the number of diblock repeats in the polymer provided discrete control over the number of coupled motors, which in the present series of experiments ranged from one to three. The C-terminus of each scaffold was labelled with biotin to allow the motor assemblies to be tethered to streptavidin coated surfaces. These results indicated that the artificial proteins provided a structural framework that allowed motors to push and pull on one another to enhance activity through control of the supramolecular architecture of multimotor assemblies, providing a means to reconfigure mechanisms of collective biomotor transport.
 | ||
| Fig. 14 Engineered multimotor assemblies. (A) Schematic representation of the synthesis of the engineered multimotor assemblies. As a complex, the zippers (ZE and ZR) form a rigid linker approximately 6.5 nm long (assuming 6.3 heptad repeats in a zipper and 1.03 nm per heptad). The length of the flexible ELF block can be approximated by assuming a β-spiral conformation. In this conformational state, elastin proteins possess a spiral pitch of 1 nm, where each turn contains three VPGVαG pentapeptide units. Repeating this ELF motif, (VPGVG)2VPGFG(VPGVG)2, five times gives a length of 8 nm for the ELF5 domain. Considering that four amino acids (KASK) form linkers between adjacent ZR–ELF5 diblock units, the total intermotor spacing set by the polymer is approximately 16 nm when bound to a microtubule (shown in red). (B) Matrix-assisted laser desorption/ionization mass spectra of polymer scaffolds containing one, two, and three repeats of the ZR–ELF5 diblock. The splitting of the main peaks (M+) is due to a 525 Da shift in mass that arises from biotin functionalization at the C-terminal cysteine positions of the polymers. A tris-tricine gel of all three polymers is shown in the inset. From ref. 216. Reprinted with permission from AAAS. | ||
5. Final remarks
Supramolecular chemistry in general aims to construct defined structures based on non-covalent interactions. The manner in which molecules self-assemble and their interaction energies, shapes and ultimate functions can be programmed at the molecular level. Examples illustrated herein have shown that amphiphilic peptide-based hybrids can be used to construct nanometre-scale structures. Challenging is to a priori design molecules that self-assemble in a predictive manner into targeted three-dimensional supramolecular structures. With this ability in hand, the next step is to incorporate function. The increasing demand for miniaturization in both academia and industry necessitates progress and addresses questions to all scientific disciplines, from chemistry and physics to biology, medicine, materials science, and engineering, pointing out the cross-disciplinary role of peptide-based amphiphiles.Acknowledgements
We thank Hugo Simões, Jan-Willem Handgraaf and Zan Peeters for the help in the realization of some of the figures. This work was partially supported by CICYT (CTQ2006-03794/BQU), the Instituto de Salud Carlos III (CB06_01_0074), the Generalitat de Catalunya (2005SGR 00662), the Institute for Research in Biomedicine, and the Barcelona Science Park.Notes and references
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS.
- J. M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS.
- R. Feynman, Engineering and Science, 1960 Search PubMed.
- E. H. C. Bromley, K. Channon, E. Moutevelis and D. N. Woolfson, ACS Chem. Biol., 2008, 3, 38–50 CrossRef CAS.
- K. Channon, E. H. C. Bromley and D. N. Woolfson, Curr. Opin. Struct. Biol., 2008, 18, 491–498 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS.
- R. Fairman and K. S. Åkerfeldt, Curr. Opin. Struct. Biol., 2005, 15, 453–463 CrossRef CAS.
- K. Rajagopal and J. P. Schneider, Curr. Opin. Struct. Biol., 2004, 14, 480–486 CrossRef CAS.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- P. Chen, Colloids Surf., A, 2005, 261, 3–24 CrossRef CAS.
- X. Zhao and S. Zhang, Trends Biotechnol., 2004, 22, 470–476 CrossRef CAS.
- J. Venkatraman, S. C. Shankaramma and P. Balaram, Chem. Rev., 2001, 101, 3131–3152 CrossRef CAS.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- T. Muraoka, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2008, 130, 2946–2947 CrossRef CAS.
- K. Pagel, T. Vagt and B. Koksch, Org. Biomol. Chem., 2005, 3, 3843–3850 RSC.
- J. Tulla-Puche and F. Albericio, The Power of Functional Resins in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim (Germany), 2008 Search PubMed.
- N. L. Benoiton, Chemistry of Peptide Synthesis, CRC, Taylor & Francis, Boca Raton, FL, 2006 Search PubMed.
- P. Lloyd-Williams, F. Albericio and E. Giralt, Chemical Approaches to the Synthesis of Peptides and Proteins, CRC, Boca Raton, FL, USA, 1997 Search PubMed.
- J. J. L. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS.
- X. Gao and H. Matsui, Adv. Mater., 2005, 17, 2037–2050 CrossRef CAS.
- S. Zhang, D. M. Marini, W. Hwang and S. Santoso, Curr. Opin. Chem. Biol., 2002, 6, 865–871 CrossRef CAS.
- D. W. P. M. Löwik and J. C. van Hest, Chem. Soc. Rev., 2004, 33, 234–245 RSC.
- S. G. Zhang, T. C. Holmes, C. M. DiPersio, R. O. Hynes, X. Su and A. Rich, Biomaterials, 1995, 16, 1385–1393 CrossRef CAS.
- S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CAS.
- K. Rajagopal, B. Ozbas, D. J. Pochan and J. P. Schneider, Eur. Biophys. J., 2006, 35, 162–169 CrossRef CAS.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS.
- C. Whitehouse, J. Fang, A. Aggeli, M. Bell, R. Brydson, C. W. G. Fishwick, J. R. Henderson, C. M. Knobler, R. W. Owens, N. H. Thomson, D. A. Smith and B. N, Angew. Chem., Int. Ed., 2005, 44, 1965–1968 CrossRef CAS.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS.
- A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS.
- A. Aggeli, M. Bell, N. Boden, L. M. Carrick and A. E. Strong, Angew. Chem., Int. Ed., 2003, 42, 5603–5606 CrossRef CAS.
- H. Rapaport, K. Kjaer, T. R. Jensen, L. Leiserowitz and D. A. Tirrell, J. Am. Chem. Soc., 2000, 122, 12523–12529 CrossRef CAS.
- H. Rapaport, G. Möller, C. M. Knobler, T. R. Jensen, K. Kjaer, L. Leiserowitz and D. A. Tirrell, J. Am. Chem. Soc., 2002, 124, 9342–9343 CrossRef CAS.
- R. Sneer, M. J. Weygand, K. Kjaer, D. A. Tirrell and H. Rapaport, ChemPhysChem, 2004, 5, 747–750 CrossRef CAS.
- N. Ashkenasy, W. S. Horne and M. R. Ghadiri, Small, 2006, 2, 99–102 CrossRef CAS.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS.
- D. A. D. Parry, R. D. B. Fraser and J. M. Squire, J. Struct. Biol., 2008, 163, 258–269 CrossRef CAS.
- J. M. Mason and K. M. Arndt, ChemBioChem, 2004, 5, 170–176 CrossRef CAS.
- W. D. Kohn, C. M. Kay and R. S. Hodges, J. Mol. Biol., 1998, 283, 993–1012 CrossRef CAS.
- D. Papapostolou, E. H. C. Bromley, C. Bano and D. N. Woolfson, J. Am. Chem. Soc., 2008, 130, 5124–5130 CrossRef CAS.
- C. Gribbon, K. J. Channon, W. Zhang, E. F. Banwell, E. H. Bromley, J. B. Chaudhuri, O. C. Oreffo and D. N. Woolfson, Biochemistry, 2008, 47, 10365–10371 CrossRef CAS.
- Y. S. Velichko, S. I. Stupp and M. O. de la Cruz, J. Phys. Chem. B, 2008, 112, 2326–2334 CrossRef CAS.
- S. Tsonchev, K. L. Niece, G. C. Schatz, M. A. Ratner and S. I. Stupp, J. Phys. Chem. B, 2008, 112, 441–447 CrossRef CAS.
- L. S. Li, H. Z. Jiang, B. W. Messmore, S. R. Bull and S. I. Stupp, Angew. Chem., Int. Ed., 2007, 46, 5873–5876 CrossRef CAS.
- J. C. Stendahl, M. S. Rao, M. O. Guler and S. I. Stupp, Adv. Funct. Mater., 2006, 16, 499–508 CrossRef CAS.
- P. Forns, J. L. Lauer-Fields, S. Gao and G. B. Fields, Biopolymers, 2000, 54, 531–546 CrossRef CAS.
- Y.-C. Yu, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1998, 120, 9979–9987 CrossRef CAS.
- J. Y. Su, R. S. Hodges and C. M. Hay, Biochemistry, 1994, 33, 15501–15510 CrossRef CAS.
- D. W. P. M. Löwik, J. G. Linhardt, P. J. H. M. Adams and J. C. M. van Hest, Org. Biomol. Chem., 2003, 1, 1827–1829 RSC.
- B. Ludolph, F. Eisele and H. Waldmann, J. Am. Chem. Soc., 2002, 124, 5954–5955 CrossRef CAS.
- K. Kuhn, D. J. Owen, B. Bader, A. Wittinghofer, J. Kuhlmann and H. Waldmann, J. Am. Chem. Soc., 2001, 123, 1023–1035 CrossRef CAS.
- B. Bader, K. Kuhn, D. J. Owen, H. Waldmann, A. Wittinghofer and J. Kuhlmann, Nature, 2000, 403, 223–226 CrossRef CAS.
- J. Fernández-Carneado, M. J. Kogan, N. Van Mau, S. Pujals, C. López-Iglesias, F. Heitz and E. Giralt, J. Pept. Res., 2005, 65, 580–590 CrossRef CAS.
- D. Stroumpoulis, H. Zhang, L. Rubalcava, J. Gliem and M. Tirrell, Langmuir, 2007, 23, 3849–3856 CrossRef CAS.
- L. You and G. W. Gokel, Chem.–Eur. J., 2008, 14, 5861–5870 CrossRef CAS.
- M. S. Arnold, M. O. Guler, M. C. Hersam and S. I. Stupp, Langmuir, 2005, 21, 4705–4709 CrossRef CAS.
- P. J. Casey, Science, 1995, 268, 221–225 CrossRef CAS.
- P. J. Casey and J. E. Buss, Methods in Enzymology: Lipid Modification of Proteins, Elsevier, New York, 1995 Search PubMed.
- B. Kellam, P. A. De Bank and K. M. Shakesheff, Chem. Soc. Rev., 2003, 32, 327–337 RSC.
- H.-J. Musiol, S. Dong, M. Kaiser, R. Bausinger, A. Zumbusch, U. Bertsch and L. Moroder, ChemBioChem, 2005, 6, 625–628 CrossRef CAS.
- R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984, vol. 1, pp. 1–176 Search PubMed.
- P. E. Dawson, T. W. Muir, I. Clarklewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS.
- A. Otaka, S. Ueda, K. Tomita, Y. Yano, H. Tamamura, K. Matsuzaki and N. Fujii, Chem. Commun., 2004, 1722–1723 RSC.
- L. V. Chernomordik and M. M. Kozlov, Nat. Struct. Mol. Biol., 2008, 15, 675–683 CrossRef CAS.
- H. Robson Marsden, N. A. Elbers, P. H. H. Bomans, N. A. J. M. Sommerdijk and A. Kros, Angew. Chem., Int. Ed., 2009, 48, 2330–2333.
- S. Cavalli, J.-W. Handgraaf, E. E. Tellers, D. C. Popescu, M. Overhand, K. Kjaer, V. Vaiser, N. A. J. M. Sommerdijk, H. Rapaport and A. Kros, J. Am. Chem. Soc., 2006, 128, 13959–13966 CrossRef CAS.
- S. Cavalli, D. C. Popescu, E. E. Tellers, M. R. J. Vos, B. P. Pichon, M. Overhand, H. Rapaport, N. A. J. M. Sommerdijk and A. Kros, Angew. Chem., Int. Ed., 2006, 45, 739–744 CrossRef CAS.
- H. Robson Marsden and A. Kros, Macromol. Biosci., 2009, 9, 939–951 CrossRef CAS.
- M. A. Gauthier and H. A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
- A. Rösler, H.-A. Klok, I. W. Hamley, V. Castelletto and O. O. Mykhaylyk, Biomacromolecules, 2003, 4, 859–863 CrossRef.
- J. M. Smeenk, M. B. J. Otten, J. Thies, D. A. Tirrell, H. G. Stunnenberg and J. C. M. van Hest, Angew. Chem., Int. Ed., 2005, 44, 1968–1971 CrossRef CAS.
- T. J. Deming, Adv. Mater., 1997, 9, 299–311 CrossRef CAS.
- A. Kros, W. Jesse, G. A. Metselaar and J. J. L. M. Cornelissen, Angew. Chem., Int. Ed., 2005, 44, 4349–4352 CrossRef CAS.
- T. J. Deming and S. A. Curtin, J. Am. Chem. Soc., 2000, 122, 5710–5717 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS.
- C. Wang, R. J. Stewart and J. Kopeček, Nature, 1999, 397, 69–420 CrossRef CAS.
- L. Mi, S. Fischer, B. Chung, S. Sundelacruz and J. L. Harden, Biomacromolecules, 2006, 7, 38–47 CrossRef CAS.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–391 CrossRef CAS.
- W. Shen, K. Zhang, J. A. Kornfield and D. A. Tirrell, Nat. Mater., 2006, 5, 153–158 CrossRef CAS.
- H. Robson Marsden, A. V. Korobko, E. N. M. van Leeuwen, E. M. Pouget, S. J. Veen, N. A. J. M. Sommerdijk and A. Kros, J. Am. Chem. Soc., 2008, 130, 9386–9393 CrossRef CAS.
- H.-A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1509–1513 CrossRef CAS.
- A. A. Zompra, A. S. Galanis, O. Werbitzky and F. Albericio, Future Med. Chem., 2009, 1, 361–377 Search PubMed.
- S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS.
- M. Mutter, A. Chandravarkar, C. Boyat, J. Lopez, S. Dos Santos, B. Mandal, R. Mimna, K. Murat, L. Patiny, L. Saucede and G. Tuchscherer, Angew. Chem., Int. Ed., 2004, 43, 4172–4178 CrossRef.
- A. Taniguchi, Y. Sohma, M. Kimura, T. Okada, K. Ikeda, Y. Hayashi, T. Kimura, S. Hirota, K. Matsuzaki and Y. Kiso, J. Am. Chem. Soc., 2006, 128, 696–697 CrossRef CAS.
- I. Coin, R. Doelling, E. Krause, M. Bienert, M. Beyermann, C. D. Sferdean and L. A. Carpino, J. Org. Chem., 2006, 71, 6171–6177 CrossRef.
- J. Hentschel, E. Krause and H. G. Borner, J. Am. Chem. Soc., 2006, 128, 7722–7723 CrossRef CAS.
- J. C. M. van Hest and D. A. Tirrell, Chem. Commun., 2001, 1897–1904 RSC.
- D. A. Dougherty, Curr. Opin. Chem. Biol., 2000, 4, 645–652 CrossRef CAS.
- T. Hohsaka, D. Kajihara, Y. Ashizuka, H. Murakami and M. Sisido, J. Am. Chem. Soc., 1999, 121, 34–40 CrossRef CAS.
- S. A. Maskarinec and D. A. Tirrell, Curr. Opin. Biotechnol., 2005, 16, 422–426 CrossRef CAS.
- H. R. Kricheldorf, α-Amino acid N-carboxyanhydrides and related heterocycles, Springer Verlag, Berlin-Heidelberg-New York, 1987 Search PubMed.
- T. J. Deming, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3011–3018 CrossRef CAS.
- K. Hashimoto, Prog. Polym. Sci., 2000, 25, 1411–1462 CrossRef CAS.
- I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944–2945 RSC.
- T. J. Deming, Macromol. Biosci., 1999, 32, 4500–4502 CAS.
- V. T. Kung and C. T. Redemann, Biochim. Biophys. Acta, Biomembr., 1986, 862, 435–439 CrossRef CAS.
- M. Fleiner, P. Benzinger, T. Fichert and U. Massing, Bioconjugate Chem., 2001, 12, 470–475 CrossRef CAS.
- F. J. Martin and D. Papahadjopoulos, J. Biol. Chem., 1982, 257, 286–288 CAS.
- Y. Nakano, M. Mori, S. Nishinohara, Y. Takita, S. Naito, H. Kato, M. Taneichi, K. Komuro and T. Uchoda, Bioconjugate Chem., 2001, 12, 391–395 CrossRef CAS.
- L. Bourel-Bonnet, E.-I. Pecheeur, C. Grandjean, A. Blanpain, T. Baust, O. Melnyk, B. Hoflack and H. Gras-Masse, Bioconjugate Chem., 2005, 16, 450–457 CrossRef CAS.
- A. Dirksen and P. E. Dawson, Curr. Opin. Chem. Biol., 2008, 12, 760–766 CrossRef CAS.
- R. Huisgen, Pure Appl. Chem., 1989, 61, 613–628 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS.
- Z.-X. Wang and H.-L. Qin, Chem. Commun., 2003, 2450–2451 RSC.
- W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radic, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CAS.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and M. Adhya, J. Org. Chem., 1989, 54, 5302–5308 CrossRef CAS.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3062 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS.
- V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- L. Ayres, H. H. M. Adams, D. W. P. M. Löwik and J. C. M. van Hest, Biomacromolecules, 2005, 6, 825–831 CrossRef CAS.
- L. Ayres, K. Koch, H. H. M. Adams and J. C. M. van Hest, Macromolecules, 2005, 38, 1699–1704 CrossRef CAS.
- L. Ayres, M. R. J. Vos, H. H. M. Adams, I. O. Shklyarevskiy and J. C. M. van Hest, Macromolecules, 2003, 36, 5967–5973 CrossRef CAS.
- Y. Mei, K. L. Beers, H. C. M. Byrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS.
- L. Ayres, G. M. Grotenbreg, G. A. van der Marel, H. S. Overkleeft, M. Overhand and J. C. M. van Hest, Macromol. Rapid Commun., 2005, 26, 1336–1340 CrossRef CAS.
- E. J. Prenner, R. Lewis and R. N. McElhaney, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 201–221 CrossRef CAS.
- N. Izuyima, T. Kato, H. Aoyagi, M. Waki and M. Kondo, Synthetic Aspects of Biologically Active Cyclic Peptides: Gramicidin S and Tyrocidines, Halstead (Wiley), New York, 1979 Search PubMed.
- S. E. Hull, R. Karlsson, P. Main, M. M. Woolfson and E. J. Dodson, Nature, 1978, 275, 206–207 CAS.
- A. Stern, W. A. Gibbons and L. C. Craig, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 734–741 CAS.
- R. Issac, Y.-W. Ham and J. Chmielewski, Curr. Opin. Struct. Biol., 2001, 11, 458–463 CrossRef CAS.
- I. Ghosh and J. Chmielewski, Curr. Opin. Chem. Biol., 2004, 8, 640–644 CrossRef CAS.
- X. Li and J. Chmielewski, J. Am. Chem. Soc., 2003, 125, 11820–11821 CrossRef CAS.
- X. Li and J. Chmielewski, Org. Biomol. Chem., 2003, 1, 901–904 RSC.
- M. G. Ryadnov, B. Ceyhan, C. M. Niemeyer and D. N. Woolfson, J. Am. Chem. Soc., 2003, 125, 9388–9394 CrossRef CAS.
- G. Ashkenasy, R. Jagasia, M. Yadav and M. R. Ghadiri, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10872–10877 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS.
- S. Cavalli and A. Kros, Adv. Mater., 2008, 20, 627–631 CrossRef CAS.
- E. Kokkoli, A. Mardilovich, A. Wedekind, E. L. Rexeisen, A. Garga and J. A. Craig, Soft Matter, 2006, 2, 1015–1024 RSC.
- M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082–12083 CrossRef CAS.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, Acc. Chem. Res., 2009, 42, 681–692 CrossRef CAS.
- K. Velonia, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2002, 124, 4224–4225 CrossRef CAS.
- D. Lensen, D. M. Vriezema and J. C. M. van Hest, Macromol. Biosci., 2008, 8, 991–1005 CrossRef CAS.
- D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS.
- F. Chécot, S. Lecommandoux, H.-A. Klok and Y. Gnanou, Eur. Phys. J. E, 2003, 10, 25–35 CrossRef CAS.
- F. Chécot, S. Lecommandoux, Y. Gnanou and H.-A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1339–1343 CrossRef CAS.
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772–776 CrossRef CAS.
- M. G. Ryadnov, Angew. Chem., Int. Ed., 2007, 46, 969–972 CrossRef CAS.
- M. D. Pierschbacher and E. Ruoslahti, Nature, 1984, 309, 30–33 CrossRef CAS.
- L. Gentilucci, A. Tolomelli and F. Squassabia, Curr. Med. Chem., 2006, 13, 2449–2466 CrossRef CAS.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385 CrossRef CAS.
- N. Oku, C. Koike, Y. Tokudome, S. Okada, N. Nishikawa, H. Tsukada, M. Kiso, A. Hasegawa, H. Fujii, J. Murata and I. Saiki, Adv. Drug Delivery Rev., 1997, 24, 215–223 CrossRef.
- T. Asai and N. Oku, Methods Enzymol., 2005, 391, 163–176 CAS.
- N. Yagi, Y. Ogawa, M. Kodaka, T. Okada, T. Tomohiro, T. Konakahara and H. Okuno, Chem. Commun., 1999, 1687–1688 RSC.
- T. Pakalns, K. L. Haverstick, G. B. Fields, J. B. McCarthy, D. L. Mooradian and M. Tirrell, Biomaterials, 1999, 20, 2265–2279 CrossRef CAS.
- V. Marchi-Artzner, B. L. U. Hellerer, M. Kantlehner, H. Kessler and E. Sackmann, Chem.–Eur. J., 2001, 7, 1095–1101 CrossRef CAS.
- V. Marchi-Artzner, B. Lorz, C. Gosse, L. Jullien, R. Merkel, H. Kessler and E. Sackmann, Langmuir, 2003, 19, 835–841 CrossRef CAS.
- J. P. Jung, J. L. Jones, S. A. Cronier and J. H. Collier, Biomaterials, 2008, 29, 2143–2151 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- A. Mata, L. Hsu, R. Capito, C. Aparicio, K. Henriksonc and S. I. Stupp, Soft Matter, 2009, 5, 1228–1236 RSC.
- A. Abbott, Nature, 2003, 424, 870–872 CrossRef CAS.
- H. K. Kleinman, M. L. McGarvey, J. R. Hassell, V. L. Star, F. B. Cannon, G. W. Laurie and G. R. Martin, Biochemistry, 1986, 25, 312–318 CrossRef CAS.
- H. K. Kleinman, M. L. McGarvey, L. A. Liotta, P. G. Robey, K. Tryggvason and G. R. Martin, Biochemistry, 1982, 21, 6188–6193 CrossRef CAS.
- T. C. Holmes, S. de Lacalle, X. Su, G. Liu, A. Rich and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6833 CrossRef CAS.
- J. Kisiday, M. Jin, B. Kurz, H. Hung, C. Semino, S. G. Zhang and A. J. Grodzinsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9996–10001 CrossRef CAS.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS.
- M. E. Davis, P. C. H. Hsieh, T. Takahashi, Q. Song, S. Zhang, R. D. Kamm, A. J. Grodzinsky, P. Anversa and R. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8155–8160 CrossRef CAS.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS.
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS.
- G. von Maltzahn, S. Vauthey, S. Santoso and S. Zhang, Langmuir, 2003, 19, 4332–4337 CrossRef CAS.
- S. Keller, I. Sauer, H. Strauss, K. Gast, M. Dathe and M. Bienert, Angew. Chem., Int. Ed., 2005, 44, 5252–5255 CrossRef CAS.
- D. Missirlis, H. Khant and M. Tirrell, Biochemistry, 2009, 48, 3304–3314 CrossRef CAS.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS.
- C. Galanth, F. Abbassi, O. Lequin, J. Ayala-Sanmartin, A. Ladram, P. Nicolas and M. Amiche, Biochemistry, 2009, 48, 313–327 CrossRef CAS.
- A. F. Chu-Kung, K. N. Bozzelli, N. A. Lockwood, J. R. Haseman, K. H. Mayo and M. V. Tirrell, Bioconjugate Chem., 2004, 15, 530–535 CrossRef CAS.
- C. Mas-Moruno, L. Cascales, L. J. Cruz, P. Mora, E. Pérez-Payá and F. Albericio, ChemMedChem, 2008, 3, 1748–1755 CrossRef CAS.
- E. D. Sone and S. I. Stupp, J. Am. Chem. Soc., 2004, 126, 12756–12757 CrossRef CAS.
- L.-S. Li and S. I. Stupp, Angew. Chem., Int. Ed., 2005, 44, 1833–1836 CrossRef CAS.
- H. Bekele, J. H. Fendler and J. W. Kelly, J. Am. Chem. Soc., 1999, 121, 7266–7267 CrossRef CAS.
- D. Volkmer, M. Fricke, T. Huber and N. Sewald, Chem. Commun., 2004, 1872–1873 RSC.
- E. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Science, 2009, 323, 1455–1458 CrossRef CAS.
- D. C. Popescu, M. M. J. Smulders, B. P. Pichon, N. Chebotareva, S.-Y. Kwak, O. L. J. van Asselen, R. P. Sijbesma, E. DiMasi and N. A. J. M. Sommerdijk, J. Am. Chem. Soc., 2007, 129, 14058–14067 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- Z. Huang, T. D. Sargeant, J. F. Hulvat, A. Mata, P. Bringas, C.-Y. Koh, S. I. Stupp and M. L. Snead, J. Bone Miner. Res., 2008, 23, 1995–2006 CrossRef.
- S. Froidevaux and A. N. Eberle, Biopolymers, 2002, 66, 161–183 CrossRef CAS.
- H. Tanisaka, S. Kizaka-Kondoh, A. Makino, S. Tanaka, M. Hiraoka and S. Kimura, Bioconjugate Chem., 2008, 19, 109–117 CrossRef CAS.
- A. Morisco, A. Accardo, E. Gianolio, D. Tesauro, E. Benedettia and G. Morelli, J. Pept. Sci., 2009, 15, 242–250 CrossRef CAS.
- S. R. Bull, M. O. Guler, R. E. Bras, T. J. Meade and S. I. Stupp, Nano Lett., 2005, 5, 1–4 CrossRef CAS.
- D. Ryan, B. A. Parviz, V. Linder, V. Semetey, S. K. Sia, J. Su, M. Mrksich and G. M. Whitesides, Langmuir, 2004, 20, 9080–9088 CrossRef CAS.
- C. B. Herbert, T. L. McLernon, C. L. Hypolite, D. N. Adams, L. Pikus, C.-C. Huang, G. B. Fields, P. C. Letourneau, M. D. Distefano and W. S. Hu, Chem. Biol., 1997, 4, 731–737 CrossRef CAS.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Biotechnol. Prog., 1998, 14, 356–363 CrossRef CAS.
- S. Takayama, J. C. McDonald, E. Ostuni, M. N. Liang, P. J. A. Kenis, R. F. Ismagilov and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5545–5548 CrossRef CAS.
- E. Delamarche, A. Bernard, H. Schmid, B. Michel and H. Biebuyck, Science, 1997, 276, 779–781 CrossRef CAS.
- K. Y. Suh, J. Seong, A. Khademhosseini, P. E. Laibinis and R. Langer, Biomaterials, 2004, 25, 557–563 CrossRef CAS.
- A. Khademhosseini, S. Jon, K. Y. Suh, T.-N. T. Tran, G. Eng, J. Yeh, J. Seong and R. Langer, Adv. Mater., 2003, 15, 1995–2000 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS.
- R. S. Kane, S. Takayama, E. Ostuni, D. E. Ingber and G. M. Whitesides, Biomaterials, 1999, 20, 2363–2376 CrossRef.
- J. Bredenbeck, J. Helbing, J. R. Kumita, G. A. Woolley and P. Hamm, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2379–2384 CrossRef CAS.
- K. Tajima and T. Aida, Chem. Commun., 2000, 2399–2412 RSC.
- A. Kros, R. J. M. Nolte and N. A. J. M. Sommerdijk, Adv. Mater., 2002, 14, 1779–1782 CrossRef CAS.
- J.-E. Osterholm, Y. Cao, F. Klavetter and P. Smith, Synth. Met., 1993, 55, 1034–1039 CrossRef CAS.
- N. Kuramoto, J. C. Michaelson, A. J. McEvoy and M. Grätzel, J. Chem. Soc., Chem. Commun., 1990, 1478–1480 RSC.
- E. Ruckenstein and L. Hong, Synth. Met., 1994, 66, 249–256 CrossRef CAS.
- N. Sakmeche, S. Aeiyach, J.-J. Aaron, M. Jouini, J. C. Lacroix and P.-C. Lacaze, Langmuir, 1999, 15, 2566–2574 CrossRef CAS.
- N. Sakmeche, J.-J. Aaron, M. Fall, S. Aeiyach, M. Jouini, J. C. Lacroix and P.-C. Lacaze, Chem. Commun., 1996, 2723–2724 RSC.
- A. Kros, J. G. Linhardt, H. K. Bowman and D. A. Tirrell, Adv. Mater., 2004, 16, 723–727 CrossRef CAS.
- J. D. Tovar, B. M. Rabatic and S. I. Stupp, Small, 2007, 3, 2024–2028 CrossRef CAS.
- R. Djalali, Y. F. Chen and H. Matsui, J. Am. Chem. Soc., 2002, 124, 13660–13661 CrossRef CAS.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS.
- T. Scheibel, R. Parthasarathy, G. Sawicki, X.-M. Lin, H. Jaeger and S. L. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4527–4532 CrossRef CAS.
- J. Ryu and C. B. Park, Adv. Mater., 2008, 20, 3754–3758 CrossRef CAS.
- J. Ryu and C. B. Park, Angew. Chem., Int. Ed., 2009, 48, 4820–4823 CrossRef CAS.
- J. Ryu, S. Y. Lim and C. B. Park, Adv. Mater., 2009, 21, 1577 CrossRef CAS.
- D. Gottlieb, S. A. Morin, S. Jin and R. T. Raines, J. Mater. Chem., 2008, 18, 3865–3870 RSC.
- E. Braun, Y. Eichen, U. Sivan and G. Ben-Yoseph, Nature, 1998, 391, 775–778 CrossRef CAS.
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377–1400 CrossRef CAS.
- M. R. Diehl, K. Zhang, H. J. Lee and D. A. Tirrell, Science, 2006, 311, 1468–1471 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |