Lanthanide luminescence for functional materials and bio-sciences†
Svetlana V. Eliseevaa and Jean-Claude G. Bünzli*ab
aLaboratory of Lanthanide Supramolecular Chemistry, Swiss Federal Institute of Technology, Lausanne (EPFL). E-mail: jean-claude.bunzli@epfl.ch; Fax: +41 21 693 9825; Tel: +41 21 6939821
bWorld Class University (WCU) Professor, Department of Advanced Materials Chemistry, Korea University, Chungnam 339-700, South Korea
First published on 11th September 2009
Abstract
Recent startling interest for lanthanide luminescence is stimulated by the continuously expanding need for luminescent materials meeting the stringent requirements of telecommunication, lighting, electroluminescent devices, (bio-)analytical sensors and bio-imaging set-ups. This critical review describes the latest developments in (i) the sensitization of near-infrared luminescence, (ii) “soft” luminescent materials (liquid crystals, ionic liquids, ionogels), (iii) electroluminescent materials for organic light emitting diodes, with emphasis on white light generation, and (iv) applications in luminescent bio-sensing and bio-imaging based on time-resolved detection and multiphoton excitation (500 references).
 Svetlana V. Eliseeva | Svetlana V. Eliseeva graduated from Lomonosov Moscow State University (MSU), earning a degree in chemistry with honors in 2003 and a PhD degree in inorganic chemistry in 2006 under the supervision of Professor Natalia P. Kuzmina. After two years of a post-doctoral fellow under a joint program between the Department of Material Sciences at MSU and Saint-Gobain company (France), she joined the group of Professor Jean-Claude G. Bünzli at École Polytechnique Fédérale de Lausanne (EPFL). Her current research interests mainly focus on the development of luminescent lanthanide-containing coordination compounds and nanoparticles suitable for bio-analysis and lighting applications, as well as on multiphoton excitation measurements. |
 Jean-Claude Bünzli | Jean-Claude Bünzli is an active researcher in the field of coordination, supramolecular and biological chemistry of the lanthanide ions. He earned a degree in chemical engineering in 1968 and a PhD in 1971 from the École Polytechnique Fédérale de Lausanne (EPFL). He spent two years at the University of British Columbia (Canada) and one year at the Swiss Federal Institute of Technology in Zürich before being appointed at the University of Lausanne in 1974 and at EPFL in 2001 as a full professor of inorganic chemistry. Since September 2009 he also holds a professorship at Korea University. His research focuses on the self-assembly of building blocks for photoluminescent materials and of lanthanide luminescent bioprobes. |
1. Scope of the review
The present critical review aims at describing selected themes of current interest in science and technology and dealing with lanthanide luminescence. It can be considered as being a follow up and an extension of a 2005 review published in this journal.1 To keep it self-consistent however, the general description of lanthanide luminescence and its applications is maintained and expanded. In view of the current trends in lanthanide luminescence, and reflecting the proportions of the data published recently, the topics discussed below refer to NIR luminescence, special “soft” luminescent materials with technological potentials, lighting devices such as light emitting diodes, and bio-analysis and imaging. The literature is selectively covered from late 2005 (late 2006 for NIR luminescence) to March 2009.2. Lanthanide luminescence: why, how and what?
In 1937, J. H. van Vleck2 wrote an article titled The Puzzle of Rare-Earth Spectra in Solids which perfectly reflects the fascination exerted by the intricate optical properties of the trivalent lanthanide ions, hereafter LnIII. The electronic [Xe]4fn configurations (n = 0–14) indeed generate a rich variety of electronic levels, the number of which is given by 14!/n!(14 −n)!, translating to 3432 for GdIII, for instance (see Fig. 1). The energies of these levels are well defined due to the shielding of the 4f orbitals by the filled 5s25p6 sub-shells and, in addition, they do not vary much with the chemical environments in which the lanthanide ions are inserted. As a corollary, inner-shell 4f–4f transitions are sharp and easily recognizable. This has been a definite advantage in the discovery of the lanthanide elements between 1803 and 1907 during which all of the lanthanides have been identified, barring artificial Pm (1947). | ||
| Fig. 1 Partial energy level diagram for LnIII ions doped in a low-symmetry crystal (LaF3). Redrawn from G. K. Liu.21 | ||
One of the landmarks in lanthanide luminescence is the discovery of the highly emissive Y2O3:EuIII material,3 which is at the origin of the phosphors for cathode-ray tubes and fluorescent lamps, still in heavy use today.4 Other milestones for purely inorganic compounds are the findings of neodymium YAG (Yttrium Aluminium Garnet) lasers in 19645 and Er-doped optical fibres for telecommunications in 1987.6 Attention on luminescent coordination compounds started in the mid-seventies when Finnish researchers proposed EuIII and TbIII (later also SmIII and DyIII) polyaminocarboxylates and β-diketonates as bioprobes in time-resolved luminescent (TRL) immunoassays.7,8 This new technology generated a large interest and further developments, such as homogeneous TRL assays,9 optimization of bioconjugation methods for lanthanide luminescent chelates,10 and time-resolved luminescence microscopy (TRLM)11 resulted in applications of lanthanide luminescent bioprobes (LLBs)12 in many fields of biological and medicinal analyses, including tissue13 and cell imaging,14 as well as monitoring drug delivery.15 These bio-applications have been a major factor in the unprecedented expansion of lanthanide coordination chemistry during the past 20 years, together with the design of contrast agents for magnetic resonance imaging (MRI)16 and more efficient separation techniques.17 Other drives for lanthanide coordination chemistry arise from the design of lanthanide-doped OLEDs (Organic Light Emitting Diodes)18 emitting either in the NIR, such as ErIII tris(8-hydroxyquinolinate),19 or in the visible.20
2.1 Lanthanide photophysics22
In addition, when the LnIII ion is inserted into a chemical environment, the (2J + 1)-degenerate J-levels are split by ligand-field effects into so-called Stark sub-levels, the number of which depends on the site symmetry of the metal ion.22 Transitions between these sub-levels are governed by symmetry-related selection rules which can be obtained from:
| Γop∈Γ(Ψi) ×Γ(Ψf) | (1) |
The selection rules are derived under several hypotheses which are not always completely fulfilled in reality (in particular 4f wavefunctions are not totally pure), so that the terms “forbidden” and “allowed” transitions cannot be taken too rigidly. A more correct wording would be that a forbidden transition has a low probability and an allowed transition a high probability of occurring. The intensities of the induced electric dipole f–f transitions can be derived from Judd–Ofelt (JO) theory, in which the dipole strength Ded in 1036 debye2 is expressed by:
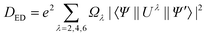 | (2) |
For LnIII ions, intensities of ED transitions have the same order of magnitude than those of MD transitions, so that both are seen in the optical spectra, while EQ transitions are much weaker and have usually not been identified.
| Symmetry | Site symmetry | Integer J | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| Cubic | T, Td, Th, O, Oh | 1 | 1 | 2 | 3 | 4 | 4 | 6 | 6 | 7 |
| Hexagonal | C3h, D3h, C6, C6h, C6v, D6, D6h | 1 | 2 | 3 | 5 | 6 | 7 | 9 | 10 | 11 |
| Trigonal | C3, S6, C3v, D3, D3d | 1 | 2 | 3 | 5 | 6 | 7 | 9 | 10 | 11 |
| Tetragonal | C4, S4, C4h, C4v, D4, D2d, D4h | 1 | 2 | 4 | 5 | 7 | 8 | 10 | 11 | 13 |
| Low | C1, CS, C2, C2h, C2v, D2, D2h | 1 | 3 | 5 | 7 | 9 | 11 | 13 | 15 | 17 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1, λ < 200 nm), except for CeIII (>32000 cm−1, λ < 312 nm), PrIII and TbIII (>40000 cm−1, λ < 250 nm), so that they are rarely observed in coordination compounds.25,26 Similarly, charge-transfer transitions (e.g. ligand-to-metal charge transfer, LMCT) are parity allowed, their energy is large, and they appear usually at wavelengths smaller than 200 nm. Exceptions are EuIII and YbIII (possibly SmIII and TmIII) which can be more easily reduced than the other LnIII ions.1
000 cm−1, λ < 200 nm), except for CeIII (>32000 cm−1, λ < 312 nm), PrIII and TbIII (>40000 cm−1, λ < 250 nm), so that they are rarely observed in coordination compounds.25,26 Similarly, charge-transfer transitions (e.g. ligand-to-metal charge transfer, LMCT) are parity allowed, their energy is large, and they appear usually at wavelengths smaller than 200 nm. Exceptions are EuIII and YbIII (possibly SmIII and TmIII) which can be more easily reduced than the other LnIII ions.1Important parameters characterizing the emission of light from a LnIII ion are (i) the quantum yield Q, which is equal to the ratio between the number of emitted photons divided by the number of absorbed photons, and (ii) the lifetime of the excited state τobs = 1/kobs with kobs being the rate constant (in s−1) of the de-population of the excited state. If the metal ion is directly excited into a 4f level, these two quantities are related by:
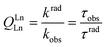 | (3) |
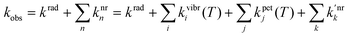 | (4) |
In absence of non-radiative deactivation processes, kobs = krad and the quantum yield is equal to 1, which seldom happens. Examples are, in solid state and under excitation at 254 nm, Y2O3:EuIII (3%) with Q = 0.99 and terbium benzoate with Q = 1;28 in solution, a terbium complex with a dipyrazolylpyridine bearing aminocarboxylate coordinating groups was reported having Q = 0.95.29 To date, the largest quantum yield recorded for an EuIII complex, [Eu(tta)3DBSO] is 85% (tta is thenoyltrifluoroacetylacetonate and DBSO dibenzyl sulfoxide).30 The latter quantum yields have been obtained by excitation into the metal–ion surroundings and are termed overall quantum yields, with ηsens, the sensitization efficiency, defined as the efficacy with which energy is transferred from the feeding levels of the metal-ion surroundings onto the LnIII excited states:
| QLLn = ηsensQLnLn | (5) |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ) using Einstein’s rates of spontaneous emission A from an initial state |ΨJ〉, characterized by a quantum number J, to a final state |Ψ′J′〉:
) using Einstein’s rates of spontaneous emission A from an initial state |ΨJ〉, characterized by a quantum number J, to a final state |Ψ′J′〉: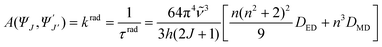 | (6) |
is the mean energy of the transition, h Planck’s constant, and n the refractive index; DED is given by eqn (2) and DMD by eqn (7):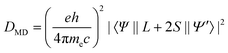 | (7) |
) is known, which may be the case when the luminescence transitions terminate onto the ground level, the radiative lifetime can be extracted from the following equation with NA being Avogadro’s number (6.023 × 1023):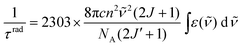 | (8) |
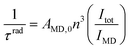 | (9) |
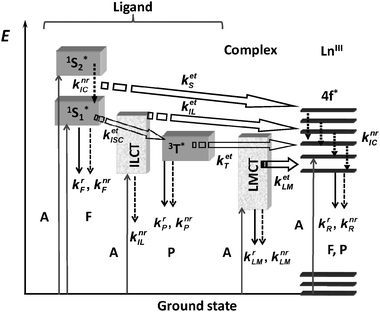 | ||
| Fig. 2 Schematic representation of energy absorption, migration, emission (plain arrows) and dissipation (dotted arrows) processes in a lanthanide complex. 1S* or S = singlet state, 3T* or T = triplet state, A = absorption, F = fluorescence, P = phosphorescence, k = rate constant, r = radiative, nr = non-radiative, IC = internal conversion, ISC = intersystem crossing, ILCT (indices IL) = intra-ligand charge transfer, LMCT (indices LM) = ligand-to-metal charge transfer. Back transfer processes are not drawn for the sake of clarity. | ||
One of the main energy migration path implies Laporte- and spin-allowed ligand-centred absorptions followed by intersystem crossing (1S* →3T*, kISC), 3T* →Ln* (ket) transfer, and metal-centred emission. It is noteworthy that although important, this energy transfer path is by far not the only operative one. For instance, Kleinerman reached the conclusion, based on a systematic study of over 600 lanthanide chelates, that excited singlet states contribute to the transfer and may even be the privileged donor states, depending on the relative values of the rate constants for the various intervening processes.35 Such cases are documented for both EuIII,36 and TbIII.37
Photophysical requirements for highly emissive compounds are related to the two parameters intervening in eqn (5): energy transfer (ηsens) and minimization of non-radiative processes (QLnLn). The first one is difficult to master in view of its intricate mechanisms. Some authors have nevertheless established phenomenological rules which ought to be used with caution: they rely on the simplified idea that the 1S*–3T*–Ln* energy transfer path is the only one operative and on considering that the sole parameter of importance is the energy difference between 3T* and the emitting LnIII level. The following lessons can be drawn from these data.38–40
• The largest values of quantum yields occur when the triplet state energy is close to the energy of one of the higher excited states of the metal ion; if the energy of the feeding state becomes closer to the energy of the emitting state, back transfer operates. For EuIII and TbIII, a “safe” energy difference minimizing this process is around 2500–3500 cm−1 and this probably applies to the other LnIII ions too.
• For EuIII, the energy of the triplet state corresponding to the largest quantum yields depends on the type of ligand: it is close to the energy of the 5D0 level for Schiff-base complexes,405D1 level for β-diketonates,38 and 5D2 level for polyaminocarboxylates.39
• When comparing complexes with EuIII and TbIII with the same polyaminocarboxylate ligands, the maximum quantum yield values reached for TbIII are larger than those for EuIII: this reflects the smaller Eu(5D0–7F6) energy gap compared to Tb(5D4–7F0).
Since efficient ISC transfers take place when the energy difference between the singlet and triplet states is around 5000 cm−1, ligand designers try to keep to the following rules: ΔE(1S*–3T*) ≈ 5000 cm−1 and ΔE(3T*–Ln* emissive level) in the bracket 2500–3500 cm−1. However, minute energy differences in the ligand states sometimes lead to large differences in overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor, an essential parameter of the overall energy transfer process, thus resulting in large differences in quantum yield. When bound to soft-donor ligands, influence of the LMCT states becomes important: in complexes with diethyl-dithiocarbamate, [Ln(Et2dtc)3(bpy)] for instance, emission of EuIII is quenched (ECT = 19300 cm−1) but not of YbIII (ECT = 28300 cm−1).41
2.2 Information gained from lanthanide luminescence
Any luminescent lanthanide ion may act as a probe, but some ions bring more information (e.g. EuIII), or are more luminescent (e.g. TbIII) than others, which explains their preferential use. The following section will mainly deal with EuIII. Generally speaking a lanthanide luminescent tag functions either as a structural probe deciphering the symmetry of the chemical environment and, partly, the composition of the inner-coordination sphere, or as simple analytical (bio)marker, the switching on (or off) or the modulation of its luminescence representing the analytical signal.
•Number of metal-ion sites, N. High resolution of the Eu(5D0→7F0) transition, which is unique for a given chemical environment associated with spectral decomposition with Lorentzian–Gaussian shape functions42,43 gives a direct access to N. Experimentally, laser-excited excitation spectra of the 5D0←7F0 transition yield more sensitive results.
•Composition of the first coordination sphere. The energy of the 5D0←7F0 transition at 298 K, calc in cm−1, is correlated with the nephelauxetic effect δi generated by each coordinated group:44
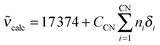 | (10) |
•Population analysis. When several LnIII sites are present, their relative populations Pi can be determined by analysis of the multi-exponential luminescence decay:
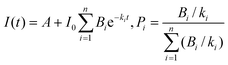 | (11) |
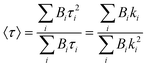 | (12) |
•Site symmetry. This determination is best performed by analysing the Eu(5D0) emission, deciphering the crystal field splitting of the 7FJ levels and comparing them to theoretical predictions.23,45 To avoid wrong conclusions, vibronic contributions should be identified.46
•Strength of the Ln–L bond. The intensity of vibronic satellites, which are particularly intense when associated with hypersensitive transitions, is proportional to the ligand-to-metal bond strength and constitutes a useful measure of the latter.47
•Solution state of the LnIII ion. Lanthanide luminescence is very sensitive to the quenching by high-energy vibrations, particularly O–H. This quenching can be turned into an advantage for calculating the number q of inner-sphere bonded water molecules by measuring the lifetime in both water and deuterated water. Provided that the O–H quenching is the main non-radiative process operating (i.e. in absence of other temperature-dependent processes such as photo-induced electron transfer or back transfer), phenomenological equations can be worked out (Table 3):
| q = A(Δkobs−B) −C |
| Δkobs = kH2O−kD2O = 1/τH2O− 1/τD2O | (13) |
| q = Akobs−C | (14) |
| Ln | Solvent | Ligands | A | B | Ca | Δkobs units | Ref. |
|---|---|---|---|---|---|---|---|
| a qN is the number of N–H oscillators in the second coordination sphere. | |||||||
| Eu | H2O | Various | 1.11 | 0.31 | 0 | ms | 48 |
| Eu | H2O | Cyclen derivatives | 1.2 | 0.25 | 0.075qN | ms | 27 |
| Tb | H2O | Cyclen derivatives | 5.0 | 0.06 | 0 | ms | 27 |
| Yb | H2O | Cyclen derivatives | 1.0 | 0.2 | 0 | μs | 27 |
| Yb | MeOH | Cyclen derivatives | 2.0 | 0.1 | 0 | μs | 27 |
| Nd | H2O | Polyaminocarboxylates | 0.36 | 0 | 2.0 | μs | 49 |
| Sm | H2O | Polyaminocarboxylates | 25.4 | 0 | 0.37 | μs | 50 |
| Dy | H2O | Polyaminocarboxylates | 21.1 | 0 | 0.60 | μs | 50 |
•Donor–acceptor distances. Distances between two metal-ion sites or between a chromophore and an ion, RDA, may be determined by measuring the lifetimes of the donor (D) in presence (τobs) and in absence (τ0) of the acceptor (A):
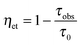 | (15) |
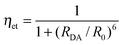 | (16) |
When it comes to bio-analysis, time-resolved detection enhances considerably the signal-to-noise ratio and additional selectivity can be obtained by using FRET (Förster resonant energy transfer) methodology,54 a common practice in homoimmunoassays55 or high-throughput screening.56
3. Near-infrared luminescence
Compounds exhibiting lanthanide near-infrared (NIR) luminescence mainly fall into two categories: (i) purely inorganic substances, mostly oxides or doped semiconductor materials (e.g. GaN); they find use in optical fibres, lasers, and planar amplifiers for telecommunications, as well as in light-emitting diodes (LEDs), security inks, or bio-analysis; chalcogenide clusters with low-energy phonon density of states may be classified in this group as well; (ii) complexes with organic ligands, developed in the hope of producing electroluminescent materials for the same applications, but more economical and more versatile; this field has been tremendously stimulated by the discovery in 1990 of electroluminescence in conjugated polymers (namely poly(para-phenylene vinylene), PPV).57 Since our last review on lanthanide NIR luminescence covering the literature until September 2006,58 more than two hundred papers have appeared. In this section, we mainly focus on molecular compounds of NdIII, ErIII, YbIII, and to a lesser extent PrIII, TmIII, and classify them according to the sensitization mode. Furthermore attention is given to reports containing quantitative data (quantum yields, lifetimes) or describing innovative systems and/or energy transfer paths. Quantitative data remain scarce because few laboratories are equipped for quantum yield measurement in the NIR range; as an alternative, intrinsic quantum yields are often calculated from lifetimes, with a “literature value” of τrad, therefore they have to be considered with extreme care.3.1 Sensitization by organic ligands
Despite the dramatic limitations inherent to the use of organic ligands for the sensitization of NIR luminescence,58 namely the presence of high-energy vibrators such as C–H59 or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, considerable efforts have been undertaken recently to test several classes of ligands.60–100 A listing of the corresponding photophysical properties is provided in Table S1 (ESI†) while complexes with the best photophysical properties are listed in Table 4. Altogether, the top overall quantum yields obtained for NdIII, ErIII and YbIII lie in the ranges 0.1–0.4, 0.01–0.03 and 0.6–1.4%, respectively, for solid-state samples and 0.01–0.07, 0.01–0.02 and 0.5–3.8%, respectively, for solutions in non-deuterated organic solvents. Data for aqueous solutions are much scarcer and are around 0.03% for NdIII and 0.14% for YbIII. For the latter ion, the maximum overall quantum yield in water remains that reported by Korovin et al. for a dinuclear macrocyclic complex (0.53%).101
C, considerable efforts have been undertaken recently to test several classes of ligands.60–100 A listing of the corresponding photophysical properties is provided in Table S1 (ESI†) while complexes with the best photophysical properties are listed in Table 4. Altogether, the top overall quantum yields obtained for NdIII, ErIII and YbIII lie in the ranges 0.1–0.4, 0.01–0.03 and 0.6–1.4%, respectively, for solid-state samples and 0.01–0.07, 0.01–0.02 and 0.5–3.8%, respectively, for solutions in non-deuterated organic solvents. Data for aqueous solutions are much scarcer and are around 0.03% for NdIII and 0.14% for YbIII. For the latter ion, the maximum overall quantum yield in water remains that reported by Korovin et al. for a dinuclear macrocyclic complex (0.53%).101
| Complex | Sample | QLLn (%) | τ/μs | QLnLn (%) | Ref. |
|---|---|---|---|---|---|
| a Average lifetimes: see eqn (17).b Recalculated from τav assuming the radiative lifetime found for Er-doped silica: 14 ms. | |||||
| [Nd(2c)3]·MeOH | Solid | 0.40 | 1.57 | n.a. | 62 |
| [Nd(4h)3] | Solid | 0.33 | 1.82 | 0.67 | 64 |
| [Nd(8)3] | CD3CN | n.a. | 44 | n.a. | 71 |
| [Nd(21b)3(bpy)] | Toluene | 0.072 | n.a. | n.a. | 83 |
| [Nd(H36a)] | H2O (pH 7.4) | 0.027 | 0.15 | n.a. | 67 |
| [Er(7b)3] | Nanorods | n.a. | 300a | 2.1b | 69 |
| [Er0.5Yb0.5(7b)3] | Nanorods | n.a. | 733a | 5.2b | 69 |
| [Er(2c)3] | Solid | 0.033 | 4.05 | n.a. | 62 |
| [Er(8)3] | CD3CN | n.a. | 741 | n.a. | 71 |
| [Er(22)4]− | MeCN | 0.021 | n.a | n.a. | 98 |
| [Yb(2c)3]·3H2O | Solid | 1.40 | 20.6 | n.a. | 62 |
| [KAlYb(5b)3](OTf) | Solid | 1.17 | 22.6 | n.a. | 66 |
| [Yb(8)3] | CD3CN | n.a. | 1111 | n.a. | 71 |
| DMSB[Yb(21f)4] | MeCN | n.a. | 46.4 | n.a. | 99 |
| [Yb(22)4]− | MeCN | 3.8 | 24.6 | n.a. | 98 |
| [Yb(21b)3(phen)] | Toluene | 1.28 | n.a. | n.a. | 83 |
| [Yb(3)2](NO3) | MeCN | 1.1 | n.a. | n.a. | 63 |
| [Yb(6c)] | H2O (pH 7.4) | 0.14 | 2.05 | n.a. | 100 |
Since erbium tris(8-hydroxyquinolinate) was shown to be brightly electroluminescent at 1.5 μm and compatible with silicon technology,102 8-hydroxyquinolinate has been a chromophore commonly incorporated in ligands designed for sensitising LnIII NIR luminescence (Scheme 1). As a matter of fact, the simple tridentate ligands H2, which lead to neutral, well defined tris(complexes), perform quite well in populating the LnIII excited states, particularly when substituted by two bromine atoms in H2c. Indeed in going from H2a to H2c, the overall quantum yields increase by a factor 2.5–2.9 while the lifetimes are lengthened two- to four-fold. The main factor here is the removal of C–H vibrators on the ligand core, in addition to an increased heavy-atom effect.62 These building blocks have successfully been incorporated into ditopic ligands self-assembling into trimetallic helicates with overall formulae [KLn2(5a)3]OTf or [KAlLn(5b)3]OTf (OTf is the triflate anion) in which the YbIII ions retain most of the photophysical properties of the mononuclear precursors.66
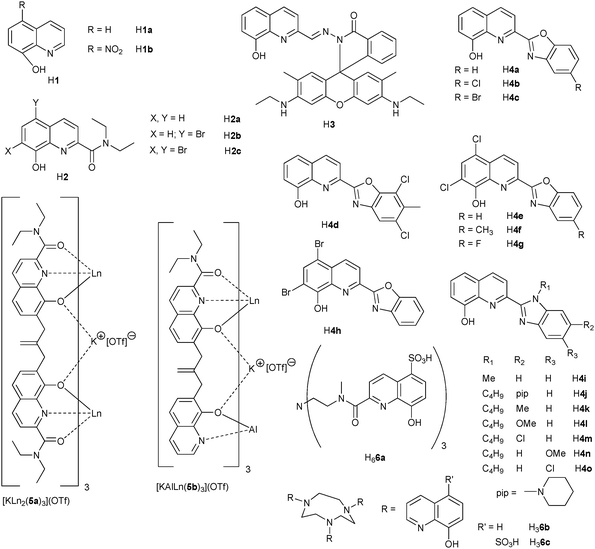 | ||
| Scheme 1 Ligands incorporating 8-hydroxyquinoline moieties.60–64,66,67,100 | ||
Another way of benefiting from the chromophoric properties of 8-hydroxyquinolinate together with a large chelate effect is to incorporate these units in tripodal67 or tetrapodal103 ligands fitted with sulfonate groups for ensuring water solubility. The tripod H66a performs less well than its tetrapodal counterpart, but the sensitization of the YbIII luminescence remains noticeable in aerated water (QLLn = 0.13%). An additional advantage of the resulting podates is their stability which is comparable to that of edta chelates so that they may be envisaged as probes for in vitro analysis.67 When the pivotal amine group is replaced by an 1,4,7-triazacyclononane core, similar results are obtained with quantum yields of 0.60 and 0.14% for the solid state sample [Yb(6b)] and an aqueous solution of [Yb(6c)], respectively.
Substituting the 2-position of 8-hydroxyquinoline by a benzoxazole moiety results in N,N,O-chelating units H4 bearing an extended chromophore, which can be modulated by grafting diverse substituents. Large absorption in the visible (508–527 nm, ε = 7.5–9.6 × 103 M−1 cm−1 in the tris complexes) is another plus for these systems which proved to be particularly efficient for NdIII luminescence with quantum yields up to 0.33%. Similarly to the chelates with H2, 5,7-dihalogenation of the 8-hydroxyquinoline core is beneficial, the QLNd increasing by a factor of two.64 When benzoxazole is replaced by benzimidazole, yielding a tridentate N,N,N ligand, similar effects are observed for NdIII, with [Nd(4n)3]·3H2O having QLNd = 0.34%.65 Finally, an alternative approach to sensitising YbIII luminescence has been recently proposed in which 8-hydroxyquinoline is decorated with a rhodamine chromophore (triplet state energy ≈17000 cm−1) via a carboxy-hydrazone linker (H3). Two tetradentate ligands provide a saturated coordination environment for YbIII and the reported overall quantum yield amounts to 1.1% in acetonitrile.63
In an effort to minimize C–H quenching in ErIII complexes meant for applications in telecommunications, bis(phenyl)phosphinic acid (H7a), has been fully fluorinated (H7b, Scheme 2).68,69 The corresponding [Er(7)3] salts simply obtained by reacting the metal chloride with the free acid in water, are thermally stable, can be easily dehydrated, and possess a polymeric structure. While the luminescence decay of the Er(4I13/2→4I15/2) transition at ≈1.5 μm is a single exponential function (τ = 4.9 μs) for the unfluorinated salt [Er(7a)3], the perfluorinated species exhibit a more complicated behaviour and the decay had to be analyzed in terms of a “stretched exponential” function, β representing an empirical factor and 〈τ〉 an average lifetime:
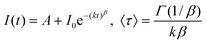 | (17) |
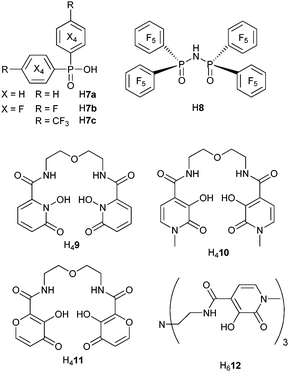 | ||
| Scheme 2 Phosphinic acid- and hydroxypyridine-containing ligands.68–74 | ||
A slightly modified synthetic procedure of [Er(7b)3] gives rise to nanorods with double-exponential luminescence decays. The average Er(4I13/2) lifetime is equal to 300 μs, in perfect agreement with the above-mentioned value. It is considerably lengthened when the Er(4I11/2) level is populated by the long-lived Yb(2F5/2) level in [Er0.5Yb0.5(7b)3] nanorods, reaching 733 μs.69 In a similar attempt to eliminate the detrimental effect of C–H bonds, the imidodiphosphinate chelating unit was fitted with perfluorinated phenyl groups. Not only does the ligand H8 lacks C–H bonds, but it also features a low-lying πσ* (≈14000 cm−1) state mainly located on the perfluorobenzene units and very favourable to NIR sensitisation; as a matter of fact, the NdIII, ErIII and YbIII ions have among the longest lifetimes recorded for complexes with organic ligands, up to 1.1 ms for [Yb(8)3] in deuterated acetonitrile for instance.71 After testing successfully the 1-hydroxypyridin-2-one chromophore (H49) for stimulating EuIII emission, and considering the large stability of the 1:2 complexes (pEu = 18.6),104 Raymond et al. have investigated the ability of the similar ligands H410 and H411 to sensitise NIR luminescence (Scheme 2). In Tris buffer (pH 7.4), the ≈1-μm lines of [Pr(H210)2]− (1D2→3F4) and [Ho(H210)2]− (5F5→5I7) are indeed seen, in addition to visible emission; the corresponding lifetimes are τ(1D2) = 8.0(4) ns and τ(5F5) = 6.5(3) ns, the latter being a very rare example of lifetime determination for a HoIII complex in aqueous solution. Stability constants for the complexes with (H211)2−, log β1i0 are sizeable, 11 (i = 1) and 19 (i = 2) and correspond to pLn = 14.7; the NdIII and YbIII bis(complexes) display NIR luminescence in MeOH and Tris buffer (pH 7.4).74 The hexadentate tripodal ligand (H612) encapsulates the YbIII ion into a coordinative environment completed by two water molecules, as determined at pH 7.4 from eqn (13) with τH2O = 0.37 and τD2O = 8.06 μs.74
Reaction of sodium benzoate derivatives (Scheme 3) with LnCl3 in THF and in presence of phenanthroline (phen) afforded well characterized dimeric compounds [Ln(13a)3(phen)]2 (LnIII = Er, Yb) in which the metal ions lie at a short distance (Er–Er = 406 pm).75 The ErIII emission is enhanced in co-crystals of ErIII and YbIII in the ratio 7 : 3 due to Yb-to-Er energy transfer, the efficiency of which reaches 55% as calculated from the lifetimes of Yb(2F7/2), 58.9 μs in the homodinuclear compound and 26.7 μs in the co-crystallized dimers, see eqn (15). The energy transfer process from a benzothiazole chromophore grafted onto a p-substituted benzoic acid to LnIII ions in [Ln(H13x)(tpy)] (x = b, c, LnIII = Nd, Er, Yb; tpy is terpyridine) depends both on the solvent (toluene or ethanol) and on the para substituent of the benzoic acid moiety: it is less efficient in ethanol (except for YbIII and x = b) and more efficient for x = b compared to x = c because the hydroxyphenyl derivative exhibits excited state intramolecular proton transfer. The estimated rate of energy transfer in methanol amounts to 5.72 × 109 s−1 for NdIII and 3.36 × 109 s−1 for ErIII but the sensitisation efficiencies remain very modest (0.2–0.9 × 10−4 for ErIII in toluene).76 Naphthalene substitution in H14 leads to a noticeable improvement of the latter parameter (2.5–2.8 × 10−4 in chlorobenzene).78 Among the other carboxylates tested,77,79–81,97 [Yb(15)3] displays a modest 0.7% overall quantum yield in aerated acetonitrile77 while QYbYb = 0.15% for [Yb(17)]− in water (pH 7.0);79 on the other hand, [Eu(15)3] is highly luminescent (QLEu = 60%).77
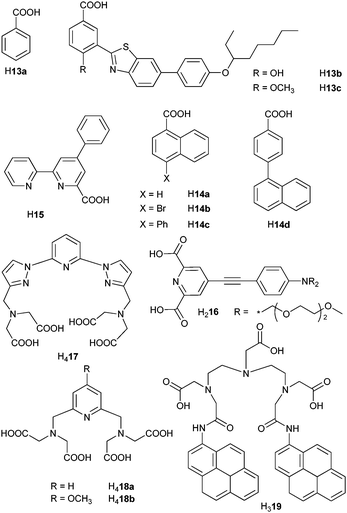 | ||
| Scheme 3 Carboxylic and polyaminocarboxylic acids.75,77–81,91,97 | ||
β-Diketonates remain much studied luminescent complexes. Ligand-to-metal energy transfer in [Er(tta)3(tpy)] has been proved to involve the excited triplet state of the ligand and since oxygen does not quench the NIR luminescence, its rate is faster than 107 s−1.105 In toluene solution, β-diketonate ternary complexes with phen82,83 and bpy83 have sizeable overall quantum yields, up to 1.28% for ligand H21b and YbIII (Scheme 4),83 but fail to yield highly luminescent complexes with NdIII and ErIII. Noteworthy features are the visible excitation of [Ln(20a)3(phen)] via an ILCT state82 and the observation of an appreciable quantum yield for the bis(hydrate) [Yb(21b)3(H2O)2]83 as also pointed out for [Nd(18)(H2O)2]−.80 Moreover, fluorination of the diketonate core allied with complexation by fluorinated phosphoryl ternary ligands in [Er(21e)3(OP(C6F5)3)2] results in a complex with a lifetime of 16.8 μs and a quantum yield larger by one order of magnitude with respect to [Er(21d)3(OP(C6F5)3)2], 0.17%. The radiative lifetime is up to 4 ms and the threshold intensity as low as 3 W cm−2, an encouraging result for designing planar waveguides pumped by LEDs.106
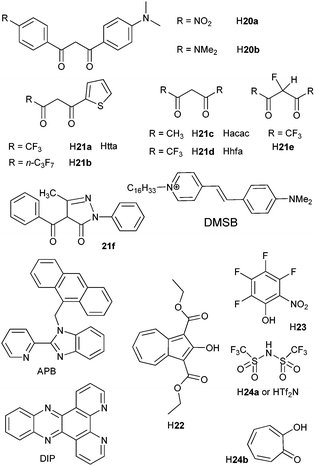 | ||
| Scheme 4 β-Diketones and miscellaneous ligands.82,83,85,86,98,99 | ||
Intense NIR luminescence from NdIII, ErIII and YbIII is detected when anionic tetrakis(diketonates) [Ln(21f)4]− are associated with the hemicyanine cationic chromophore DMSB (Scheme 4). Lifetimes of the excited states in acetonitrile are as long as 1.0, 3.2 and 46.4 μs for 4F3/2, 4I13/2 and 2F5/2, respectively.99
The azulene derivative H22 (Scheme 4) is a versatile ligand with a low-energy triplet state (14300 cm−1); it transfers energy onto several NIR-emitting ions, NdIII, ErIII, TmIII and YbIII, the quantum yield for the latter being up to 3.8% in acetonitrile.98 The CH-devoid ligand H23 forms crystalline Cs2[Ln(23)5·nEt2O] complexes which have particularly long lifetimes: τ = 22.9 (ErIII, n = 1) and 〈τ〉 = 159 μs (YbIII, n = 0.5).85 In a similar attempt, H24a was used as chelating unit with low-energy vibrations in conjunction with phen and DIP (dipyridophenazine) as antennae and the intrinsic quantum yields of [Yb(24a)3(phen)2] and [Yb(24a)3(DIP)] in dmso-d6, measured upon excitation at 940 nm, amount to 7.4 and 9.1%, respectively.86 Inserting lanthanide ions into nanoparticles is a way of decreasing radiationless deactivation: the Yb(2F5/2) lifetime increases from 12.4 μs in [Yb(24b)4]− (Scheme 4) to 68 μs for the ions inserted into the core of tropolonate-decorated NaY0.8Ln0.2F4 (LnIII = Nd, Yb) nanocrystals; a similar effect is observed for Nd(4F3/2).84,107
Unusual sensitisation of NIR luminescence has been demonstrated in [Ln(hfa)3(APB)] (LnIII = Nd, Er, Yb),108 in which the anthracene antenna (A) proceeds by electron transfer: [1A*–PB–Ln] → [A˙+–PB˙−–Ln] → [3A*–PB–Ln] → [A–PB–Ln*] (PB is a chelating unit of the APB, 2-(2-pyridyl)benzimidazole, Scheme 4).
Finally, a dendrimeric YbIII complex with ethylenediamine core and quinoline chromophores proved to be a useful NIR sensor of thiocyanate.109
3.2 Sensitisation by macrocyclic ligands
A neutral bioprobe combining visible-light excitation (488 nm), strong coordinating units, and NIR emission was obtained by grafting fluorescein on the cyclen (1,4,7,10-tetrazacyclododecane) framework (Scheme 5); as a result, luminescence of [Nd(25)] can be detected in time-resolved mode in water at pH 8 (τ = 2.3 μs).87 Quinoxaline is an alternative chromophore which has the advantage of sensitising both NdIII, EuIII and YbIII, but the lifetimes recorded for [Ln(26)]3+ in methanol remain short, the less coordinating amide groups allowing interaction with the solvent (q = 0.4 and 0.7 for NdIII and YbIII, respectively).88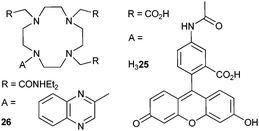 | ||
| Scheme 5 Cyclen derivatives.87,88 | ||
Most of the work with macrocyclic ligands has concentrated on porphyrinate derivatives. Indeed porphyrins have well defined absorptions (Q and Soret bands at ≈420 and 550–590 nm) and emission bands (650, 710 nm) and their low excited states are convenient for populating excited states of NIR-emitting LnIII ions. Interest in lanthanide porphyrinates is due to (i) their ability to serve both as charge-transport, electron hole recombinant, and NIR emitter materials in electroluminescent devices, (ii) their potentiality in photodynamic therapy of cancer, and (iii) their optical limiting properties. Optical limiters are transparent at normal light intensities and opaque to very bright light, henceforth they help avoiding damages caused to human eyes or optical components by sudden and intense laser pulses. Their characteristics are low ground state absorption and strong excited state absorption, which is the case for porphyrinates (see ref. 110 for a summary on lanthanide porphyrinates as optical limiters). For efficient NIR sensitisation, the coordination of the LnIII ion in monoporphyrinates has to be completed by a capping ligand to avoid solvent interaction; these ligands include β-diketonates and tripodal anions such as LOR− (cyclopentadienyl-tris(dialkylphosphito)cobaltate) or TpR− (hydridotris(pyrazolyl)borate). Tetraphenylporphyrins are the most used ligands and the influence of substitution on the phenyl or on the five-membered cycles has been investigated (Scheme 6).
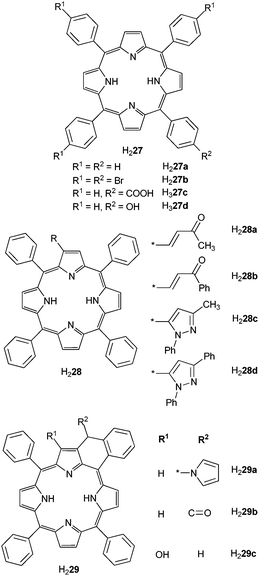 | ||
| Scheme 6 Tri- and tetra-phenylporphyrins.89–91 | ||
The monoporphyrinates [Ln(27)(H2O)3] react with cyanometallates K2M(CN)4 (MII = Ni, Pt) and K3Fe(CN)6 to form trimetallic species with a linear Ln–CN–M–CN–Ln core in which the NIR luminescence is partially quenched.89 Preparation of monoporphyrinate is often difficult and a new method has been proposed, starting from the free base and LnIII acetate in 1,2,4-trichlorobenzene. The obtained [Yb(27a)(O2CCH3)(MeOH)2] complex is a starting material for the preparation of highly functionalized porphyrinates. For instance, methanol can easily be replaced by a 4-methylphenanthroline chromophore and the corresponding eight-coordinated complex has QYbYb = 0.86%.90
Metalloporphyrinates are also viable chromophores for LnIII NIR luminescence. For instance, three [Pt(27c)]− and one terpyridine molecule assemble around an ErIII ion to form fully saturated nine-coordinate complex {Er[Pt(27c)]3(tpy)} with noticeable NIR emission. Modelling of the energy transfer process pointed to the Er(4F9/2) state receiving 83% of the energy transferred from the ligand triplet state, the other receiving level being Er(4I9/2).91 The quantum yield of the YbIII luminescence in [Yb(28)(acac)] and [Yb(29)(acac)] can be altered by varying the substituents on the porphyrins and [Yb(28b)(acac)] was found to be the best emissive complex, with QLYb = 0.47%.92 No quantum yields have been measured for Yb(LOR−) complexes with derivatised tetraphenylporphyrins (Scheme S1, ESI†), but Yb(2F5/2) lifetimes in the range 30–40 μs are reported in toluene93 and around 10 μs in water.94 On the other hand, YbIII chelates with N-confused porphyrins display short lifetimes in toluene (0.2–0.4 μs).95
3.3 Sensitisation by metal-containing Schiff bases
In addition to metalloporphyrins, authors have made use of Schiff-base complexes with zinc111 and cadmium112 to sensitise NIR luminescence. Quantitative data are very rare, except for [Ln(NO3)3Zn(32)] (LnIII = Nd, Yb, Scheme 7) in acetonitrile for which lifetimes are available: 1.23–1.27 and 13.40–15.89 μs for NdIII and YbIII derivatives, respectively.96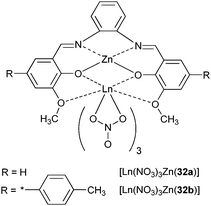 | ||
| Scheme 7 Zinc complexes with Schiff bases. | ||
3.4 Sensitisation by d–f energy transfer
The intricate energy transfer processes leading to population of 4fn excited states in which broad excited states from the ligands interact with narrow, long-lived metal-ion states render the design of suitable molecular systems very experimental. A more controlled approach is to take advantage of intermetallic communication between two (or more) metal ions inserted into polymetallic edifices so that directional energy transfer becomes feasible.Such a strategy has been mostly used for sensitising NIR emitting LnIII ions;1 long-lived 3MLCT states of d-transition metals (e.g. RuII, ReI, OsII, AuI, PtII, IrIII) can be excited by visible light and transfer efficiently their energy onto the 4fn manifolds, thus providing an effective pathway for energy migration within heterometallic complexes. The structures of the latter are shown on Scheme 8 and their photophysical properties are listed in Table S2 (ESI†).
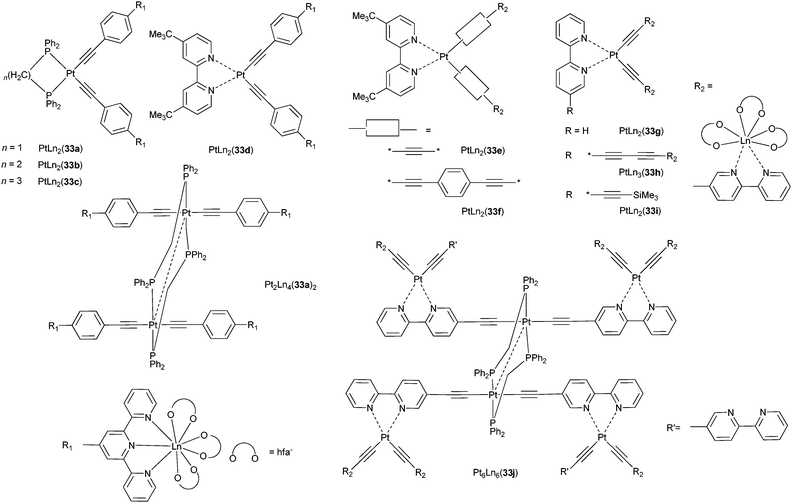 | ||
| Scheme 8 Pt–Ln complexes with bpy and tpy.113–117 | ||
The most studied systems are those containing PtII and their design is described in Fig. 3: PtII is chelated by a bidentate ligand and linked to two LnIII complexes, usually [Ln(hfa)3] (hfa is hexafluoroacetylacetonate) or [Ln(tta)3], via an electronic relay featuring alkyl and aromatic units.113–118 Alternatively, both PtII and LnIII ions can be electronically connected by a bis(bidentate) ligand such as bppz (2,3-bis(2-pyridyl)pyrazine).118
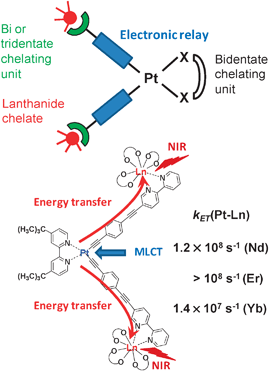 | ||
| Fig. 3 Design of a PtLn2 edifice for Pt–Ln transfer (top) and practical example of transfer rates in PtLn2(33f)115 (bottom). | ||
In the complexes PtxLn2x(33a)x (x = 1, 2; Scheme 8) the bidentate ligand is 1,2-bis(diphenylphosphino)methane (dppm), which is bridging in the complexes with x = 2. Excitation into the 3MLCT (x = 1) or 3MMLCT (x = 2) states of the PtII alkynyl chromophores occurs in a spectral range (360–500 nm) in which the model complex [Ln(hfa)3(alkynyl-tpy)] does not absorb. The Pt-to-Ln energy transfer is incomplete, as ascertained by the observation of an 3ILCT luminescence in solution, as well as a weak 3MMLCT emission. The rates of transfer in the hexanuclear complexes could be estimated from the lifetimes of the residual Pt–ligand phosphorescence: kET(NdIII), 6.07 × 107 s−1, is 286-fold larger than kET(YbIII), a fact attributed to the numerous NdIII acceptor levels in the energy range of the 3MMLCT state.113
Similar energy transfers from mixed 3MMLCT and 3ILCT states are operative in PtLn2(33b,c) (LnIII = Nd, Yb), in which dppm has been replaced by the longer molecules dppe (1,2-bis(diphenylphosphino)ethane) and dppp (1,2-bis(diphenylphosphino)propane).114 The rate of the Pt-to-Ln energy transfer is modulated by the nature and length of the electronic relay as deciphered in comparing PtLn2(33d,e,f) for which the bridge is (i) an acetylide and the coordinating unit bpy (33e) leading to a Pt–Ln distance of ≈8.6 Å, (ii) an acetylide–phenylene–tpy unit (33d, Pt–Ln ≈ 14.1 Å), or (iii) an acetylide–phenylene–acetylide–bpy moiety (33f, Pt–Ln ≈ 14.9 Å). As expected, the longer bridge results in a slower energy transfer, 1.4 × 107 s−1 for YbIII as compared to >108 s−1 for the two other complexes. Here again, the rate of transfer in the NdIII edifice with 33f is faster (1.24 × 108 s−1) than in the YbIII chelate and this is also true for ErIII (>108 s−1) which possesses several electronic levels in the range of the 3MLCT state.115
Analogous data are extracted from a comparison between PtLn2(33g) with Pt–Ln ≈ 8.4 Å and PtLn3(33h) with Pt–Ln ≈ 13.3 Å: energy migration across the longer Pt–bpyC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CCbpy–Ln array is less efficient, 2.82 × 106 s−1 versus >108 s−1, and kET(NdIII) ≈20 kET(YbIII) in the tetranuclear species.116 The corresponding transfer rates are somewhat reduced in the higher nuclearity complexes Pt6Ln6(33j): in contrast to a seemingly complete transfer from the Pt(bpy)(acetylide)2 chromophore evidenced in the PtLn2(33i) building blocks, the transfer from the Pt(bpy)(CCR)2 unit is incomplete and rate constants amount to 1.02 × 107 and 1.83 × 105 s−1 for NdIII and YbIII, respectively.117 Following a similar strategy as for the Pt–Ln assemblies, Chen et al. have come up with Au–Ln polymetallic complexes (Scheme S2, ESI†) which display ILCT, 3MMLCT and f–f emission (LnIII = Nd, Er, Yb).119
C–CCbpy–Ln array is less efficient, 2.82 × 106 s−1 versus >108 s−1, and kET(NdIII) ≈20 kET(YbIII) in the tetranuclear species.116 The corresponding transfer rates are somewhat reduced in the higher nuclearity complexes Pt6Ln6(33j): in contrast to a seemingly complete transfer from the Pt(bpy)(acetylide)2 chromophore evidenced in the PtLn2(33i) building blocks, the transfer from the Pt(bpy)(CCR)2 unit is incomplete and rate constants amount to 1.02 × 107 and 1.83 × 105 s−1 for NdIII and YbIII, respectively.117 Following a similar strategy as for the Pt–Ln assemblies, Chen et al. have come up with Au–Ln polymetallic complexes (Scheme S2, ESI†) which display ILCT, 3MMLCT and f–f emission (LnIII = Nd, Er, Yb).119
The bppz ditopic ligand provides two advantages over other bridging ligands: the association constants between the 5d-transition chromophore and the LnIIIβ-diketonates are usually large and the 5d-LnIII separation remains substantial (≈7.4 Å in ReGd(35b) for instance). It has been used to compare PtII- and ReI-chromophores (see Scheme 9). In the PtLn(33l) complexes, the Pt-to-Ln rate of energy transfer decreases from 109 s−1 (NdIII) to 1.4 × 108 s−1 (PrIII). Additionally, the 3MLCT luminescence is also quenched in the PtGd and PtLu adducts in which none of the LnIII ions has suitable accepting level. The effect is ascribed to an increase in vibrational quenching upon complex formation. This effect is very large in ReI adducts, so that the additional quenching by the NIR-luminescent LnIII ion does not affect further the lifetime of the 3MLCT level, thus no 5d–4f rate constant could be estimated.118 A Re(CO)3(bpy)(py) chromophore has also been appended to a cyclen framework to produce a dual imaging agent and YbIII luminescence was observed in ReYb(35c) upon energy transfer from the 3MLCT state.120
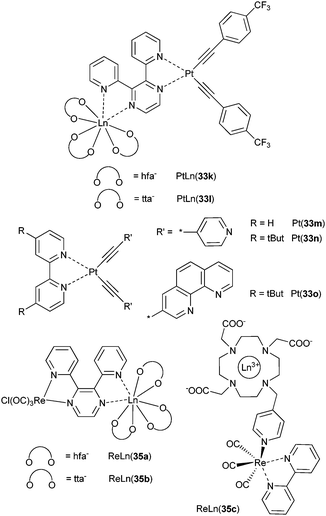 | ||
| Scheme 9 Bppz-, bpy- and cyclen-based complexes for studying Pt–Ln and Re–Ln energy transfers.118,120,121 | ||
The IrIII chromophores in the tetranuclear neutral complex Ir3Yb(36a) (Scheme 10) transfer energy onto Yb(2F5/2) with an efficiency of 65% relative to the efficacy of the energy transfer in the reference YbIII chelate (i.e. without the appended IrIII moieties) and the overall quantum yield is up to 0.7% in methylene chloride while the Yb(2F5/2) lifetime amounts to 17.7 μs.122 An analogous lifetime (22.1 μs) has been recorded for the Ir2Yb(36b) array and sensitised NdIII and ErIII luminescence could be observed with this ligand system.123
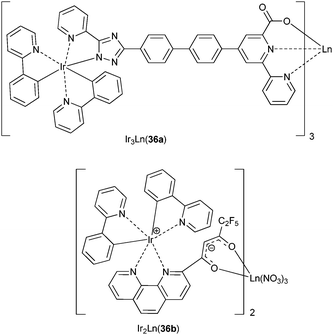 | ||
| Scheme 10 Ir–Ln complexes.122,123 | ||
Ruthenium and osmium [M(tBut2bpy)(bpym)]X2 (MII = Ru, Os; bpym = 2,2′-bipyrimidine; X = Cl, NCS, PF6) chromophores have low-energy MLCT states because of the presence of bpym and have consequently been tested as antennae for f-metal excitation. In fact, only RuLn(37a) and RuLn(37b) (see Scheme 11) with PF6− as counterion (3MLCT at ≈13500 cm−1) display LnIII-sensitised luminescence for NdIII and YbIII, respectively.124 An important issue is to determine which mechanism is operating in d–f energy transfer. The detailed study of MLn(37c–e) (MII = Ru, Os; LnIII = Nd, Er) by Ward et al. sheds light on this aspect.125 Rate constants for the energy transfer have been calculated from
| ket = τq−1−τu−1 | (18) |
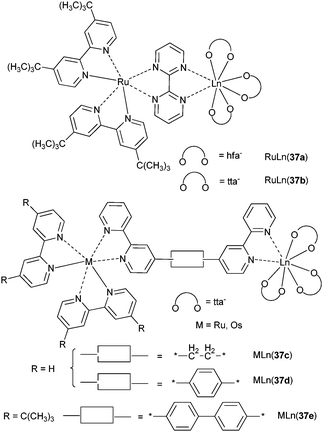 | ||
| Scheme 11 Ru–Ln and Os–Ln complexes.124,125 | ||
Analyzing the data in terms of Förster’s and Dexter’s mechanisms, the authors came to the conclusion that the first type of transfer is precluded because the critical distances extracted from the data modelled with this mechanism are small (≈8.5 Å in RuNd(37c)) and would correspond to an improbable, highly folded conformation of the chromophore. Therefore they concluded that energy is transferred via a Dexter’s super-exchange interaction mediated by the conjugated bridging ligands. Dexter’s mechanism requires direct orbital overlap and therefore is typically operating at short ranges. But when the donor and the acceptor are linked by conjugated bridging ligands, coupling occurs via the orbitals of the latter and Dexter-type energy transfer operates at longer distances, up to 20 Å in MLn(37d,e).125
The increase in transfer yield from 46% for RuNd(37a) to 91% for RuNd(37d) despite an increase in Ru–Nd separation illustrates the role of the electronic relay provided by the phenyl group. On the other hand, the efficiency decreases to 44% in RuNd(37e) because of the increased Ru–Nd distance. Ru–Er (63%) and Ru–Yb (52%) transfers are smaller in RuLn(37d) due to reduced availability of f-states. Os–Nd energy transfer is much smaller, 3 and 36% for OsNd(37c) and OsNd(37d), respectively.
Finally, large efficiency for Ru–Nd energy transfer (90%) has been reported for a cyclam-based dendrimer. (Scheme S3, ESI†).126
000 cm−1) and which transfer energy onto LnIII (LnIII = Pr, Nd, Er and Yb). In solid state, the transfer rate constants for the two coordination polymers decrease from 108 s−1 (NdIII) to 2–4 × 107 s−1 (PrIII, ErIII), and 1–2 × 106 s−1 (YbIII).121To promote Ru-to-Ln energy transfer, heterometallic polynuclear assemblies and coordination networks have been prepared from d-block cyanometallates and lanthanide salts. A rich variety of structures have been characterized, discrete {[Ru(CN)4(phen)]4[Ln(phen)2(H2O)]2}2−, honeycomb sheets, ladders, single-stranded chains, and chains of squares.127 Rates of Ru-to-Ln energy transfer in the honeycomb sheets range from 2.5 × 107 s−1 for YbIII to 5 × 108 s−1 for NdIII.
In cyanide-bridged 3d–4f discrete [Ln(dmf)4(H2O)3(μ-CN)Co(CN)5] species and {[Ln(dmf)4(H2O)2(μ-CN)2Cr(CN)4]}∞ infinite chains, the red emission of the [M(CN)6]3− moieties is completely quenched when LnIII = Nd, Yb, the quenching rate constants being >108 s−1.128
Rare infinite triple-stranded helical motifs {[ErAg3(dpa)3(H2O)]}∞ or 3D porous sandwich-type frameworks {[ErAg3(L)2(H2O)]}∞ (L = 4-hydroxypyridine-2,6-dicarboxylate) display ErIII emission in the NIR.129
3.6 Chalcogen-bound clusters and aryloxide complexes
As evidenced in the above description, non-radiative processes remain the main operative de-excitation paths in complexes with organic ligands, the intrinsic quantum yields being rarely larger than 1% for YbIII while values for NdIII and ErIII are orders of magnitude lower. Improving this situation requires designing edifices with much smaller vibrational energies and inorganic chalcogen-containing clusters with phonon frequencies in the range 450–700 cm−1 are good candidates. In 2005, Brennan et al. reported an amazing chalcogenide-bound ErIII cluster, (thf)14Er10S6Se12I6, with a record intrinsic quantum yield in the range 75–78%.130 Recent investigations of related NdIII and TmIII chalcogen-bound clusters and their mononuclear precursors,131–134 as well as mononuclear complexes with fluorinated phenoxide ligands OC6F5 (Table 5),135 point to intrinsic quantum yields up to 41% for [(py)24Nd28F68(SePh)16], that is only a factor two or so smaller than in Nd-doped silica materials,133 16% for [(dme)2Er(OC6F5)3],135 and 11% for [(dme)4Tm4(SPh)12].134 The intrinsic yields are calculated from eqn (3) with τrad estimated from eqn (2), (6), (7) and Judd–Ofelt parameters extracted from the absorption spectra; they can therefore be considered as being “experimental” data. In order to take into account energy migration among LnIII ions, back transfer and potential up-conversion processes, luminescence decays were modelled by Monte Carlo simulations, from which averaged τobs data were calculated.131 The [(py)24Nd28F68(SePh)16] cluster, prepared by reaction of Nd(SePh)3 with NH4F in pyridine and subsequently crystallized upon layering with hexane or slow cooling, is shown on Fig. 4. It contains a central set of four 12-coordinate NdIII ions and twenty four 8- or 9-coordinate NdIII ions located on the surface.![Fluoride cluster [(py)24Ln28F68(SePh)16] with C and H atoms removed for clarity. Reproduced with permission from ref. 133, © Wiley-VCH Verlag GmbH & Co 2008.](/image/article/2010/CS/b905604c/b905604c-f4.gif) | ||
| Fig. 4 Fluoride cluster [(py)24Ln28F68(SePh)16] with C and H atoms removed for clarity. Reproduced with permission from ref. 133, © Wiley-VCH Verlag GmbH & Co 2008. | ||
| Compound | Transition | λem/nm | τobs/μs | τrad/msb | QLnLn (%) | Ref. |
|---|---|---|---|---|---|---|
| a Excitation at 800 nm for Nd and Tm and at 980 nm for Er samples; dme = 1,2-dimethoxyethane, thf = tetrahydrofuran, py = pyridine.b Calculated from Judd–Ofelt theory (two decimal digits).c Calculated from the reported values of τobs and QLnLn. | ||||||
| (dme)2Nd(SC6F5)3 | 4F3/2→4I11/2 | 1071 | 110 | 1.40 | 7.9 | 131 |
| 4F3/2→4I13/2 | 1347 | 130 | 1.40 | 9.3 | 131 | |
| (dme)2Nd(OC6F5)3 | 4F3/2→4I11/2 | 1059 | 170 | 8.60 | 2.0 | 135 |
| (thf)8Nd8O2Se2(SePh)16 | 4F3/2→4I11/2 | 1078 | 186 | 1.19 | 15.7 | 131 |
| 4F3/2→4I9/2 | 927 | 78 | 1.19 | 6.6 | 131 | |
| [(py)18Nd12O6Se4(Se2)4(SePh)4(Se2Ph)2Hg2(SePh)4][(Hg(SePh)3]2 | 4F3/2→4I11/2 | 1082 | 141 | 1.16 | 12.2 | 132 |
| [(py)24Nd28F68(SePh)16] | 4F3/2→4I11/2 | 1076 | 303 | 12.4c | 41 | 133 |
| (dme)2Tm(SC6F5)3 | 3H4→3F4 | 1470 | 70 | 3.20 | 2.2 | 134 |
| (dme)2Tm(OC6F5)3 | 3H4→3F4 | 1458 | 145 | 7.52 | 1.9 | 135 |
| 3F4→3H6 | 1759 | 127 | 2.78 | 4.6 | 135 | |
| (dme)4Tm4(SPh)12 | 3H4→3F4 | 1470 | 111 | 1.00 | 11.1 | 134 |
| 3F4→3H6 | 1772 | 120 | 18.73 | 0.64 | 134 | |
| (thf)6Tm4Se9(SeC6F5)2 | 3H4→3F4 | 1470 | 80 | 1.66 | 4.8 | 134 |
| 3F4→3H6 | 1772 | 110 | 35.71 | 0.31 | 134 | |
| (dme)2Er(OC6F5)3 | 4I13/2→4I15/2 | 1550 | 1000 | 6.16 | 16.2 | 135 |
On the other hand, the self-assembled H10[Yb9(hesa)16(μ-O)10(NO3)] cluster which features an organic sensitiser (hesa is hexylsalicylate) is a less efficient light-converter compared to the inorganic clusters, with τ(2F5/2) = 0.6 and 2 μs for the two species present in methanol, that is an order of magnitude smaller than those listed for molecular compounds (Table 5).136
3.7 Extended structures
In an effort toward the design of practical applications, NIR-emitting molecules and compounds have been inserted into extended structures such as polymers, silica matrices,137 sol–gel glasses or mesoporous materials; the latter two classes of compounds are often referred to as inorganic-organic hybrid materials or metal–organic frameworks (MOFs).138,139 In view of the limitations discussed above, no frontier-breaking achievements have been recorded recently and only selected examples are presented below.Polymethylmethacrylate (PMMA) is a widespread polymer which easily forms thin films, thus it is often used for designing various optical materials. For instance, the DIP (dipyridophenazine) adduct of YbIII bis(perfluoromethanesulfonyl)imide salt, Yb(24a)3(H2O)8(DIP) (Scheme 4) displays overall quantum yields of 0.18 and 0.26% when inserted into PMMA or PMMA/DMSO thin films respectively; these figures are enhanced to 0.72–0.73% upon annealing the films at 80 °C during 1 h,140 but they remain modest (see Table 4 for comparison). Similarly, the TPPO (triphenylphosphine oxide) adduct of ErIII pentafluorobenzoate co-polymerized with PMMA displays a modest intrinsic yield of 0.12% (τrad from JO parameters = 12.66 ms).141
Tetraethoxysilane (TEOS) is a common starting material for the synthesis of doped glasses by sol–gel procedures,142 despite of the presence of O–H groups potentially detrimental for the Ln-centred NIR luminescence. Several complexes have been inserted in such xerogels. In [Er1.4Yb0.6(benzoate)6(phen)2]-doped glass75 for instance, τobs(4I13/2) of ErIII populated through intramolecular transfer from YbIII decreases to 12.7 μs from 19.3 μs in the bulk complex and this has been ascribed to O–H quenching; with τrad = 8.95 ms, calculated from JO parameters, the intrinsic quantum yield amounts to only 0.14%.143 An example of HoIII and TmIII luminescence in the same xerogel is provided by the β-diketonate complexes [Ln(tta)3(L)] with L = phen, bpy or TPPO. Similarly to the previous example, the Ho(5F5) and Tm(3H4) lifetimes are substantially reduced in the xerogel in comparison with the bulk complexes: for the TPPO adducts, τ(5F5) decreases from 24.3 to 18.3 ns and τ(3H4) from 43.3 to 25.4 ns. The radiative lifetime of Ho(5F5) calculated from the JO parameters amounts to 5.39 ms, therefore QHoHo = 3.4 × 10−4% in the xerogel.144 Complexes can also be covalently attached to the xerogel for instance through an adequately derivatised phen molecule (Fig. 5); in this way, NIR emission bands 4G5/2→6FJ (J = 5/2, 7/2, 9/2) of a SmIII fluorinated β-diketonate ternary complex were observed at 0.95, 1.03 and 1.18 μm. The bi-exponential decay corresponds to lifetimes of 4.6 (37%) and 20 (63%) μs.145 Luminescence from NdIII, ErIII and YbIII is sensitised as well146 and similar complexes were covalently attached to the mesoporous material MCM-41; in the latter τ(4G5/2) is substantially longer than in the covalently bound xerogel: 12 (26%) and 57 (74%) μs.147 On the other hand, doping simple 8-hydroxyquinolinates (LnIII = Nd, Er, Yb) into mesoporous SBA-15 results in shorter lifetimes than in the corresponding bulk complexes.
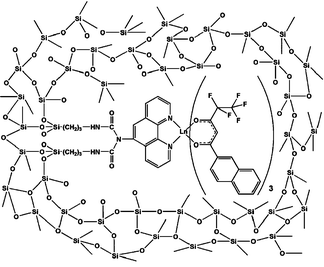 | ||
| Fig. 5 β-Diketonate ternary complex covalently linked to a xerogel. Reproduced with permission from ref. 145, © Royal Society of Chemistry 2009. | ||
4. Luminescent materials
In this section we present selected examples of various classes of materials with the emphasis on “soft” and electroluminescent materials. Polyoxometallates, which feature O → W or O → Mo LMCT states able to sensitise lanthanide luminescence have been reviewed recently and are not dealt with here.151,1524.1 Nanomaterials
The fascination for nanosciences is reflected in the continuously expanding number of papers dealing with one aspect or another of this new field of investigation. As far as rare earths are concerned, two major applications are attracting most of the attention, namely (i) visible- and/or NIR-emitting phosphors for lighting devices and solar cells, as well as (ii) up-converting nanoparticles for bio-analysis. The former are the subject of intense research most of it concentrating on microparticles. But recently, authors have doped a combination of ErIII, HoIII, or TmIII, together with YbIII as activator, in Y2O3,153,154 YAlO3,155 Gd2(MoO4)3,156 LuGaO,157 LaF3,158,159 nanoparticles or nanocrystals, as well as in SiO2–Al2O3–NaF–YF3 nanocomposites160 or in oxyfluoride nano-glasses.161 Upon photodiode excitation into the Yb(2F5/2) level, a combination of red, green and blue luminescence results in white light emission. Alternatively, red and green emission from EuIII and TbIII combined with blue emission of Ga2O3 nanoparticles also leads to white light emission.162 The same result is obtained by mixing the blue emission from the substrate of GdGaO nanoparticles163 or GdAl3(BO3)4164 nanorods with the yellow DyIII emission (activated by CeIII in the latter material).Harsh synthetic conditions such as the non-aqueous route proposed by Karmaoui et al.165 starting from lanthanide sesquioxides reacted at 275 °C in presence of 4-biphenylmethane result in lamellar nanostructures into which doped luminescent LnIII ions present an intense emission. In particular, the EuIII nanohybrid in Gd2O3 has a luminance value larger than the conventional Y2O3:EuIII phosphor.166 Colour-tuning has been obtained with LaF3 nanoparticles coated with a benzoic acid derivative and combining the emission of both the ligand and a LnIII ion (e.g. EuIII).167
Other evolving fields are (i) the production of luminescent security coatings168–170 and inks for which nanoparticles seem to be promising,171 as well as (ii) coatings for optical applications such as white light emitting diodes, low refractive index layers for antireflective devices, or silica binders in photocatalysis.172
Nanomaterials for bio-applications have been reviewed very recently,173,174 both from their design, synthesis, and application points of view, so that we do not discuss them here.
4.2 Ionic liquids and ionogels
Ionic liquids are salts with a low melting point (<100 °C) and several of them are liquid at room temperature (RTIL). The cation is generally an organic moiety (e.g. 1-alkyl-3-methylimidazolium, see Scheme 12) and the anion modulates the hydrophilicity of the liquid.175 RTILs find uses in catalysis,176 in the design of inorganic materials,177 in extraction processes,178 particularly within the context of nuclear fuel reprocessing,179 in electrolytes for batteries and photovoltaic devices,180 and in the electro-deposition of zero-valent lanthanide metals.181 Investigations of spectroscopic properties of LnIII ions in RTILs, described in Binnemans’ recent review,175 revealed that (i) the ionic liquid may participate in the sensitisation process, (ii) non-radiative deactivation processes are minimized compared to solid state or organic solutions, at least as long as the (otherwise highly hygroscopic) RTIL is reasonably anhydrous, (iii) therefore ionic liquids are attractive media for NIR luminescence, and (iv) lanthanide luminescence is helpful for investigating structural aspects of these solvents.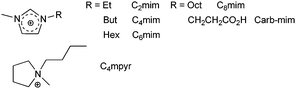 | ||
| Scheme 12 Cations of main RTILs used in spectroscopic studies. | ||
When compared to EuIII, the visible emission of SmIII is usually much less intense, at least in aqueous solvents. However, luminescence of this ion also occurs in the NIR portion of the electromagnetic spectrum, a duality that may be helpful for analytical sensors. The photophysics of three SmIII anionic tetrakis(β-diketonates) as well as of [Sm(dpa)3]3− has been elucidated in [C6mim]Tf2N. The imidazolium cation has been chosen as the cationic counterpart of the complexes, an elegant way of circumventing solubility problems.182 Moreover, while β-diketonates are known to be photo-degraded under UV excitation, their photostability is enhanced in ionic liquids.183 This is for instance the case of [C6mim][Sm(tta)4] the absorbance of which decreases by 2–6% in acetonitrile, ethanol or chloroform after 1h of irradiation at 340 nm, and to more than 10% in N,N-dimethylformamide after 10 min, while it remains practically constant in the RTIL or in methylene chloride. However, both lifetimes and quantum yields are smaller by ∼25–35% in the RTIL compared to acetonitrile; this is ascribed to quenching by O⋯H bonding between the imidazolium cations and the O atoms of the diketonate.182
Most of the latest investigations on photophysical properties in RTIL deal with EuIII. For instance, while the interaction of EuIII and CmIII with CuII leads to the quenching of the luminescence of both f ions in water, only EuIII emission is quenched in [C4mim]Tf2N (kSV = 1.54 × 106 M−1 s−1 compared to 1.20 × 104 M−1 s−1 in water), this points to different types of chemistry between the 4f and 5f elements in RTIL and opens perspectives for their separation.184 This conclusion is supported by the complexation of azide ion to EuIII in the same RTIL which results in both static and dynamic fast quenching of the 5D0 emission while the interaction kinetics is much slower for CmIII and AmIII; additionally, azide complexation seems to be stronger with EuIII triflate compared to perchlorate, an effect which may be traced back to a stronger electrostatic repulsion of N3− by the perchlorate anion.185 When water is added to [C4mim]Cl containing europium chloride, a whole range of mixed aqua–chloro complexes form, including the full aquated species when the molar ratio of water reaches 5, as shown by 5D0←7F0 excitation spectra and corresponding site-selectively excited emission spectra.186 Derivatization of the 3-position of the imidazolium cation by a carboxylic acid in Carb-mim gives rise to a so-called “task-specific” RTIL which can dissolve lanthanide oxides. Complexes such as [Eu(tta)3(phen)] are directly obtained by reacting Eu2O3, the β-diketone and phenanthroline in [Carb-mim]Br;187 such a procedure is easily extendable to polynuclear d-transition metal complexes.188 A series of low-melting europium-containing ionic liquids of composition (IL)x[Eu(Tf2N)3+x] (IL = C3mim, C4mim and x = 1; or IL = C4mpyr and x = 2) represent the first Ln-based ILs obtained without the use of any co-ligand. Melting points are between 10 and 53 °C and the crystal structures of the solids show the EuIII ion lying in a distorted tricapped trigonal prismatic, nine-coordinate environment. 5D0 lifetimes are around 1.7–1.9 ms for the C3,4mim compounds, somewhat shorter than in the solid state, due to enhanced deactivation by collisions.189
A facile procedure for the synthesis of LnF3 and Ln-doped YF3 (LnIII = Eu, Tb) rhombic-shaped nanoparticles involves precipitation from [Ln(acac)3] or Ln(OAc)3 solutions in ethanol containing [C4mim]BF4 as the source of fluoride. Varying the nature and concentration of the LnIII precursor allows one to tune the size of the particles from 130 to 340 nm (long axis) and addition of Ln nitrate (LnIII = Eu, Tb) results in doped luminescent particles.190 A large-scale synthesis of similar nanoparticles (LnIII = La, Ce, Pr, Nd, Sm, Eu and Er), although with different shapes, starts from lanthanide nitrates in ethanol, to which are added adequate amounts of the RTIL, [Cxmim]PF6 (x = 4, 8) or [C8mim]BF4. Eu-containing nanocrystals are quite luminescent and Tb-doped CeF3 nanocrystals display both CeIII and TbIII emission in the RTIL.191
Hybrid materials consisting in an ionic liquid confined inside nano-sized pores of a silica matrix, termed ionogels, feature the advantages of both the optical transparency of silica and the ionic conductivity of ionic liquids.192 Ionogels doped with [C6mim][Ln(tta)4] (LnIII = Nd, Sm, Eu, Tb, Ho, Er, Yb) and [choline]3[Tb(dpa)3] are highly luminescent inorganic-organic hybrids. This is ascertained by the lifetimes in the ionogel which are very comparable to those in the ionic liquid, for instance, 1.2 vs. 1.3 μs for NdIII, 86 vs. 81 μs for SmIII, 1.80 vs. 1.65 ms for TbIII, 1.6 vs. 1.9 μs for ErIII, and 16 versus 17 μs for YbIII. Thus the emission colour of these gels can easily be tuned by changing the emissive ion and their mechanical properties make them ideal luminophores.193
Some ionic liquids behave as liquid crystals as well, for instance [C12mim]Cl.194 The potential of the EuIII structural probe has been exploited to evidence the crystal to smectic A, Cr → SmA (0 °C) and SmA → I (100 °C) transitions of this RTIL doped with 5 mol% of europium nitrate by monitoring the intensity ratio of two components of the hypersensitive transition 5D0→7F2.195 The luminescent properties of adducts of lanthanide chlorides (LnIII = Eu, Tb) with phen and bpy have also been analyzed in this IL, particularly with respect to energy transfer mechanisms.196 Finally, highly luminescent ionic liquid crystalline phases can be engineered by coupling one or two mesogenic units (cholesterol or cyanobiphenyl) to an imidazolium cation itself associated with an anionic tetrakis(β-diketonate) such as [Eu(tta)4]−.197
4.3 Liquid crystals: lanthanidomesogens
The predictive electronic, optical and magnetic metal-centred properties of lanthanide ions make them particularly attractive for being inserted into switchable macroscopic materials responding to external electric and magnetic stimuli such as thermotropic liquid crystals (LC) which are then termed lanthanidomesogens.198 Interest for these materials dates back in the mid 1980s and an amazing variety of structures with specific properties have been synthesized,199 especially with respect to luminescent compounds,200 but rational synthesis only developed recently.201,202 Lately, efforts have essentially been focused on developing new ligands for d- or f-metallomesogens, e.g. imidazo[4,5-f]1,10-phenanthrolines,203 and for unravelling the thermodynamics of LC phase formation,204,205 their alignment under magnetic field,206 or their intimate structure. The field has been regularly reviewed.198–201 Only new developments involving luminescence data are therefore presented here.Luminescence is a highly sensitive analytical technique able to sense small differences generated by phase transitions, not only in the inner-coordination sphere but also in the outer environment of the emissive LnIII ion. Following the initial demonstration that indeed phase transitions in LC can be detected by variations in luminescence intensity and lifetime,204,207 or in relative band intensity of transitions to specific ligand-field (Stark) sub-levels,195 photoacoustic spectroscopy (PA) was tested for the same purpose in LC phases doped with EuIII, TbIII and HoIIIβ-diketonates.208 Although interpretation of data requires a relatively involved theoretical treatment, this technique proved to be helpful, principally when it comes to evaluating energy transfer efficiencies in luminescent lanthanidomesogens.209 Pressure-induced phase transitions between (i) the SmA phase of [Eu(bta)3(38)2] and its lamellar structure and (ii) the latter and an amorphous state (Scheme 13) are clearly evidenced by changes in the Raman spectrum and in the photophysical properties. For instance, the energy of the Eu(5D0→7F0) transition first decreases until the transition to the amorphous state is completed and then increases again. This blue shift is indicative of a smaller nephelauxetic effect, due to a weaker Eu–O(phenol) bond compared to Eu–O(phenolate), a consequence of the proton transfer.210
![Pressure-induced phase transition from lamellar (left) to amorphous (right) states in [Eu(bta)3(38)2].210](/image/article/2010/CS/b905604c/b905604c-s13.gif) | ||
| Scheme 13 Pressure-induced phase transition from lamellar (left) to amorphous (right) states in [Eu(bta)3(38)2].210 | ||
One of the rationale for synthesizing luminescent LC phases is the development of luminescent displays. For this purpose, luminescent lanthanide complexes are usually dissolved in nematic phases such as MBBA (N-(methoxybenzylidene)-4-butylaniline), 5CB (4-n-pentyl-4′-cyanobiphenyl), or 6CHBT (Scheme 14). When doped into the latter, the luminescent complexes [Ln(39)3(bpy)], with quantum yields of 5 (EuIII) and 19% (TbIII) in CHCl3, retain their characteristics. For instance the Eu(5D0) observed and radiative lifetimes amounts to 1.69 and 5.3 ms, as compared to 1.37 and 4.1 ms in chloroform. In addition, the intensities of the hypersensitive transition Eu(5D0→7F2) and of the Tb(5D4→7F5) transition sustain a ≈2-fold increase when the electric field is varied from 0 to ±30 V, reflecting the development of asymmetric environments in the LC phase. A sizeable hysteresis of this phenomenon is associated with the ion transport through the LC layer and to adsorption of residual charged molecules at the surface.211 Similarly, introduction of a small amount (0.1 wt%) of [Nd(tta)3(phen)] into a commercially available chiral nematic LC phase to which cholesteryl nonanoate is added results in a decrease in the NIR emission of NdIII (contrast ratio 3 : 1) and YbIII (1.5 : 1) when an AC voltage between 50 and 220 V is applied to the cell for inducing the cholesterol-to-nematic phase transition. This effect is due to the better scattering of the excitation light by the chiral nematic (cholesteric) phase.212
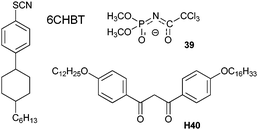 | ||
| Scheme 14 Ligands for synthesising luminescent liquid crystalline phases.211,213 | ||
Ligand H40 bears two mesogenic C12 and C16 chains grafted in the para positions of diphenylacetylacetone (Scheme 14) and the [Ln(40)3(phen)] complexes with LnIII = Eu, Er and Yb are luminescent at room temperature; all of the investigated complexes with the heavier lanthanides (LnIII = Eu–Dy, Er, Yb) exhibit a monotropic smectic A phase with I→SmA transition in the range 115–135 °C.213
4.4 Up-converting materials
Up-conversion is a non-linear, sequential absorption phenomenon of two or more photons via long-lived excited states, followed by emission of light with a shorter wavelength than the pump light source (red-to-blue conversion). Three types of absorption mechanisms have been recognised: excited state absorption (ESA) by a single ion, energy transfer up-conversion between two neighbouring ions (ETU), and photon avalanche (PA), a more complex process also involving two neighbouring ions. ETU is by far the most efficient process and is, in addition, independent of the pump power.214 These excitation mechanisms are different from so-called multiphoton excitation in which photons are absorbed simultaneously, and not sequentially, by the chromophore and which necessitate the use of powerful lasers operating in the femtosecond range (see section 5.5). Inorganic, usually lanthanide-containing, phosphors with up-conversion characteristics215 are used in light-emitting diodes, solar cells (see section 4.6), lasers, optical communications, data storage, security inks, and flat-panel displays.216 More recently, they have found applications in immunoassays58 and other biomedical analyses173 (see section 5.3). The materials are habitually used under the form of doped glasses,217 single crystals, or nanoparticles.218 White-light generation is an often sought-for property, displayed by tri-doped up-converting phosphors (Tm/Er/Yb)219 or glasses (Tm/Ho/Yb,220,221 Pr/Er/Yb222). The subject is vast and a bit too technical to fit into this overview; more information can be found in recent review articles.223,224An exciting new development is the preparation of coordination polymers from mixed carboxylic acids (oxalic acid, H2ox and 4,4′-oxybis(benzoic acid), H2oba) under hydrothermal conditions, [Ln(oba)(ox)0.5(H2O)2]∞, LnIII = Y, Er, Yb or co-doped Er, Yb into Y. Typical visible emission of ErIII in the latter is seen upon excitation by a continuous diode laser at 975 nm and both two- and three-photon processes have been evidenced.225 The high intensity of up-conversion, compared to the previously reported coordination polymer with pyrazine dicarboxylic acid226 or 1,4-benzenedicarboxylic acid227 is attributed to the presence of oxalate, which is devoid of high-energy vibrations. Some simple complexes, e.g. dipicolinates (LnIII = Nd, Er, Tm) also display up-conversion properties and have been characterized in an effort to develop luminescent biolabels.228
4.5 Electroluminescent materials
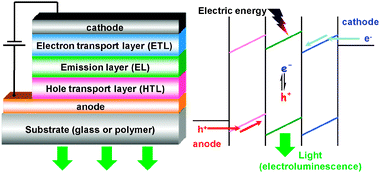 | ||
| Fig. 6 Typical set-up of an OLED and simplified scheme of injection, transport and recombination of charge carriers. | ||
To meet the first requirement the use of low work-function metals adjusted to the lowest unoccupied molecular orbital (LUMO) level of the next layer is necessary, as well as appropriate surface treatment of anodes (usually indium tin oxide, ITO). Charge recombination and emission are mostly determined by the nature of the emission layer material and can be reasonably optimized by a proper design while a balance of electrons and holes is difficult to achieve in a single layer. That is why usually additional layers are introduced into the OLED structure with high hole (HTL) or electron (ETL) mobility, their energy levels being tuned to the highest occupied molecular orbital (HOMO) or LUMO of the emissive layer, respectively (chemical formulae of some typical layers are drawn on Scheme 15). It is worth noting that encapsulation of OLEDs plays an important role in device stability. More detailed information about possible ways of OLED structure optimization, mechanisms of injection, transport, and recombination can be found in refs. 215 and 229–231.
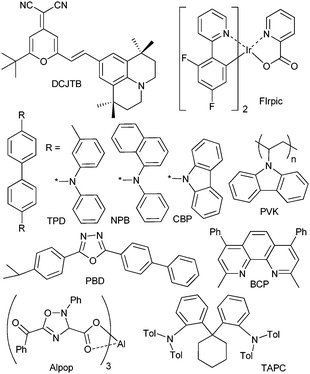 | ||
| Scheme 15 Examples of hosts and/or hole and electron transporting or blocking materials used in OLEDs. | ||
For describing precisely OLED operation, current–voltage and luminance (or brightness)–voltage characteristics have to be measured. The so called turn-on voltage (Ui), defined as the voltage at which brightness (L) exceeds 1 candela per square metre (cd m−2) can then be extracted from these data. Several quantitative parameters characterize OLED efficiency.
The luminous efficacy (ηv [lm W−1]) is the ratio of luminous flux (lumen) emitted by the source to the input electrical power (watt) and is determined by two factors:
| ηv = ηeK | (19) |
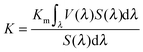 | (20) |
The external quantum efficiency (ηext) is the ratio between the number of photons emitted and the number of electrons injected to the device; it can be decomposed into the following terms:
| ηext = ηrecηELηextr | (21) |
When it comes to practical lighting applications of OLEDs, such an important characteristic as the colour rendering index (CRI) has to be estimated, because CRI shows how natural colours of the objects look under given illumination. Other parameters such as CIE (Commission Internationale de l’éclairage) coordinates and correlated colour temperature (CCT) should be determined as well.232
From the above discussion it is quite obvious that designing efficient OLEDs is a complex problem. Here, we concentrate mostly on the elaboration of emission layer materials which should meet the following criteria: (i) high photoluminescent efficiency; (ii) good carrier injection and transportability; (iii) large thermal and electrochemical stability; (iv) adequate solubility or volatility in order to obtain high-quality thin films. Different classes of compounds have been tested as electroluminescent materials for OLEDs, from conjugated polymers,57 small molecular compounds, either fluorescent229 or phosphorescent,233,234 to inorganic–organic hybrids (MOFs).230,235 Lanthanide complexes with organic ligands present two major advantages in view of their application in OLEDs: (i) quasi monochromatic emission and (ii) an efficiency ηrec (see eqn (21)) significantly improved in comparison with materials based on pure fluorescent compounds because both singlet (25%) and triplet (75%) excitons formed as a result of electron-hole recombination can be utilized for emission.233 A starting point for tailoring lanthanide-based materials for OLEDs was the demonstration in 1990 by Kido et al. of a characteristic green electroluminescence of the ternary complex terbium tris(acetylacetonate) with o-phenanthroline.236 The corresponding OLED with a simple structure ITO|TPD|Tb(acac)3(phen)|Al had a brightness of 7 cd m−2 at a current density of 0.4 mA cm−2. After this pioneer work many efforts have been devoted towards both the synthesis of new lanthanide-containing compounds with improved characteristics and the optimization of OLED architectures.18,58,215,233,234,237–240
Three main classes of anionic organic ligands have been under test for potential applications to visible-emitting OLEDs: β-diketonates and their derivatives,241–266 pyrazolonates,267–270 (Scheme 16 and 17) and carboxylates (Scheme 18).271–279 In addition, recent reports describe OLEDs made of a TbIII cluster with a benzamide derivative280 or of a CeIII complex with a polybenzimidazole-based tripodal ligand.281 Selected examples of visible-emitting monochromatic OLEDs reported recently are presented in Table 6 (those with the best performances have been selected).
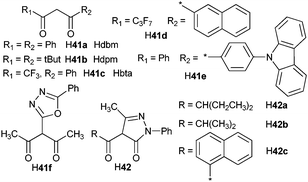 | ||
| Scheme 16 β-Diketones, pyrazolones, and their derivatives as ligands for lanthanide-based emissive layers in OLEDs. | ||
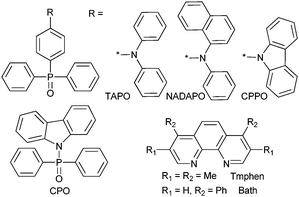 | ||
| Scheme 17 Ancillary ligands used for improving the transport properties of lanthanide-containing edifices in OLEDs. | ||
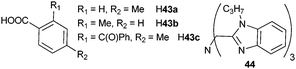 | ||
| Scheme 18 Carboxylic acids and polybenzimidazole-based ligand for emissive layers in OLEDs. | ||
| OLED structurea | Ui/V | Lmax/cd m−2 (U/V) | ηv/lm W−1 | ηext (%) | Ref. |
|---|---|---|---|---|---|
| a CuPc = copper phthalocyanine; MADN = 2-methyl-9,10-di(2-naphthyl)anthracene; PEDOT:PSS = poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate).b Brightness and luminous efficacy at 9.1 V, not maximum value. | |||||
| EuIII-emitting OLEDs | |||||
| ITO|TPD|Eu(dbm)2(41e)(Bath)–PBD (1 : 1 mol ratio)|BCP|Alq3|Mg0.9Ag0.1|Ag | n.a. | 2797 (14.0) | 0.27 | n.a. | 233 |
| ITO|TAPC|Eu(dpm)3–BCP (1 : 1)|BCP|Alq3|Mg0.9Ag0.1|Ag | n.a. | 2123 (10.0) | n.a. | ∼1.0 | 241 |
| ITO|TPD|Eu(dbm)3(Tmphen) (5.5%)–DCJTB (0.2%)–CBP|BCP|Alq3|LiF|Al | 4.6 | 2450 (20.4) | n.a. | n.a. | 242 |
| ITO|TPD|Eu(dbm)3(Tmphen) (5.5%)–CBP|BCP|Alq3|LiF|Al | 4.8 | 492 (18.6) | n.a. | n.a. | 242 |
| ITO|TPD|Eu(dbm)3(Tmphen)|BCP|Alq3|LiF|Al | 8.4 | 160 (20.2) | n.a. | n.a. | 242 |
| ITO|NPB|Eu(tta)3(CPPO)–CBP|BCP|Alq3|Mg0.9Ag0.1|Ag | 7.4 | 1271 (19.6) | 2.35 | 3.60 | 243 |
| ITO|NPB|Eu(tta)3(CPPO)|BCP|Alq3|Mg0.9Ag0.1|Ag | 8.0 | 852 (22.2) | 1.23 | 2.08 | 244 |
| ITO|NPB|Eu(tta)3(TAPO)|BCP|Alq3|Mg0.9Ag0.1|Ag | 4.4 | 1195 (19.6) | 3.62 | 3.2 | 244 |
| ITO|NPB|Eu(tta)3(NADAPO)|BCP|Alq3|Mg0.9Ag0.1|Ag | 4.8 | 1158 (18.0) | 3.69 | 3.71 | 244 |
| ITO|NPB|Eu(tta)3(CPO)|BCP|Alq3|Mg0.9Ag0.1|Ag | 8.4 | 399 (20.0) | 0.99 | 1.68 | 243 |
| ITO|PEDOT:PSS|Eu(43c)3(phen) (10 wt%)–DCJTB (3 wt%)–PVK|ZnS|LiF|Al | n.a. | 650 (20) | n.a. | n.a. | 272 |
| ITO|PEDOT:PSS|Eu(43c)3(phen) (10 wt%)–DCJTB (3 wt%)–PVK|BCP|Alq3|LiF|Al | n.a. | 420 (20) | n.a. | n.a. | 272 |
| ITO|NPB|Eu(42c)3(TPPO)(H2O) (15%)–CPB|BCP|Alq3|Mg0.9Ag0.1 | ∼12 | 247 (18.5) | n.a. | n.a. | 270 |
| TbIII-emitting OLEDs | |||||
| ITO|NPB|Tb(42a)3(TPPO)|BCP|Alq3|Mg0.9Ag0.1|Ag | 7.0 | 12000 (18) | 11.3 | n.a. | 268 |
| ITO|Tb(41a)3(H2O)2 (5 wt%)–PVK–PBD|Alpop|CsF|Al | 8.0 | 550 (20.0) | n.a. | 1.1 | 247 |
| ITO|Tb(43c)3(phen)–PVK (1 : 3 wt ratio)|ZnS|Al | n.a. | 326 (18.0) | n.a. | n.a. | 271 |
| ITO|Tb(43b)3(phen)–PVK (1 : 5 wt ratio)|Alq3|LiF|Al | 7.0 | 180 (15.0) | n.a. | n.a. | 273 |
| ITO|TbY(43a)6(phen)2–PVK (1 : 5 wt ratio)|Alq3|LiF|Al | ∼12 | 334 (23.0) | n.a. | n.a. | 274 |
| SmIII-emitting OLEDs | |||||
| ITO|PVK|Sm(bfa)3(phen)|PBD|Al | 10.0 | 135 (21.0) | n.a. | n.a. | 245 |
| ITO|TPD|Sm(41d)3(phen)|BCP|Alq3|LiF|Al | 10.4 | 82 (19.4) | 0.007 | n.a. | 246 |
| DyIII-emitting OLEDs | |||||
| ITO|CuPc|Dy(42b)3(TPPO)2|BCP|Alq3|LiF|Ala | ∼10 | 524 (19.0) | 0.16 | n.a. | 269 |
| Blue-emitting OLEDs | |||||
| ITO|PVK|Tm(acac)3(phen)|Al | n.a. | 6.0 (16.0) | n.a. | n.a. | 298 |
| ITO|NPB|[Ce(44)2](OTf)3 (5%)–MADN|Alq3|LiF|Alb | n.a. | 1.5 (9.1)b | 0.52c | n.a. | 281 |
For NIR-emitting lanthanide ions, mainly NdIII, ErIII and YbIII, the most efficient OLEDs are built from complexes with 8-hydroxyquinolinates,19,102,282–287β-diketonates288–293 or porphyrin-containing ligands.293–297 The maximum achieved values of external quantum efficiency are 7 × 10−3% for NdIII,290 10−4% for ErIII,290 and 0.1% for YbIII.294 However, to our knowledge, since the latest reviews appeared,18,58,233 no report on this subject has been published in periodical journals, so that they are not discussed further.
The majority of the investigations dealing with visible-emitting OLEDs utilize EuIII- or TbIII-containing materials as emission layers. However, some examples of OLEDs based on SmIIIβ-diketonates with o-phenanthroline, [Sm(bfa)3(phen)]245 and [Sm(41d)3(phen)],246 or DyIII pyrazolonate with triphenylphosphine oxide, [Dy(42b)3(TPPO)2]269 are documented, but their efficiencies are usually much lower. It is worth noting that the DyIII complex could be a candidate for white-emitting OLEDs because its CIE coordinates (0.35; 0.40) lie at the edge of the white region.269 There is one example of an OLED made of a ternary complex of TmIII acetylacetonate with phen and displaying a characteristic blue electroluminescence centred at 482 nm but in addition to low brightness, 6 cd m−2, the device exhibits an irreversible change of emission colour to orange when voltage bias values higher than 18 V are applied.298 The CeIII 5d → 4f emission in [Ce(44)2](OTf)3 is also promising in the design of blue-emitting OLEDs.281
The best red-emitting OLEDs contain EuIIIβ-diketonates: the maximum brightness (2797 cd m−2) and the highest external quantum yield (7.5%) with corresponding luminous efficacy of 10 lm W−1 are reached for [Eu(dbm)2(41e)(Bath)]233 and [Eu(dbm)3(Bath)],251 respectively (Bath is 4,7-diphenylphenanthroline). OLEDs with EuIII carboxylates as emissive layers have lower brightness values272 and devices made of EuIII pyrazolonate, e.g. [Eu(42c)3(TPPO)(H2O)],270 are even less efficient. On the other hand, Lmax as high as 12000 cd m−2 and luminous efficacy of 11.3 lm W−1 were recorded for a green-emitting OLED incorporating a TbIII pyrazolonate ternary complex with triphenylphosphine oxide.268 Conversely, the maximum brightness of an OLED containing the TbIII oxadiazole-functionalized β-diketonate [Tb(41a)3(H2O)2]247 is only 550 cd m−2 and the corresponding values for TbIII carboxylates do not exceed 350 cd m−2. 271,273,274
These results reflect, firstly, the suitability of the corresponding classes of ligands for sensitising either EuIII or TbIII photoluminescence: the triplet level of pyrazolonates is usually too energetic for an efficient energy transfer to EuIII,270 while that of β-diketonates with conjugated substituents has too low energy for TbIII.299 In addition, home-built solution processable OLEDs generally exhibit lower efficiencies than the ones obtained by vacuum deposition;258 this is for instance the case for lanthanide carboxylates because of their polymeric structure and thus low volatility.300
Several approaches towards the improvement of the emissive layer properties can be sketched:
• Doping of lanthanide compounds into polymers or small molecular compounds with high hole and/or electron mobility. This approach, along with an enhancement in the transportability of the emission layer, improves film-forming properties and leads to high-quality thin films. Moreover, doping EuIIIβ-diketonates into blue-emitting polymers such as poly(9,9-dioctylfluorene) and their derivatives is promising in the design of colour-tuneable OLEDs.301,302 The most efficient host material for the enhancement of EuIII electroluminescence appears to be 4,4′-bis(carbazol-9-yl)biphenyl (CBP, Scheme 15). For example, OLEDs relying on [Eu(dbm)3(Tmphen)]242 or [Eu(tta)3(CPPO)]243,244 doped into CBP have ∼3- and 1.5-fold increases in values of Lmax, respectively, in addition for the former device to a ∼2-fold decrease in turn-on voltage. Doping [Eu(dbm)3(DIP)] (Scheme 4) into Bath is beneficial as well: the ITO|TPD|Eu(dbm)3(DIP) (25–30%)–Bath|Bath|LiF|Al OLED has a brightness of 1320 cd m−2 and ηext∼2.5%.250 Co-doping the emission layer with assistant compounds such as the organic dye DCJTB242,272 or the IrIII complex FIrpic255 (Scheme 15) also helps optimizing OLED performances. However, one major drawback of the doping approach is the danger of phase separation during device operation.233
• Grafting hole and/or electron-transporting groups on anionic and/or ancillary ligands. Examples of modification of phen, bpy and anionic β-diketonates, mainly dibenzoylmethanates, can be found in ref. 233. Covalent attachment of lanthanide complexes to polymers is reviewed in ref. 18. According to recent publications, derivatives of triphenylphosphine oxide such as TAPO, NADAPO, CPPO, CPO (Scheme 17) appear to be promising neutral ligands for lanthanide-based electroluminescent materials.234,244 Grafting phen or bpy with calix[4]arene derivatives is also helpful.249 However, these strategies usually result in an increased molecular weight and thus in a loss of volatility. To overcome this disadvantage Adachi et al. have suggested a vacuum co-deposition method of volatile LnIII compound (e.g. [Eu(dpm)3]) and BCP, a material with good transport properties.241 The obtained OLED exhibits a brightness of 2123 cd m−2 and ηext∼1%, which positions it in the best three devices reported so far. The problem of low volatility and solubility is also real for polymeric lanthanide aromatic carboxylates which feature high photoluminescence quantum yields (TbIII benzoate has 100% efficiency).28 A possible way out could be a vacuum deposition method based on an exchange reaction between a volatile LnIIIβ-diketonate and benzoic acid.303
• Another way to boost OLED efficiency is the introduction of inorganic layers such as TiO2264 or ZnS271,272,278,279 which act as electron-transporting and hole-blocking compounds. Thus, at least 1.5-fold improvement in brightness was observed for OLEDs containing [Ln(43c)3(phen)] when BCP and Alq3 (EuIII) or Alq3 and LiF (TbIII) layers were substituted with ZnS.271,272 Enhancement of TbIII or EuIII electroluminescence also occurs upon partial substitution of LnIII with YIII in devices incorporating [Tb(43a)3(phen)2]274 or [Eu(tta)3(bpy)].265 For the latter complex, a similar effect happens when GdIII264 or TbIII266 are introduced.
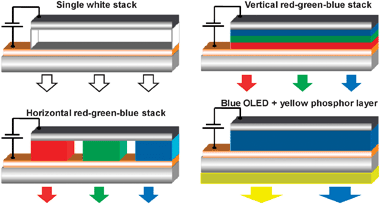 | ||
| Fig. 7 Simplified scheme of four general approaches for construction of white OLEDs.304–306 | ||
In the case of inorganic LEDs, two approaches have been privileged: (i) combination of individual red, green and blue LEDs and (ii) conversion of blue or near UV light to lower energy emission by means of yellow-emitting phosphors, or by combinations of blue, green and red phosphors coated on the semiconductor chip (Fig. 8).215,307
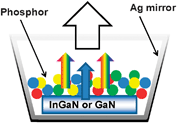 | ||
| Fig. 8 Principle of white-light generation in phosphor-coated LEDs.215 | ||
The number of publications dealing with white-emitting OLEDs relying on lanthanide coordination compounds remains quite scarce.308–312 Early works describe near-white OLEDs built by stacking green- and red-emitting layers containing TbIII or EuIII ternary β-diketonates, respectively.308,310 Alternatively, a layer doped with mixed [TbxEu1−x(acac)3(phen)] complexes is also adequate.309 Use of a dendritic EuIII complex grafted with carbazole units as a single dopant in a device with composition ITO|NPB|EuL–CBP|BCP|Mg0.9Ag0.1 resulted in white light emission with CIE coordinates (0.333; 0.348) at 16.2 V.311 The maximum brightness of this OLED reached 229 cd m−2 at 20.5 V with ηext = 1.1%. Another very efficient white-emitting OLED was built by combining red emission from a [Eu(tta)3(Tmphen)] (Scheme 17) layer co-doped with an IrIII phosphorescent complex in CBP host and blue emission from a layer consisting in the dye p-bis(p-N,N-diphenylaminostyryl) benzene.312 Once optimized, the device had a maximum brightness of 19000 cd m−2 at 17 V and luminous efficacy of 4.5 lm W−1, while CIE coordinates changed from (0.39; 0.32) at 6 V to (0.33; 0.30) at 14 V.
White light generation in a single layer is feasible with polymers incorporating EuIII, TbIII and DyIII complexes in appropriate ratios,313,314 by combining a blue-emitting DyIII polymer with a red-emitting RuII complex,315 or by relying on heterobimetallic EuIII–IrIII complexes.316 The design of OLEDs based on the latter complexes was patented in 2006.317
A combination of a blue-emitting OLED, containing either a fluorescent polymer or a phosphorescent IrIII complex, with down-conversion layers is efficient in generating white light too (Fig. 7).215,306 When the down-conversion layer consists in a nitridosilicate phosphor ([Sr,Ba,Ca]2Si5N8:Eu2+), cool white light with CIE coordinates (0.26; 0.40) and luminous efficacy of 25 lm W−1 is emitted.306 Following the same idea, a white OLED with CCT = 5000 K and CRI = 88% was built from a combination of perylene orange, red dyes and the Y(Gd)Al5O12:Ce3+ phosphor.215 This is one of several examples of white-light generation relying on inorganic phosphors. Garnets, silicates, sulfides, oxynitrides or nitrides doped with an intricate mix of lanthanides are commonly found in so-called phosphor-converted LEDs (pc-LEDs, Fig. 8).307,318–324 According to the working principle of these devices, the phosphors should possess strong absorption in the blue- or near UV and have high photochemical stability. In fact, pc-LEDs are already on the market, and further developments in this area, not taking into account technological aspects such as light out-coupling efficiency and LED package gain, should be targeted at improving the colour quality, i.e. CTT in the range 2500–6500 K.215 Recent developments deal with M2Si5N8:Eu2+ (MII = Sr, Ba)325 or SrAlSi4N7:Eu2+.326 Another approach is the use of near-UV semiconductor chips instead of blue-emitting ones, with a combination of red, green and blue phosphors.215 One of the challenging doping ions is Eu3+, however: weak absorption is a real problem. To overcome this, some recent achievements use ternary EuIIIβ-diketonates with phen,20,327 or CdO-Al2O3-3SiO2:Eu3+ which displays an emission ∼3-fold more intense than the well-known commercial Y2O3:Eu3+ phosphor,328 or Ca3Ln2W2O12:Eu3+ (Ln = La, Gd, Y).329 In parallel, generation of white light from single-emitting inorganic phosphors is developing, for instance, Eu2+-activated (Ba, Sr)13−xAl22−2xSi10+2xO66330 or incorporation of Si4+–N3− into Ce3+-doped garnets.331
The growing interest in Ln-doped polymeric optical waveguides can be traced back from their better flexibility and a large core diameter which enable efficient coupling.332,333 However, insolubility of lanthanide inorganic salts in organic systems creates problems. One of the feasible solutions is the use of lanthanide complexes with organic ligands, for example, ternary ErIII dibenzoylmethanate with phen,346,347 ErIII nitrate with derivatives of triphenylphosphine oxide343 or ErIII/YbIII co-doped tris(4-pentylbenzoate) with phen.345 An embedded 15-mm long waveguide based on the latter complex with polymer SU-8 as a cladding layer showed an optical gain of 5.2 dB at 1550 nm for a signal power of 0.3 mW. When PMMA-glycidylmethacrylate was used, the optical gain reached 6.5 dB in a 12-mm long waveguide with an input power of 1 mW. Another way out takes advantage of nanosystems such as ErIII/YbIII co-doped lanthanum fluoride351,352 or phosphate353 nanoparticles modified with organic ligands or dispersed in sol–gel. In addition, the feasibility of using Y2O3:ErIII nanotubes for the same purpose has been established.354 A 22-mm long waveguide based on a hybrid inorganic–organic system with LaF3:ErIII,YbIII nanoparticles demonstrated a relative optical gain of 5 dB at 1535 nm.352 Detailed description about problems dealing with NIR polymer optical fibres and their possible solutions, as well as earlier achievements in this area are summarized in ref. 58.
Recent examples describing improvements in waveguide performance are to be mentioned. For instance, (i) a new pumping scheme at 477 nm for Si-nanocluster sensitised ErIII-doped waveguide amplifiers has been suggested355 and (ii) metal-catalyzed growth via self-organized ErIII or AuI:ErIII implantation was exploited to produce SiOx nanowires on silicon surfaces.356 These nanostructures exhibit characteristic ErIII photoluminescence at 1530 nm with a remarkably long lifetime of 24 ms and could find wide application in optical systems.
Some researchers have also demonstrated the feasibility of optical amplification in the visible range in polymer waveguides doped with ternary EuIIIβ-diketonates with phen346,347 or LnIII–Al nanoclusters (Ln = Eu, Tb).357–359 For the latter system the highest optical gain coefficients were estimated to be 0.56 and 1.28 mm−1 for TbIII–Al and EuIII–Al nanoclusters, respectively.
4.6 Solar energy conversion
To meet the steadily rising energy requirements of modern civilization and take into account the ongoing energy crisis and global warming problems the quest for harvesting renewable energy sources such as sunlight is becoming a vital issue. A theory based on quantum electrodynamics has been developed which establishes correlations between structures and fundamental mechanisms operating in energy harvesting systems, with emphasis on dendrimers and lanthanide-doped solid compounds.360Sunlight can be directly converted into electricity in so-called solar (SC) or photovoltaic (PV) cells. The most widely used inorganic semiconductor for SC is silicon (Si, Eg = 1.12 eV).361 The two major loss mechanisms originating from the discrete band-gap structure of semiconductors have to be overcome for any single-junction SC, to enhance device efficiency: (i) lattice thermalisation and (ii) transparency to sub-bandgap losses. One of possible ways out is photon conversion through: (i) down-shifting, (ii) down-conversion (known also as quantum cutting), or (iii) up-conversion mechanisms. The main principle of this concept is the conversion of the solar spectrum via luminescence for matching the absorption properties of the semiconductor device. The up-conversion layer is usually located on the rear reflector to capture sub-bandgap light, while the down-conversion layer is deposited on top of the solar cell to convert each high-energy photon into multiple low-energy photons (Fig. 9). Once optimised both up- and down-conversion layers can be combined in one solar cell leading to increased efficiency.362–366
 | ||
| Fig. 9 Simplified schemes of bifacial and monofacial solar cells with up- and down-converting layers, respectively. | ||
Down-shifting is similar to down-conversion, except that the luminescence quantum efficiency of this process is less than unity. However, it can still result in significant SC efficiency improvement.367 Interest in lanthanide-based materials for photon converting layers in SC again stems from their unique luminescent properties with emission from UV to NIR spectral ranges (Section 2), while the main drawback to overcome, i.e. low absorption coefficients, remains. In earlier studies materials incorporating EuIII or TbIII nitrates with phen or bpy were tested as down-shifting layers;368–369 presently, however, more attention is paid to lanthanide-containing inorganic phosphors.217,370–379 Recent examples are down-shifting layers consisting in YbIII-doped glass ceramics containing Ba2TiSi2O8 nanocrystals.370 In this material the conversion of near-UV radiation (250–350 nm) into NIR emission of YbIII (970–1100 nm) occurs through energy transfer from the silicon–oxygen-related defects in SiO4 units to the 2F5/2 level, thus overcoming the problem of low absorptivity.
Down-conversion in phosphors can be achieved via cooperative energy transfer when the inorganic matrix is co-doped with appropriate lanthanide ions.363 In transparent glass ceramics with embedded PrIII/YbIII:β-YF3 nanocrystals an efficient YbIII NIR luminescence with optimum quantum efficiency close to 200% is achieved.371 Following the same principle, combination of YbIII with TbIII in polyborate La0.99−xYbxBaB9O16:Tb0.01372 or doped into Zn2SiO4 thin films,377 as well as TmIII/YbIII co-doping into YPO4373 or oxyfluoride glass ceramics containing LaF3 nanocrystals376 also lead to conversion of UV-blue emission into NIR with quantum efficiency higher than 150%. Benefiting from the strong 5d ← 4f absorption of CeIII in the range 250–380 nm, high cooperative energy transfer to YbIII with efficiency of 74% and corresponding quantum efficiency of 174% was measured in CeIII/YbIII co-doped borate glasses.374 An interesting example of three-photon infrared quantum-cutting material is Er0.3Gd0.7VO4: when the 2H11/2 level is excited at 523.5 nm, NIR emission at 1532.5 nm (4I13/2→4I15/2 transition) is stimulated, the estimated efficiency being 178.6%.375
Up-converting materials for solar cells often contain ErIII-doped compounds364,365 which convert absorbed light in the range 1480–1580 nm to emission at ∼545, 665, 800 and 980 nm, as recently demonstrated with the Gd2(MoO4)3:ErIII nanophosphor.379 Moreover, up-conversion of 1170 nm excitation into 650- and 910-nm emission in HoIII-doped oxyfluoride nanophase glass ceramics has been recently achieved.378 This material, in combination with Er-doped up-converting phosphors, could increase absorption of the solar spectrum and the efficiency of silicon SCs.
Luminescent solar concentrators which are able to concentrate sunlight onto a small area represent an alternative for boosting the efficiency of silicon-based SCs. An overview of potential compounds for this purpose, including lanthanide-based systems is given in ref. 380.
Apart from energy problems, optimization of sunlight conversion efficiency is also welcome in the photosynthesis of plants, in order to improve crop yield. Usually, plant pigments such as chlorophyll, carotenes and xanthophylls, harvest primarily the blue and red parts of sunlight. Thus, the efficiency of photosynthesis can be optimized if the unused portions of the solar spectrum are converted into blue and red light. Following this, the concept of dual-excitation dual-emission phosphors (DE2) has been suggested.381 For instance, Ca0.6Sr0.4S:0.005Cu+, 0.001Eu2+ has an optimal efficiency for converting ultraviolet into blue and green into red. In this way, more than 20% increase in cabbage and pimento production was achieved by using plastic films covered with the suggested phosphor.
5. Applications in bio-sciences
The biological chemistry of rare earths has roots in the 19th century when cerium oxalate was widely prescribed as an antiemetic drug to cure sickness due to pregnancy. Moreover, the in vitro antimicrobial properties of several lanthanide complexes stimulated clinical trials in the treatment of tuberculosis and leprosy although their impact was minimal.382 Lanthanide ions have also anticoagulant properties but their main therapeutic applications lie presently in the radioactive treatment of cancers. The toxicity of free lanthanide ions vary considerably, IC50 being reported between 1 and 1000 μM for GdIII for instance,383–385 while under given experimental conditions they even enhance cell proliferation.386 Gadolinium chelates are ubiquitous contrast agents for magnetic resonance imaging387–389 and despite some recently reported problems with one class of compounds390 they are well tolerated and considered as being harmless.Since the mid-1970s, time-resolved capability of lanthanide luminescent bioprobes (LLBs)12 and the availability of adequate bioconjugation protocols10 allowed the development of highly sensitive immunoassays,391,392 protein staining assays,393 nucleic acid analyses,55 or the determination of enzyme activities.56 More recently, time-resolved microscopy11,394 has also benefited from the intrinsic advantages of LLBs and cell-penetrating optical probes allow not only the visualisation of live cells14 but also carrying out targeted analyses of key biochemical metabolites.395–397 Extension to immunocytochemical analyses and immunohistochemical detection of cancerous tissues are now at hand.13,398
The field is measureless and hundreds of articles are published yearly in photophysical, chemical, biochemical, pharmaceutical, and medicinal journals. In the following we therefore restrict ourselves to highlighting only a few characteristics of these analyses, particularly those related to fundamental aspects of photophysics and to the latest development in time-resolved imaging; the reader is referred to the abundant reviews published recently on these subjects for further information.55,56,104,392,393,395,396,399–402
5.1 General considerations
A LLB may be used in various ways taking advantage of different properties of the LnIII ion. Initial applications have dealt with (i) simple substitution of CaII or ZnII by LnIII in proteins to gain information on the composition of metal-binding sites (e.g. hydration number) or metal-to-metal and/or metal-to-chromophore distances by Förster resonant energy transfer experiments (FRET),403 and (ii) titration of a bio-compound with LnIII salts to determine the number of metal-binding sites.45,404 Nowadays more astute techniques are at hand based on carefully tailored chelates. The latter are either used directly, e.g. in analytical responsive probes and simple cellular imaging experiments without specific targeting, or conjugated to a protein or to an antibody which specifically recognizes a targeted biomolecule.Requirements for efficient LLBs are challenging: (i) water solubility, (ii) large thermodynamic stability, (iii) kinetic inertness, (iv) intense absorption above 330 nm, (v) efficient energy transfer onto the LnIII ion, (vi) coordination cavity minimizing non-radiative deactivations, (vii) long excited-state lifetime, (viii) when relevant, ability to conjugate with bioactive molecules while retaining its photophysical properties and not altering the bio-affinity of the host.
Generally speaking a LLB can be used in a direct way or indirect way. In the first case, the LLB luminescence (or its enhancement, or its quenching) is simply detected, preferably in time-resolved (TR) mode, after specific interaction with the analyte. In turn, these responsive systems can be applied directly, the chelate interacting itself with the analyte, such as in the cases of e.g. oxygen, pH, ATP, or anion sensors, or indirectly, after bioconjugation to a suitable antibody or peptide405 which performs the recognition of the analyte via a specific biochemical reaction (Fig. 10, top). Interaction of the probe with the analyte often relies on the strong and specific avidin (resp. streptavidin)—biotin interaction (Kas≈1015 M−1); to increase the number of LLB per analyte and thus to boost the sensitivity, the LLBs are sometimes additionally attached to bovine or human serum albumin (BSA, or HSA); alternatively, two LnIII ions are grafted onto the same peptide, yielding double lanthanide binding tags (dlbt).406
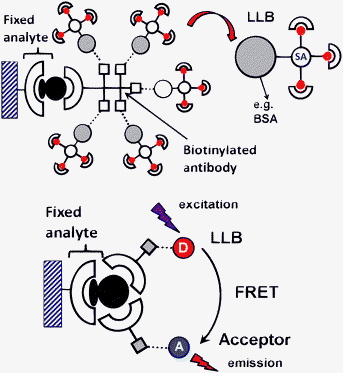 | ||
| Fig. 10 Direct (top) and indirect (bottom) use of a LLB. | ||
Bioconjugation is most commonly performed in solution by allowing a chelate with an activated group to react with a functional group of a bioactive molecule. Derivatization of synthetic biomolecules can be conveniently achieved in solid phase with standard peptide and nucleotide synthesizers, but solution-phase labelling of proteins is also common. In all cases, the labelling reaction must be as selective and effective as possible. The various protocols for efficient covalent bioconjugation of luminescent chelates have been reviewed recently.10
In the second indirect method, the excitation energy is transferred via the LnIII ion onto an organic acceptor by FRET (Fig. 10, bottom). FRET occurs when the donor (D) and the acceptor (A) lie at distances larger than 40 pm, the corresponding mechanism being dipole–dipolar (through space) and its yield is defined by eqn (15) and (16). FRET eliminates the need for washing unreacted reagents since the transfer only occurs when the two entities, the LLB donor and the organic acceptor, are linked together via some kind of conjugation. The essential point is that the organic fluorophore emits light with an apparent lifetime equal to the lifetime of the donor, its excited state being populated via the LnIII ion. LLBs perform better than organic dyes in that the distance at which the transfer may be detected is larger (typically up to 800–1000 pm, as compared to 500–600 pm for organic dyes). Applications of FRET are far reaching, ranging from homogeneous immunoassays to DNA hybridization assays, or analyte imaging in cells (such as calcium or phosphorylated molecules).407
5.2 Responsive luminescent probes
One emerging area of investigation is the determination of enzyme activity such as glucose oxidase, catalase and peroxidase. Enzymes catalyzing phosphorylation or de-phosphorylation reactions are also under investigation in view of their high relevance to drug targets: protein kinases, adenyl cyclases, phosphodiesterases, phosphatases, ATPases, to name a few.As an example, tetracycline (Scheme 19) complexes with 1 : 1 or 3 : 1 Eu : L stoichiometry are among the most convenient EuIII probes for quantifying phosphate, ATP and hydrogen peroxide. For instance, [Eu(45a)] luminescence increases 15-fold upon interaction with H2O2 which is the product of the activity of almost all oxidases and therefore this enhancement may be used to quantify the latter. On the other hand the luminescence of [Eu(45a)] is quenched by the presence of phosphate, leading to an assay of this anion on microliter plates with a detection limit of 3 μmol L−1.408 Citrate and oxytetracycline (45b) can by conveniently probed by EuIII in complex media such as drug tablets or serum with detection limits in the range 0.1–0.2 μg mL−1.409 Alkaline phosphatase enhances the luminescence of [Eu3(45a)] and the corresponding time-resolved luminescence assay has a detection limit of 4 μmol L−1.410 Applications of europium tetracycline complexes in assays for phosphate, malate, lecithin, heparin, HSA, citrate, low density lipoproteins (LDL), and so on are detailed in the review by Courrol and Samad,400 while more EuIII probes are described and compared in Spangler et al.56 Nucleotides and phosphate probes often rely on TbIII luminescence,411 for instance the 4.5 : 1 complex of TbIII with norfloxacin (46) exhibits a five-fold increase in luminescence upon interaction with ATP.
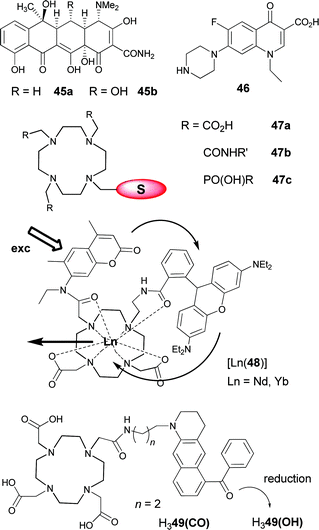 | ||
| Scheme 19 Ligands used for Ln luminescent probes. From top to bottom: tetracycline (left) and norflaxin (right); cyclen core structures (S = sensitising unit); cascade energy transfer in a coumarin-rhodamine cyclen complex; ligands for reporters of redox metabolism. | ||
Many probes for DNA analysis have been proposed,55,412,413 some of them exploiting the time-resolved capability of the LnIII ions leading to detection limits up to 50-fold larger than those reached with the organic dye FITC,414–416 their propensity to be easily entrapped into nanoparticles,402 their up-conversion ability,417 or their NIR luminescence.418 Examples of the latter are the cyclen complexes [Ln(48)] (LnIII = Nd, Yb; Scheme 19): the cascade energy transfers which populates the LnIII excited states is interrupted when the coumarin substituent interacts with ds-DNA, making the complexes DNA probes; however, no detection limit is given.418
A series of successful lanthanide responsive probes for quantifying analytes of biological relevance are based on the cyclen framework (Scheme 19).52,395,396,419 The latter usually features three coordinating units (carboxylates, substituted amides, phosphinates) and one sensitising pendant which often simultaneously binds the central metal ion. Signalling is achieved by enhancing or quenching the metal-centred luminescence. Typical analytes which can be quantified are pH, p(O2) and various anions (halides, HPO42−, SO42−, acetate, oxalate, malonate, succinate, lactate, citrate)420–422 or amino acids.52 One of the most prominent results reported recently is the ratiometric method for the determination of lactate or citrate in samples of human serum, urine or prostate fluids for diagnosis of prostate cancer.423 Luminescent reporter substrates for redox metabolism, such as [Nd(49(CO))], see their metal-centred NIR luminescence switched on upon reduction of the ketone group into alcohol, leading to [Nd(49(OH))]; these sensors are able to detect human aldo-ketoreductases, which are involved in steroid hormone metabolism and stress response mechanisms.424
Alternate substrates have been used for similar purposes, for instance chiral tripodal ligands for the analysis of anions,425 polyaminocarboxylates immobilized on sensory chips for detection and separation of histidine-tagged ubiquitin proteins,426 transferrin and lactoferrin complexes of TbIII for pH sensing,427 or dendrimeric complexes for NIR detection of anions.109
5.3 Up-converting phosphors
Among recent developments in lanthanide-aided bio-analysis, nanoparticles,174 quantum dots,54 and up-converting phosphors (UCPs),417,428,429 attract much attention. UCPs are rare-earth doped ceramic-type materials (oxides, oxysulfides, fluorides, oxyfluorides for instance) which convert red light into visible light. They appear as microspheres or nanospheres, were proposed for bioassays at the beginning of the 1990s and represent now a well established class of bio-probes. They bear many advantages, such as low sensitivity to photobleaching, high optical sensitivity due to the presence of many emitting centres per particle, capability for multiplex analyses in view of the possibility of doping different LnIII ions, as well as cheap diode laser excitation (e.g. at 980 nm). Most of the UCPs feature ErIII as the emitter since it has two emission lines in the green (540 nm) and in the red (654 nm), along with YbIII as activator, but other pairs such as TmIII/HoIII can also be envisaged.The synthesis of the microspheres involves several steps. For instance for (Y0.86Yb0.08Er0.06)2OS,430 the lanthanide salts are first mixed in stoichiometric ratios and the particles are precipitated by added urea before being thermally converted to oxides and treated with H2S. Activation of the particles follows by fluidization under argon at elevated temperature. Aggregation is minimized and monodispersed spheres with average diameter of 0.4 μm are obtained. Homogeneity of the sample is important to ensure constancy in the emission intensity.431 Secondly, the hydrophilicity of the microspheres is increased by coating them with a 20–30 nm thick layer of silica. Finally the surface is functionalized by covalent linkage of the adequate antibody (Fig. 11, top) using known bioconjugation methods. Another important experimental point is to ensure that the excitation light from the diode laser is focused tightly. A weakly focused beam may lead to 10-fold smaller emission intensities.432
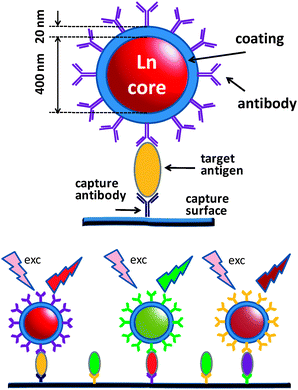 | ||
| Fig. 11 (Top) Coated and derivatised up-converting phosphor (UCP) microsphere for targeting a specific antigen. (Bottom) Scheme of a multiplex assay. | ||
The microspheres are excellent reporters for the detection of antigens in tissue sections or cell membranes; presently they are often obtained as nano-objects, with diameters of 2–20 nm. They have been initially tested on the prostate-specific antigen in tissues and the CD4 membrane antigen on human lymphocytes433 and then used in several homogeneous immunoassays, e.g. for estradiol.434 Up-converting phosphors are also able to transfer energy onto “tandem dyes” comprised of an absorber (B-phycoerythrin) and an emitter (an Alexa Fluor 680 dye); in this way, low-power NIR excitation results in specific dye-centred visible emission devoid of autofluorescence, even without time-resolved detection, thus allowing very sensitive homogeneous immunoassays.435 In view of these successes, various types of up-converting nanophosphors have been synthesized and studied for their spectroscopic properties.428,436–440 For example, NaYF4 nanoparticles (diameter ≈11 nm) doped with YbIII, TmIII or YbIII, HoIII have been derivatised with carboxylic acid functions; after classical treatment with EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride) and sulfo-NHS (N-hydroxysuccinimide)12 conjugation with streptavidin, and coupling with a biotinylated peptide functioning as capture-DNA they have proved to be adequate FRET sensors for reporter DNA.441 The performances of UCPs for bio-assays have been compared to those of EuIII-based labels442 while their usefulness for these assays is summarized in two recent review articles.417,429
5.4 Cell imaging and sensing
Given that many lanthanide complexes are cell-permeable and that techniques of time-resolved detection in microscopy are well mastered,11,443 scientists have used the unique spectroscopic properties of LnIII ions to get images of cells, for instance in the context of the follow-up of cancer therapy. Additionally, LLBs are also able to signal the presence of many important analytes (e.g. CaII, bicarbonate, ascorbate, urate) so that their fluctuations are tractable with high spatial resolution and yield information of utmost importance on cellular metabolism. Optical probes are unique in doing this type of job since acquisition of signals is fast. Specific modification of the LLBs, or their bioconjugation, furthermore opens the door to deciphering where analytes accumulate in live cells and what are their concentrations in the various cell organelles, mitochondria, for example. Furthermore, analysis of cancerous tissues is crucial to both medical diagnosis and monitoring of a therapeutic treatment13,398 and here again the time-resolved (TR) capability of LLBs decreases the detection limits of tumour cell biomarkers.444 Curiously, despite that TR detection has made the success of lanthanide-based immunoassays for decades, few studies on cell imaging took advantage of it until recently.445–448In the course of an extensive and seminal investigation of over 60 different cyclen-based LLBs fitted with various chromophores (Scheme 19), the group of Parker has established that the nature of the chromophore and its attachment mode to the macrocycle primarily determines the cell uptake and localization and not the charge of the complex or its lipophilicity. The cell lines studied were mouse skin fibroblasts (NIH-T3 cells), Chinese hamster ovarian (CHO), or cervix carcinoma HeLa cells. The reason invoked is that polycyclic sensitiser units are recognized by protein association. About 80% of the macrocyclic complexes have endosomal/lysosomal localization; in these cases the rates of uptake and egress are fast. Half a dozen of these complexes are also found to display fast uptake, but slower egress, and are found to shuttle between mitochondria and endosomal/lysosomal compartments. Complexes staying in the mitochondria have low IC50 values and cause cell apoptosis while those internalized in lysosomes are non-toxic and therefore can act as responsive probes. Finally a few complexes display slow uptake and egress and enter the ribosomes and the nucleoli. Their IC50 values are in the range 40–90 μM.396 The nature of the substituent on the sensitiser unit of the macrocyclic ligand is a key factor with respect to the sensitivity of the LLB; it also strongly affects protein affinity.449
Another class of cell-imaging LLBs are binuclear helicates [Ln2L3] which self-assemble in water at pH 7.4 and room temperature. The polyoxyethylene pendants ensure water solubility and higher hydrophilicity of the helicates (Scheme 20). The latter are thermodynamically stable and kinetically inert.14,450 Several ligands have been tested451–455 and the best ones turned out to be (50a)2− which sensitises the luminescence of several lanthanides, primarily EuIII (QLLn = 21%) and TbIII (11%), but also SmIII (0.4%) and YbIII (0.15%), thus allowing multiplex analyses, as well as (50b)2− amenable to excitation at the beginning of the visible range. A sensitive and versatile method of DNA and polymerase chain reaction (PCR) product analysis has been devised with [Eu2(50a)3].414 All the chelates are non-cytotoxic for HeLa, MCF-7 (human breast cancer), Jurkat (human T leukaemia), 5D10 (mouse hybridoma) and HaCat (non-malignant epithelial human cells) cell lines, with IC50 > 500 μM. The uptake in the cells (Fig. 12) is slow and proceeds by endocytosis, with a localization in the endoplasmatic reticulum, irrespective of the substituent or nature of the ligand core,454,455 and an average number of helicates per cell in the range 2–5 × 108, much as for the cyclen-based complexes. Egress is also slow and almost no leakage is seen after 24 h. Microscopy images recorded in TR mode have a sensitivity two- to three-fold higher than those measured conventionally.456
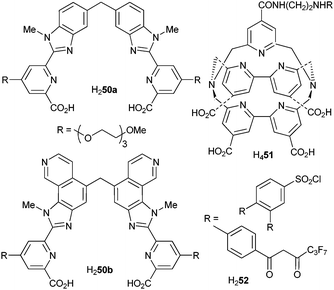 | ||
| Scheme 20 Left: examples of ditopic, hexadentate ligands for the self-assembly of binuclear helicates. Right: cryptand and bis(β-diketone) for luminescent EuIII complexes. | ||
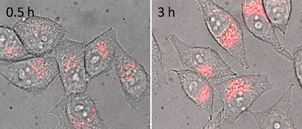 | ||
| Fig. 12 Merged bright-field and time-resolved luminescence microscopy of HeLa cells incubated 0.5 and 3 h with 200 mM {Eu2(50b)3}. Adapted from ref. 455. | ||
In view of their advantages, nano-materials are also being tested for cell imaging. For instance, EuIII-doped nanorods (20 × 500 nm) coated with chromophore-grafted silica stain the cytoplasm of living human lung carcinoma cells. They are internalized and maximum emission is seen after 2 h.457 Nanoparticles entrapping the EuIII complex with H252 and with diameter < 50 nm can distinguish the pathogen Giardia lamblia in environmental water when detected under TR mode.458
The homogeneous time-resolved luminescence method designed for immunoassays459 and relying on FRET can be extended to in vivo cellular imaging by labelling specific molecules within the cells. A proof of concept has been given with an assay on transfected living HEK cells expressing a tagged membrane receptor (GABAB2) which was detected by a specific monoclonal antibody labelled with the chelate [Eu(51)]−.460
One of the first reports on the suitability of lanthanide NIR luminescence for imaging tumour cells dates back to 1989461,462 when Russian workers detected tumours in mice with tetraphenylporphyrinate derivatives of YbIII with contrast ratios up to 45, the porphyrinate binding specifically to cancer cells. The field has then been silent until a recent revival when these macrocyclic complexes were coupled to BSA for enhanced emission.463 A more elaborate probe was proposed lately for both NIR tumour imaging and photodynamic treatment of cancer: an amphiphilic YbIII bis(porphyrinate) complex which is up-taken by rat Sarcoma 180 cells, localizes in their lysosome and displays a large cytotoxicity (light dose required for 50% death: 6.2 J cm−2).94 NdIII emission from core/shell NdF3/SiO2 nanoparticles has been recorded after their injection into thigh and abdominal cavities of mice at a depth of 0.3 and 1 cm, respectively, confirming the interest of NIR luminescence for imaging in view of its penetration depth in tissues.464
5.5 The advent of multiphoton excitation
Several of the probe systems experimented so far either for analyte sensing in vitro or in vivo or for cell and tissue imaging have the disadvantage of necessitating a relatively short wavelength of excitation (typically 300–360 nm). Since UV light damages cells and tissues, it is desirable to extend this wavelength into the visible. However, there are intrinsic limits to the design of Ln-containing luminescent molecules which severely limit this approach, at least as far as visible-emitting probes are concerned; for instance, EuIII can rarely be excited above 400–420 nm because of back-transfer455,465 problems. For instance, when dipicolinic acid is substituted to give H253e–g (Scheme 21) with tuneable charge-transfer states (push–pull ligands), the corresponding EuIII complexes have absorption maxima around 320 nm, compared to 280 nm, and they display sizeable quantum yields, up to 43%. However, when the excitation wavelength is shifted to 427 nm in [Eu(53h)3]3−, no EuIII emission is detected at room temperature.97 A way out are the up-converting phosphors described above. Another solution, similar and sometimes confused with up-conversion, is multiphoton absorption (MPA), principally two- or three-photon absorption (2PA or 3PA). Multiphoton absorption allows non-invasive 3D imaging of biological tissues without creating collateral damages. For MPA to occur with reasonable probability, high-power femto lasers with output in the range 700–800 nm are required. Microscopes fitted with such an excitation source are available,466 so that efforts are being devoted toward the design of systems displaying suitable MPA cross sections. An extensive and comprehensive review on multiphoton absorbing materials, their design, characterization and applications has been published recently in what has to be considered as a reference work.467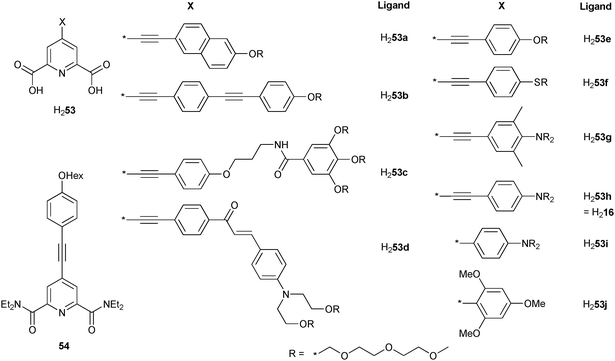 | ||
| Scheme 21 Chemical structures of organic ligands for MPA derived from dipicolinic acid. | ||
The possibility of a chromophore to simultaneously absorb two (or more) photons has been mentioned for the first time by Maria Göppert-Mayer in 1931. To describe this optical phenomenon in quantum mechanical terms, let us consider a laser beam of frequency ν passing through a non-linear absorbing medium. The occurrence of 2PA can be visualized as one photon being absorbed first to promote the molecule into an “intermediate state” from which absorption of a second photon props it up into the excited state. Contrary to up-conversion, the “intermediate state” needs not to be a real state; in this situation, the molecule may be in any of its eigenstates (except the ground and final excited state) with a distribution between them. Because the uncertainty of the distribution is large, the lifetime of the “intermediate state” is extremely short according to the uncertainty principle, so that the two absorption steps occur quasi-simultaneously. While most of the experiments are conducted with single-frequency laser light (degenerate MPA), simultaneous absorption of several photons of different energies is also feasible. MPA is related to non-linear optical (NLO) properties of the chromophore under consideration and the cross section is controlled by its molecular structure. There is an evident correlation between intramolecular charge-transfer processes and MPA: the permanent ground-state dipole moment and the transition dipole moments are essential factors in these phenomena. Regarding 2PA, the extent of conjugation between the π-donor and the π-acceptor is critical as is co-planarity between the two poles of the molecule. Increasing the number of conjugation paths to form two- or three-dimensional structures greatly increases the 2PA cross-section.467
The 2PA (σ(2)) and 3PA (σ(3)) cross sections are expressed in GM (Göppert-Mayer) units: 1 GM = 10−50 cm4 s photon−1 (2PA) or 10−85 cm6 s2 photon−2 (3PA). They are difficult to determine in an absolute way and experimental uncertainties are often on the order of ±25%. Measurements of 2PA are usually made by comparison with a standard:
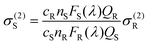 | (22) |
Determination of the level of MPA is simply made by plotting the logarithm of the luminescence intensity versus the logarithm of incident power. The slope of the straight line (1, 2 or 3) reflects 1PA, 2PA or 3PA processes. This experiment is important to carry out when lanthanide complexes are measured in view of the high power laser excitation needed for MPA. For instance, [Eu(dpa)3]3− emission can be excited at 532 nm, but the intensity/power plot has slope 1: excitation goes through the faint 5D1←7F1 transition (7F1 population at room temperature: ≈35%; estimated ε at 532 nm: 0.015 M−1 cm−1) and corresponds to 1PA.469 We note that 2PA is allowed for f–f transitions but is rarely seen in complexes with organic ligands while it is well documented for inorganic compounds.24,470
Quantitative data for 2PA in lanthanide complexes are still scarce and most of them deal with EuIII. They are listed in Table 7 along with other photophysical data. Several classes of ligands have been tested for 2PA, the more extensive series being derivatives of dipicolinic acid (Scheme 21) since the conjugation criterion can easily be met and since the complexes have idealized octupolar D3 symmetry, two favourable assets for MPA. A parameter to be carefully tuned is the energy of the (intraligand) charge transfer state; for dipicolinic acid derivatives the ideal energy appears to be around 23000 cm−1, that is slightly higher than the Eu(5D1) level.97 The best chromophore is ligand (53h)2− bearing a stronger amino donor group compared with alkoxy groups in (53e)2− with, to our knowledge, the largest σ(2) reported to date for a lanthanide complex, 775 GM at 740 nm. This is close to the maximum that one may hope to reach for such molecules. Reducing the conjugation either by twisting or by shortening the π-skeleton in (53g)2− and (53i)2−, respectively, leads to substantial decreases in σ(2) (96 GM at 740 nm and 193 GM at 730 nm, respectively).471
| Compound | State | λmaxabs/nma | 103εmax/M−1 cm−1a | τobs/ms | QLLn (1PA) (%) | λmax2PA/nmb | σ(2)max/GMc | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Value for the lowest-energy absorption band.b Wavelength at which maximum value of multiphoton absorption cross-section was detected.c 1 GM = 10−50 cm4 s photon−1.d Emission from charge transfer state at 565 nm.e Wavelength at which cross-section was determined; maximum value of σ(2) not yet reached.f In cetyltrimethyl ammonium bromide (CTAB) micelles (dav = 33.1 nm).g Maximum value under excitation at 402–420 nm, measured at 283 K.h Action cross section σ(2)Q per nanoparticle; the same value per [Eu(tta)3(dpbt)] molecule is 37.4 GM.i Values for solution in H2O, pH = 7.4.j Action cross section σ(2)Q. | ||||||||
| [NBu4]3[Eu(53a)3] | CH2Cl2 | 335 | 86.0 | 0.90 | 9.0 | 700 | 110 | 471 |
| [NBu4]3[Eu(53b)3] | CH2Cl2 | 335 | 131.0 | 0.45 | 3.6 | 700 | 218 | 471 |
| Na3[Eu(53c)3] | H2O | 332 | 78.7 | 1.06 | 15.7 | 700 | 92 | 490 |
| [NBu4]3[Eu(53d)3] | CH2Cl2 | 427 | 94.0 | n.a. | 7.0d | 840 | 173 | 471 |
| [NBu4]3[Eu(53e)3] | CH2Cl2 | 321 | 92.0 | 1.90 | 15.0 | 700 | 14 | 471 |
| [NBu4]3[Eu(53f)3] | CH2Cl2 | 322 | 79.0 | 1.42 | 43.0 | 700 | 53 | 471 |
| [NBu4]3[Eu(53g)3] | CH2Cl2 | 318 | 58.5 | 1.81 | 27.0 | 740 | 96 | 471 |
| [NBu4]3[Eu(53h)3] | CH2Cl2 | 370 | 89.2 | 0.85 | 7.0 | 740 | 775 | 471 |
| [NBu4]3[Eu(53i)3] | CH2Cl2 | 364 | 63.6 | 1.75 | 28.0 | 730 | 193 | 471 |
| [Tb(53j)3]3− | H2O (pH 8.3) | 314 | 23.0 | n.a. | 31.0 | 705e | >26e | 473 |
| [Eu(54)3][OTf]3 | MeCN | 360 | 75.0 | 0.27 | 5.6 | 720 | 96 | 500 |
| Na2[Eu(55)] | H2O (pH 7.0) | 334 | 22.8 | 1.1 | 15 | 705 | 28.6 | 472 |
| [Eu(tta)3(dpbt)] | Toluene | 406 | 63.0 | n.a. | 58.0 | 808 | 141 | 475 |
| Toluene | 406 | 55.0 | 0.48 | 52.0 | 808 | 157 | 36, 477 | |
| [Eu(tta)3(dpbt)]f | H2O-MeOH/CTAB | 420 | 43.7 | 0.42, 0.22, 0.07 | 27.0g | 832 | 3.2 × 105h | 474 |
| [Eu(tta)3(dmpbt)] | Toluene | 409 | 11.0 | n.a. | 59.0 | 812 | 149 | 475 |
| [Eu(fod)3(Mk)] | Toluene | 413 | 31.0 | n.a. | 17.0 | 810 | 253 | 473 |
| [Tb(57a)(NO3)3] | MeOH | ∼295 | n.a. | 11.0 | 690 | 3.1 | 487 | |
| [Eu(60a)(H2O)]3+ | D2O (pD 7.8) | 384i | 4.07i | 0.90 | 8.0 | 760e | 1.7e | 486 |
| [Eu(60b)(H2O)]3+ | D2O (pD 7.8) | n.a. | n.a. | n.a. | 1.6 | 760 | 0.4 | 486 |
| [Eu(60b)(H2O)]3+ + 20 mM HCO3− | D2O (pD 7.8) | n.a. | n.a. | n.a. | 5.0 | 760 | 0.4 | 486 |
| Tb2-Transferrin | H2O (pH 9.0) | n.a. | n.a. | n.a. | n.a. | 500 | 7.4j | 489 |
Carboxylates and aminocarboxylates complexes (Scheme 22) perform less well as seen for Na2[Eu(55)] for instance (Table 7).472 On the other hand, β-diketonates have larger σ(2) values, the fod ternary complex with Michler’s ketone reaching σ(2) = 253 GM at 810 nm.473 Interestingly, while [Eu(tta)3(dpbt)] has an action cross section σ(2)Q = 37.4 GM at 832 nm, its insertion into colloidal nanoparticles (cetyltrimethyl ammonium bromide micelles) results in σ(2)Q = 3.2 × 105 GM per particle,474 a value comparable to the largest ones reported for organic molecules.467 Other complexes, for instance with benzamide-containing chelating ligands (Scheme 23), have much lower values of σ(2).
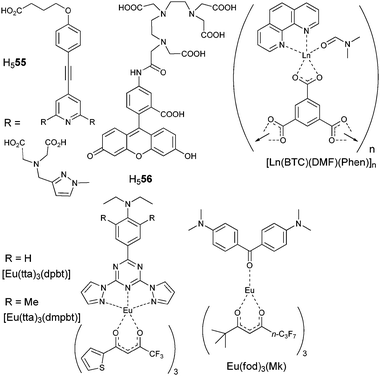 | ||
| Scheme 22 Carboxylate-containing ligands and complexes (top) and EuIIIβ-diketonate ternary complexes (bottom); Mk is Michler’s ketone.36,472–478 | ||
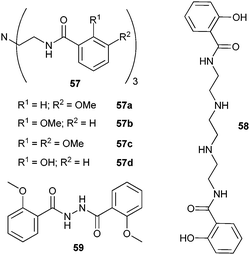 | ||
| Scheme 23 Benzamide-containing chelating agents.480,481,486–488 | ||
Less ligand optimization has been done for TbIII complexes, and the cross sections determined to date are only between 3 and 26 GM.
Some reports describe 3PA LnIII complexes, but no absolute cross-section is reported to our present knowledge. For instance, polymeric [Ln(BTC)(DMF)(phen)]n carboxylate ternary complexes (Scheme 22) with phen acting as the antenna (LnIII = Nd, Eu, Tb) are luminescent under excitation at 800 nm, the NdIII complex representing a rare example of 3PA-induced NIR luminescence in LnIII complexes with organic ligands.476 Power dependence plots for the polymeric tripodal complexes [Ln(NO3)3(57b)]n (LnIII = Eu, Tb, see Scheme 23) clearly point to 3PA mechanism when excited at 845 nm.479 The [Tb(NO3)3(H2O)3](59)2 complex is particular in that the azido ligand lies in the second coordination sphere, being only linked by H-bonds to the bound water molecules; both ligand and TbIII emission are generated by 3PA.480 To close this short overview of MPA phenomena in lanthanide complexes, we would like to mention the polymeric TbIII complex [Tb(NO3)3(57c)]n, which upon laser excitation at 1.26 μm exhibits green TbIII emission from a 4PA process as well as blue and red emission resulting from single and third harmonic generation, the complex playing the role of the gain medium.481
Two-photon excitation luminescence microscopy is a relatively new technique which started in the 1990s.482 Besides using a “cell-friendly” NIR excitation wavelength, it allows the imaging of difficult samples with good resolution. The laser beam is spatially focused and signals from the out-of-focused zone are reduced to practically zero, enhancing the contrast. The technique is suited for micro-volume bioaffinity assays and microarray platforms;483 probes are available for the determination of intracellular free metal ions, acidic vesicles, and lipid rafts in live tissues.484 3PA is also feasible, as demonstrated by the measurement of serotonin distribution in live cells.485
Applications involving LLBs are still in their infancy despite their potential. For instance, the 2PA action cross section for TbIII bound to transferrin at 500 nm, Tb2Trf, (Table 7) is comparable to those of the strongly DNA-binding fluorescent dye DAPI (4′,6-diamidino-2-phenylindole) or of Coumarin 307.489 And cross sections for EuIII complexes are much larger.
The first 2PA microscopy images involving an LnIII tag relied on [Tb(dpa)3]3− as LLB and were those of single crystals of a lysozyme derivative of the tris(dipicolinate) complex.469 The same authors subsequently obtained luminescence images of T24 cancer cells, fixed in ethanol and loaded with [Eu(53c)3]3−, upon 2PA excitation at 760 nm; the cross section for the LLB in the cell is σ(2) = 19 GM.490 After demonstrating 2PA phenomena in three EuIII complexes with derivatised cyclens, [Eu(60a,b)(H2O)]3+ (Scheme 24) having cross sections up to 2 GM,486 Parker et al. obtained 2PA images of NIH-3T3 and HeLa cells upon excitation at 720 nm after loading the cells with the macrocyclic complexes [Tb(61a)]3+ bioconjugated to arginine ([Tb(61b)]3+) or guanidinium ([Tb(61c)]3+). The guanidinium conjugate localises in mitochondria and is cytotoxic (IC50 = 12 μM).491 Another TbIII probe, [Tb(NO3)3(57a)] was loaded into human lung carcinoma (A549), HeLa and HONE1 cells which could be imaged under 800-nm irradiation, that is upon 3PA excitation.487
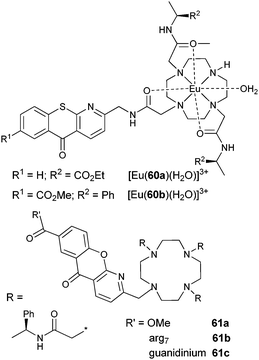 | ||
| Scheme 24 Cyclen derivatives and tripodal ligand for MPA experiments.486,491 | ||
Near-infrared MPA excitation of NIR-emitting LnIII ions has also been demonstrated, for instance for [Ln(56)]2− (Scheme 22) with LnIII = Nd, Yb in D2O,492 but no practical imaging experiment has been tempted to date. However, although based on a slightly different excitation mechanism, we would like to mention here the use of up-converting nanophosphors (see section 5.3) for imaging purposes. For instance, the digestive system of live C. elegans has been visualized with UC Y2O3 nanoparticles doped with ErIII and YbIII,493 while polyethyleneimine-coated NaYF4:YbIII,ErIII nanoparticles have been injected into the body of rats and their luminescence could be detected upon 980-nm excitation.494 However the wide-field images recorded were of moderate quality and in order to improve the situation, Yu et al. have proposed a new method of laser scanning up-conversion luminescence microscopy eliminating interferences. In this way, sharp images of HeLa cells dual-labelled with the organic dye DiIC18 (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate salt) and the same fluoride nanophosphor as above could be recorded. The technique is little sensitive to photobleaching.495
6. Hopes for a bright future
The diversity of the subjects dealt with in this review, despite its limited scope, allied to the wealth of original papers and review articles published monthly on these subjects speak for themselves: lanthanide luminescence has still an incommensurable, untouched space to expand and yield exciting results, both at the fundamental and applied levels.One striking example of contribution to fundamental aspects of photophysics is the d–f edifices tailored for sensitising NIR luminescence. The rich variety of polynuclear structures which can be generated by mixing the sterically demanding d-block elements with the versatile spherical lanthanides opens a vast playground for investigating and quantifying energy transfer mechanisms in which broad ligand or d-metal centred states populate energetically well defined f states. Similarly the advances in organic synthesis facilitate studying the influence of electronic effects from substituents grafted onto basic ligand cores, henceforth allowing to understand the fine-tuning of lanthanide luminescence. In this respect, one starts to discover the role played by non-covalent intermolecular interactions (particularly H-bonds, but also π-stacking) in funnelling energy from the metal-ion surrounding into the f states, thanks to electron density maps;47,496 the latter becoming more accessible thanks to improvements in the precision of X-ray diffraction measurements. Following the same line of thinking, reports are now published in which sizeable luminescence is found for LnIII ions bound to highly quenching water molecules;47,497 the key to this apparent mystery is again tractable to non-covalent H-bonding involving these molecules and which considerably weaken the Ln–O(H2) bonds, henceforth decreasing the quenching effect. All these outcomes will have to be incorporated in the design of new, highly luminescent lanthanide edifices.
Sensitisation of NIR luminescence remains a real challenge when organic ligands are used and the numerous studies described in section 3 clearly point to an unavoidable solution: decreasing the energy of the vibrational oscillators located in the surrounding of the emitting ion, not only in the first coordination sphere, but also much further away.59 Fluorination is beneficial, but involves more complicated syntheses of the ligands. Another way out would be to find how to decrease the radiative lifetime (see eqn (3)) but although one example is now documented,498 we have little clue as how to reach this goal.
Electroluminescent materials, particularly for OLEDs and solar energy conversion, are a bonanza for lanthanide chemists. A lot remains to be done in order to fully optimize these devices which have to feature good optical quality, easy tuning of the refractive index and the emitted colour, facile processing and adaptability to various surfaces, for instance. Replacement of the highly luminescent β-diketonates by complexes less sensitive to UV degradation is an important goal. The field has also to take full advantage of nanomaterials in which luminescent lanthanide centres can be introduced at will, often without loss of optical properties, on the contrary.
Life sciences are bound to highly benefit from lanthanide luminescent bioprobes. Although unavoidable since more than 20 years in immunoassays, a field boosted by the advent of up-converting phosphors, they have still to gain ground in the sensing of metabolites and deciphering of signalling processes in live cells. The definitive advantage of time-resolved detection should help rendering these probes more ubiquitous both for in vitro and in vivo sensing and imaging. From this point of view, structure–activity relationships are still missing for understanding how metal complexes penetrate into live cells and why they locate into given organelles. More efficient NIR emitting probes are also needed to improve the penetration depth of imaging,499 a very important aspect when monitoring, for instance a chirurgical ablation of tumour. Bifunctional agents acting both as cell-imaging probes and therapeutic agents, such as YbIII porphyrinates aimed at photodynamic treatment of cancer94 should also be the focus of much attention, as well as dual imaging (MRI/optical) probes.
Almost every field of our present technological society needs the help of lanthanide luminescence. More particularly it seems that this luminescence will be of invaluable help in solving the two major technological problems the world is facing, namely supplying enough sustainable energy and food for a rapidly expanding population, in addition to providing its assistance in medicine. This should incite more researchers to join the club!
Acknowledgements
This research is supported through grants from the Swiss National Science Foundation and the Swiss Office for Science and Education (within the frame of COST Action D38 from the European Science Foundation). JCB also thanks the World Class University program from the National Research Foundation of Korea (Ministry of Education, Science and Technology) for support (grant R31-1003r).Notes and references
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC
.
- J. H. Van Vleck, J. Phys. Chem., 1937, 41, 67 CrossRef CAS
- G. Urbain, C. R. Acad. Sci. Paris, 1906, 142, 205 Search PubMed
- S. Shionoya and W. M. Yen, Phosphor Handbook, CRC Press Inc., Boca Raton, FL 33431, USA, 1999 Search PubMed
- J. E. Geusic, H. M. Marcos and L. G. Vanuitert, Appl. Phys. Lett., 1964, 4, 182 CAS
- R. J. Mears, L. Reekie, I. M. Jauncey and D. N. Payne, Electron. Lett., 1987, 23, 1026 CrossRef
- E. Soini and I. Hemmilä, Clin. Chem., 1979, 25, 353 CAS
- I. Hemmilä, T. Ståhlberg and P. Mottram, Bioanalytical Applications of Labelling Technologies, Wallac Oy, Turku, 1995 Search PubMed
- G. Mathis, in Rare Earths, ed. R. Saez Puche and P. Caro, Editorial Complutense, Madrid, 1998, pp. 285–97 Search PubMed
- J. Hovinen and P. M. Guy, Bioconjugate Chem., 2009, 20, 404 CrossRef CAS
- R. E. Connally and J. A. Piper, Ann. N. Y. Acad. Sci., 2008, 1130, 106 CrossRef CAS
- J.-C. G. Bünzli, Chem. Lett., 2009, 38, 104 CrossRef CAS
- B. A. Hess, A. Kedziorski, L. Smentek and D. J. Bornhop, J. Phys. Chem. A, 2008, 112, 2397 CrossRef CAS
- J.-C. G. Bünzli, A.-S. Chauvin, C. D. B. Vandevyver, B. Song and S. Comby, Ann. N. Y. Acad. Sci., 2008, 1130, 97 CrossRef CAS
- Y. Fan, P. Yang, S. Huang, J. Jiang, H. Lian and J. Lin, J. Phys. Chem. C, 2009, 113, 7628
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512 RSC
- A. Roca-Sabio, M. Mato-Iglesias, D. Esteban-Gomez, E. Toth, A. de Blas, C. Platas-Iglesias and T. Rodriguez-Blas, J. Am. Chem. Soc., 2009, 131, 3331 CrossRef CAS
- A. de Bettencourt-Dias, Dalton Trans., 2007, 2229 RSC
- O. M. Khreis, R. J. Curry, M. Somerton and W. P. Gillin, J. Appl. Phys., 2000, 88, 777 CrossRef CAS
- P. He, H. H. Wang, S. G. Liu, W. Hu, J. X. Shi, G. Wang and M. L. Gong, J. Electrochem. Soc., 2009, 156, E46 CrossRef CAS
- G. K. Liu, in Spectroscopic Properties of Rare Earths in Optical Materials, ed. G. K. Liu and B. Jacquier, Springer Verlag, Berlin, 2005, vol. 83, ch. 1, pp. 1–94 Search PubMed
- J.-C. G. Bünzli and S. V. Eliseeva, in Springer Series on Fluorescence, Vol. 7, Lanthanide Spectroscopy, Materials, and Bio-applications, ed. P. Hänninen and H. Härmä, Springer Verlag, Berlin, 2010, ch. 2, vol. 7 Search PubMed
- C. Görller-Walrand and K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, Jr and L. Eyring, Elsevier Science B.V., Amsterdam, 1996, ch. 155, vol. 23 Search PubMed
- C. Görller-Walrand and K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, Jr and L. Eyring, Elsevier Science B.V., Amsterdam, 1998, ch. 167, vol. 25 Search PubMed
- P. Dorenbos, J. Lumin., 2000, 91, 91 CrossRef CAS
- K. Ogasawara, S. Watanabe, H. Toyoshima and M. G. Brik, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B.V., Amsterdam, 2007, ch. 231, vol. 37 Search PubMed
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC
- M. Bredol, U. Kynast and C. Ronda, Adv. Mater., 1991, 3, 361 CrossRef CAS
- E. Brunet, O. Juanes, R. Sedano and J.-C. Rodriguez-Ubis, Photochem. Photobiol. Sci., 2002, 1, 613 RSC
- O. L. Malta, H. F. Brito, J. F. S. Menezes, F. R. Gonçalves e Silva, C. D. Donega and S. Alves, Chem. Phys. Lett., 1998, 282, 233 CrossRef CAS
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC
- A. Aebischer, F. Gumy and J.-C. G. Bünzli, Phys. Chem. Chem. Phys., 2009, 11, 1346 RSC
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214 CrossRef CAS
- G. F. de Sá, O. L. Malta, C. D. Donega, A. M. Simas, R. L. Longo, P. A. Santa-Cruz and E. F. da Silva, Coord. Chem. Rev., 2000, 196, 165 CrossRef CAS
- M. Kleinerman, J. Chem. Phys., 1969, 51, 2370 CrossRef
- C. Yang, L. M. Fu, Y. Wang, J. P. Zhang, W. T. Wong, X. C. Ai, Y. F. Qiao, B. S. Zou and L. L. Gui, Angew. Chem., Int. Ed., 2004, 43, 5010 CrossRef CAS
- R. Rodriguez-Cortinas, F. Avecilla, C. Platas-Iglesias, D. Imbert, J.-C. G. Bünzli, A. de Blas and T. Rodriguez-Blas, Inorg. Chem., 2002, 41, 5336 CrossRef CAS
- S. Sato and M. Wada, Bull. Chem. Soc. Jpn., 1970, 43, 1955 CAS
- M. Latva, H. Takalo, V. M. Mukkala, C. Matachescu, J.-C. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149 CrossRef CAS
- R. D. Archer, H. Y. Chen and L. C. Thompson, Inorg. Chem., 1998, 37, 2089 CrossRef CAS
- G. K. Liu, M. P. Jensen and P. M. Almond, J. Phys. Chem. A, 2006, 110, 2081 CrossRef CAS
- J.-C. G. Bünzli, P. D. Morand and J.-M. Pfefferlé, J. Physique, 1987, 48, C7/625 Search PubMed
- G. Muller, S. D. Kean, D. Parker and J. P. Riehl, J. Phys. Chem. A, 2002, 106, 12349 CrossRef CAS
- S. T. Frey and W. D. Horrocks, Jr, Inorg. Chim. Acta, 1995, 229, 383 CrossRef CAS
- J.-C. G. Bünzli, in Lanthanide Probes in Life, Chemical and Earth Sciences. Theory and Practice, ed. J.-C. G. Bünzli and G. R. Choppin, Elsevier Science Publ. B.V., Amsterdam, 1989, ch. 7, pp. 219–93 Search PubMed
- J.-C. G. Bünzli, B. Klein, D. Wessner and N. W. Alcock, Inorg. Chim. Acta, 1982, 59, 269 CrossRef
- L. N. Puntus, K. A. Lyssenko, M. Y. Antipin and J.-C. G. Bünzli, Inorg. Chem., 2008, 47, 11095 CrossRef CAS
- R. M. Supkowski and W. D. Horrocks, Jr, Inorg. Chim. Acta, 2002, 340, 44 CrossRef CAS
- T. Kimura and Y. Kato, J. Alloys Compd., 1998, 271–273, 867 CrossRef CAS
- T. Kimura and Y. Kato, J. Alloys Compd., 1998, 275–277, 806 CrossRef CAS
- M. S. Tremblay, M. Halim and D. Sames, J. Am. Chem. Soc., 2007, 129, 7570 CrossRef CAS
- D. Parker and J. A. G. Williams, in Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker Inc., New York, 2003, vol. 40 Search PubMed
- D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977 CrossRef CAS
- L. J. Charbonnière and N. Hildebrandt, Eur. J. Inorg. Chem., 2008, 3241 CrossRef CAS
- T. Nishioka, K. Fukui and K. Matsumoto, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, Jr, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B.V., Amsterdam, 2007, vol. 37 Search PubMed
- C. M. Spangler, C. Spangler and M. Schaerling, Ann. N. Y. Acad. Sci., 2008, 1130, 138 CrossRef CAS
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS
- S. Comby and J.-C. G. Bünzli, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B.V., Amsterdam, 2007, vol. 37, ch. 235 Search PubMed
- L. Winkless, R. H. C. Tan, Y. Zheng, M. Motevalli, P. B. Wyatt and W. P. Gillin, Appl. Phys. Lett., 2006, 89, 111115 CrossRef
- L. N. Sun, H. J. Zhang, J. B. Yu, S. Y. Yu, C. Y. Peng, S. Dang, X. M. Guo and J. Feng, Langmuir, 2008, 24, 5500 CrossRef CAS
- R. Van Deun, P. Fias, P. Nockemann, K. Van Hecke, L. Van Meervelt and K. Binnemans, Eur. J. Inorg. Chem., 2007, 302 CrossRef CAS
- M. Albrecht, O. Osetska, J. Klankermayer, R. Fröhlich, F. Gumy and J.-C. G. Bünzli, Chem. Commun., 2007, 1834 RSC
- W. Huang, D. Wu, D. Guo, X. Zhu, C. He, Q. Meng and C. Duan, Dalton Trans., 2009, 2081 RSC
- N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. G. Bünzli, Inorg. Chem., 2009, 48, 2908 CrossRef CAS
- N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. G. Bünzli, Inorg. Chem., 2008, 47, 9055 CrossRef CAS
- M. Albrecht, R. Fröhlich, J.-C. G. Bünzli, A. Aebischer, F. Gumy and J. Hamacek, J. Am. Chem. Soc., 2007, 129, 14178 CrossRef CAS
- S. Comby, D. Imbert, C. D. B. Vandevyver and J.-C. G. Bünzli, Chem.–Eur. J., 2007, 13, 936 CrossRef CAS
- Y. Zheng, J. Pearson, R. H. C. Tan, W. P. Gillin and P. B. Wyatt, J. Mater. Sci.: Mater. Electron., 2009, 20, 430 CrossRef CAS
- L. M. Song, J. Hu, J. S. Wang, X. H. Liu and Z. Zhen, Photochem. Photobiol. Sci., 2008, 7, 689 RSC
- R. H. C. Tan, J. M. Pearson, Y. Zheng, P. B. Wyatt and W. P. Gillin, Appl. Phys. Lett., 2008, 92, 103303 CrossRef
- P. B. Glover, A. P. Bassett, P. Nockemann, B. M. Kariuki, R. Van Deun and Z. Pikramenou, Chem.–Eur. J., 2007, 13, 6308 CrossRef CAS
- C. J. Jocher, E. G. Moore, J. D. Pierce and K. N. Raymond, Inorg. Chem., 2008, 47, 7951 CrossRef CAS
- E. G. Moore, G. Szigethy, J. Xu, L. O. Palsson, A. Beeby and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 9500 CrossRef CAS
- E. G. Moore, M. Seitz and K. N. Raymond, Inorg. Chem., 2008, 47, 8571 CrossRef CAS
- L. Song, Q. Wang, D. Tang, X. Liu and Z. Zhen, New J. Chem., 2007, 31, 506 RSC
- M. K. Nah, S. G. Rho, H. K. Kim and J. G. Kang, J. Phys. Chem. A, 2007, 111, 11437 CrossRef CAS
- G. S. Kottas, M. Mehlstaubl, R. Froehlich and L. De Cola, Eur. J. Inorg. Chem., 2007, 3465 CrossRef CAS
- N. S. Baek, Y. H. Kim and H. K. Kim, Bull. Korean Chem. Soc., 2006, 27, 1729 CAS
- M. Mato-Iglesias, T. Rodriguez-Blas, C. Platas-Iglesias, M. Starck, P. Kadjane, R. Ziessel and L. Charbonniere, Inorg. Chem., 2009, 48, 1507 CrossRef CAS
- L. Pellegatti, J. Zhang, B. Drahos, S. Villette, F. Suzenet, G. Guillaumet, S. Petoud and E. Toth, Chem. Commun., 2008, 6591 RSC
- S. J. A. Pope, Polyhedron, 2007, 26, 4818 CrossRef CAS
- N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. G. Bünzli, Eur. J. Inorg. Chem., 2008, 1523 CrossRef CAS
- L. N. Sun, J. B. Yu, G. L. Zheng, H. J. Zhang, Q. G. Meng, C. Y. Peng, L. S. Fu, F. Y. Liu and Y. N. Yu, Eur. J. Inorg. Chem., 2006, 3962 CrossRef CAS
- J. Zhang, P. D. Badger, S. J. Greib and S. Petoud, Angew. Chem., Int. Ed., 2005, 44, 2508 CrossRef CAS
- Y. X. Zheng, M. Motevalli, R. H. C. Tan, I. Abrahams, W. P. Gillin and P. B. Wyatt, Polyhedron, 2008, 27, 1503 CrossRef CAS
- Y. Hasegawa, T. Yasuda, K. Nakamura and T. Kawai, Jpn. J. Appl. Phys., 2008, 47, 1192 CrossRef CAS
- K. Aita, T. Temma, Y. Kuge and H. Saji, Luminescence, 2007, 22, 455 CrossRef CAS
- M. Andrews, R. H. Laye, L. P. Harding and S. J. A. Pope, Polyhedron, 2008, 27, 2365 CrossRef CAS
- X. J. Zhu, W. K. Wong, J. P. Guo, W. Y. Wong and J. P. Zhang, Eur. J. Inorg. Chem., 2008, 3515 CrossRef CAS
- H. He and A. G. Sykes, Inorg. Chem. Commun., 2008, 11, 1304 CrossRef CAS
- M. K. Nah, J. B. Oh, H. K. Kim, K. H. Choi, Y. R. Kim and J. G. Kang, J. Phys. Chem. A, 2007, 111, 6157 CrossRef CAS
- S. Zhuravlyov, N. Rusakova and Y. Korovin, J. Alloys Compd., 2008, 451, 334 CrossRef CAS
- F. L. Jiang, W. K. Wong, X. J. Zhu, G. J. Zhou, W. Y. Wong, P. L. Wu, H. L. Tam, K. W. Cheah, C. Ye and Y. Liu, Eur. J. Inorg. Chem., 2007, 3365 CrossRef CAS
- F. L. Jiang, C. T. Poon, W. K. Wong, H. K. Koon, N. K. Mak, C. Y. Choi, D. W. J. Kwong and Y. Liu, ChemBioChem, 2008, 9, 1034 CrossRef CAS
- X. J. Zhu, F. L. Jiang, C. T. Poon, W. K. Wong and W. Y. Wong, Eur. J. Inorg. Chem., 2008, 3151 CrossRef CAS
- W. K. Lo, W. K. Wong, W. Y. Wong, J. P. Guo, K. T. Yeung, Y. K. Cheng, X. Yang and R. A. Jones, Inorg. Chem., 2006, 45, 9315 CrossRef CAS
- A. D’Aleo, A. Picot, A. Beeby, J. A. Gareth Williams, B. Le Guennic, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10258 CrossRef CAS
- J. Zhang and S. Petoud, Chem.–Eur. J., 2008, 14, 1264 CrossRef CAS
- R. Van Deun, P. Nockemann, T. N. Parac-Vogt, K. Van Hecke, L. Van Meervelt, C. Gorller-Walrand and K. Binnemans, Polyhedron, 2007, 26, 5441 CrossRef CAS
- A. Nonat, D. Imbert, J. Pécaut, M. Giraud and M. Mazzanti, Inorg. Chem., 2009, 48, 4207 CrossRef CAS
- Y. V. Korovin, N. V. Rusakova, Y. A. Popkov and V. P. Dotsenko, J. Appl. Spectrosc., 2002, 69, 841 CrossRef CAS
- W. P. Gillin and R. J. Curry, Appl. Phys. Lett., 1999, 74, 798 CrossRef CAS
- S. Comby, D. Imbert, A.-S. Chauvin and J.-C. G. Bünzli, Inorg. Chem., 2006, 45, 732 CrossRef CAS
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542 CrossRef CAS
- N. S. Baek, M. K. Nah, Y. H. Kim and H. K. Kim, J. Lumin., 2007, 127, 707 CrossRef CAS
- A. Monguzzi, R. Tubino, F. Meinardi, A. O. Biroli, M. Pizzotti, F. Demartin, F. Quochi, F. Cordella and M. A. Loi, Chem. Mater., 2009, 21, 128 CrossRef CAS
- J. Zhang, C. M. Shade, D. A. Chengelis and S. Petoud, J. Am. Chem. Soc., 2007, 129, 14834 CrossRef CAS
- T. Lazarides, M. A. H. Alamiry, H. Adams, S. J. A. Pope, S. Faulkner and M. D. Ward, Dalton Trans., 2007, 1484 RSC
- H. Tsukube, Y. Suzuki, D. Paul, Y. Kataoka and S. Shinoda, Chem. Commun., 2007, 2533 RSC
- W. K. Wong, X. Zhu and W. Y. Wong, Coord. Chem. Rev., 2007, 251, 2386 CrossRef CAS
- W. Y. Bi, X. Q. Lu, W. L. Chai, J. R. Song, W. K. Wong, X. P. Yang and R. A. Jones, Z. Anorg. Allg. Chem., 2008, 634, 1795 CrossRef CAS
- Y. X. Chi, S. Y. Niu, Z. L. Wang and J. Jin, Eur. J. Inorg. Chem., 2008, 2336 CrossRef CAS
- X. L. Li, L. X. Shi, L. Y. Zhang, H. M. Wen and Z. N. Chen, Inorg. Chem., 2007, 46, 10892 CrossRef CAS
- X. L. Li, F. R. Dai, L. Y. Zhang, Y. M. Zhu, Q. Peng and Z. N. Chen, Organometallics, 2007, 26, 4483 CrossRef CAS
- H. B. Xu, L. Y. Zhang, X. M. Chen, X. L. Li and Z. N. Chen, Cryst. Growth Des., 2009, 9, 569 CrossRef CAS
- H. B. Xu, L. Y. Zhang, Z. H. Chen, L. X. Shi and Z. N. Chen, Dalton Trans., 2008, 4664 RSC
- H.-B. Xu, L.-Y. Zhang, Z.-L. Xie, E. Ma and Z.-N. Chen, Chem. Commun., 2007, 2744 RSC
- F. Kennedy, N. M. Shavaleev, T. Koullourou, Z. R. Bell, J. C. Jeffery, S. Faulkner and M. D. Ward, Dalton Trans., 2007, 1492 RSC
- H. B. Xu, L. Y. Zhang, J. Ni, H. Y. Chao and Z. N. Chen, Inorg. Chem., 2008, 47, 10744 CrossRef CAS
- T. Koullourou, L. S. Natrajan, H. Bhavsar, S. J. A. Pope, J. H. Feng, J. Narvainen, R. Shaw, E. Scales, R. Kauppinen, A. M. Kenwright and S. Faulkner, J. Am. Chem. Soc., 2008, 130, 2178 CrossRef CAS
- T. K. Ronson, T. Lazarides, H. Adams, S. J. A. Pope, D. Sykes, S. Faulkner, S. J. Coles, M. B. Hursthouse, W. Clegg, R. W. Harrington and M. D. Ward, Chem.–Eur. J., 2006, 12, 9299 CrossRef CAS
- M. Mehlstäubl, G. S. Kottas, S. Colella and L. De Cola, Dalton Trans., 2008, 2385 RSC
- F. F. Chen, Z. Q. Bian, B. Lou, E. Ma, Z. W. Liu, D. B. Nie, Z. Q. Chen, J. Bian, N. Z. Chen and C. H. Huang, Dalton Trans., 2008, 5577 RSC
- T. Lazarides, H. Adams, D. Sykes, S. Faulkner, G. Calogero and M. D. Ward, Dalton Trans., 2008, 691 RSC
- T. Lazarides, D. Sykes, S. Faulkner, A. Barbieri and M. D. Ward, Chem.–Eur. J., 2008, 14, 9389 CrossRef CAS
- C. Giansante, P. Ceroni, V. Balzani and F. Vögtle, Angew. Chem., Int. Ed., 2008, 47, 5422 CrossRef CAS
- S. G. Baca, H. Adams, D. Sykes, S. Faulkner and M. D. Ward, Dalton Trans., 2007, 2419 RSC
- T. Lazarides, G. M. Davies, H. Adams, C. Sabatini, F. Barigelletti, A. Barbieri, S. J. A. Pope, S. Faulkner and M. D. Ward, Photochem. Photobiol. Sci., 2007, 6, 1152 RSC
- B. Zhao, X. Q. Zhao, Z. Chen, W. Shi, P. Cheng, S. P. Yan and D. Z. Liao, CrystEngComm, 2008, 10, 1144 RSC
- G. A. Kumar, R. E. Riman, L. A. D. Torres, O. B. Garcia, S. Banerjee, A. Kornienko and J. G. Brennan, Chem. Mater., 2005, 17, 5130 CrossRef
- G. A. Kumar, R. E. Riman, L. A. D. Torres, S. Banerjee, A. D. Romanelli, T. J. Emge and J. G. Brennan, Chem. Mater., 2007, 19, 2937 CrossRef CAS
- S. Banerjee, G. A. Kumar, R. E. Riman, T. J. Emge and J. G. Brennan, J. Am. Chem. Soc., 2007, 129, 5926 CrossRef CAS
- M. Romanelli, G. A. Kumar, T. J. Emge, R. E. Riman and J. G. Brennan, Angew. Chem., Int. Ed., 2008, 47, 6049 CAS
- S. Banerjee, G. A. Kumar, T. J. Emge, R. E. Riman and J. G. Brennan, Chem. Mater., 2008, 20, 4367 CrossRef CAS
- K. Norton, G. A. Kumar, J. L. Dilks, T. J. Emge, R. E. Riman, M. G. Brik and J. G. Brennan, Inorg. Chem., 2009, 48, 3573 CrossRef CAS
- K. Manseki, Y. Hasegawa, Y. Wada, H. Ichida, Y. Kanematsu and T. Kushida, J. Lumin., 2007, 122–123, 262 CrossRef CAS
- B. H. Tong, S. J. Wang, Y. Z. Meng and B. Wang, Photochem. Photobiol. Sci., 2007, 6, 519 RSC
- C. Molina, R. A. S. Ferreira, G. Poirier, L. Fu, S. J. L. Ribeiro, Y. Messsaddeq and L. D. Carlos, J. Phys. Chem. C, 2008, 112, 19346 CrossRef CAS
- M. D. Allendorf, C. A. Bauer, R. B. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC
- S. I. Kishimoto, A. Tanaka, Y. Hasegawa and T. Kawai, Thin Solid Films, 2008, 517, 1497 CrossRef CAS
- L. M. Song, J. S. Wang, J. Hu, X. H. Liu and Z. Zhen, J. Alloys Compd., 2009, 473, 201 CrossRef CAS
- S. Comby, F. Gumy, J.-C. G. Bünzli, T. Saraidarov and R. Reisfeld, Chem. Phys. Lett., 2006, 432, 128 CrossRef CAS
- L. M. Song, J. S. Wang, X. H. Liu and Z. Zhen, J. Non-Cryst. Solids, 2008, 354, 3375 CrossRef CAS
- S. Dang, L. N. Sun, H. J. Zhang, X. M. Guo, Z. F. Li, J. Feng, H. D. Guo and Z. Y. Guo, J. Phys. Chem. C, 2008, 112, 13240 CrossRef CAS
- J. Feng, J. B. Yu, S. Y. Song, L. N. Sun, W. S. Fan, X. M. Guo, S. Dang and H. J. Zhang, Dalton Trans., 2009, 2406 RSC
- L. N. Sun, H. J. Zhang, J. B. Yu, Q. G. Meng, F. Y. Liu and C. Y. Peng, J. Photochem. Photobiol., A, 2008, 193, 153 CrossRef CAS
- L. N. Sun, J. B. Yu, H. J. Zhang, Q. G. Meng, E. Ma, C. Y. Peng and K. Y. Yang, Microporous Mesoporous Mater., 2007, 98, 156 CrossRef CAS
- B. Chen, Y. Yang, F. Zapata, G. Qian, Y. Luo, J. Zhang and E. B. Lobkovsky, Inorg. Chem., 2006, 45, 8882 CrossRef CAS
- H. S. Wang, B. Zhao, B. Zhai, W. Shi, P. Cheng, D. Z. Liao and S. P. Yan, Cryst. Growth Des., 2007, 7, 1851 CrossRef CAS
- G. Zucchi, O. Maury and M. Ephritikhine, Inorg. Chem., 2008, 47, 10398 CrossRef CAS
- M. T. Pope, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B.V., Amsterdam, 2008, ch. 240, vol. 38 Search PubMed
- T. Yamase, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, Jr, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science BV, Amsterdam, 2009, ch. 242, vol. 39 Search PubMed
- G. Glaspell, J. Anderson, J. R. Wilkins and M. S. El-Shall, J. Phys. Chem. C, 2008, 112, 11527 CrossRef CAS
- G. Y. Chen, Y. Liu, Y. G. Zhang, G. Somesfalean, Z. G. Zhang, Q. Sun and F. P. Wang, Appl. Phys. Lett., 2007, 91, 133103 CrossRef
- W. Lü, X. Ma, H. Zhou, G. Chen, J. Li, Z. Zhu, Z. You and C. Tu, J. Phys. Chem. C, 2008, 112, 15071 CrossRef CAS
- Y. X. Pan and Q. Y. Zhang, Mater. Sci. Eng. B, Solid State Mater. Adv. Technol., 2007, 138, 90 CrossRef CAS
- V. Mahalingam, F. Mangiarini, F. Vetrone, V. Venkatramu, M. Bettinelli, A. Speghini and J. A. Capobianco, J. Phys. Chem. C, 2008, 112, 17745 CrossRef CAS
- S. Sivakumar, J. C. Boyer, E. Bovero and F. C. J. M. Van Veggel, J. Mater. Chem., 2009, 19, 2392 RSC
- D. Q. Chen, Y. S. Wang, K. L. Zheng, T. L. Guo, Y. L. Yu and P. Huang, Appl. Phys. Lett., 2007, 91, 251903 CrossRef
- D. Q. Chen, Y. S. Wang, Y. L. Yu, P. Huang and F. Y. Weng, J. Solid State Chem., 2008, 181, 2763 CrossRef CAS
- C. Liu and J. Heo, Mater. Lett., 2007, 61, 3751 CrossRef CAS
- G. Sinha and A. Patra, Chem. Phys. Lett., 2009, 473, 151 CrossRef CAS
- W. Lü, H. Zhou, G. Chen, J. Li, Z. Zhu, Z. You and C. Tu, J. Phys. Chem. C, 2009, 113, 3844 CrossRef
- Q. Y. Zhang, C. H. Yang and Y. X. Pan, Nanotechnology, 2007, 18, 145602 CrossRef
- M. Karmaoui, R. A. S. Ferreira, A. T. Mane, L. D. Carlos and N. Pinna, Chem. Mater., 2006, 18, 4493 CrossRef CAS
- M. Karmaoui, L. Mafra, R. A. SaFerreira, J. Rocha, L. D. Carlos and N. Pinna, J. Phys. Chem. C, 2007, 111, 2539 CrossRef CAS
- B. Kokuoz, J. R. DiMaio, C. Kucera, D. D. Evanoff and J. Ballato, J. Am. Chem. Soc., 2008, 130, 12222 CrossRef CAS
- S. D. Maind, S. A. Kumar, N. Chattopadhyay, C. Gandhi and M. Sudersanan, Forens. Sci. Int., 2006, 159, 32 CrossRef CAS
- S. D. Maind, N. Chattopadhyay, C. Gandhi, S. C. Kumar and M. Sudersanan, Sci. Justice, 2008, 48, 61 Search PubMed
- D. F. Reardon, US Pat., 2008305243 A1 2008121, 2008.
- R. Steiger, R. Beer, J. F. Fernandez-Sanchez and U. E. Spichiger-Keller, Nanosci. Technol. 1&2, 2007, 121–123, 1193 Search PubMed
- A. L. Pénard, T. Gacoin and J. P. Boilot, Acc. Chem. Res., 2007, 40, 895 CrossRef CAS
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976 RSC
- J. Shen, L. D. Sun and C. H. Yan, Dalton Trans., 2008, 5687 RSC
- K. Binnemans, Chem. Rev., 2007, 107, 2592 CrossRef CAS
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS
- A. Taubert and Li Zhonghao, Dalton Trans., 2007, 723 RSC
- K. Shimojo, K. Kurahashi and H. Naganawa, Dalton Trans., 2008, 5083 RSC
- I. Billard, C. Gaillard and C. Hennig, Dalton Trans., 2007, 4214 RSC
- M. Grätzel, Philos. Trans. R. Soc. London, Ser. A, 2007, 365, 993 CrossRef CAS
- A. I. Bhatt, I. May, V. A. Volkovich, D. Collison, M. Helliwell, I. B. Polovov and R. G. Lewin, Inorg. Chem., 2005, 44, 4934 CrossRef CAS
- K. Lunstroot, P. Nockemann, K. Van Hecke, L. Van Meervelt, C. Görller-Walrand, K. Binnemans and K. Driesen, Inorg. Chem., 2009, 48, 3018 CrossRef CAS
- P. Nockemann, E. Beurer, K. Driesen, R. Van Deun, K. Van Hecke, L. Van Meervelt and K. Binnemans, Chem. Commun., 2005, 4354 RSC
- S. Stumpf, I. Billard, P. J. Panak and S. Mekki, Dalton Trans., 2007, 240 RSC
- S. Stumpf, I. Billard, C. Gaillard, P. J. Panak and K. Dardenne, Inorg. Chem., 2008, 47, 4618 CrossRef CAS
- S. Samikkanu, K. Mellem, M. Berry and P. S. May, Inorg. Chem., 2007, 46, 7121 CrossRef CAS
- H. R. Li, H. F. Shao, Y. G. Wang, D. S. Qin, B. Y. Liu, W. J. Zhang and W. D. Yan, Chem. Commun., 2008, 5209 RSC
- P. Nockemann, B. Thijs, K. Van Hecke, L. Van Meervelt and K. Binnemans, Cryst. Growth Des., 2008, 8, 1353 CrossRef
- S. Tang, A. Babai and A.-V. Mudring, Angew. Chem., Int. Ed., 2008, 47, 7631 CrossRef CAS
- N. O. Nuñez and M. Ocana, Nanotechnology, 2007, 18, 455606 CrossRef
- C. Zhang, J. Chen, Y. Zhou and D. Li, J. Phys. Chem. C, 2008, 112, 10083 CrossRef CAS
- K. Lunstroot, K. Driesen, P. Nockemann, C. Gorller-Walrand, K. Binnemans, S. Bellayer, J. Le Bideau and A. Vioux, Chem. Mater., 2006, 18, 5711 CrossRef CAS
- K. Lunstroot, K. Driesen, P. Nockemann, K. Van Hecke, L. Van Meervelt, C. Gorller-Walrand, K. Binnemans, S. Bellayer, L. Viau, J. Le Bideau and A. Vioux, Dalton Trans., 2009, 298 RSC
- K. Binnemans, Chem. Rev., 2005, 105, 4148 CrossRef CAS
- J. Kocher, F. Gumy, A.-S. Chauvin and J.-C. G. Bünzli, J. Mater. Chem., 2007, 17, 654 RSC
- L. N. Puntus, K. P. Zhuravlev, I. S. Pekareva, K. A. Lyssenko and V. F. Zolin, Opt. Mater., 2008, 30, 806 CrossRef CAS
- K. Goossens, P. Nockemann, K. Driesen, B. Goderis, C. Goerller-Walrand, K. Van Hecke, L. Van Meervelt, E. Pouzet, K. Binnemans and T. Cardinaels, Chem. Mater., 2008, 20, 157 CrossRef CAS
- C. Piguet, J.-C. G. Bünzli, B. Donnio and D. Guillon, Chem. Commun., 2006, 3755 RSC
- K. Binnemans and C. Görller-Walrand, Chem. Rev., 2002, 102, 2303 CrossRef CAS
- K. Binnemans, J. Mater. Chem., 2009, 19, 448 RSC
- E. Terazzi, S. Suárez, S. Torelli, H. Nozary, D. Imbert, O. Mamula, J.-P. Rivera, E. Guillet, J.-M. Benech, G. Bernardinelli, R. Scopelliti and B. Donnio, et al., Adv. Funct. Mater., 2006, 16, 157 CrossRef CAS
- E. Terazzi, L. Guénée, P.-Y. Morgantini, G. Bernardinelli, B. Donnio, D. Guillon and C. Piguet, Chem.–Eur. J., 2007, 13, 1674 CrossRef CAS
- T. Cardinaels, J. Ramaekers, P. Nockemann, K. Driesen, K. Van Hecke, L. Van Meervelt, S. B. Lei, S. De Feyter, D. Guillon, B. Donnio and K. Binnemans, Chem. Mater., 2008, 20, 1278 CrossRef CAS
- A. Escande, L. Guénée, H. Nozary, G. Bernardinelli, F. Gumy, A. Aebischer, J.-C. G. Bünzli, B. Donnio, D. Guillon and C. Piguet, Chem.–Eur. J., 2007, 13, 8696 CrossRef
- T. B. Jensen, E. Terazzi, B. Donnio, D. Guillon and C. Piguet, Chem. Commun., 2008, 181 RSC
- Y. G. Galyametdinov, W. Haase, B. Goderis, D. Moors, K. Driesen, R. VanDeun and K. Binnemans, J. Phys. Chem. B, 2007, 111, 13881 CrossRef CAS
- S. Suárez, D. Imbert, F. Gumy, C. Piguet and J.-C. G. Bünzli, Chem. Mater., 2004, 16, 3257 CrossRef CAS
- Y. T. Yang, J. J. Li, X. J. Liu and S. Y. Zhang, Eur. Phys. J. Spec. Top., 2008, 153, 49 Search PubMed
- Y. T. Yang, J. J. Li, X. Liu, S. Y. Zhang, K. Driesen, P. Nockemann and K. Binnemans, ChemPhysChem, 2008, 9, 600 CrossRef CAS
- Y. Yang, X. Liu, A. Nakamura, K. Binnemans and J. Liu, J. Phys. Chem. B, 2008, 112, 5291 CrossRef CAS
- K. Palewska, A. Miniewicz, S. Bartkiewicz, J. Legendziewicz and W. Strek, J. Lumin., 2007, 124, 265 CrossRef CAS
- K. Driesen, D. Moors, J. Beeckman, K. Neytsb, C. Gorller-Walrand and K. Binnemans, J. Lumin., 2007, 127, 611 CrossRef CAS
- A. A. Knyazev, Y. G. Galyametdinov, B. Goderis, K. Driesen, K. Goossens, C. Gorller-Walrand, K. Binnemans and T. Cardinaels, Eur. J. Inorg. Chem., 2008, 756 CrossRef CAS
- F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS
- C. Ronda, Luminescence: From Theory to Applications, WILEY-VCH, Weinheim, 2008 Search PubMed
- M. P. Hehlen, M. L. F. Phillips, N. J. Cockroft and H. U. Güdel, in Encyclopedia of Materials: Science and Technology, ed. K. H. J. Buschow, R. W. Cahn, M. C. Flemings, B. Ilschner, E. J. Kramer and S. Mahajan, Elsevier Science Ltd, Oxford, 2001, pp. 9456–9458 Search PubMed
- D. Chen, Y. Yu, Y. Wang, P. Huang and F. Weng, J. Phys. Chem. C, 2009, 113, 6406 CrossRef CAS
- P. Ghosh, J. Oliva, E. D. l. Rosa, K. K. Haldar, D. Solis and A. Patra, J. Phys. Chem. C, 2008, 112, 9650 CrossRef CAS
- C. Cao, W. Qin, J. Zhang, Y. Wang, G. Wang, G. Wei, P. Zhu, L. Wang and L. Jin, Opt. Commun., 2008, 281, 1716 CAS
- A. S. Gouveia-Neto, L. A. Bueno, R. F. do Nascimento, E. A. da Silva, E. B. da Costa and V. B. do Nascimento, Appl. Phys. Lett., 2007, 91, 091114 CrossRef
- N. K. Giri, D. K. Rai and S. B. Rai, J. Appl. Phys., 2008, 104, 113107 CrossRef
- Y. Dwivedi, A. Rai and S. B. Rai, J. Appl. Phys., 2008, 104, 043509 CrossRef
- J. F. Suyver, A. Aebischer, D. Biner, P. Gerner, J. Grimm, S. Heer, K. W. Kraemer, C. Reinhard and H. U. Guedel, Opt. Mater., 2005, 27, 1111 CrossRef CAS
- A. Rapaport, J. Milliez, M. Bass, A. Cassanho and H. Jenssen, J. Display Technol., 2006, 2, 68 CrossRef CAS
- C. Y. Sun, X. J. Zheng, X. B. Chen, L. C. Li and L. P. Jin, Inorg. Chim. Acta, 2009, 362, 325 CrossRef CAS
- D. F. Weng, X. J. Zheng, X. B. Chen, L. Li and L. P. Jin, Eur. J. Inorg. Chem., 2007, 3410 CrossRef CAS
- D. F. Weng, X. J. Zheng and L. P. Jin, Eur. J. Inorg. Chem., 2006, 4184 CrossRef CAS
- X. D. Xiao, J. P. Haushalter and G. W. Faris, Opt. Lett., 2005, 30, 1674 Search PubMed
- L. S. Hung and C. H. Chen, Mater. Sci. Eng., R, 2002, 39, 143 CrossRef
- K. Müllen and U. Scherf, Organic Light Emitting Devices. Synthesis, Properties and Applications, WILEY-VCH, Weinhein, 2006 Search PubMed
- J. Kalinowski, Organic Light-Emitting Diodes: Principles, Characteristics, and Processes, Marcel Dekker, USA, 2005 Search PubMed
- Y. Ohno, Proc. SPIE, 2004, 5530, 88
- H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, WILEY-VCH, Weinheim, 2008 Search PubMed
- R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093 CAS
- L. D. Carlos, R. A. S. Ferreira, V. D. Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509 CrossRef CAS
- J. Kido, K. Nagai and Y. Ohashi, Chem. Lett., 1990, 657 CAS
- J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357 CrossRef CAS
- M. A. Katkova, A. Vitukhnovsky and M. N. Bochkarev, Russ. Chem. Rev., 2005, 74, 1089 Search PubMed
- N. P. Kuz’mina and S. V. Eliseeva, Russ. J. Inorg. Chem., 2006, 51, 73 CrossRef
- W. C. H. Choy and D. Tao, in Solid State Chemistry Research Trends, ed. R. W. Buckley, Nova Science Publishers Inc., 2007, ch. 2, pp. 23–62 Search PubMed
- T. Oyamada, Y. Kawamura, T. Koyama, H. Sasabe and C. Adachi, Adv. Mater., 2004, 16, 1082 CrossRef CAS
- J. Fang, H. You, J. Gao and D. Ma, Chem. Phys. Lett., 2004, 392, 11 CrossRef CAS
- H. Xu, K. Yin and W. Huang, ChemPhysChem, 2008, 9, 1752 CrossRef CAS
- H. Xu, K. Yin and W. Huang, Chem.–Eur. J., 2007, 13, 10281 CrossRef
- E. Stathatos, P. Lianos, E. Evgeniou and A. D. Keramidas, Synth. Met., 2003, 139, 433 CrossRef CAS
- J. Yu, H. Zhang, L. Zhou, R. Deng, Z. Peng, Z. Li, L. Fu and Z. Guo, J. Lumin., 2007, 122–123, 678 CrossRef CAS
- J. F. Wang, R. Y. Wang, J. Yang, Z. P. Zheng, M. D. Carducci, T. Cayou, N. Peyghambarian and G. E. Jabbour, J. Am. Chem. Soc., 2001, 123, 6179 CrossRef CAS
- H. He, W. Li, Z. Su, T. Li, W. Su, B. Chu, D. Bi, L. Han, D. Wang, L. Chen, B. Li and Z. Zhang, et al., Solid-State Electron., 2008, 52, 31 CrossRef CAS
- X. Q. Wei, G. Yang, J. B. Cheng, Z. Y. Lu and M. G. Xie, Mater. Chem. Phys., 2007, 102, 214 CrossRef CAS
- Q. Xin, W. L. Li, W. M. Su, T. L. Li, Z. S. Su, B. Chu and B. Li, J. Appl. Phys., 2007, 101, 044512 CrossRef
- T. W. Canzler and J. Kido, Org. Electron., 2006, 7, 29 CrossRef CAS
- X. Y. Sun, W. L. Li, Z. R. Hong, Q. Xin, B. Chu, B. Li, Z. Q. Zhang and Z. Z. Hu, J. Phys. D: Appl. Phys., 2006, 39, 1363 CrossRef CAS
- K. Yin, H. Xu, G. Y. Zhong, G. Ni and W. Huang, Appl. Phys. A: Mater. Sci. Process., 2009, 95, 595 CrossRef CAS
- L. Rino, W. Simões, G. Santos, F. J. Fonseca, A. M. Andrade, V. A. F. Deichmann, L. Akcelrud and L. Pereira, J. Non-Cryst. Solids, 2008, 354, 5326 CrossRef CAS
- H. You, H. Z. Li, Q. Wang, L. X. Wang and D. Ma, J. Phys. D: Appl. Phys., 2007, 40, 1363 CrossRef CAS
- H. You, J. Fang, L. Wang, X. Zhu, W. Huang and D. Ma, Opt. Mater., 2007, 29, 1514 CrossRef CAS
- Y. Zhang, C. Li, H. H. Shi, B. Du, W. Yang and Y. Cao, New J. Chem., 2007, 31, 569 RSC
- J. Fang, C. Chan Choy, D. Ma and E. C. W. Ou, Thin Solid Films, 2006, 515, 2419 CrossRef CAS
- H. Jang, C. H. Shin, B. J. Jung, D. H. Kim, H. K. Shim and Y. Do, Eur. J. Inorg. Chem., 2006, 718 CrossRef CAS
- M. Guan, L. Gao, S. Wang, C. Huang and K. Wang, J. Lumin., 2007, 127, 489 CrossRef CAS
- W. G. Quirino, R. D. Adati, S. A. M. Lima, C. Legnani, M. Jafelicci, Jr, M. R. Davolos and M. Cremona, Thin Solid Films, 2006, 515, 927 CrossRef CAS
- Y. Lv, Q. Li, Y. Zhang, Y. Han, H. Liu, G. Wang, J. Li, D. Wu, Z. Yang, L. Wang and J. Xie, Solid-State Electron., 2008, 52, 1149 CrossRef CAS
- C. R. De Silva, F. Li, C. Huang and Z. Zheng, Thin Solid Films, 2008, 517, 957 CrossRef CAS
- Y. Lv, J. Zhang, Y. Fu, W. Cao, L. Song and Z. Xu, Mater. Lett., 2008, 62, 1107 CrossRef CAS
- Y. Lv, J. Zhang, W. Cao, J. C. Juan, F. Zhang and Z. Xu, Spectrochim. Acta, Part A, 2007, 68, 382 CrossRef
- Y. Lv, J. Zhang, L. Wang, W. Cao and Z. Xu, J. Lumin., 2008, 128, 117 CrossRef CAS
- W. Zhu, Q. Jiang, Z. Lu, X. Wei, M. Xie, D. Zou and T. Tsutsui, Synth. Met., 2000, 111–112, 445 CrossRef CAS
- H. Xin, F. Y. Li, M. Shi, Z. Q. Bian and C. H. Huang, J. Am. Chem. Soc., 2003, 125, 7166 CrossRef CAS
- Z. F. Li, L. Zhou, J. B. Yu, H. J. Zhang, R. P. Deng, Z. P. Peng and Z. Y. Guo, J. Phys. Chem. C, 2007, 111, 2295 CrossRef CAS
- M. Shi, F. Y. Li, T. Yi, D. Zhang, H. Hu and C. H. Huang, Inorg. Chem., 2005, 44, 8929 CrossRef CAS
- F. Zhang, Z. Xu, S. Zhao, L. Liu, B. Sun and J. Pei, Physica B: Condens. Matter, 2006, 381, 256 CAS
- F. Zhang, Z. Xu, S. Zhao, L. Wang and L. Lu, Solid-State Electron., 2008, 52, 1806 CrossRef CAS
- Y. Zhang, Z. Deng and R. Wang, J. Lumin., 2007, 122–123, 690 CrossRef CAS
- Z. Chen, Z. Deng, Y. Shi, Y. Xu, J. Xiao, Y. Zhang and R. Wang, J. Lumin., 2007, 122–123, 671 CrossRef CAS
- F. J. Zhang, S. L. Zhao, Z. Xu, J. Z. Huang, G. C. Yuan, Y. Li, Y. Wang and X. R. Xu, Opt. Mater., 2007, 30, 427 CrossRef CAS
- D. Guo, Z. Deng, C. Liang, P. Lin, Y. Li and Y. Xu, J. Lumin., 2007, 122–123, 683 CrossRef CAS
- Y. Shi, Z. Deng, J. Xiao, D. Xu, Z. Chen and R. Wang, J. Lumin., 2007, 122–123, 272 CrossRef CAS
- Y. Lv, J. Zhang, W. Cao, L. Song and Z. Xu, Spectrochim. Acta, Part A, 2008, 70, 253 CrossRef
- Y. G. Lv, J. C. Zhang, W. L. Cao, J. C. Juan, F. J. Zhang and Z. Xu, J. Photochem. Photobiol., A, 2007, 188, 155 CrossRef CAS
- H. B. Xu, X. F. Li and D. Yan, Inorg. Chem. Commun., 2008, 11, 1187 CrossRef CAS
- X. L. Zheng, Y. Liu, M. Pan, X. Q. Lu, J. Y. Zhang, C. Y. Zhao, Y. X. Tong and C. Y. Su, Angew. Chem., Int. Ed., 2007, 46, 7399 CrossRef CAS
- O. M. Khreis, W. P. Gillin, M. Somerton and R. J. Curry, Org. Electron., 2001, 2, 45 CrossRef CAS
- R. J. Curry and W. P. Gillin, Synth. Met., 2000, 111, 35 CrossRef
- R. J. Curry, W. P. Gillin, A. P. Knights and R. Gwilliam, Appl. Phys. Lett., 2000, 77, 2271 CrossRef CAS
- R. J. Curry, W. P. Gillin, A. P. Knights and R. Gwilliam, Opt. Mater., 2001, 17, 161 CrossRef CAS
- R. J. Curry and W. P. Gillin, Curr. Opin. Solid State Mater. Sci., 2001, 5, 481 CrossRef CAS
- H. Suzuki, J. Photochem. Photobiol., A, 2004, 166, 155 CrossRef CAS
- Y. Kawamura, Y. Wada, Y. Hasegawa, M. Iwamuro, T. Kitamura and S. Yanagida, Appl. Phys. Lett., 1999, 74, 3245 CrossRef CAS
- Y. Kawamura, Y. Wada, M. Iwamuro, T. Kitamura and S. Yanagida, Chem. Lett., 2000, 280 CrossRef CAS
- Y. Kawamura, Y. Wada and S. Yanagida, Jpn. J. Appl. Phys., 2001, 40, 350 CrossRef CAS
- R. G. Sun, Y. Z. Wang, Q. B. Zheng, H. J. Zhang and A. J. Epstein, J. Appl. Phys., 2000, 87, 7589 CrossRef
- Z. R. Hong, C. J. Liang, R. G. Li, D. Zhao, D. Fan and W. L. Li, Thin Solid Films, 2001, 391, 122 CrossRef CAS
- T. S. Kang, B. S. Harrison, M. Bouguettaya, T. J. Foley, J. M. Boncella, K. S. Schanze and J. R. Reynolds, Adv. Funct. Mater., 2003, 13, 205 CrossRef CAS
- T. S. Kang, B. S. Harrison, T. J. Foley, A. S. Knefely, J. M. Boncella, J. R. Reynolds and K. S. Schanze, Adv. Mater., 2003, 15, 1093 CrossRef CAS
- K. S. Schanze, J. R. Reynolds, J. M. Boncella, B. S. Harrison, T. J. Foley, M. Bouguettaya and T. S. Kang, Synth. Met., 2003, 137, 1013 CrossRef CAS
- B. S. Harrison, T. J. Foley, A. S. Knefely, J. K. Mwaura, G. B. Cunningham, T. S. Kang, M. Bouguettaya, J. M. Boncella, J. R. Reynolds and K. S. Schanze, Chem. Mater., 2004, 16, 2938 CrossRef CAS
- B. S. Harrison, T. J. Foley, M. Bouguettaya, J. M. Boncella, J. R. Reynolds, K. S. Schanze, J. Shim, P. H. Holloway, G. Padmanaban and S. Ramakrishnan, Appl. Phys. Lett., 2001, 79, 3770 CrossRef CAS
- Z. R. Hong, W. L. Li, D. X. Zhao, C. J. Liang, X. Y. Liu, J. B. Peng and D. Zhao, Synth. Met., 1999, 104, 165 CrossRef CAS
- K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B.V., Amsterdam, 2005, ch. 225, vol. 35 Search PubMed
- A. Ouchi, Y. Suzuki, Y. Ohki and Y. Koizumi, Coord. Chem. Rev., 1988, 92, 29 CrossRef CAS
- U. Giovanella, M. Pasini, C. Freund, C. Botta, W. Porzio and S. Destri, J. Phys. Chem. C, 2009, 113, 2290 CrossRef CAS
- Y. Liu, J. Li, C. Li, J. Song, Y. Zhang, J. Peng, X. Wang, M. Zhu, Y. Cao and W. Zhu, Chem. Phys. Lett., 2007, 433, 331 CrossRef CAS
- O. V. Kotova, V. V. Utochnikova, S. V. Eliseeva, S. V. Samoilenkov and N. P. Kuz'mina, Russ. J. Coord. Chem., 2007, 33, 454 CrossRef CAS
- B. W. D’Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585 CrossRef
- M. S. Arnold, G. J. Mcgraw, S. R. Forrest and R. R. Lunt, Appl. Phys. Lett., 2008, 92, 053301 CrossRef
- B. C. Krummacher, V. E. Choong, M. K. Mathai, S. A. Choulis, F. So, F. Jermann, T. Fiedler and M. Zachau, Appl. Phys. Lett., 2006, 88, 113506 CrossRef
- R. J. Xie and N. Hirosaki, Sci. Technol. Adv. Mater., 2007, 8, 588 CrossRef CAS
- J. Kido, W. Ikeda, M. Kimura and K. Nagai, Jpn. J. Appl. Phys., 1996, 35, L394 CrossRef CAS
- D. Zhao, W. Li, Z. Hong, X. Liu, C. Liang and D. Zhao, J. Lumin., 1999, 82, 105 CrossRef
- S. Woo Pyo, S. Phil Lee, H. Sung Lee, O. Kwau Kwon, H. Sue Hoe, S. Hee Lee, Y. K. Ha, Y. Kwan Kim and J. Soo Kim, Thin Solid Films, 2000, 363, 232 CrossRef CAS
- S. Li, G. Zhong, W. H. Zhu, F. Li, J. Pan, W. Huang and H. Tian, J. Mater. Chem., 2005, 15, 3221 RSC
- H. You and D. G. Ma, J. Phys. D: Appl. Phys., 2008, 41, 155113 CrossRef
- R. Shunmugam and G. N. Tew, Macromol. Rapid Commun., 2008, 29, 1355 CrossRef CAS
- R. Shunmugam and G. N. Tew, Polym. Adv. Technol., 2007, 18, 940 CrossRef CAS
- R. Shunmugam and G. N. Tew, Polym. Adv. Technol., 2008, 19, 596 CrossRef CAS
- P. Coppo, M. Duati, V. N. Kozhevnikov, J. W. Hofstraat and L. De Cola, Angew. Chem., Int. Ed., 2005, 44, 1806 CrossRef CAS
- J. W. Hofstraat, K. Brunner, R. T. Wegh, L. De Cola, E. A. Plummer and P. Coppo, Int. Pat., WO 2006/024997 A1, 2006.
- S. J. Rak, Y. C. Soo, K. C. Hoon and P. I. Woo, US Pat. Appl., 20080315752, 2008.
- H. S. Jang and D. Y. Jeon, US Pat. Appl., 20090085467, 2009.
- H. S. Jang, H. Yang, S. W. Kim, J. Y. Han, S. G. Lee and D. Y. Jeon, Adv. Mater., 2008, 20, 2696 CrossRef CAS
- H. S. Jang and D. Y. Jeon, Opt. Lett., 2007, 32, 3444 Search PubMed
- H. He, R. L. Fu, H. Wang, X. F. Song, Z. W. Pan, X. R. Zhao, X. L. Zhang and Y. G. Cao, J. Mater. Res., 2008, 23, 3288 CrossRef CAS
- C. C. Yang, C. M. Lin, Y. J. Chen, Y. T. Wu, S. R. Chuang, R. S. Liu and S. F. Hu, Appl. Phys. Lett., 2007, 90 Search PubMed
- Y. H. Won, H. S. Jang, K. W. Cho, Y. S. Song, D. Y. Leon and H. K. Kwon, Opt. Lett., 2009, 34, 1 Search PubMed
- M. Zeuner, F. Hintze and W. Schnick, Chem. Mater., 2009, 21, 336 CrossRef CAS
- C. Hecht, F. Stadler, P. J. Schmidt, J. Schmedt auf der Gunne, V. Baumann and W. Schnick, Chem. Mater., 2009, 21, 1595 CrossRef CAS
- N. J. Xiang, Y. Xu, Z. L. Wang, X. X. Wang, L. M. Leung, J. Wang, Q. Su and M. L. Gong, Spectrochim. Acta, Part A, 2008, 69, 1150 CrossRef
- Y. Cong, B. Li, S. Yue, Y. Liu, W. Li and X. J. Wang, J. Phys. Chem. C, 2009, 113, 493 CrossRef CAS
- Q. H. Zeng, H. B. Liang, M. L. Gong and Q. Sua, J. Electrochem. Soc., 2008, 155, H730 CrossRef CAS
- G. Denis, P. Deniard, E. Gautron, F. Clabau, A. Garcia and S. Jobic, Inorg. Chem., 2008, 47, 4226 CrossRef CAS
- A. A. Setlur, W. J. Heward, M. E. Hannah and U. Happek, Chem. Mater., 2008, 20, 6277 CrossRef CAS
- L. H. Slooff, A. van Blaaderen, A. Polman, G. A. Hebbink, S. I. Klink, F. C. J. M. Van Veggel, D. N. Reinhoudt and J. W. Hofstraat, J. Appl. Phys., 2002, 91, 3955 CrossRef CAS
- K. Kuriki, Y. Koike and Y. Okamoto, Chem. Rev., 2002, 102, 2347 CrossRef CAS
- F. Ondracek, J. Jagerska, L. Salavcova, M. Mika, J. Spirkova and J. Ctyroky, IEEE J. Quantum Electron., 2008, 44, 536 CrossRef CAS
- R. Salas-Montiel, L. Bastard, G. Grosa and J. E. Broquin, Mater. Sci. Eng. B, Solid State Mater. Adv. Technol., 2008, 149, 181 CAS
- X. Z. Zhang, K. Liu, S. K. Mu, C. Z. Tan, D. Zhang, E. Y. B. Pun and D. M. Zhang, Opt. Commun., 2006, 268, 300 CrossRef CAS
- N. D. Psaila, R. R. Thomson, H. T. Bookey, A. K. Kar, N. Chiodo, R. Osellame, G. Cerullo, A. Jha and S. Shen, Appl. Phys. Lett., 2007, 90, 131102 CrossRef
- C. Armellini, A. Chiappini, A. Chiasera, M. Ferrari, Y. Jestin, M. Mortier, E. Moser, R. Retoux and G. C. Righini, J. Nanomater., 2007 Search PubMed
- A. Kahn, H. Kuhn, S. Heinrich, K. Petermann, J. D. B. Bradley, K. Worhoff, M. Pollnau, Y. Kuzminykh and G. Huber, J. Opt. Soc. Am. B, 2008, 25, 1850 Search PubMed
- A. Kahn, S. Heinrich, H. Kuhn, K. Petermann, J. D. B. Bradley, K. Worhoff, M. Pollnau and G. Huber, Opt. Express, 2009, 17, 4412 CrossRef CAS
- S. Bär, H. Scheife, K. Petermann and G. Huber, Top. Appl. Phys., 2006, 106, 401
- L. Tsonev, Opt. Mater., 2008, 30, 892 CrossRef CAS
- A. Q. Le Quang, E. Besson, R. Hierle, A. Mehdi, C. Reyé, R. Corriu and I. Ledoux, Opt. Mater., 2007, 29, 941 CrossRef
- A. Q. L. Quang, V. G. Truong, A. M. Jurdyc, B. Jacquier, J. Zyss and I. Ledoux, J. Appl. Phys., 2007, 101, 023110-1
- C. Chen, D. Zhang, T. Li, D. M. Zhang, L. M. Song and Z. Zhen, Appl. Phys. Lett., 2009, 94, 041119-1
- S. Moynihan, R. Van Deun, K. Binnemans, J. Krueger, G. von Papen, A. Kewell, G. Crean and G. Redmond, Opt. Mater., 2007, 29, 1798 CrossRef CAS
- S. Moynihan, R. Van Deun, K. Binnemans and G. Redmond, Opt. Mater., 2007, 29, 1821 CrossRef CAS
- F. Chen, Crit. Rev. Solid State Mat. Sci., 2008, 33, 165 Search PubMed
- M. Malinowski, M. Nakielska, R. Piramidowicz and J. Sarnecki, Spectrosc. Lett., 2007, 40, 271 CrossRef CAS
- F. Auzel, Spectrosc. Lett., 2007, 40, 197 CrossRef CAS
- S. Bo, J. Wang, H. Zhao, H. Ren, Q. Wang, G. Xu, X. Zhang, X. Liu and Z. Zhen, Appl. Phys. B: Lasers Opt., 2008, 91, 79 CrossRef CAS
- D. Zhang, C. Chen, C. M. Chen, C. S. Ma, D. M. Zhang, S. Bo and Z. Zhen, Appl. Phys. Lett., 2007, 91 Search PubMed
- H. Y. Gan, L. Li, C. T. Derose, R. A. Norwood, C. R. De Silva, Z. P. Zheng and N. Peyghambarian, Proc. SPIE, 2007, 6469, B4690
- Y. Mao, J. Y. Huang, R. Ostroumov, K. L. Wang and J. P. Chang, J. Phys. Chem. C, 2008, 112, 2278 CrossRef CAS
- V. Toccafondo, S. Faralli and F. Di Pasquale, J. Lightwave Technol., 2008, 26, 3584 CrossRef CAS
- P. K. Sekhar, A. R. Wilkinson, R. G. Elliman, T. H. Kim and S. Bhansali, J. Phys. Chem. C, 2008, 112, 20109 CrossRef CAS
- H. Mataki, K. Tsuchii, J. Sun, H. Taniguchi, K. Yamashita and K. Oe, Jpn. J. Appl. Phys., 2007, 46, L83 CrossRef CAS
- H. Mataki, K. Tsuchii, N. Mibuka, A. Suzuki, J. S. H. Taniguchi, K. Yamashita and K. Oe, J. Photopolym. Sci. Technol., 2007, 20, 67 CrossRef CAS
- K. Yamashita, N. Takeuchi, H. Taniguchi, S. Yuyama, K. Oe, N. Mibuka, A. Suzuki and H. Mataki, J. Lumin., 2009, 129, 526 CrossRef CAS
- D. L. Andrews, J. Nanophoton., 2008, 2, 022502 Search PubMed
- S. R. Wenham and M. A. Green, Progr. Photovolt.: Res. Appl., 1996, 4, 3 Search PubMed
- B. S. Richards, Sol. Energy Mater. Sol. Cells, 2006, 90, 2329 CrossRef CAS
- B. S. Richards, Sol.
Energy Mater. Sol. Cells, 2006, 90, 1189 CrossRef CAS
- A. Shalav, B. S. Richards and M. A. Green, Sol. Energy Mater. Sol. Cells, 2007, 91, 829 CrossRef CAS
- B. S. Richards and A. Shalav, IEEE Trans. Electron Devices, 2007, 54, 2679 CrossRef CAS
- C. Strümpel, M. McCann, G. Beaucarne, V. Arkhipov, A. Slaoui, V. Svrcek, C. del Canizo and I. Tobias, Sol. Energy Mater. Sol. Cells, 2007, 91, 238 CrossRef
- K. R. McIntosh, G. Lau, J. N. Cotsell, K. Hanton, D. L. Batzner, F. Bettiol and B. S. Richards, Progr. Photovolt.: Res. Appl., 2009, 17, 191 Search PubMed
- S. Marchionna, F. Meinardi, M. Acciarri, S. Binetti, A. Papagni, S. Pizzini, V. Malatesta and R. Tubino, J. Lumin., 2006, 118, 325 CrossRef CAS
- T. Jin, S. Inoue, K. Machida and G. Adachi, J. Electrochem. Soc., 1997, 144, 4054 CrossRef CAS
- S. Ye, B. Zhu, J. Luo, Y. Teng, J. X. Chen, G. Lakshminarayana, G. D. Qian and J. R. Qiu, Appl. Phys. Lett., 2008, 93, 181110 CrossRef
- D. Q. Chen, Y. S. Wang, Y. L. Yu, P. Huang and F. Y. Weng, Opt. Lett., 2008, 33, 1884 Search PubMed
- Y. H. Wang, L. C. Xie and H. J. Zhang, J. Appl. Phys., 2009, 105, 023528-1
- L. C. Xie, Y. H. Wang and H. J. Zhang, Appl. Phys. Lett., 2009, 94, 061905-1
- D. Q. Chen, Y. S. Wang, Y. L. Yu, P. Huang and F. Y. Weng, J. Appl. Phys., 2008, 104, 116105-1
- X. B. Chen, J. G. Wu, X. L. Xu, Y. Z. Zhang, N. Sawanobori, C. L. Zhang, Q. H. Pan and G. J. Salamo, Opt. Lett., 2009, 34, 887 Search PubMed
- S. Ye, B. Zhu, J. Luo, J. X. Chen, G. Lakshminarayana and J. R. Qiu, Opt. Express, 2008, 16, 8989 CrossRef CAS
- X. Y. Huang and Q. Y. Zhang, J. Appl. Phys., 2009, 105, 053521 CrossRef
- F. Lahoz, Opt. Lett., 2008, 33, 2982 Search PubMed
- X. F. Liang, X. Y. Huang and Q. Y. Zhang, J. Fluoresc., 2009, 19, 285 CrossRef CAS
- B. C. Rowan, L. R. Wilson and B. S. Richards, IEEE J. Sel. Top. Quantum Electron., 2008, 14, 1312 CrossRef CAS
- S. Lian, C. Rong, D. Yin and S. Liu, J. Phys. Chem. C, 2009, 113, 6298 CrossRef CAS
- C. H. Evans, Biochemistry of the Lanthanides, Plenum Press, New York, 1990 Search PubMed
- R. A. Bulman, in Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker Inc., New York, 2003, ch. 17, vol. 40 Search PubMed
- I. Kostova, W. Kiefer and G. Momekov, Arch. Pharm. Chem. Life Sci., 2007, 340, 642 Search PubMed
- Y. Kubota, S. Takahashi, I. Takahashi and G. Patrick, Toxicol. Vitro, 2000, 309 Search PubMed
- K. Wang, in Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker Inc., New York, 2003, vol. 40, ch. 17 Search PubMed
- E. J. Werner, A. Datta, C. J. Jocher and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 8568 CrossRef CAS
- P. Hermann, J. Kotek, V. Kubicek and I. Lukes, Dalton Trans., 2008, 3027 RSC
- M. Woods, E. W. C. Donald and A. D. Sherry, Chem. Soc. Rev., 2006, 35, 500 RSC
- J. M. Idée, M. Port, I. Raynal, M. Schaefer, S. Le Greneur and C. Corot, Fundam. Clin. Pharmacol., 2006, 20, 563 CrossRef CAS
- I. Hemmilä and V. Laitala, J. Fluoresc., 2005, 15, 529 CrossRef CAS
- J. Yuan and G. Wang, TrAC, Trends Anal. Chem., 2006, 25, 490 CrossRef CAS
- T. Matsuya, N. Hoshino and T. Okuyama, Curr. Anal. Chem., 2006, 2, 397 CrossRef CAS
- G. Vereb, E. Jares-Erijman, P. R. Selvin and T. M. Jovin, Biophys. J., 1998, 74, 2210 CrossRef CAS
- S. Pandya, J. H. Yu and D. Parker, Dalton Trans., 2006, 2757 RSC
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS
- B. Song, G. Wang, M. Tan and J. Yuan, J. Am. Chem. Soc., 2006, 128, 13442 CrossRef CAS
- H. C. Manning, S. M. Smith, M. Sexton, S. Haviland, M. F. Bai, K. Cederquist, N. Stella and D. J. Bornhop, Bioconjugate Chem., 2006, 17, 735 CrossRef CAS
- S. Faulkner and J. L. Matthews, in Comprehensive Coordination Chemistry II, ed. M. D. Ward, Elsevier Pergamon, Amsterdam, 2004, ch. 9.21, vol. 9, pp. 913–44 Search PubMed
- L. C. Courrol and R. E. Samad, Curr. Pharm. Anal., 2008, 4, 238 Search PubMed
- S. Shinoda, H. Miyake and H. Tsukube, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, Jr, J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science B.V., Amsterdam, 2005, ch. 226, vol. 35 Search PubMed
- P. Escribano, B. Pez, A. Planelles, E. Cordoncillo, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23 RSC
- W. D. Horrocks, Jr and M. Albin, Prog. Inorg. Chem., 1984, 31, 1
- J.-C. G. Bünzli and J.-M. Pfefferlé, Helv. Chim. Acta, 1994, 77, 323 CrossRef
- A. M. Reynolds, B. R. Sculimbrene and B. Imperiali, Bioconjugate Chem., 2008, 19, 588 CrossRef CAS
- L. J. Martin, M. J. Hahnke, M. Nitz, J. Wohnert, N. R. Silvaggi, K. N. Allen, H. Schwalbe and B. Imperiali, J. Am. Chem. Soc., 2007, 129, 7106 CrossRef CAS
- P. R. Selvin, Nat. Struct. Biol., 2000, 7, 730 CrossRef CAS
- A. Duerkop, M. Turel, A. Lobnik and O. S. Wolfbeis, Anal. Chim. Acta, 2006, 555, 292 CrossRef CAS
- A. Yegorova, E. Vityukova, S. Beltyukova and A. Duerkop, Microchem. J., 2006, 83, 1 CrossRef CAS
- P. Schrenkhammer, I. C. Rosnizeck, A. Duerkop, O. S. Wolfbeis and M. Schaferling, J. Biomol. Screening, 2007, 13, 9 CrossRef
- A. Duerkop, D. Aleksandrova, Y. Scripinets, A. Yegorova and E. Vityukova, Ann. N. Y. Acad. Sci., 2008, 1130, 172 CrossRef CAS
- J. L. Worlinsky and S. Basu, J. Phys. Chem. B, 2009, 113, 865 CrossRef CAS
- T. Nishioka, J. Yuan, Y. Yamamoto, K. Sumitomo, Z. Wang, K. Hashino, C. Hosoya, K. Ikawa, G. Wang and K. Matsumoto, Inorg. Chem., 2006, 45, 4088 CrossRef CAS
- B. Song, C. D. B. Vandevyver, E. Deiters, A.-S. Chauvin, I. A. Hemmilä and J.-C. G. Bünzli, Analyst, 2008, 133, 1749 RSC
- V. Laitala, A. Ylikoski, H. M. Raussi, P. Ollikka and I. Hemmilä, Anal. Biochem., 2007, 361, 126 CrossRef CAS
- Y. Chen and Z. H. Lu, Anal. Chim. Acta, 2007, 587, 180 CrossRef CAS
- T. Soukka, T. Rantanen and K. Kuningas, Ann. N. Y. Acad. Sci., 2008, 1130, 188 CrossRef CAS
- A. Bodi, K. E. Borbas and J. I. Bruce, Dalton Trans., 2007, 4352 RSC
- A. Thibon and V. C. Pierre, Anal. Bioanal. Chem., 2009, 394, 107 CrossRef CAS
- S. E. Plush and T. Gunnlaugsson, Dalton Trans., 2008, 3801 RSC
- J. Massue, S. J. Quinn and T. Gunnlaugsson, J. Am. Chem. Soc., 2008, 130, 6900 CrossRef CAS
- C. M. G. dos Santos, A. J. Harte, S. J. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512 CrossRef CAS
- R. Pal, D. Parker and L. C. Costero, Org. Biomol. Chem., 2009, 7, 1525 RSC
- M. Halim, M. S. Tremblay, S. Jockusch, N. J. Turro and D. Sames, J. Am. Chem. Soc., 2007, 129, 7704 CrossRef CAS
- Y. Kataoka, D. Paul, H. Miyake, T. Yaita, E. Miyoshi, H. Mori, S. Tsukamoto, H. Tatewaki, S. Shinoda and H. Tsukube, Chem.–Eur. J., 2008, 14, 5258 CrossRef CAS
- H. Tsukube, K. Yano, A. Ishida and S. Shinoda, Chem. Lett., 2007, 36, 554 CrossRef CAS
- Y. Kataoka, S. Shinoda and H. Tsukube, J. Nanosci. Nanotechnol., 2009, 9, 655 CrossRef CAS
- I. Hyppänen, J. Holsa, J. Kankare, M. Lastusaari and L. Pihlgren, Ann. N. Y. Acad. Sci., 2008, 1130, 267 CrossRef CAS
- D. E. Cooper, A. D’Andrea, G. W. Faris, B. MacQueen and W. H. Wright, in Immunoassay and Other Bioanalytical Techniques, ed. J. M. Van Emon, CRC Press Taylor & Francis group, Boca Raton, FL, 2007, ch. 9, pp. 217–247 Search PubMed
- A. Sanjurjo, K.-H. Lau, D. Lowe, A. Canizales, N. Jiang, V. M. Wong, L. Jiang, L. V. Schneider, N. Mufti, R. T. Rewick, M. Johansson and K. Kardos, US Pat., 6039894, 2000.
- C. G. Morgan, S. Dad and A. C. Mitchell, J. Alloys Compd., 2008, 451, 526 CrossRef CAS
- C. G. Morgan and A. C. Mitchell, Biosens. Bioelectron., 2007, 22, 1769 CrossRef CAS
- H. J. M. A. Zijlmans, J. Bonnet, J. Burton, K. Kardos, T. Vail, R. S. Niedbala and H. J. Tanke, Anal. Biochem., 1999, 267, 30 CrossRef CAS
- K. Kuningas, T. Ukonaho, H. Pakkila, T. Rantanen, J. Rosenberg, T. Lovgren and T. Soukka, Anal. Chem., 2006, 78, 4690 CrossRef CAS
- T. Rantanen, H. Pakkila, L. Jamsen, K. Kuningas, T. Ukonaho, T. Lovgren and T. Soukka, Anal. Chem., 2007, 79, 6312 CrossRef CAS
- G. A. Kumar, C. W. Chen and R. E. Riman, Appl. Phys. Lett., 2007, 90, 093123 CrossRef
- Y. P. Du, Y. W. Zhang, L. D. Sun and C. H. Yan, J. Phys. Chem. C, 2008, 112, 405 CrossRef CAS
- Z. Y. Liu, G. S. Yi, H. T. Zhang, J. Ding, Y. W. Zhang and J. M. Xue, Chem. Commun., 2008, 694 RSC
- G. Chen, H. Liu, H. Liang, G. Somesfalean and Z. Zhang, J. Phys. Chem. C, 2008, 112, 12030 CrossRef CAS
- Z. Chen, W. Bu, N. Zhang and J. Shi, J. Phys. Chem. C, 2008, 112, 4378 CrossRef CAS
- Z. Chen, H. Chen, H. Hu, M. Yu, F. Li, Q. Zhang, Z. Zhou, T. Yi and C. Huang, J. Am. Chem. Soc., 2008, 130, 3023 CrossRef CAS
- T. Ukonaho, T. Rantanen, L. Jamsen, K. Kuningas, H. Pakkila, T. Lovgren and T. Soukka, Anal. Chim. Acta, 2007, 596, 106 CrossRef CAS
- N. Sergent, J. A. Levitt, M. A. Green and K. Suhling, Proc. SPIE, 2008, 6861, 68610K
- B. Song, V. Sivagnanam, C. D. B. Vandevyver, I. A. Hemmilä, H.-A. Lehr, M. A. M. Gijs and J.-C. G. Bünzli, Analyst, 2009 10.1039/b911301k
- S. Phimphivong and S. S. Saavedra, Bioconjugate Chem., 1998, 9, 350 CrossRef CAS
- J. H. Yu, D. Parker, R. Pal, R. A. Poole and M. J. Cann, J. Am. Chem. Soc., 2006, 128, 2294 CrossRef CAS
- L. J. Charbonnière, N. Hildebrandt, R. F. Ziessel and H. G. Lohmannsroben, J. Am. Chem. Soc., 2006, 128, 12800 CrossRef CAS
- K. Hanaoka, K. Kikuchi, S. Kobayashi and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13502 CrossRef CAS
- F. Kielar, G. L. Law, E. J. New and D. Parker, Org. Biomol. Chem., 2008, 6, 2256 RSC
- J.-C. G. Bünzli, S. Comby, A.-S. Chauvin and C. D. B. Vandevyver, J. Rare Earths, 2007, 25, 257 CrossRef
- C. D. B. Vandevyver, A.-S. Chauvin, S. Comby and J.-C. G. Bünzli, Chem. Commun., 2007, 1716 RSC
- A.-S. Chauvin, S. Comby, B. Song, C. D. B. Vandevyver and J.-C. G. Bünzli, Chem.–Eur. J., 2007, 13, 9515 CrossRef
- A.-S. Chauvin, S. Comby, B. Song, C. D. B. Vandevyver and J.-C. G. Bünzli, Chem.–Eur. J., 2008, 14, 1726 CrossRef
- E. Deiters, B. Song, A.-S. Chauvin, C. D. B. Vandevyver and J.-C. G. Bünzli, New J. Chem., 2008, 32, 1140 RSC
- E. Deiters, B. Song, A.-S. Chauvin, C. Vandevyver and J.-C. G. Bünzli, Chem.–Eur. J., 2009, 15, 885 CrossRef CAS
- B. Song, C. D. B. Vandevyver, A.-S. Chauvin and J.-C. G. Bünzli, Org. Biomol. Chem., 2008, 6, 4125 RSC
- K. L. Wong, G. L. Law, M. B. Murphy, P. A. Tanner, W. T. Wong, P. K.-S. Lam and M. H.-W. Lam, Inorg. Chem., 2008, 47, 5190 CrossRef CAS
- J. Wu, Z. Ye, G. Wang, D. Jin, J. Yuan, Y. Guang and J. Piper, J. Mater. Chem., 2009, 19, 1258 RSC
- H. Bazin, M. Preaudat, E. Trinquet and G. Mathis, Spectrochim. Acta, Part A, 2001, 57, 2197 CrossRef CAS
- S. Ghose, E. Trinquet, M. Laget, H. Bazin and G. Mathis, J. Alloys Compd., 2008, 451, 35 CrossRef CAS
- M. I. Gaiduck, V. V. Grigoryants, A. F. Mironov, L. D. Roitman, V. I. Chissov, V. D. Rumiantseva and G. M. Sukhin, Dokl. Acad. Nauk. SSSR, 1989, 309, 980
- M. I. Gaiduck, V. V. Grigoryants, A. F. Mironov, V. D. Rumyantseva, V. I. Chissov and G. M. Sukhin, J. Photochem. Photobiol., B, 1990, 7, 15 CrossRef CAS
- M. Tsvirko, Y. Korovin and N. Rusakova, J. Phys. Conf. Ser., 2007, 79, 012025 CrossRef
- X. F. Yu, L. D. Chen, M. Li, M. Y. Xie, L. Zhou, Y. Li and Q. Q. Wang, Adv. Mater., 2008, 20, 4118 CrossRef CAS
- A. Dadabhoy, S. Faulkner and P. G. Sammes, J. Chem. Soc., Perkin Trans. 2, 2002, 348 RSC
- C. Xu, W. Zipfel, J. B. Shear, R. M. Williams and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10763 CrossRef CAS
- G. S. He, L. S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029 CrossRef CAS
- A. D'Aléo, G. Pompidor, B. Elena, J. Vicat, P. L. Baldeck, L. Toupet, R. Kahn, C. Andraud and O. Maury, ChemPhysChem, 2007, 8, 2125 CrossRef CAS
- R. G. Denning, Eur. J. Solid State Inorg. Chem., 1991, 28, 33 CAS
- A. D’Aleo, A. Picot, P. L. Baldeck, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10269 CrossRef CAS
- P. Kadjane, M. Starck, F. Camerel, D. Hill, N. Hildebrandt, R. Ziessel and L. J. Charbonniere, Inorg. Chem., 2009, 48, 4601 CrossRef CAS
- M. H. V. Werts, N. Nerambourg, D. Pelegry, Y. Le Grand and M. Blanchard-Desce, Photochem. Photobiol. Sci., 2005, 4, 531 RSC
- X. F. Wen, M. Y. Li, Y. Wang, J. P. Zhang, L. M. Fu, R. Hao, Y. Ma and X. C. Ai, Langmuir, 2008, 24, 6932 CrossRef CAS
- R. Hao, M. Li, Y. Wang, J. P. Zhang, Y. Ma, L. M. Fu, X. F. Wen, Y. S. Wu, X. C. Ai, S. Zhang and Y. Wei, Adv. Funct. Mater., 2007, 17, 3663 CrossRef CAS
- G. L. Law, K. L. Wong, Y. Y. Yang, H. L. Yang, W. T. Wong, M. H. W. Lam, H. L. Tam and K. W. Cheah, J. Fluoresc., 2008, 18, 749 CrossRef CAS
- L. M. Fu, X. F. Wen, X. C. Ai, Y. Sun, Y. S. Wu, J. P. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2005, 44, 747 CrossRef CAS
- X. Lu, W. Bi, W. Chai, J. Song, J. Meng, W. Y. Wong, W. K. Wong, X. Yang and R. A. Jones, Polyhedron, 2009, 28, 27 CrossRef
- K. L. Wong, W. M. Kwok, W. T. Wong, D. L. Phillips and K. W. Cheah, Angew. Chem., Int. Ed., 2004, 43, 4659 CrossRef CAS
- G. L. Law, W. M. Kwok, W. T. Wong, K. L. Wong and P. A. Tanner, J. Phys. Chem. B, 2007, 111, 10858 CrossRef CAS
- K. L. Wong, G. L. Law, W. M. Kwok, W. T. Wong and D. L. Phillips, Angew. Chem., Int. Ed., 2005, 44, 3436 CrossRef CAS
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73 CrossRef CAS
- P. Hänninen, J. Soukka and J. T. Soini, Ann. N. Y. Acad. Sci., 2008, 1130, 320 CrossRef CAS
- H. M. Kim and B. R. Cho, Acc. Chem. Res., 2009, 42, 863 CrossRef CAS
- S. Maiti, J. B. Shear, R. M. Williams, W. R. Zipfel and W. W. Webb, Science, 1997, 275, 530 CrossRef CAS
- L. O. Palsson, R. Pal, B. S. Murray, D. Parker and A. Beeby, Dalton Trans., 2007, 5726 RSC
- G. L. Law, K. L. Wong, C. W. Y. Man, W. T. Wong, S. W. Tsao, M. H. W. Lam and P. K. S. Lam, J. Am. Chem. Soc., 2008, 130, 3714 CrossRef CAS
- W. P. W. Lai, W. T. Wong, B. K. F. Li and K. W. Cheah, New J. Chem., 2002, 26, 576 RSC
- G. F. White, K. L. Litvinenko, S. R. Meech, D. L. Andrews and A. J. Thomson, Photochem. Photobiol. Sci., 2004, 3, 47 RSC
- A. Picot, A. D’Aleo, P. L. Baldeck, A. Grichine, A. Duperray, C. Andraud and O. Maury, J. Am. Chem. Soc., 2008, 130, 1532 CrossRef CAS
- F. Kielar, A. Congreve, G. L. Law, E. J. New, D. Parker, K. L. Wong, P. Castreno and J. de Mendoza, Chem. Commun., 2008, 2435 RSC
- G. Piszczek, I. Gryczynski, B. P. Maliwal and J. R. Lakowicz, J. Fluoresc., 2002, 12, 15 CrossRef CAS
- S. F. Lim, R. Riehn, W. S. Ryu, N. Khanarian, C. K. Tung, D. Tank and R. H. Austin, Nano
Lett., 2006, 6, 169 CrossRef CAS
- D. K. Chatterjee, A. J. Rufaihah and Y. Zhang, Biomaterials, 2008, 29, 937 CrossRef CAS
- M. X. Yu, F. Y. Li, Z. G. Chen, H. Hu, C. Zhan, H. Yang and C. H. Huang, Anal. Chem., 2009, 81, 930 CrossRef CAS
- L. N. Puntus, K. A. Lyssenko, I. Pekareva and J.-C. G. Bünzli, J. Phys. Chem. B, 2009, 113, 9265 CrossRef CAS
- C. Daiguebonne, N. Kerbellec, O. Guillou, J.-C. G. Bünzli, F. Gumy, L. Catala, T. Mallah, N. Audebrand, Y. Gérault, K. Bernot and G. Calvez, Inorg. Chem., 2008, 47, 3700 CrossRef CAS
- N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. G. Bünzli, Inorg. Chem., 2009, 48, 5611 CrossRef CAS
- S. Petoud, Annual meeting, COST Action D38, Warsaw, April 25–27, 2009, abstract O1.
- A. Picot, F. Malvolti, B. LeGuennic, P. L. Baldeck, J. A. G. Williams, C. Andraud and O. Maury, Inorg. Chem., 2007, 46, 2659 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1 and S2, and Schemes S1–S3 (photophysical properties of NIR emitting NdIII, ErIII and YbIII molecular compounds). See DOI: 10.1039/b905604c |
| ‡ For integration, spectra must be corrected for the instrumental function and expressed in terms of photons/s versus wavelength. |
| § A gamma function calculator can be found at: http://functions.wolfram.com/webMathematica/FunctionEvaluation.jsp?name=Gamma |
| ¶ The value of 441 μs reported in ref. 68 is erroneous (see J. Mater. Sci: Mater. Electron., 2009, 20, 788). |
| This journal is © The Royal Society of Chemistry 2010 |