Recent advances in crystal engineering
Christer B.
Aakeröy
*a,
Neil R.
Champness
*b and
Christoph
Janiak
*c
aDepartment of Chemistry, Kansas State University, Manhattan, KS 66506, USA. E-mail: aakeroy@ksu.edu
bSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, UK. E-mail: neil.champness@ nottingham.ac.uk; Fax: +44 (0)115 9513563; Tel: +44 (0)115 9513490
cInstitut für Anorganische und Analytische Chemie, Universität Freiburg, Albertstr. 21, 79104, Freiburg, Germany. E-mail: janiak@uni-freiburg.de; Fax: +49 761 2036147; Tel: +49 761 2036127
First published on 16th November 2009
Abstract
The articles published in the tenth anniversary issue of CrystEngComm are reviewed. The issue highlighted the state-of-the-art of crystal engineering and new trends and developing areas in crystal engineering. In particular, the following article emphasises developments in the areas of intermolecular interactions, notably hydrogen and halogen bonds; metal–organic frameworks or coordination polymers; polymorphism and solvates.
 Christer B. Aakeröy | Christer Aakeröy obtained an MSc in Chemistry from Uppsala University, Sweden, and a PhD (1990) from the University of Sussex, UK with K. R. Seddon. In 1992, Aakeröy won a Fellowship from the Swedish National Academy of Science that allowed him to go to the University of Minnesota and spend time in the laboratories of Peggy Etter and Jan Almlöf. In 1993 he became a Lecturer in Inorganic Chemistry at the Queen's University of Belfast. In 1996 he accepted a position as an Assistant Professor at Kansas State University. He was promoted to Associate Professor in 2001, and to Full Professor in 2006. Aakeröy was an invited visiting Professor at Universite Louis Pasteur, Institute Le Bel, Strasbourg in 2000 and 2006. Christer is also the CrystEngComm Associate Editor for the Americas. |
 Neil R. Champness | Neil Champness is Professor of Chemical Nanoscience at the University of Nottingham. He obtained a BSc in Chemistry (1989) at the University of Southampton and PhD (1993) under the supervision of Prof. W. Levason. Following postdoctoral studies in Southampton with Dr G. Reid, he moved to the University of Nottingham in 1995 as a Teaching Fellow in Inorganic Chemistry. He took up a Lectureship in Inorganic Chemistry in 1998 and was promoted to Reader in Chemistry in 2003. He was appointed to the Chair of Chemical Nanoscience in 2004. Neil is currently chair of the editorial board of CrystEngComm having served on the editorial board since 2003. He is a Senior Advisor to the British Council on matters relating to Science and Engineering. His research achievements have been recognised by the award of the Royal Society of Chemistry Corday-Morgan medal in 2006 and the Bob Hay lectureship by the RSC UK Macrocycles and Supramolecular Chemistry Group in 2005. |
 Christoph Janiak | Christoph Janiak studied chemistry at the TU Berlin and the University of Oklahoma in Norman with his MSc (1984) and PhD degree (1987) under the supervision of Prof. Jerold J. Zuckerman and Prof. Herbert Schumann, respectively. After postdoctoral work with Prof. Roald Hoffmann at Cornell University and with the polyolefin division of BASF AG at Ludwigshafen, he returned to the TU Berlin for his “habilitation” which was concluded in 1995. In 1996 he moved to the University of Freiburg as a professor of inorganic and analytical chemistry. |
1. Introduction
In 2008 CrystEngComm celebrated its tenth anniversary. Over the preceding ten years the journal had expanded from its earliest days as one of the first “electronic only” journals to become the primary journal for those publishing in the field of crystal engineering.1 The journal's tenth birthday was celebrated with an issue that contained articles by leading researchers in the field from across the world and across the many facets of the research discipline. This article reviews those articles and puts them in the wider context of the growing area of crystal engineering. In many ways this Highlight is a partner, or perhaps younger sibling, of the article published in 2005 in CrystEngComm by Braga, Brammer and Champness,2 which highlighted the development of the discipline as discussed and published on the occasion of CrystEngComm Discussion 2 (CECD2) held in Nottingham at the end of 2004.In many ways crystal engineering has grown since the inception of CrystEngComm notably in the development of various areas of application, for example gas storage in metal–organic frameworks,3–8 and in the development of our understanding of polymorphism.9 Other areas remain matters for ongoing discussion, not least in terms of topology and the debate surrounding co-crystals.10
Running through all the areas of crystal engineering remain the themes surrounding intermolecular interactions or as they have become commonly known, supramolecular synthons.11 Although hydrogen bonding and coordination bonds still remain at the forefront of crystal engineering strategies other interactions have received increasing attention over recent years, notably halogen bonds12–14 and π–π interactions.15 This article endeavours to highlight the latest developments in the use of all of these interactions and show how they have been used in disparate fields.
Another significant area of progress in crystal engineering has been the development of new techniques that are being used to evaluate structure and function of the systems from high pressure crystallography16 through to the use of solid-state NMR spectroscopy.17 The utilization of new techniques to understand crystalline-state phenomena further support the realization that crystal engineering has expanded in the last decade. An area which may once have been criticized as ‘just’ looking at crystal structures has clearly developed into one of the most vibrant areas of modern chemical research.
The expansion of crystal engineering is amply demonstrated by an area that has received perhaps the greatest level of attention, that of coordination polymers or as they have commonly become known metal–organic frameworks (MOFs).3,18 Over the last ten years MOFs have become one of the most studied areas of chemistry and materials science, underpinned by a fascination with their properties, notably in gas storage3–8 but also in many other applications.19–29 Although many groups direct their research on MOFs towards materials applications there is still considerable and invaluable research into the “design” of such compounds18,30 and synthetic developments are still a major focus for coordination polymer research. This article will highlight developments in this field and illustrate how far crystal engineering has developed over the last ten years.
This Highlight article brings together many of the recent developments in crystal engineering and tries to emphasise the directions which have been taken over recent years. From this collection of articles it is abundantly clear that crystal engineering is still in it's infancy with many developments still emerging.
2. Intermolecular interactions at the heart of crystal engineering
The expansion of crystal engineering as a research field has been accompanied by significant interest in the nature of intermolecular interactions and their subsequent use in the formation of solid-state structures. The arrangement of molecules in the solid-state underpins much of modern crystal engineering and thus the subject continues to be driven by the understanding of intermolecular interactions and their use in trying to control solid-state structures. Of the interactions typically encountered two still dictate much of the research reported, hydrogen bonds and coordination bonds. The uses of both of these interactions are covered in this Highlight but attention is also given to other interactions including those that may be considered “up and coming” interactions such as the halogen bond.2.1 Hydrogen bonds in crystal engineering
The question whether interchanging similarly sized substituents (e.g. chloro/methyl) will affect a crystal structure in a significant way has been a long-standing issue at the core of crystal engineering. Since the days of Kitaigorodskii,31 close-packing has been viewed as an important driving force for achieving the most stable lattice of an organic solid. Although about 30% (out of 105) of the pairs of molecules where a chlorine/methyl substitution has been made are isostructural,32 (based upon data from the CSD33) much more work is needed to establish the balance between “close-packing effects” and more directional and anisotropic intermolecular interactions.In order to examine the structural balance between chloro/methyl substituents, Polito et al.34 presented a structural and theoretical analysis of 2-methylbenzoic acid, 2-chlorobenzoic acid, and of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-crystal containing both components. The structure determinations show that the primary hydrogen-bond interactions leading to an infinite ribbon, Fig. 1, are present in all three structures, although they are not isostructural as there are differences in the way in which the ribbons are arranged within each lattice.
:1 co-crystal containing both components. The structure determinations show that the primary hydrogen-bond interactions leading to an infinite ribbon, Fig. 1, are present in all three structures, although they are not isostructural as there are differences in the way in which the ribbons are arranged within each lattice.
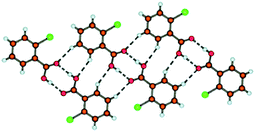 | ||
| Fig. 1 The infinite ribbon present in 2-chloro-benzoic acid, 2-methylbenzoic acid and in their 1:1 co-crystal. The green spheres represent both methyl and chloro substituents.34 | ||
Powder X-ray diffraction studies also indicated that there may be an additional polymorph of 2-methylbenzoic acid. The crystal structures were subsequently compared with hypothetical structures obtained from lattice energy minimizations (allowing for an interchange of substituents). The authors show that the energies of all structures are close, although the observed structures are slightly more favoured (1–2 kJ mol−1) compared with the hypothetically generated isomorphous analogues. The computer simulations do produce other plausible low-energy structures, which is consistent with the experimental observation of polymorphism in 2-methylbenzoic acid. This study underscores how difficult it is to translate information about molecular structure into predictions regarding crystal structure. Primary structural motifs, governed by relatively strong interactions, often display considerable consistencies (this is the practical foundation for “supramolecular synthons”11), whereas weaker and less well-defined interactions can dramatically alter the 3D arrangement of the dominating structural motifs. One of the true challenges in crystal engineering is to improve our understanding of how the balance between strong/weak forces determines the precise result of crystallization and solid-state assembly.
Structural similarities and synthon equivalencies are also addressed in a paper by Nangia and co-workers.35 The basis for their study is a set of crystal structures of Ag/urea-pyridyl complexes. The ligand, Scheme 1, is systematically varied such that the relative structural influence of one substituent can be examined in detail.
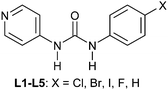 | ||
| Scheme 1 35 | ||
The Ag-complexes with L1–L3 are essentially isostructural and the geometry around the metal ion is shown in Fig. 2.
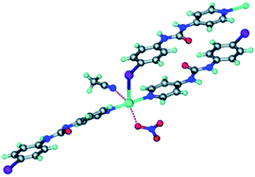 | ||
| Fig. 2 The Ag(I) ion is coordinated by one anion, one MeCN solvent molecule, two pyridyl moieties and is in close proximity of a halogen atom from the ligand.35 | ||
As expected, the Ag–X distance decreases with increasing polarizability of the halogen atom Cl < Br < I, and the authors suggest that the iodine atom is coordinated to the silver ion, whereas the R–Br and R–Cl are merely in close contact with the metal ion. Regardless of the precise nature of the metal-halogen interactions, all three structures are isostructural. In each case, the halogen atom is playing two different structural roles simultaneously. The electron-rich equatorial region of the halogen atom is oriented towards the metal cation, whereas the electron-deficient ‘tip’ of the atom is in close proximity of a neighbouring oxygen atom from the nitrate counterion. The three remaining structures were obtained with L4 (two with different chemical composition) and L5, and neither of the two former compounds contain a coordination complex with a short contact between Ag(I) and the fluorine atom on the ligand.
In what may be considered a complementary study to that of Nangia and co-workers,35 Swift et al. elegantly demonstrate the use of halogen bonds in a study of the structural arrangements of a range of meta-substituted diphenylurea derivatives, 1,3-bis(m-di-X-phenyl)ureas where X is Cl, Br or I.36 Interestingly growth of crystals of this family of compounds using a variety of growth conditions leads to isostructural and isomorphous phases of the compounds, including mono- and di-tolyl analogues, which crystallize in the space group P21212. The structural arrangement is a direct result of a number of intermolecular arrangements, inter-urea [R12(6)] hydrogen bonds forming chain structures, accompanied by π–π interactions between the phenyl substituents reinforcing the formation of the chains. Adjacent chains interact in an antiparallel fashion through halogen⋯halogen contacts (Fig. 3). The conformation of the compounds observed in these structures was found to be close in energy to that of the conformation found from ab initio calculations and the orthorhombic phase observed was also found to be the dominant crystalline form for all the compounds studied using powder X-ray diffraction, although small amounts of additional peaks were found to be present under some crystallization conditions indicating the formation of either other polymorphs or solvates.
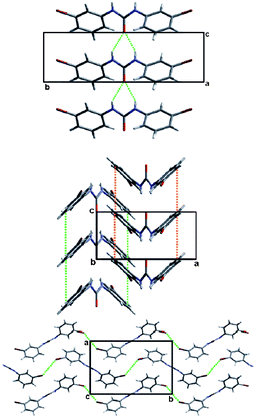 | ||
| Fig. 3 View of the structure of 1,3-bis(m-di-Br-phenyl)urea along the (top) a-, (middle) b- or (bottom) c-axis. Significant intermolecular interactions are shown using dotted lines showing hydrogen-bonds (top), π–π interactions (middle) or halogen bonds (bottom).36 | ||
Although the hydrogen bond is undoubtedly the most effective and widely used tool for supramolecular assembly, it is by no means the only one available for crystal engineering. Rissanen and co-workers37 have demonstrated the importance of cation⋯π interactions for the formation of molecular capsules constructed from cavitand-based building blocks. Resorcinarenes are well-known bowl-shaped molecules that have been used extensively as host molecules for electron-deficient guests due to their electron-rich, concave interior.38,39 In order to bring together two cavitands into a discrete molecular capsule, the cavitands need to be functionalized with groups capable of directional and complementary intermolecular interactions, or a templating guest may be required.40 In this new study,37 the role of the cation (structure/shape) in affecting cavitand assembly was examined, and five new crystal structures were presented. An outline of the different components examined is presented in Scheme 2.
 | ||
| Scheme 2 37 | ||
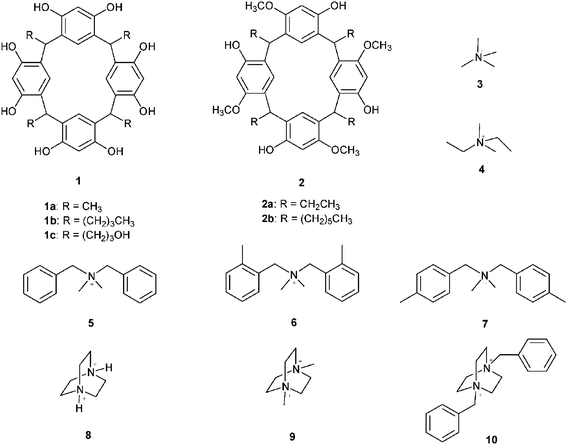
In two of the five structures, a dimeric capsule was obtained. In the first capsule, composed of 1b and tetramethylammonium bromide (3Br) (Fig. 4), solvent molecules act as the glue between the resorcinarene halves, with the cation encapsulated within the host, and the bromide ion located between the alkyl ‘feet’ of one of the cavitands, Fig. 3a–b.
 | ||
| Fig. 4 The molecular capsule in the crystal structure of 1b/3Br shown in schematic (a) and space-filling (b) mode to the left.37 | ||
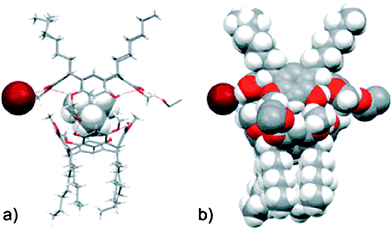
The second capsule was produced in the reaction between 2b and 3Br but this time, solvent molecules were not acting as bridges between the two halves. The anion is located closer to the equatorial part of the capsule, but it does not bridge two halves. Again, the cation is completely encapsulated by the host, and despite the fact that the host has no opportunity for strong intermolecular hydrogen bonds that can facilitate capsule formation, the dimeric assembly is accomplished by several cation⋯π/CH⋯π interactions.
The remaining three structures contain larger cations that are unable to fit within the interior of a capsule resulting in an ‘open complex’, which is illustrated in the crystal structure obtained on the product from the reaction between 1c and 7Br, Fig. 5.
 | ||
| Fig. 5 The size of the cation in the crystal structure of 1c/7Br prevents the assembly and formation of dimeric capsules.37 | ||
It is clear that directed assembly of large host molecules is made even more complicated in the presence of isotropic anions that can interact effectively with cations, neutral hosts, and solvent molecules in an unpredictable manner.
Solid-state host–guest chemistry remains an important and highly productive aspect of crystal engineering, and Bishop and co-workers41 examined the inclusion behaviour of a dialcohol, endo-4-,endo-8-dimethylbicyclo[3,3,1]nonane-endo-2,endo-6-diol, D1, which can be viewed as a supramolecular relative of Dianin's compound. Dianin's compound42 is a well-known clathrand capable of hosting a variety of guests, typically in ratios of 6:1 or 3:1 (host–guest). The ‘waist’ of the hour-glass shape of the host cage43 is a result of an R66(12) motif of O–H⋯O hydrogen bonds, Scheme 3.
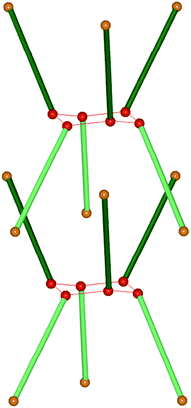 | ||
| Scheme 3 One cage in the Dianin's compound where each molecule is represented by a solid green rod (opposite enantiomers light or dark) with a solid sphere at each end (red, hydroxy group; orange, ether oxygen).41 | ||
The authors now demonstrate that D1 is also an excellent supramolecular host with an accessible void space of approximately 78 Å3. These host cages are part of a complex 3D network dominated by cyclic (O–H)6 hydrogen bonds, Fig. 6.
 | ||
| Fig. 6 One cage in the crystal structure of D13·(benzene).41 | ||
A comparison of the structure of Dianin's compound and inclusion compounds based on D1 shows that the six cyclic hydrogen-bonded motifs in D1 have taken on the same role as the six equatorially located rings in the former. This study provides an educational example of how very different molecular species can, through complementary hydrogen-bond interactions, produce similar supramolecular entities. Dianin's compound, D1, as well as hydroquinone,44 all display similar solid-state behaviour, which is best appreciated by employing nodal points as a tool for examining and classifying complex crystal structures.45
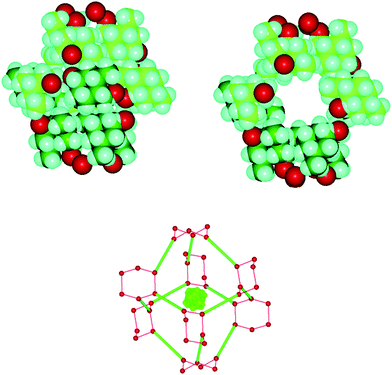
Hardie and co-workers have provided considerable information about the synthesis and host–guest properties of various CTV-derivatives (CTV = cyclotriveratrylene), and in a recent paper, they explored the ability of cyclotriguaiacylen (CTG)-based scaffolds to act as metal-binding ligands.46 Much work on cavitand-containing molecules such as calixarenes, calixresorcinarenes, CTV, and CTG derivatives is geared towards the construction of discrete capsules comprising two, or more, cavitands. The assembly of such constructions has to be achieved through non-covalent means and, consequently, hydrogen bonds and reversibly-formed metal–ligand bonds are the most common synthetic tools for such supramolecular targets. The CTG derivative examined in this new contribution47 has been modified in such a way that both hydrogen-bond based, as well as metal–ligand driven assembly is possible thanks to the presence of three pyridyl moieties at the rim of the CTG backbone, L6, Scheme 4.
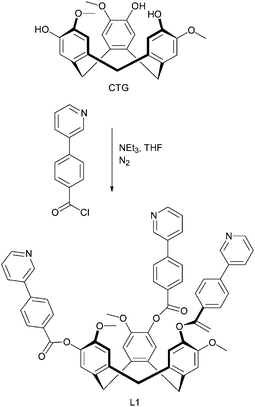 | ||
| Scheme 4 Synthesis of L6.47 | ||
The crystal structure determination of L6 itself reveals a capsule-like arrangement of molecules belonging to different assemblies which, in turn, are held together by C–H⋯N(py) hydrogen bonds. Each capsule contains two CHCl3 molecules, Fig. 7. In an attempt to create metallo-capsules L6 was subsequently allowed to react with a variety of Ag(I) salts, which produced three new crystals that were nearly identical albeit with different counterions, BF4−, AsF6−, and ClO4−, respectively.
2·3(MeCN) (a) One trigonal bipyramidal coordination cage. (b) Three cages bridged by μ3-Cl− with disordered MeCN guests shown in green.47](/image/article/2010/CE/b919819a/b919819a-f7.gif) | ||
| Fig. 7 From the crystal structure of [Ag3(MeCN)3(L6)2Cl](BF4)2·3(MeCN) (a) One trigonal bipyramidal coordination cage. (b) Three cages bridged by μ3-Cl− with disordered MeCN guests shown in green.47 | ||
The key feature in all three structures is a trigonal bipyramidal coordination capsule composed of two ligands, three silver ions, and one molecule of MeCN, Fig. 7. Each silver ion also acts as a μ3-bridge between three capsules, which overall produces a 2D layer with 63 topology. The inherent symmetry in the ligand is propagated throughout the lattice, and it is also interesting to note that both the ligand itself and the metallo-complex display similar dimeric capsules. Although coordination polymers that contain dimeric metallo-capsule hosts are quite rare,48 they are likely to become more important as we gain better control over the way in which such architectures with predictable metrics and topologies can be assembled.
Although most multi-component solids are assembled through the use of hydrogen bonds, there is a long history of charge-transfer (CT) complexes49 that can also be viewed as co-crystals; the driving force for assembly is typically a π⋯π* transition between an electron-rich and an electron-poor compound. Kuroda et al. have examined both solution synthesis and solid-state grinding in the preparation of a series of colourful CT complexes involving benzoquinone (BQ) racemic bis-β-naphthol (rac-BN), BQ and biphenyl (BF).50 In addition to one new crystal structure, several previously reported compounds are re-examined,51 and the authors demonstrate that the outcome of a reaction is heavily dependant upon the method of preparation. Colour changes take place as compounds are ground together, and the reactions can be monitored using powder X-ray diffraction, Fig. 8.
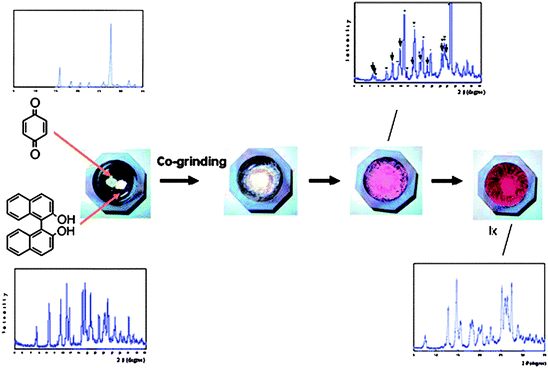
The authors offer a detailed structural analysis of two-component and three-component systems and show how the method of preparation influences (and indeed controls) the stoichiometry and structure of the product. For example, five of nine three-component charge-transfer systems yielded different products in solution vs. solid-state based synthesis.
The concept of the supramolecular synthon, introduced by Desiraju in 1995,11 remains a key aspect in crystal engineering, and provides a focal point for the recognition of complementary intermolecular interactions that can be used as synthetic tools in the reliable construction of complex supramolecular architectures. Bourne and co-workers51 have examined the crystal structures of a series of achiral benzylammonium benzoates, in order to establish if they contain either of the two most common motifs that are known to be present in chiral analogues,52Scheme 5.
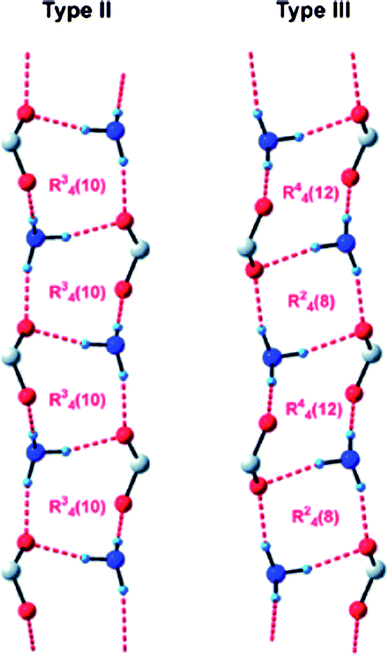 | ||
| Scheme 5 52 | ||
In addition, since in all five structures reported, 3-iodobenzoate was employed as the anion, the authors also decided to examine what, if any, halogen-bond based synthons53 could be identified in this series, Scheme 6. In three of the five cases, a type II interaction was found, whereas in two structures a type I halogen bond appeared (there was also one case of a C–N⋯I interaction). As for the hydrogen-bond based motifs, three structures contained the type II ladder like arrangement, whereas two compounds, benzylammonium 3-iodobenzoate and (3-pyridylmethyl)ammonium 3-iodobenzoate form structures that contain 2D sheets and a different 1D column, respectively.
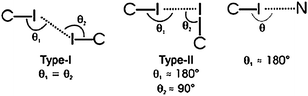 | ||
| Scheme 6 52 | ||
Another concept that has become of considerable practical importance in crystal engineering is the ‘tecton’. Wuest54 has defined tectons as “molecules whose interactions are dominated by specific attractive forces that induce the assembly of aggregates with controlled geometries” and, consequently, tectons offer a very useful complement to ‘synthons’ in supramolecular parlance.55,56 Champness and co-workers57 examined the structural chemistry of three new tectons (11–13), Scheme 7, in order to generate infinite 2D hydrogen-bond based networks by combining 11–13 with the supramolecularly complementary melamine, Scheme 8.
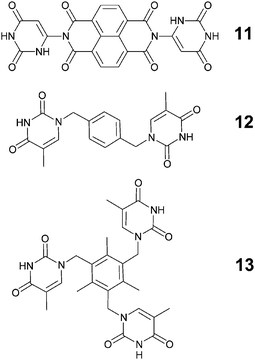 | ||
| Scheme 7 57 | ||
 | ||
| Scheme 8 57 | ||
Due to the low solubility of 11–13, co-crystallization reactions were limited to using DMSO to obtain crystals suitable for single-crystal X-ray diffraction. The crystal structures of 11–13 and the two co-crystals 11:melamine and 12:melamine, (13 did not form co-crystals with melamine) are somewhat difficult to interpret in terms of recurring patterns as all resulting structures appeared as solvates, typically with DMSO and benzene. However, the hydrogen-bond based supramolecular complementarity between uracil/thymine moieties and melamine was present to some extent in both 11:M and 12:M and those interactions are undoubtedly responsible for driving the assembly of these co-crystals. In 11:M, there is a 1:1 stoichiometry of the two main components, and they combine to produce a 2D layer despite the presence of DMSO molecules, Fig. 9.
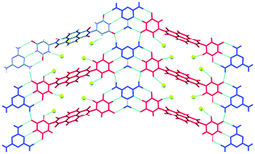 | ||
| Fig. 9 Hydrogen-bonded layer in the crystal structure of 11:M. The assembly is driven by complementary triple hydrogen bonds between 11 (red) and melamine (blue).57 | ||
In the crystal structure of 12:M, there is also a 1:1 stoichiometry of the main components, and again, the overall result of the complementary hydrogen bonds between tecton 12 and melamine, is a 2D layer, Fig. 10.
 | ||
| Fig. 10 A hydrogen-bonded sheet in the crystal structure of 12:M with the complementary triple hydrogen bonds adopted between tecton 12 (red or orange) and melamine (blue). Oxygen atoms of DMSO molecules are shown as yellow spheres with the rest of the DMSO molecule omitted for clarity.57 | ||
The results presented in this paper illustrate the importance of identifying complementary moieties for the directed assembly of complex multi-component architectures, but it is also clear that the physical properties, notably solubility, of the individual components need to be comparable otherwise the solution-based synthetic techniques may not be accessible. The fact that the structures form distinctive 2D motifs may also make them suitable for surface-based assembly.58
One practical route towards generating solids capable of guest inclusion is based upon the use of large molecular building blocks with shapes that preclude effective close-packing.59 Bacchi and co-workers60 have demonstrated that such building blocks can be constructed by combining metal ions and suitable ligands. These “wheel-and-axle-diols” (waad), Scheme 9, are also modular and may be used to ‘dial in’ void spaces within a lattice that is suitable for a specific guest molecule.
 | ||
| Scheme 9 60 | ||
The modularity in this system can be affected by changing either the wheel or the nature of the axle (which in this case comprises a metal ion centre). In this contribution, ten crystal structures were obtained on compounds based upon Pd(II) and a new ligand, L7, Scheme 10, and eight of them include solvent in the lattice.
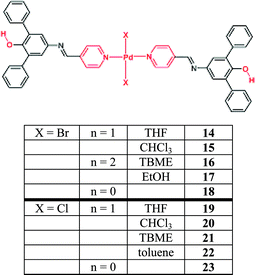 | ||
| Scheme 10 60 | ||
Four of the five bromide complexes are solvates, and all four of the chloride compounds contain solvent in their crystal structures. There is a striking similarity between the molecular structures of the former complexes as illustrated in Fig. 11.
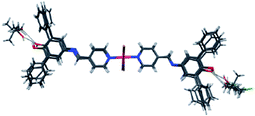 | ||
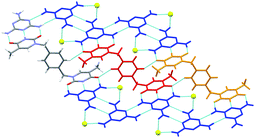
| Fig. 11 The molecular structure of 14–17, with guests occupying the same sites. 1:2 solvates (16 and 17) interact with two guest molecules on both sides, while 1:1 solvates (14 and 15) interact only to one guest at the site here on the right.60 | ||
As the chloride ions are better hydrogen-bond acceptors, they produce a different set of motifs where complex molecules are arranged in centrosymmetric pairs, Fig. 12.
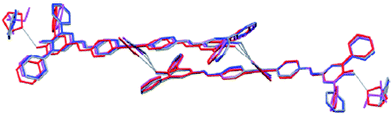
These wheel-and-axle complexes are effective at creating solid-state inclusion compounds, but the precise nature of the substituents does make a big difference, because if the two phenyl rings are replaced with –CH3, and –H, no solvent inclusion is observed.60
As crystal engineering is driven by non-covalent interactions, it should be noted that a reliable synthetic strategy for complex architectures is governed primarily by molecular recognition events. Thus, the nature of the building block may, in many cases be of secondary importance which is illustrated by the fact that there can be great structural similarities between purely organic structures and coordination networks. This was noted by Braga and co-workers61 as a result of a structural study of trans-1,4-diammoniocyclohexane (daceH2) dichloride dimethylsulfoxide 24, trans(ammonio-4-amino)cyclohexane monochloride 25, and piperazinium dichloride hemihydrate 26. A comparison of the crystal structure of 24 with that of CuCl2(daceH2)(DMSO)2,62 show a striking similarity, Fig. 13.
![Crystal structure of [daceH2]Cl2·2(DMSO) (left) and [CuCl2(dace)(DMSO)2].61](/image/article/2010/CE/b919819a/b919819a-f13.gif) | ||
| Fig. 13 Crystal structure of [daceH2]Cl2·2(DMSO) (left) and [CuCl2(dace)(DMSO)2].61 | ||
The authors also presented powder diffraction and thermal data to elucidate decomposition pathways and subsequently found a non-solvent based method for the preparation of 26.
Mechanochemical reactions can offer many advantages over conventional solution-based synthetic pathways63 and Orpen and co-workers64 recently demonstrated how coordination compounds can be readily obtained by combining metal hydroxides/carbonates with chloride salts of the appropriate N-heterocyclic ligand, Scheme 11.
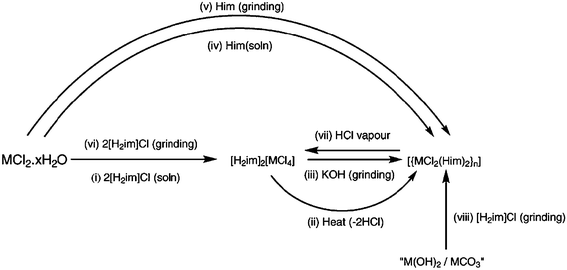 | ||
| Scheme 11 64 | ||
The outcome of the mechanochemical reactions was analyzed using powder X-ray diffraction and in all three cases reported herein, the powder data match the diffraction pattern calculated from single-crystal data, Fig. 14.
![Powder X-ray diffraction patterns of [{CuCl2(Him)2}n]. Bottom: calculated from single crystal data; centre: grinding imidazole and CuCl2·2H2O (Scheme 11, reaction (v)); top: grinding imidazolium chloride with basic copper(ii) carbonate (Scheme 11, reaction (viii)).64](/image/article/2010/CE/b919819a/b919819a-f14.gif) | ||
| Fig. 14 Powder X-ray diffraction patterns of [{CuCl2(Him)2}n]. Bottom: calculated from single crystal data; centre: grinding imidazole and CuCl2·2H2O (Scheme 11, reaction (v)); top: grinding imidazolium chloride with basic copper(II) carbonate (Scheme 11, reaction (viii)).64 | ||
These results demonstrate that metal hydroxides/carbonates provide viable starting materials for mechanochemical synthesis of a series of transition-metal compounds, including Cu(II) salts and that dry HCl gas can be used for hydrochlorination under a range of reaction conditions. Furthermore, in addition to providing a synthetic complement to conventional routes, solid-state based methods can also lead to new polymorphs compared to those obtained from solution.
Despite the fact that crystal engineering has come a long way in the last twenty years and we have developed a much better understanding of how supramolecular synthons can offer useful and versatile tools for the non-covalent solid-state assembly of new architectures, the synthetic target is not always reached and chance and serendipity still affect the outcome. Nangia and co-workers65 present two crystal structures that both illustrate the unpredictability of crystal engineering. First, the product of the reaction between 1,3-cis-cyclohexanetricarboxylic acid (H3cta) and 4(3H)-pyrimidinone in a 1:3 ratio produced a 1:2 co-crystal with unexpected interactions. The pyrimidinone was intended to act as a mimic of 4,4′-bipyridine, but since the necessary dimer did not form, Scheme 12, the assembly strategy was hampered.
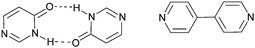 | ||
| Scheme 12 Dimer of 4(3H)-pyrimidinone (left) and 4,4′-bipyridine (right).65 | ||
In addition the structure also contained an acid⋯acid dimer, which further complicated the supramolecular synthesis (the primary intermolecular interactions are shown in Fig. 15a).
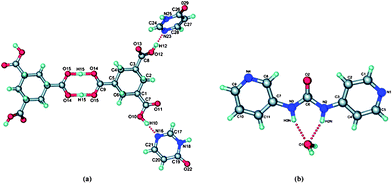 | ||
| Fig. 15 The primary intermolecular interactions in (a) the co-crystal of H3cta (4(3H-pyrimidonone)2 and in (b) 3,4-pyrU hydrate.65 | ||
The overall assembly produces interpenetrated 2D hexagonal sheets. Second, the crystal structure of N-3-pyridyl-N′-4-pyridyl urea (3,4-pyrU) is dominated by the unpredictable structural behaviour of the incorporated water molecule, Fig. 15b. Although urea hydrates are not unusual,66 the fact that the water molecule acts as a tetrahedral node in this case, could not readily be inferred from other urea-based crystal structures.
Aakeröy et al.,67 offered crystal structures of seven new co-crystals accompanied, in each case, by a description of the intended supramolecular target as well as of the rationale behind the synthesis.68 The co-crystal formation was driven by halogen-bond interactions involving 2,3,5,6-tetrafluoro-diiodobenzene 27, and the subsequent organization of supermolecules affected by hydrogen bonds. An example of where the proposed assembly strategy produced the desired extended network can be seen in the co-crystal of 27 and 3-acetamidopyridine, Fig. 16.
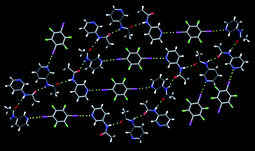 | ||
| Fig. 16 An infinite 2D layer in the crystal structure of 27 and 3-acetamidopyridine.67 | ||
The assembly is driven by an I⋯N halogen bond and the resulting trimers are subsequently arranged into 2D sheets through self-complementary amide⋯amide, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N, hydrogen bonds. Another successful co-crystal synthesis was observed when 4-(pyrazol-3-yl) pyridine was co-crystallized with 27, Fig. 17.
O⋯H–N, hydrogen bonds. Another successful co-crystal synthesis was observed when 4-(pyrazol-3-yl) pyridine was co-crystallized with 27, Fig. 17.
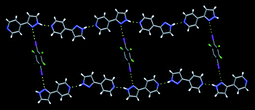 | ||
| Fig. 17 The primary intermolecular interactions in the crystal structure of 2,3,5,6-tetrafluoro-diiodobenzene and 4-(pyrazol-3-yl) pyridine.67 | ||
The only hydrogen-bond donor in these two molecules interacts with the best acceptor site (pyridine is better than pyrazole), which leaves the N-pyrazole to form a halogen bond with diiodobenzene. Problems arose when solvent molecules capable of forming hydrogen bonds were incorporated into the lattice or when a ‘free’ molecule of either reagent found its way into the lattice. In some cases, the expected synthons did not form which meant that the synthetic goal was not realized. For example, the authors were attempting to synthesize a ternary co-crystal assembled using acid⋯(acetamido)py and I⋯py containing supermolecules with a 1:2:4 stoichiometry.
However, although the acid⋯(acetamido) complementary hydrogen bonds did appear, the rest of the assembly did not follow the plan, Fig. 18.
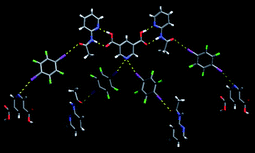 | ||
| Fig. 18 The main non-covalent interactions present in a ternary 2:1:2 co-crystal.67 | ||
A highly unusual bifurcated halogen bond scuppers the remaining construction, and although the result was a ternary co-crystal, neither the stoichiometry, nor the desired synthons were present.
An area of increasing focus in crystal engineering has been the study of water clusters in the solid-state. Extensive interest in this area over recent years has been driven by a desire to improve understanding of water clusters in a biological context as well as to provide insight into some of the anomalous properties of water.69 In this context Ruiz-Pérez et al.70 have reported an interesting example of unusual water tetramers hosted within a coordination polymer, [Co2(bta)(H2O)8]n·4nH2O [H4bta=1,2,4,5-benzenetetracarboxylic acid]. Two distinct tetramers are observed with either S4 or D2h symmetry (Fig. 19). The S4 symmetric cluster exhibits the same hydrogen bonding connectivity and planarity as those in the more common C4 symmetric cluster but the free hydrogen atoms in the S4 arrangement are alternatively oriented above and below the ring giving an uudd arrangement (Fig. 19). The arrangements of the clusters reported are the first experimentally well-resolved examples of such cluster conformations and arise due to cooperative interactions between the host coordination polymers and the tetrameric clusters.
![Tetrameric water clusters observed in [Co2(bta)(H2O)8]n·4nH2O revealing either S4 or D2h symmetry.70](/image/article/2010/CE/b919819a/b919819a-f19.gif) | ||
| Fig. 19 Tetrameric water clusters observed in [Co2(bta)(H2O)8]n·4nH2O revealing either S4 or D2h symmetry.70 | ||
2.2 The development of the halogen bond
Although crystal engineering is still generally dominated by hydrogen-bonding and metal–ligand interactions, see elsewhere in this article, halogen bonds have become increasingly studied and exploited, with a major renaissance over the last ten years, as evidenced by some of the studies discussed above. Many of these studies have focused on the study of purely organic compounds13 but there have also been increasing range of studies associated with the use of metal-containing species12 and anions14 as covered in recent Highlight articles in CrystEngComm. The timely article by Brammer et al.12 summarises developments in the understanding of C–X⋯X′–M halogen bonds and particularly their use in forming network systems. The article also assesses the strength of such halogen bonds in light of the fact that their energy can be tuned by variations in the organic (C–X) or inorganic (M–X′) halogen. The article also provides valuable insight into the nature of halogen bonds where the acceptor is not a halogen but rather a metal oxo or nitride group (C–X⋯A–M) or a metal carbonyl, cyanide or another related π-acceptor ligand. A nice example of this latter unconventional halogen bond is shown in Fig. 20 where C–I⋯OC–Mn halogen bonds [I⋯O 3.395 Å; C–I⋯O 178.2°; I⋯O≡C 91.7°] lead to assembly of [Mn(CO)4(PPh2C(I)PPh2)] complexes into one-dimensional chains.71 Brammer et al. also highlight the existence of other unusual halogen bonds (C–X⋯A–M) where the metal is a main group element typically from groups 13–15 with a few rare examples from group 16. The article summarises the breadth of examples of halogen bonds where at least one of the components of the interactions is a metal and illustrates that the range of species capable of adopting halogen bonding interactions indicates a prevalent future for halogen bonds in crystal engineering.![One-dimensional chains of [Mn(CO)4(PPh2C(i)PPh2)] complexes formed by C–I⋯OC–Mn halogen bonds.12,71](/image/article/2010/CE/b919819a/b919819a-f20.gif) | ||
| Fig. 20 One-dimensional chains of [Mn(CO)4(PPh2C(I)PPh2)] complexes formed by C–I⋯OC–Mn halogen bonds.12,71 | ||
3. Polymorphism, solvates and crystal-to-crystal transformations
Areas of significant and long-standing interest in crystal engineering have included crystal-to-crystal transformations72 and polymorphism.9 These fields continue to receive much attention with both areas requiring in-depth investigations of crystal systems. Such studies have recently been exemplified by a study of two polymorphic forms, monoclinic and orthorhombic, of all-trans-1,6-diphenyl-1,3,5-hexatriene by Ogawa et al.73 The study demonstrates via the use of detailed difference electron density maps, that at elevated temperatures, room temperature or higher, a minor component of all-trans-1,6-diphenyl-1,3,5-hexatriene undergoes a conformational change (Fig. 21).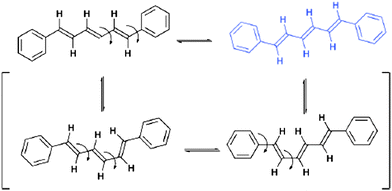 | ||
| Fig. 21 Conformational changes observed in crystals of all-trans-1,6-diphenyl-1,3,5-hexatriene at elevated temperatures. The conformers shown in brackets are not observed in the crystal structures.73 | ||
The study of polymorphism has quite rightly maintained its significance in crystal engineering and has benefited from the increasing development and use of advanced techniques beyond crystallography, as demonstrated by a study of the polymorphs of the organic semiconductor dibenzotetrathiafulvalene by Brillante and co-workers.74 They found that dibenzotetrathiafulvalene exhibits four polymorphs at room temperature, three in addition to the previously known α-form. A second monoclinic β-form has been identified by single crystal X-ray diffraction and two further polymorphs, γ and δ, have been established by lattice phonon confocal Raman microscopy. The formation of the polymorphs is significantly affected by the growth process and a range of studies were reported, including vapour phase or solution deposition, and studies of thin films versus bulk phases samples. A number of transformations between phases and phase mixing occur depending on the growth conditions. For example, vapour deposition of dibenzotetrathiafulvalene onto a cold glass tip results in the initial formation of the γ-polymorph as a thin film but deposition of further sublimed material results in the formation of needle-shaped crystals of the α-polymorph. This growth behaviour was characterised by lattice phonon Raman spectroscopy illustrating the utility of this technique (Fig. 22).
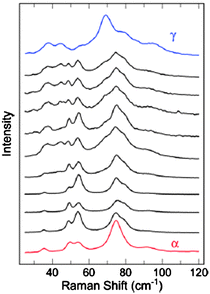 | ||
| Fig. 22 Lattice phonon Raman spectroscopy of the growth of α- and γ-polymorphs of dibenzotetrathiafulvalene on a cold glass tip.74 | ||
The issue of mixtures of phases, and particularly amorphous components, is a problem faced by many researchers in the field of crystal engineering, notably those working on polymorphism or materials chemistry where bulk purity is crucial. A study by Stephens, Miller and co-workers75 illustrates an elegant approach to this problem by demonstrating the use of Rietveld refinement of high-resolution, synchrotron X-ray powder diffraction data to determine the structure of a CrIIF(NCMe)2BF4, crystalline phase in the presence of co-existing amorphous phase(s). The CrIIF(NCMe)2BF4 product prepared from the reaction of CrII(NCMe)4(BF4)2 and [NBun4][TCNE] (TCNE = tetracyanoethylene) is found in a mixture with an amorphous reduced-TCNE species (Fig. 23). The use of Rietveld refinement for such a mixture of products provides considerable hope for many crystal engineers confronted by complex mixtures of crystalline and amorphous materials.
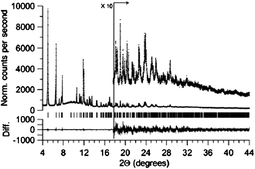 | ||
| Fig. 23 High-resolution powder X-ray diffraction data (dots) and Rietveld fit for the refined structure of CrIIF(NCMe)2BF4 (solid line). The lower trace is the difference (measured − calculated) plotted on the same vertical scale.75 | ||
Polymorphism also represents the focus of a study by Parsons et al.16 but in this case polymorphism is induced by pressure. The resulting polymorph of L-serine monohydrate is characterised using high-pressure single-crystal X-ray diffraction further expanding the use of this technique in crystal engineering.76,77 The structure of L-serine monohydrate at ambient pressure reveals extensive hydrogen bonding between zwitterionic serine moieties and H2O molecules leading to a layered structure which is further linked by additional H2O based hydrogen bonds. With increased pressure of 5.2 GPa the structure undergoes a phase transition to a high pressure polymorph, named L-serine monohydrate-II as determined by a combination of single-crystal X-ray and neutron powder diffraction studies. The structure of this high pressure polymorph is distinguished by a reduction in the interlayer separation accompanied by rotation of the H2O molecules so that both hydrogen-bond donations from the H2O molecules occur within the serine layer. In addition the serine ammonium groups also adopt a different conformation resulting in further subtle adaptations in the hydrogen bonded structure (Fig. 24). The authors relate their observations to the first step in the pressure-denaturation of proteins.
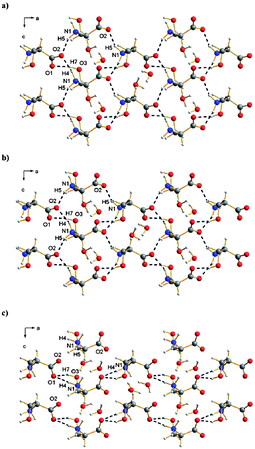 | ||
| Fig. 24 View along the crystallographic b-axis of L-serine monohydrate (a) at ambient pressure,16 (b) at 3.8 GPa and L-serine monohydrate-II (c) at 5.8 GPa. | ||
A problem highly related to that of polymorphism, that of solvates and co-crystals, is the focus of recent studies by Janiak17 and Desiraju respectively.78 Just as research into polymorphism continues to be one of the most important areas of crystal engineering so the formation of co-crystals has been an area of intense interest over recent years,79 despite some discussion of the usefulness of the term “co-crystal”.10 Ruiz, Hoffmann and Janiak et al. report the use of solid-state CPMAS 13C NMR to study the desolvation of a toluene solvate of [{(N,N-dimethylbenzylamine-κN,κC)Pd}2(μ-OAc)(μ-NCPh2)] (Fig. 25).17 Two toluene solvates of the compound are observed, [{(N,N-dimethylbenzylamine-κN,κC)Pd}2(μ-OAc)(μ-NCPh2)].1.5C6H5CH3 and [{(N,N-dimethylbenzylamine-κN,κC)Pd}2(μ-OAc)(μ-NCPh2)].0.25C6H5CH3 which have Z′ values of 1 and 2, respectively. The transformation from the solvate with Z′ = 1 to that with Z′ = 2 results from partial loss of toluene from the crystal and was followed by solid-state CPMAS NMR and was shown to proceed by a single-crystal-to-crystal transition by single-crystal X-ray diffraction studies.
![View of the asymmetric unit of [{(N,N-dimethylbenzylamine-κN,κC)Pd}2(μ-OAc)(μ-NCPh2)]·0.25C6H5CH3 which exhibits Z′ = 2. The solvate structure is formed as a result of the toluene solvent loss from a corresponding solvate with Z′ = 1.17](/image/article/2010/CE/b919819a/b919819a-f25.gif) | ||
| Fig. 25 View of the asymmetric unit of [{(N,N-dimethylbenzylamine-κN,κC)Pd}2(μ-OAc)(μ-NCPh2)]·0.25C6H5CH3 which exhibits Z′ = 2. The solvate structure is formed as a result of the toluene solvent loss from a corresponding solvate with Z′ = 1.17 | ||
Desiraju et al.78 report the use of co-crystal formation, in which one component contains a heavy atom, to enable absolute configuration determination of chiral organic compounds. Thus, co-crystals formed between pregnenolone and either 4-iodophenol or 2,4,6-trichlorophenol, and cholesterol/4-iodophenol demonstrate that incorporation of the heavy atom by the introduction of the halogenated species in formation of the co-crystal can be used to determine the absolute configuration of the chiral species, either pregnenolone or cholesterol. The investigation was extended to the co-crystal formed between two anti-HIV drugs zidovudine and lamivudine in which the latter contains a sulfur atom (Fig. 26). The strategy demonstrated by Desiraju is clearly applicable to a large number of organic compounds, and importantly is practically applicable to systems where chemical transformations are not possible or favoured for co-crystal formation. Although chemical transformation has previously been used to form co-crystals, for example by the use of acidic or basic groups to form salts, this is not always feasible and may lead to racemisation of chiral functionality. The approach outlined by Desiraju has the advantage of being essentially non-invasive and is therefore an attractive route to co-crystal formation and study.
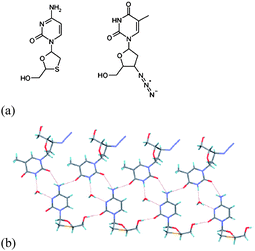 | ||
| Fig. 26 (a) Structures of lamivudine (left) and zidovudine (right); (b) crystal structure of lamivudine-zidovudine monohydrate co-crystal.78 | ||
Supramolecular chemistry and crystal engineering continue to provide progress in terms of synthetic strategies and structure-function relationships but, as illustrated above, since we are dealing with relatively weak and reversible interactions, much more work is needed before we have access to more reliable assembly processes.
4. Coordination polymers and metal–organic frameworks
Infinitely extended metal–ligand networks with bridging organic ligands are called coordination polymers or metal–organic frameworks (MOFs). The last decade has seen an almost exponential and still continuing growth in publications on their structural topologies and potential applications (reviews in ref. 3 and 18) in heterogeneous catalysis (specific examples in ref. 80), adsorption – with a special emphasis on dihydrogen and other gas storage,3–8 hosts for metal colloids or nanoparticles,19 hosts for polymerization reactions,20 luminescence,21–25 non-linear optics (NLO, frequency doubling),26 magnetism,27,28 and, recently, low temperature heating and cooling through reversible water de- and adsorption.29 While applications drive the interest in this class of compounds, we are far from being able to “design” or predict a coordination polymer structure.3,18,30 It is here where crystal engineering can contribute to the understanding of the underlying structure-directing forces and design principles. In the following we highlight recent synthetic and design aspects of coordination polymers aka metal–organic frameworks (MOFs).4.1 New methods of synthesis: from solvent-free mechanochemical conditions in a ball mill over sonochemistry to resin-assisted preparations
Nowadays solvo- or hydrothermal synthesis is one of the most popular methods to synthesize coordination networks. Yet, MOFs can also be prepared using mechanochemistry, that is, the grinding of two or more solids by using, for example, a mechanical ball mill (Fig. 27) and thereby avoid the use of solvents.81,50 | ||
| Fig. 27 Ball mills that can generate microcrystalline products within minutes. | ||
An array-based approach by Pichon and James of reacting 12 different metal salts and 5 bridging ligands in 60 potential reactions found solvent-free mechanochemical conditions (“ball-milling”) to be quite general for the synthesis of coordination polymers. By X-ray powder diffractometry (XRPD) 38 out of 60 combinations gave microcrystalline products. Six products could be identified by positively matching the measured XRPD pattern to a simulated one from single-crystal X-ray diffraction data in the Cambridge Structure Database, including the microporous MOFs [Cu(ina)2]n and [Cu3(btc)2] (HKUST-1) (ina = isonicotinate, btc = benzene-1,3,5-tricarboxylate) (Fig. 28).82
![Synthesis of [Cu(ina)2] (top) and comparison of the XRPD pattern of the product and the simulated pattern for [Cu(ina)2]·2H2O (CSD Refcode BAHGUN) based on single-crystal data.82](/image/article/2010/CE/b919819a/b919819a-f28.gif) | ||
| Fig. 28 Synthesis of [Cu(ina)2] (top) and comparison of the XRPD pattern of the product and the simulated pattern for [Cu(ina)2]·2H2O (CSD Refcode BAHGUN) based on single-crystal data.82 | ||
Scheme 13 summarizes the trends of reactivity under mechanochemical conditions which became apparent from the array-based study.
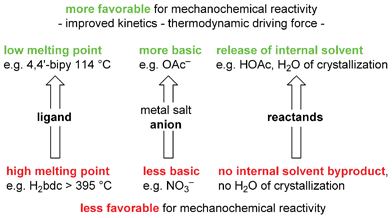 | ||
| Scheme 13 Trends revealed by studying an array of 60 potential reactions between various ligands and metal salts in ref. 82. 4,4′-bipy = 4,4′-bipyridine, H2bdc = benzene-1,4-dicarboxylic acid, OAc = O2CCH3. | ||
An interesting alternative to conventional solution synthesis may be sonochemistry: high quality MOF-5 crystals of 5–25 μm were prepared by Son et al. using a sonochemical method in substantially reduced synthesis time (ca. 30 min) compared with conventional solvothermal synthesis (24 h).83
Transition metal-exchanged polymer resin beads have been used by Du et al. as a heterogeneous controlled-release source of metal cations in high yielding, phase pure solvothermal syntheses of novel transition metal–organic frameworks (Fig. 29).84
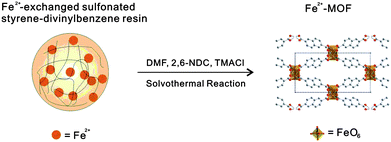 | ||
| Fig. 29 Slow release of Fe2+ ions from a polymer resin to yield a MOF in solvothermal reactions. | ||
4.2 Beyond metal nodes and bridging ligands: the role of solvents and anions
It remains a synthetic challenge to “design” metal–organic networks with porosity and zeolite-type behaviour in porous coordination polymers (PCPs) or MOFs for the reversible exchange of guest molecules in molecular sieves, gas storage and catalysis as noted by Ghosh and Kitagawa.85Paralleling the work on zeolite synthesis, structure-directing agents other than the ligands and metal atoms themselves are crucial for obtaining a 3D (potentially) porous structure (Fig. 30). The role of solvents as structure-directing agents in MOFs and the correlation of the framework structure with the solvent size, shape and non-covalent binding properties clearly need more attention for their better understanding.85,86
![Perspective view of the 3D framework of [Cd(tci)]n [where tci = tris(2-carboxyethyl)isocyanurate] with DMF solvent molecules (omitted for clarity) in the channels.85](/image/article/2010/CE/b919819a/b919819a-f30.gif) | ||
| Fig. 30 Perspective view of the 3D framework of [Cd(tci)]n [where tci = tris(2-carboxyethyl)isocyanurate] with DMF solvent molecules (omitted for clarity) in the channels.85 | ||
In addition, the anions add to this structure-directing role in the case of neutral bridging ligands, that is, cationic frameworks. Here again, the role of the anions is often overlooked in the usual narrow focus on the bridging ligand and careful studies on anion-structure correlation are rare.87 A crucial anion-influence is the competition of coordination ability between the counter anions and the organic bridging ligands to the metal. Non-coordinating anions like ClO4− or BF4− are advantageous in increasing the dimensionality of the metal–ligand framework.21,87 A counter-anion like NO3− can have a variable coordination, including bridging mode and, thereby, affect the network topology.88 Notably, the size of the anion influences the structure: The example of two Mn-bpeado frameworks by Gao et al. illustrates how the small ClO4− anion can yield a 3-fold interpenetrated α-Po-type structure (on interpenetration see also below) whereas the larger BPh4− anion results in non-interpenetrating rectangular (4,4) layers (Fig. 31).89
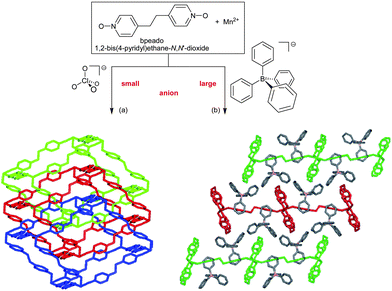 | ||
| Fig. 31 Mn(bpeado)32+ networks: (a) three-fold interpenetrated 3D networks with a ClO4− anion and (b) 2D rectangular, non-interpenetrating, (4,4) layers with a BPh4− anion. Anions not depicted for clarity with the networks.89 | ||
Similarly the ligand pypz used by Du et al. gives corrugated (4,4) layers. The small, terminal chloro anion ligand on the metal nodes allows for a 2-fold (2D → 2D) interpenetration of the (4,4) coordination layers (Fig. 32a). Whereas, elongated, larger anions as terminal ligands lead to a simple stacking of adjacent layers on top of each other (Fig. 32b and c). Such elongated anions are dicyanamide (N(CN)2−, dca) or isothiocyanate (NCS−) which point into the openings of the adjacent layers and may, thus, prevent interpenetration (Fig. 32c). The metal nodes have the anions trans in axial positions.90
![(a) Space-filling diagram of the interpenetrating (interwoven) 2D (4,4) layers of [CdCl2(pypz)2]n. (b) Single 2D (4,4) layer in [M(dca)2(pypz)2]n or (shown here) [M(NCS)2(pypz)2]n (M = Cd, Mn, Ni, Co, Cu) with metal spheres highlighted as green polyhedra. (c) Side view of adjacent layers with the central layer in a ball-and-stick model and the other two in a wire model; metal atoms in yellow. Note the mutual pointing of the axial, terminal elongated NCS anion ligands into the openings of the adjacent layers.90](/image/article/2010/CE/b919819a/b919819a-f32.gif) | ||
| Fig. 32 (a) Space-filling diagram of the interpenetrating (interwoven) 2D (4,4) layers of [CdCl2(pypz)2]n. (b) Single 2D (4,4) layer in [M(dca)2(pypz)2]n or (shown here) [M(NCS)2(pypz)2]n (M = Cd, Mn, Ni, Co, Cu) with metal spheres highlighted as green polyhedra. (c) Side view of adjacent layers with the central layer in a ball-and-stick model and the other two in a wire model; metal atoms in yellow. Note the mutual pointing of the axial, terminal elongated NCS anion ligands into the openings of the adjacent layers.90 | ||
4.3 Interpenetration, data mining and statistical analyses
One of the most fascinating aspects of crystal chemistry concerns the entanglement of individual extended motifs, observed especially in the crystal engineering of coordination polymers or MOFs. Interpenetrating networks are now numerous and extensively studied.21,89–91 The origin of interpenetration can be ascribed to the presence of large free voids in a single network, hence, interpenetration is often Nature's trick (or other option instead of solvent inclusion) to reach a densely packed structure and to circumvent potential porosity—though it has been demonstrated that interpenetration does not prevent the possibility of obtaining open porous materials.92 In the realm of crystal engineering and crystal growth it is fascinating to imagine how this intricate formation of independent, albeit symmetry related, networks is controlled by weak intermolecular forces, notably π-stacking as often pyridine-derived or other nitrogen-heterocycle bridging ligands are involved.21,93The occurrence of interpenetrating metal–organic, inorganic and supramolecular networks has been analyzed using the program package TOPOS,94 based on search results from the Cambridge or Inorganic Crystal Structural Database (CSD or ICSD, respectively).92 Following these earlier comprehensive analyses, now interpenetrated 3D networks based on hydrogen-bonded metal–organic molecular (0D) and polymeric (1D and 2D) compounds were analyzed by Baburin, Blatov, Proserpio et al.95 From 135 such motifs (71 assembled from 0D, 42 from 1D and 21 from 2D metal–organic substructures) showing the phenomenon of interpenetration, interestingly about two thirds were not detected in the original papers. For these 3D hydrogen-bonded networks the network topologies are distributed as shown in Fig. 33, with the main (overall) topology as 412.63-pcu (28.9%). Also, for the large majority of these nets the value of the degree of interpenetration is 2 (Fig. 34), with the maximum interpenetration degree being found as 5-fold (Fig. 34 and Fig. 35).
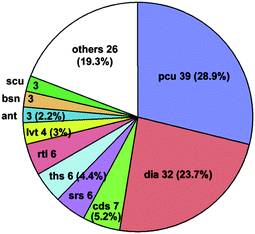 | ||
| Fig. 33 Overall distribution of the topologies within the 135 3D hydrogen-bonded metal–organic molecular (0D) and polymeric (1D and 2D) structures.95 pcu = α-Po, dia = diamond, cds = CdSO4, srs = SrSi2, ths = ThSi2, rtl = rutile, lvt = lattice complex vT, ant = anatase, bsn = β-Sn, scu = square planar and cubical nodes.96 | ||
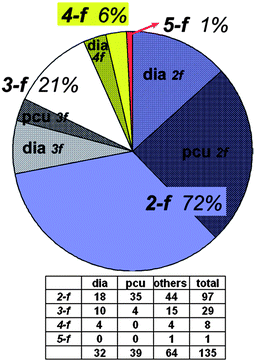 | ||
| Fig. 34 Distribution of the values of Z (degree of interpenetration). 2-f = 2-fold, 3-f = 3-fold and so on; pcu = α-Po, dia = diamond topology (cf.Fig. 33).95 | ||
![The 5-fold interpenetrated structure of NABYOF ([Ag(isonicotinamide)2]BF4),97 the record of interpenetration among hydrogen-bonded coordination compounds: (top) the standard description with three independent nodes giving the 3-connected trinodal sqc69 net and (bottom) the ring synthon description giving 4-connected 65.8-cds net.95](/image/article/2010/CE/b919819a/b919819a-f35.gif) | ||
| Fig. 35 The 5-fold interpenetrated structure of NABYOF ([Ag(isonicotinamide)2]BF4),97 the record of interpenetration among hydrogen-bonded coordination compounds: (top) the standard description with three independent nodes giving the 3-connected trinodal sqc69 net and (bottom) the ring synthon description giving 4-connected 65.8-cds net.95 | ||
Interpenetrated networks are good examples of a systematic statistical analysis in the crystal engineering of extended systems.93,96 The future will certainly see an increasing use of data banks and need for statistical analyses of coordination polymer crystal structures. This is necessary to handle the plethora of network structures and to foster the understanding of topology and underlying design principles.
Bridging benzene-di- or -tri-carboxylate ligands as sole linkers are not prone to give interpenetrating networks. This finding may be traced to the lower tendency of benzene-polycarboxylates to enter into π-stacking interactions when compared with aromatic nitrogen heterocycle containing bridging ligands. π-Stacking in turn is one of the primary weak intermolecular forces to control the intergrowth of interpenetrating networks.21,94,98 Hence, benzene-dicarboxylate ligands with tetrahedral Zn4O clusters as secondary building units were successfully used by Yaghi and Férey et al., respectively, for the construction of non-interpenetrating—despite large pore sizes—cubic isoreticular MOFs (IRMOF-n with n = 5–16 of the MOF-5 structure type series) or MILs with large tunable pore sizes ranging from 3 to 19 Å.99,100
When benzene-dicarboxylate (bdc, terephthalate) is, however, combined with other long bridging ligands in a mixed bridging-ligand22,25,101,102 coordination polymer, interpenetration becomes a possibility. The combination of benzene-dicarboxylate with pypz (cf.Scheme 14), utilized by Du et al., affords a number of interpenetrated networks (Fig. 36).
 | ||
| Scheme 14 Examples of flexible bridging ligands, see also Scheme 15. | ||
![Example of an interpenetrated network in a mixed bridging-ligand coordination polymer with benzene-dicarboxylate (bdc) and bis[(dimethyl-pyridyl)pyrazol]methane (pypz): 2D → 2D in {[Cd(bdc)(pypz)(H2O)]·1.5DMF·1.5H2O}n. (a) One hexagonal repeat unit with blue polyhedra representing the hepta-coordinated Cd atoms. (b) 2-Fold interpenetrating (interwoven) (6,3) layers in space-filling mode.90](/image/article/2010/CE/b919819a/b919819a-f36.gif) | ||
| Fig. 36 Example of an interpenetrated network in a mixed bridging-ligand coordination polymer with benzene-dicarboxylate (bdc) and bis[(dimethyl-pyridyl)pyrazol]methane (pypz): 2D → 2D in {[Cd(bdc)(pypz)(H2O)]·1.5DMF·1.5H2O}n. (a) One hexagonal repeat unit with blue polyhedra representing the hepta-coordinated Cd atoms. (b) 2-Fold interpenetrating (interwoven) (6,3) layers in space-filling mode.90 | ||
4.4 Beyond rigid spacer ligands
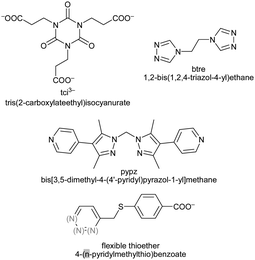
At present, work on MOFs uses largely rigid spacer ligands, like benzene-di- or –tri-carboxylate. In the future we will see more flexible ligands, like tci,85 btre,21,22 thioether,23 pypz90 and others101 in the synthesis of new PCPs or MOFs (Scheme 15).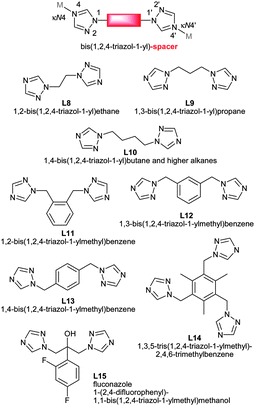 | ||
| Scheme 15 Bis- and tris(1,2,4-triazol-1-yl) derived flexible bridging ligands with spacer modification between the typically κN4:N4′ metal-coordinating triazole moieties. References: L8,103L9,104L10,104,105L11,106L12,106,107L13,106,108L14,106L15.109 | ||
Bis- and tris(1,2,4-triazol-1-yl)-alkane and -arene ligands are susceptible to a facile and wide modification of the alkane or arene inner-spacer (Scheme 15). Through their predominantly κN4:N4′ metal-coordination mode they are good bridging ligands which have recently become more popular.
The tuning from molecular to polymeric structures was illustrated by Bu et al.106 with the bis- and tris(1,2,4-triazol-1-yl) derived ligands L11–L14 and Ag(I)18,35,110 as a metal ion, which often serves well in such comparative studies on coordination polymeric structures (Fig. 37).87,106
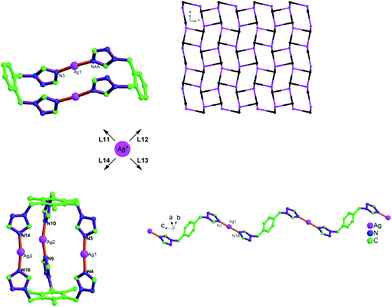 | ||
| Fig. 37 Tuning silver(I) coordination architectures by ligands design through the inner-spacer: from dinuclear, trinuclear, to 1D and 3D frameworks with bis- and tris(1,2,4-triazol-1-yl) derived flexible bridging ligands.106 For L11–L14 see Scheme 15. | ||
4.5 Magnetic interactions in coordination polymers
The O:O′-bridging mode of a carboxylate group or the N:N′-bridging mode of a triazole ring constitutes a short bridge between metal atoms which is a prerequisite for stronger magnetic coupling between paramagnetic metal centers.18,27 Magnetic interactions in MOFs are overshadowed by the recent surge in porosity work but they remain of interest.28,111 The tzdc3− ligand connects three metal atoms within 3D frameworks as shown by Chen et al. (Fig. 38).112 Spin-frustrated magnetism is a characteristic behaviour of these frameworks due to the triangular arrangement of the metal atoms.113![The tzdc3− ligand and resulting 3D framework (colour code as in formula) with Co2+ atoms of composition [(H2NMe2)[Co(tzdc)]·0.5H2O]n, exhibiting (10,3)-a topology. The channels in the framework are occupied by the protonated dimethylamine cations (not shown for clarity).112](/image/article/2010/CE/b919819a/b919819a-f38.gif) | ||
| Fig. 38 The tzdc3− ligand and resulting 3D framework (colour code as in formula) with Co2+ atoms of composition [(H2NMe2)[Co(tzdc)]·0.5H2O]n, exhibiting (10,3)-a topology. The channels in the framework are occupied by the protonated dimethylamine cations (not shown for clarity).112 | ||
4.6 Inorganic–organic hybrid materials
While coordination polymers or MOFs should refer to networks having an organic bridging ligand,18 the distinction to networks with inorganic bridging ligands and terminal organic ligands or just organic counterions initially did not exist114 and is not always made. Nowadays, for differentiation the latter compounds should better be called inorganic–organic hybrid materials.115,116From a crystal engineering perspective a common problem in metal–organic and inorganic–organic hybrid networks is the control of dimensionality (as discussed above for coordination polymers). An important subgroup in inorganic–organic hybrid materials are halometallates, [MxXy]n−,117,118 which are of continuous interest for their structural richness, semiconducting,119 magnetic,120 photo-luminescent and photo-chromic properties.121 These halometallates [MxXy]n− typically have organic counter-cations derived from alkyl or aromatic amine molecules by protonation. The ammonium cations do not coordinate to the metal atoms but serve as template molecules in the lattice of the 0D, 1D or 2D [MxXy]n− motif.
Lessons learned from counter-anions in coordination polymers (see above) can be transferred to the counter-cations in halometallates: A larger counter-cation should decrease the dimensionality. This was repeatedly shown by Beatty et al. with chlorocadmate compounds [CdxCly]n− where one can go from “expected” 2D perovskite [(CdCl4)2−]n networks with intercalated diammonium ions to a 0D linear trinuclear [Cd3Cl12]6− cluster by increasing the size of the diammonium counter-cation (Fig. 39).122
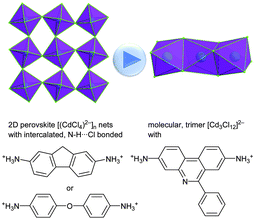 | ||
| Fig. 39 Small clusters, rather than 2D networks, can be “reversely” engineered using sterically encumbered counter-cations in halometallate networks.122 | ||
5. Conclusions
Crystal engineering has its roots in the work of Schmidt in the 1960s. The field has developed in a plethora of directions since those early studies of solid-state photocyclization reactions.123 The reactions that fascinated Schmidt still play a significant role in crystal engineering124 but crystal engineering has changed since the early stages in many ways not least through the development of structures based on hydrogen bonds125 and more recently through the development of coordination polymers.126 Ultimately the field of crystal engineering has developed to a level where it merits journals dedicated specifically to the field, including CrystEngComm.This Highlight emphasizes the great strides forward that have been taken in the field of crystal engineering. Not only are crystal engineers developing new interactions to endeavour to control and engineer solid-state structures but the field has expanded to target materials properties3–8,19–29 and to utilize previously unexploited intermolecular interactions.12–14 With new targets come new techniques27,74,75 and the number of techniques employed to solve crystal engineering problems can be expected to expand further with time. As the field continues to expand it is most certainly an exciting time to be a crystal engineer.
Acknowledgements
The authors wish to thank the remaining members of the editorial board of CrystEngComm and members of the journal's international advisory board who continually provide insight into the expanding field of crystal engineering. Additionally the authors thank the editorial team of the Royal Society of Chemistry led by Dr Jamie Humphrey, managing editor of CrystEngComm, for the continuing support of crystal engineers everywhere.References
- L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, CrystEngComm, 1999, 1, 1–4 RSC.
- D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1–19 RSC.
- M. Dinca and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef CAS; R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef; X. Lin, J. Jia, P. Hubberstey, M. Schröder and N. R. Champness, CrystEngComm, 2007, 9, 438–448 RSC; D. J. Collins and H.-C. Zhou, J. Mater. Chem., 2007, 17, 3154–3160 RSC; J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC; B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209–2214 CrossRef CAS.
- C. Gao, S. Liu, L. Xie, C. Sun, J. Cao, Y. Ren, D. Feng and Z. Su, CrystEngComm, 2009, 11, 177–182 RSC; K. Li, J. Y. Lee, D. H. Olson, T. J. Emge, W. Bi, M. J. Eibling and J. Li, Chem. Commun., 2008, 6123–6125 RSC; S. Yang, X. Lin, A. J. Blake, K. M. Thomas, P. Hubberstey, N. R. Champness and M. Schröder, Chem. Commun., 2008, 6108–6110 RSC; A. D. Burrows, K. Cassar, T. Düren, R. M. W. Friend, M. F. Mahon, S. P. Rigby and T. L. Savarese, Dalton Trans., 2008, 2465–2474 RSC; J. Jia, X. Lin, C. Wilson, A. J. Blake, N. R. Champness, P. Hubberstey, G. Walker, E. J. Cussen and M. Schröder, Chem. Commun., 2007, 840–842 RSC; Z. Wang, Y. Zhang, T. Liu, M. Kurmoo and S. Gao, Adv. Funct. Mater., 2007, 17, 1523–1536 CrossRef CAS; P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellva, Chem. Commun., 2006, 959–961 RSC; B. Panella, M. Hirscher, H. Pütter and U. Müller, Adv. Funct. Mater., 2006, 16, 520–524 CrossRef CAS; B. Panella and M. Hirscher, Adv. Mater., 2005, 17, 538–541 CrossRef.
- D.-X. Xue, J.-B. Lin, J.-P. Zhang and X.-M. Chen, CrystEngComm, 2009, 11, 183–188 RSC; C.-J. Li, Z.-J. Lin, M.-X. Peng, J.-D. Leng, M.-M. Yang and M.-L. Tong, Chem. Commun., 2008, 6348–6350 RSC; P. D. C. Dietzel, R. E. Johnsen, H. Fjellva, S. Bordiga, E. Groppo, S. Chavan and R. Blom, Chem. Commun., 2008, 5125–5127 RSC; S. Horike, S. Hasegawa, D. Tanaka, M. Higuchi and S. Kitagawa, Chem. Commun., 2008, 4436–4438 RSC; S. M. Humphrey, S. E. Oungoulian, J. W. Yoon, Y. K. Hwang, E. R. Wise and J.-S. Chang, Chem. Commun., 2008, 2891–2893 RSC.
- M. Müller, X. Zhang, Y. Wang and R. A. Fischer, Chem. Commun., 2009, 119–121 RSC; M. Müller, O. I. Lebedev and R. A. Fischer, J. Mater. Chem., 2008, 18, 5274–5281 RSC ; see also ref. 19.
- S. S. Iremonger, P. D. Southon and C. J. Kepert, Dalton Trans., 2008, 6103–6105 RSC.
- For recent examples see: M. Kitamura, CrystEngComm, 2009, 11, 949–964 Search PubMed; L. Yu, CrystEngComm, 2007, 9, 847–851 RSC; Y.-B. Men, J. Sun, Z.-T. Huang and Q.-Y. Zheng, CrystEngComm, 2009, 11, 978–979 RSC; N. K. Duggirala, S. L. Kanniah, V. K. Muppidi, R. Thaimattam and S. Devarakonda, CrystEngComm, 2009, 11, 989–992 RSC.
- A. D. Bond, CrystEngComm, 2007, 9, 833–834 RSC; G. R. Desiraju, CrystEngComm, 2003, 5, 466–467 RSC; J. D. Dunitz, CrystEngComm, 2003, 5, 506 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- L. Brammer, G. Mínguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712 RSC.
- P. Metrangolo, T. Pilati and G. Resnati, CrystEngComm, 2006, 8, 946–947 RSC; K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC; S. Samai and K. Biradha, CrystEngComm, 2009, 11, 482–492 RSC.
- P. Metrangolo, T. Pilati, G. Terraneo, S. Biella and G. Resnati, CrystEngComm, 2009, 11, 1187 RSC.
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC; C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651–669 RSC.
- R. D. L. Johnstone, D. Francis, A. R. Lennie, W. G. Marshall, S. A. Moggach, S. Parsons, E. Pidcock and J. E. Warren, CrystEngComm, 2008, 10, 1758 RSC.
- J. Ruiz, V. Rodríguez, N. Cutillas, A. Hoffmann, A.-C. Chamayou, K. Kazmierczak and C. Janiak, CrystEngComm, 2008, 10, 1928–1938 RSC.
- Reviews about coordination polymers/MOFs: C. Janiak, Dalton Trans., 2003, 2781–2804 Search PubMed; S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC; U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastré, J. Mater. Chem., 2006, 16, 626–636 RSC; N. R. Champness, Dalton Trans., 2006, 877–880 RSC; C. Janiak, Angew. Chem., Int. Ed. Engl., 1997, 36, 1431–1434 RSC; D. J. Tranchemontagne, Z. Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS; K. Biradha, M. Sarkar and L. Rajput, Chem. Commun., 2006, 4169–4179 CrossRef CAS; B.-H. Ye, M.-L. Tong and X.-M. Chen, Coord. Chem. Rev., 2005, 249, 545–565 RSC; B. Kesanli and W. Lin, Coord. Chem. Rev., 2003, 246, 305–326 CrossRef CAS; C. L. Cahill, D. T. de Lill and M. Frisch, CrystEngComm, 2007, 9, 15–26 CrossRef CAS; Y. Zhou, M. Hong and X. Wu, Chem. Commun., 2006, 135–143 RSC; R. Robson, Dalton Trans., 2008, 5113–5131 RSC; K. M. Fromm, Coord. Chem. Rev., 2008, 252, 856–885 RSC; C.-L. Chen, B.-S. Kang and C.-Y. Su, Aust. J. Chem., 2006, 59, 3–18 CrossRef CAS; J.-P. Zhang and X.-M. Chen, Chem. Commun., 2006, 1689–1699 CrossRef CAS; L. Carlucci, G. Ciani and D. Proserpio, Coord. Chem. Rev., 2003, 246, 247–289 RSC; S.-L. Zheng and X.-M. Chen, Aust. J. Chem., 2004, 57, 703–712 CrossRef CAS; D. Maspoch, D. Ruiz-Molina and J. Veciana, Chem. Soc. Rev., 2007, 36, 770–818 CrossRef CAS; D. Maspoch, D. Ruiz-Molina and J. Veciana, J. Mater. Chem., 2004, 14, 2713–2723 RSC; S. R. Batten and K. S. Murray, Coord. Chem. Rev., 2003, 246, 103–130 RSC; A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS; M. P. Suh, Y. E. Cheon and E. Y. Lee, Coord. Chem. Rev., 2008, 252, 1007–1026 CrossRef CAS; G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 CrossRef CAS; S. Bureekaew, S. Shimomura and S. Kitagawa, Sci. Technol. Adv. Mater., 2008, 9, 014108 RSC Art. No. 014108 T. K. Maji and S. Kitagawa, Pure Appl. Chem., 2007, 79, 2155–2177 CrossRef; S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 Search PubMed; S. Kitagawa, S.-I. Noro and T. Nakamura, Chem. Commun., 2006, 701–707 CrossRef CAS; C. J. Kepert, Chem. Commun., 2006, 695–700 CrossRef CAS; S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC; S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 RSC.
- F. Schröder, D. Esken, M. Cokoja, M. W. E. van den Berg, O. I. Lebedev, G. Van Tendeloo, B. Walaszek, G. Buntkowsky, H.-H. Limbach, B. Chaudret and R. A. Fischer, J. Am. Chem. Soc., 2008, 130, 6119–6130 CrossRef; M. Müller, S. Hermes, K. Kähler, M. W. E. van den Berg, M. Muhler and R. A. Fischer, Chem. Mater., 2008, 20, 4576–4587 CrossRef; S. Hermes, M.-K. Schröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS.
- T. Uemura, Y. Ono, K. Kitagawa and S. Kitagawa, Macromolecules, 2008, 41, 87–94 CrossRef CAS; T. Uemura, K. Kitagawa, S. Horike, T. Kawamura, S. Kitagawa, M. Mizuno and K. Endo, Chem. Commun., 2005, 5968–5970 RSC; T. Uemura, R. Kitaura, Yusuke Ohta, M. Nagaoka and S. Kitagawa, Angew. Chem., Int. Ed., 2006, 45, 4112–4116 CrossRef CAS.
- H. A. Habib, A. Hoffmann, H. A. Höppe, G. Steinfeld and C. Janiak, Inorg. Chem., 2009, 48, 2166–2180 CrossRef CAS.
- H. A. Habib, A. Hoffmann, H. A. Höppe and C. Janiak, Dalton Trans., 2009, 1742–1751 RSC; H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 1734–1744 RSC.
- P. Ren, M.-L. Liu, J. Zhang, W. Shi, P. Cheng, D.-Z. Liao and S.-P. Yan, Dalton Trans., 2008, 4711–4713 RSC; J. R. Black, N. R. Champness, W. Levason and G. Reid, J. Chem. Soc., Chem. Commun., 1995, 1277–9 RSC; J. R. Black, N. R. Champness, W. Levason and G. Reid, Inorg. Chem., 1996, 35, 1820–4 CrossRef CAS; J. R. Black, N. R. Champness, W. Levason and G. Reid, J. Chem. Soc., Dalton Trans., 1995, 3439–45 RSC.
- W. L. Leong, A. Y.-Y. Tam, S. K. Batabyal, L. W. Koh, S. Kasapis, V. W.-W. Yam and J. J. Vittal, Chem. Commun., 2008, 3628–3630 RSC; X.-Q. Song, W.-S. Liu, W. Dou, J.-R. Zheng, X.-L. Tang, H.-R. Zhang and D.-Q. Wang, Dalton Trans., 2008, 3582–3591 RSC; C.-J. Li, M.-X. Peng, J.-D. Leng., M.-M. Yang, Z. Lin and M.-L. Tong, CrystEngComm, 2008, 10, 1645–1652 RSC; W. Liu, L. Ye, X. Liu, L. Yuan, J. Jiang and C. Yan, CrystEngComm, 2008, 10, 1395–1403 RSC.
- S. Zang, Y. Su, Y.-Z. Li, J. Lin, X. Duan, Q. Meng and S. Gao, CrystEngComm, 2009, 11, 122–129 RSC; Y.-L. Wang, Q.-Y. Liu and L. Xu, CrystEngComm, 2008, 10, 1667–1673 RSC; J.-Y. Zhang, Q. Yue, Q.-X. Jia, A.-L. Cheng and E.-Q. Gao, CrystEngComm, 2008, 10, 1443–1449 RSC.
- P. Ayyappan, G. Sirokma, O. R. Evans, T. H. Warren and W. Lin, Inorg. Chim. Acta, 2004, 357, 3999–4004 CrossRef CAS; C. Janiak, T. G. Scharmann, P. Albrecht, F. Marlow and R. MacDonald, J. Am. Chem. Soc., 1996, 118, 6307–6308 CrossRef CAS.
- E. Pardo, R. Ruiz-García, J. Cano, X. Ottenwaelder, R. Lescouëzec, Y. Journaux, F. Lloret and M. Julve, Dalton Trans., 2008, 2780–2805 RSC.
- K. Drabent, Z. Ciunik and A. Ozarowski, Inorg. Chem., 2008, 47, 3358–3365 CrossRef CAS; D. R. Turner, S. N. Pek, J. D. Cashion, B. Moubaraki, K. S. Murray and S. R. Batten, Dalton Trans., 2008, 6877–6879 RSC; A. D. Burrows, C. G. Frost, M. F. Mahon, M. Winsper, C. Richardson, J. P. Attfield and J. A. Rodgers, Dalton Trans., 2008, 6788–6795 RSC; Z.-G. Gu, Y.-F. Xu, X.-J. Yin, X.-H. Zhou, J.-L. Zuo and X.-Z. You, Dalton Trans., 2008, 5593–5602 RSC; B.-W. Hu, J.-P. Zhao, E. C. Sanudo, F.-C. Liu, Y.-F. Zeng and X.-H. Bu, Dalton Trans., 2008, 5556–5559 RSC; H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 4877–4884 RSC; M. A. M. Abu-Youssef, A. Escuer, F. A. Mautner and L. Öhrström, Dalton Trans., 2008, 3553–3558 RSC; M.-L. Liu, W. Shi, H.-B. Song, P. Cheng, D.-Z. Liao and S.-P. Yan, CrystEngComm, 2009, 11, 102–108 RSC; V. H. Tran and B. Swiatek-Tran, Dalton Trans., 2008, 4860–4865 RSC; W. Li, Z.-F. Ju, Q.-X. Yao and J. Zhang, CrystEngComm, 2008, 10, 1325–1327 RSC; Q.-X. Jia, Y.-Q. Wang, Q. Yue, Q.-L. Wang and E.-Q. Gao, Chem. Commun., 2008, 4894–4896 RSC; W. Li, H.-P. Jia, Z.-F. Ju and J. Zhang, Dalton Trans., 2008, 5350–5357 RSC; N. Marino, T. F. Mastropietro, D. Armentano, G. De Munno, R. P. Doyle, F. Lloret and M. Julve, Dalton Trans., 2008, 5152–5154 RSC; B. Zhang, Y. Zhang, J. Zhang, J. Li and D. Zhu, Dalton Trans., 2008, 5037–5040 RSC; J.-Y. Zhang, Y. Ma, A.-L. Cheng, Q. Yue, Q. Sun and E.-Q. Gao, Dalton Trans., 2008, 2061–2066 RSC.
- S. K. Henninger, H. A. Habib and C. Janiak, J. Am. Chem. Soc., 2009, 131, 2776–2777 CrossRef CAS.
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC.
- A. I. Kitaigorodskii, “Molecular Crystals and Molecules”, Academic Press, New York, 1973 Search PubMed.
- G. R. Desiraju and J. A. R. P. Sarma, Proc.–Indian Acad. Sci., Chem. Sci., 1986, 96, 599–605 Search PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- M. Polito, E. D'Oria, L. Maini, P. G. Karamertzanis, F. Grepioni, D. Braga and S. L. Price, CrystEngComm, 2008, 10, 1848 RSC.
- S. K. Chandran, R. Thakuria and A. Nangia, CrystEngComm, 2008, 10, 1891 RSC.
- C. Capacci-Daniel, S. Dehghan, V. M. Wurster, J. A. Basile, R. Hiremath, A. A. Sarjeant and J. A. Swift, CrystEngComm, 2008, 10, 1875 RSC.
- S. Busi, H. Saxell, R. Fröhlich and K. Rissanen, CrystEngComm, 2008, 10, 1803 RSC.
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS; M. M. Conn and J. Rebek, Jr., Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS; C. A. Schalley, Adv. Mater., 1999, 11, 1535–1537 CrossRef CAS; J. Rebek, Jr., Acc. Chem. Res., 1999, 32, 278–286 CrossRef; C. A. Schalley and J. Rebek, Jr., Chemical Encapsulation in Self-Assembling Capsules, Stimulating Concepts in Chemistry, ed. J. F. Stoddart, F. Vögtle and M. Shibasaki, Wiley–VCH, Weinheim, 2000, p. 199 Search PubMed.
- J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Nature, 1994, 368, 229–231 CrossRef CAS; G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049–1052 CrossRef; J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS; J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000–1002 CrossRef CAS.
- A. Shivanyuk, K. Rissanen and E. Kolehmainen, Chem. Commun., 2000, 1107–1108 RSC; A. Shivanyuk, E. F. Paulus, K. Rissanen, E. Kolehmainen and V. Böhmer, Chem.–Eur. J., 2001, 7, 1944–1951 CrossRef CAS; H. Mansikkamäki, M. Nissinen and K. Rissanen, Chem. Commun., 2002, 1902–1903 RSC; H. Mansikkamäki, M. Nissinen, C. Schalley and K. Rissanen, New J. Chem., 2003, 27, 88–97 RSC; H. Mansikkamäki, C. A. Schalley, M. Nissinen and K. Rissanen, New J. Chem., 2005, 29, 116–127 RSC; H. Mansikkamäki, M. Nissinen and K. Rissanen, CrystEngComm, 2005, 7, 519–526 RSC; K. Murayama and K. Aoki, Chem. Commun., 1997, 119–120 RSC.
- V. T. Nguyen, R. Bishop, I. Y. H. Chan, D. C. Craig and M. L. Scudder, CrystEngComm, 2008, 10, 1810 RSC.
- W. Baker, A. J. Floyd, J. F. W. McOmie, G. Pope, A. S. Weaving and J. H. Wild, J. Chem. Soc., 1956, 2010 RSC.
- H. M. Powell and B. D. P. Wetters, Chem. Ind. (London), 1955, 256–257 CAS.
- D. D. MacNicol, Structure and design of inclusion compounds: the clathrates of hydroquinone, phenol, Dianin's compound and related systems, in Inclusion Compounds, vol. 2, ed. J. L. Atwood, J. E. D. Davies and D. D. MacNicol, Academic Press, London, 1984, ch. 1, pp. 1–45 Search PubMed.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef; S. R. Batten, CrystEngComm, 2001, 3, 67–72 RSC.
- M. J. Hardie and C. J. Sumby, Inorg. Chem., 2004, 43, 6872 CrossRef CAS; C. J. Sumby and M. J. Hardie, Angew. Chem., Int. Ed., 2005, 44, 6395 CrossRef CAS; T. K. Ronson, J. Fisher, L. P. Harding and M. J. Hardie, Angew. Chem., Int. Ed., 2007, 46, 9086 CrossRef CAS.
- T. K. Ronson and M. J. Hardie, CrystEngComm, 2008, 10, 1731 RSC.
- H. R. Webb, M. J. Hardie and C. L. Raston, Chem.–Eur. J., 2001, 7, 3616 CrossRef CAS; S. J. Dalgarno and C. L. Raston, Chem. Commun., 2002, 2216 RSC.
- J. D. Wright, Molecular Crystals, Cambridge University Press, Cambridge, 1987 Search PubMed.
- R. Kuroda, T. Sato and Y. Imai, CrystEngComm, 2008, 10, 1881–1890 RSC.
- R. Kuroda, Y. Imai and N. Tajima, Chem. Commun., 2002, 2848 RSC; R. Kuroda, Y. Imai and T. Sato, Chirality, 2001, 13, 588 CrossRef CAS; Y. Imai, N. Tajima, T. Sato and R. Kuroda, Chirality, 2002, 14, 604 CrossRef CAS; Y. Imai, N. Tajima, T. Sato and R. Kuroda, Org. Lett., 2006, 8, 2941 CrossRef CAS.
- A. Lemmerer, S. A. Bourne and M. A. Fernandes, CrystEngComm, 2008, 10, 1750 RSC.
- F. C. Pigge, V. R. Vangala and D. C. Swenson, Chem. Commun., 2006, 2123 RSC; N. Ramasubbu, R. Parthasarathay and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308 CrossRef CAS; F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem.–Eur. J., 2006, 12, 8952 CrossRef CAS; G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725 CrossRef CAS; C. B. Aakeröy, J. Desper, B. A. Helfrich, P. Metrangelo, T. Pilati, G. Resnati and A. Stevenazzi, Chem. Commun., 2007, 4236 RSC; B. K. Saha and A. Nangia, Cryst. Growth Des., 2007, 7, 393 CrossRef CAS; E. Corradi, S. V. Meille, M. T. Messina, P. Metrangelo and G. Resnati, Angew. Chem., Int. Ed., 2000, 39, 1782 CrossRef CAS; B. K. Saha, A. Nangia and M. Jaskólski, CrystEngComm, 2005, 7, 355 RSC; B. K. Saha, A. Nangia and J.-F. Nicoud, Cryst. Growth Des., 2006, 6, 1278 CrossRef CAS.
- P. Brunet, M. Simard and J. D. Wuest, J. Am. Chem. Soc., 1997, 119, 2737 CrossRef CAS.
- M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313–323 CrossRef CAS; M. W. Hosseini, CrystEngComm, 2004, 6, 318–322 RSC.
- S. A. Barnett, A. J. Blake and N. R. Champness, CrystEngComm, 2003, 5, 134–136 RSC; C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew. Chem., Int. Ed., 2001, 40, 3240 CrossRef CAS; C. B. Aakeröy, M. Fasulo, N. Schultheiss, J. Desper and C. Moore, J. Am. Chem. Soc., 2007, 129, 13772–13773 CrossRef.
- J. Hamblin, S. P. Argent, A. J. Blake, C. Wilson and N. R. Champness, CrystEngComm, 2008, 10, 1782 RSC.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS; L. M. A. Perdigão, N. R. Champness and P. H. Beton, Chem. Commun., 2006, 538–540 RSC; P. A. Staniec, L. M. A. Perdigão, A. Saywell, N. R. Champness and P. H. Beton, ChemPhysChem, 2007, 8, 2177–2181 CrossRef CAS; M. Surin, P. Samori, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Angew. Chem., Int. Ed., 2007, 46, 245–249 CrossRef CAS; M. Lackinger, S. Griessl, W. A. Heckl, M. Hietschold and G. W. Flynn, Langmuir, 2005, 21, 4984–4988 CrossRef CAS.
- A. Gavezzotti, Molecular Aggregation: Structure Analysis and Molecular Simulation of Crystals and Liquids, Oxford University Press, New York, USA, 2007 Search PubMed; E. Weber, Inclusion Compounds, ed. J. L. Atwood, J. E. D. Davies and D. D. Mac Nicol, Oxford University Press, Oxford, 1991, 4, 188–262 Search PubMed.
- A. Bacchi, M. Carcelli, T. Chiodo and F. Mezzadri, CrystEngComm, 2008, 10, 1916 RSC.
- U. Baisch, K. Rubini and D. Braga, CrystEngComm, 2008, 10, 1939 RSC.
- D. Braga, M. Curzi, A. Johansson, M. Polito, K. Rubini and F. Grepioni, Angew. Chem., Int. Ed., 2006, 45, 142–146 CrossRef CAS.
- D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002 CrossRef CAS; N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372 RSC; N. Shan, A. D. Bond and W. Jones, Cryst. Eng., 2002, 5, 9 CrossRef CAS; G. Kaupp, J. Schmeyers and J. Boy, Chemosphere, 2001, 43, 55 CrossRef CAS; J. F. Fernández-Bertrán, M. P. Hernandez, E. Reguera, H. Yee-Madeira, J. Rodriguez, A. Panaque and J. C. Llopiz, J. Phys. Chem. Solids, 2006, 67, 1612 CrossRef CAS; S. L. James, A. L. Garay and A. Pichon, Chem. Soc. Rev., 2007, 36, 846 RSC; N. Masciocchi, F. Castelli, P. M. Forster, M. M. Tafoya and A. K. Cheetham, Inorg. Chem., 2003, 42, 6147 CrossRef CAS; G. M. Espallargas, L. Brammer, J. V. D. Streek, K. Shankland, A. J. Florence and H. Adams, J. Am. Chem. Soc., 2006, 128, 9584 CrossRef; G. M. Espallargas, M. Hipler, A. J. Florence, P. Femandes, J. V. D. Streek, M. Brunelli, W. I. F. David, K. Shankland and L. Brammer, J. Am. Chem. Soc., 2007, 129, 15606 CrossRef.
- C. J. Adams, M. A. Kurawa, M. Lusi and A. G. Orpen, CrystEngComm, 2008, 10, 1790 RSC.
- B. R. Bhogala, S. K. Chandran, L. S. Reddy, R. Thakuria and A. Nangia, CrystEngComm, 2008, 10, 1735 RSC.
- M. Rafilovich, J. Bernstein, R. K. Harris, D. C. Apperley, P. G. Karamertzanis and S. L. Price, Cryst. Growth Des., 2005, 5, 2197 CrossRef CAS; L. S. Reddy, S. K. Chandran, S. George, N. J. Babu and A. Nangia, Cryst. Growth Des., 2007, 7, 2675 CrossRef CAS.
- C. B. Aakeröy, J. Desper, M. Fasulo, I. Hussain, B. Levin and N. Schultheiss, CrystEngComm, 2008, 10, 1816 RSC.
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC.
- L. Infantes and S. Motherwell, CrystEngComm, 2002, 4, 454–461 RSC; L. Infantes, J. Chisholm and S. Motherwell, CrystEngComm, 2003, 5, 480 RSC; M. Mascal, L. Infantes and J. Chisholm, Angew. Chem., Int. Ed., 2006, 45, 32 CrossRef.
- O. Fabelo, J. Pasán, L. Cañadillas-Delgado, F. S. Delgado, A. Labrador, F. Lloret, M. Julve and C. Ruiz-Pérez, CrystEngComm, 2008, 10, 1743 RSC.
- J. Ruiz, R. Arauz, V. Riera, M. Vivanco, S. García-Granda and A. Menéndez-Velázquez, Organometallics, 1994, 13, 4162 CrossRef CAS.
- R. Kuroda, K. Higashiguchi, S. Hasebe and Y. Imai, CrystEngComm, 2004, 6, 463–468 Search PubMed; Z. Duan, Y. Zhang, B. Zhang and D. B. Zhu, J. Am. Chem. Soc., 2009, 131, 6934–6935 CrossRef CAS; N. Casati, P. Macchi and A. Sironi, Chem.–Eur. J., 2009, 15, 4446–4457 CrossRef CAS.
- J. Harada, M. Harakawa and K. Ogawa, CrystEngComm, 2008, 10, 1777 RSC.
- A. Brillante, I. Bilotti, R. G. Della Valle, E. Venuti, S. Milita, C. Dionigi, F. Borgatti, A. N. Lazar, F. Biscarini, M. Mas-Torrent, N. S. Oxtoby, N. Crivillers, J. Veciana, C. Rovira, M. Leufgen, G. Schmidt and L. W. Molenkamp, CrystEngComm, 2008, 10, 1899 RSC.
- J.-H. Her, P. W. Stephens, Q. Zhu, K. J. Nelson and J. S. Miller, CrystEngComm, 2008, 10, 1728 RSC.
- I. D. H. Oswald, I. Chataigner, S. Elphick, F. P. A. Fabbiani, A. R. Lennie, J. Maddaluno, W. G. Marshall, T. J. Prior, C. R. Pulham and R. I. Smith, CrystEngComm, 2009, 11, 359 RSC; I. D. H. Oswald and C. R. Pulham, CrystEngComm, 2008, 10, 1114 RSC; A. J. Davidson, I. D. H. Oswald, D. J. Francis, A. R. Lennie, W. G. Marshall, D. I. A. Millar, C. R. Pulham, J. E. Warren and A. S. Cumming, CrystEngComm, 2008, 10, 162 RSC; F. P. A. Fabbiani and C. R. Pulham, Chem. Soc. Rev., 2006, 35, 932 RSC.
- D. R. Allan, A. J. Blake, D. Huang, T. J. Prior and M. Schröder, Chem. Commun., 2006, 4081 RSC; A. Prescimone, C. J. Milios, S. A. Moggach, J. E. Warren, A. R. Lennie, J. Sanchez-Benitez, K. Kamenev, R. Bircher, M. Murrie, S. Parsons and E. K. Brechin, Angew. Chem., Int. Ed., 2008, 47, 2828 CrossRef CAS.
- P. M. Bhatt and G. R. Desiraju, CrystEngComm, 2008, 10, 1747 RSC.
- P. Vishweshwar, J. A. McMahon and M. J. Zaworotko, Crystal Engineering of Pharmaceutical Co-crystals, in Frontiers in Crystal Engineering, ed. E. R. T. Tiekink and J. J. Vittal, Wiley, Chichester, UK, 2006, pp. 25–49 Search PubMed; C. B. Aakeroy, J. Desper and M. E. Fasulo, CrystEngComm, 2006, 8, 586–588 Search PubMed; C. B. Aakeroy, D. J. Salmon, M. M. Smith and J. Desper, CrystEngComm, 2009, 11, 439–443 RSC.
- A. Henschel, K. Gedrich, R. Kraehnert and S. Kaskel, Chem. Commun., 2008, 4192–4194 RSC; U. Ravon, M. E. Domine, C. Gaudillère, A. Desmartin-Chomel and D. Farusseng, New J. Chem., 2008, 32, 937–940 RSC; F. Gándara, B. Gomez-Lor, E. Gutiérrez-Puebla, M. Iglesias, M. A. Monge, D. M. Proserpio and N. Snejko, Chem. Mater., 2008, 20, 72–76 CrossRef CAS; B. Gómez-Lor, E. Gutiérrez-Puebla, M. Iglesias, M. A. Monge, C. Ruiz-Valero and N. Snejko, Chem. Mater., 2005, 17, 2568–2573 CrossRef CAS; S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS; C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075–1078 CrossRef CAS; C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS; J. W. Han and C. L. Hill, J. Am. Chem. Soc., 2007, 129, 15094–15095 CrossRef CAS; J. Perles, M. Iglesias, M.-A. Martín-Luengo, M. A. Monge, C. Ruiz-Valero and N. Snejko, Chem. Mater., 2005, 17, 5837–5842 CrossRef CAS; B. Xiao, H. Hou and Y. Fan, J. Organomet. Chem., 2007, 692, 2014–2020 CrossRef CAS; M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 12639–12641 CrossRef CAS; C. Di Nicola, Y. Yu. Karabach, A. M. Kirillov, M. Monari, L. Pandolfo, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2007, 46, 221–230 CrossRef; F. X. Llabrés i Xamena, O. Casanova, R. Galiasso Tailleur, H. Garcia and A. Corma, J. Catal., 2008, 255, 220–227 CrossRef CAS; S.-H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563–2565 RSC; R.-Q. Zou, H. Sakurai and Q. Xu, Angew. Chem., Int. Ed., 2006, 45, 2542–2546 CrossRef CAS; S. De Rosa, G. Giordano, T. Granato, A. Katovic, A. Siciliano and F. Tripicchio, J. Agric. Food Chem., 2005, 53, 8306–8309 CrossRef; P. Mahata, G. Madras and S. Natarajan, J. Phys. Chem. B, 2006, 110, 13759–13768 CrossRef CAS; L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem.–Eur. J., 2006, 12, 7353–7363 CrossRef CAS; C. J. Chuck, M. G. Davidson, M. D. Jones, G. Kociok-Köhn, M. D. Lunn and S. Wu, Inorg. Chem., 2006, 45, 6595–6597 CrossRef CAS.
- A. Pichon, A. Lazuem Garay and S. L. James, CrystEngComm, 2006, 8, 211 RSC.
- A. Pichon and S. L. James, CrystEngComm, 2008, 10, 1839–1847 RSC.
- W.-J. Son, J. Kim, J. Kim and W.-S. Ahn, Chem. Commun., 2008, 6336–6338 RSC.
- Y. Du, A. L. Thompson and D. O'Hare, Chem. Commun., 2008, 5987–5989 RSC.
- S. K. Ghosh and S. Kitagawa, CrystEngComm, 2008, 10, 1739–1742 RSC.
- N. N. Adarsh, D. K. Kumar and P. Dastidar, CrystEngComm, 2008, 10, 1565–1573 RSC.
- M. Kato, T. Fujihara, D. Yano and A. Nagasawa, CrystEngComm, 2008, 10, 1460–1466 RSC; H.-P. Wu, C. Janiak, G. Rheinwald and H. Lang, J. Chem. Soc., Dalton Trans., 1999, 183 RSC; C. Janiak, L. Uehlin, H.-P. Wu, P. Klüfers, H. Piotrowski and T. G. Scharmann, J. Chem. Soc., Dalton Trans., 1999, 3121 RSC; M. A. Withersby, A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li and M. Schröder, Angew. Chem., Int. Ed. Engl., 1997, 36, 2327 CrossRef CAS.
- J. L. Sague, M. Meuwly and K. M. Fromm, CrystEngComm, 2008, 10, 1542–1549 RSC; P. Byrne, G. O. Lloyd, K. M. Anderson, N. Clarke and J. W. Steed, Chem. Commun., 2008, 3720–3722 RSC.
- H.-L. Sun, Z.-M. Wang, S. Gao and S. R. Batten, CrystEngComm, 2008, 10, 1796–1802 RSC.
- M. Du, Z.-H. Zhang, X.-G. Wang, L.-F. Tang and X.-J. Zhao, CrystEngComm, 2008, 10, 1855–1865 RSC.
- Further recent examples: Z. Lin, Y. Li, A. M. Z. Slawin and R. E. Morris, Dalton Trans., 2008, 3989–3994 Search PubMed; M. Li, Z. Li and D. Li, Chem. Commun., 2008, 3390–3392 RSC; C. Volkringer, T. Loiseau, J. Marrot and G. Férey, CrystEngComm, 2009, 11, 58–60 RSC; G. Mahmoudi and A. Morsali, CrystEngComm, 2009, 11, 50–51 RSC; R. Heck, J. Bacsa, J. E. Warren, M. J. Rosseinsky and D. Bradshaw, CrystEngComm, 2008, 10, 1687–1692 RSC; K. A. Brown, D. P. Martin and R. L. LaDuca, CrystEngComm, 2008, 10, 1305–1308 RSC; J.-Q. Liu, Y.-Y. Wang, L.-F. Ma, G.-L. Wen, Q.-Z. Shi, S. R. Batten and D. M. Proserpio, CrystEngComm, 2008, 10, 1123–1125 RSC; Y. Qi, Y.-X. Che, S. R. Batten and J.-M. Zheng, CrystEngComm, 2008, 10, 1027–1030 RSC; F. Luo, Y. Yang, Y. Che and J. Zheng, CrystEngComm, 2008, 10, 981–982 RSC; R. K. Feller and A. K. Cheetham, Dalton Trans., 2008, 2034–2042 RSC.
- V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378–395 RSC; I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, J. Solid State Chem., 2005, 178, 2452 CrossRef CAS; I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, Cryst. Growth Des., 2008, 8, 519 CrossRef CAS and references therein.
- H.-P. Wu, C. Janiak, L. Uehlin, P. Klüfers and P. Mayer, Chem. Commun., 1998, 2637–2638 RSC; C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC; H. W. Roesky and M. Andruh, Coord. Chem. Rev., 2003, 236, 91 CrossRef CAS.
- http://www.topos.ssu.samara.ru ; V. A. Blatov, IUCr Compcomm. Newsletter, 2006, 7, 4 Search PubMed ; http://www.iucr.org/iucr-top/comm/ccom/newsletters.
- I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2008, 10, 1822–1838 RSC.
- The three letter symbols, proposed by M. O'Keeffe, can be retrieved with examples and further information from the Reticular Chemistry Structure Resource database, http://rcsr.anu.edu.au/. See also: O. Delgado Friedrichs, M. O'Keeffe and O. M. Yaghi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 515–525 Search PubMed.
- B. R. Bhogala, P. K. Thallapally and A. Nangia, Cryst. Growth Des., 2004, 4, 215 CrossRef CAS.
- C. Janiak, unpublished results.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef.
- G. Férey, J. Solid State Chem., 2000, 152, 37 CrossRef CAS; S. Surble, F. Millange, C. Serre, G. Férey and R. I. Walton, Chem. Commun., 2006, 1518 RSC; C. Serre, F. Millange, S. Surble and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6285 CrossRef; G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296 CrossRef CAS.
- L.-F. Ma, L.-Y. Wang, Y.-Y. Wang, M. Duc and J.-G. Wang, CrystEngComm, 2009, 11, 109–117 RSC; H. A. Habib, J. Sanchiz and C. Janiak, Inorg. Chim. Acta, 2009, 362, 2452–2460 CrossRef CAS; D. P. Martin, R. M. Supkowski and R. L. LaDuca, Dalton Trans., 2009, 514–520 RSC; L.-P. Zhang, J. Yang, J.-F. Ma, Z.-F. Jia, Y.-P. Xie and G.-H. Wei, CrystEngComm, 2008, 10, 1410–1420 Search PubMed; J. Yao, Z.-D. Lu, Y.-Z. Li, J.-G. Lin, X.-Y. Duan, S. Gao, Q.-J. Meng and C.-S. Lu, CrystEngComm, 2008, 10, 1379–1383 RSC; Y. Qi, Y.-X. Che and J.-M. Zheng, CrystEngComm, 2008, 10, 1137–1139 RSC; B. Wisser, Y. Lu and C. Janiak, Z. Anorg. Allg. Chem., 2007, 633, 1189–1192 CrossRef CAS; S. C. Manna, K.-I. Okamoto, E. Zangrando and N. R. Chaudhuri, CrystEngComm, 2007, 9, 199–292 RSC; Z.-F. Chen, S.-F. Zhang, H.-S. Luo, B. F. Abrahams and H. Liang, CrystEngComm, 2007, 9, 27–29 RSC; A. Pichon, C. M. Fierro, M. Nieuwenhuyzen and S. James, CrystEngComm, 2007, 9, 449–451 RSC; J. Pasán, J. Sanchiz, F. Lloret, M. Julve and C. Ruiz-Pérez, CrystEngComm, 2007, 9, 478–487 RSC; M. D. Stephenson and M. J. Hardie, Dalton Trans., 2006, 3407–3417 RSC; G.-H. Wei, J. Yang, J.-F. Ma, Y.-Y. Liu, S.-L. Li and L.-P. Zhang, Dalton Trans., 2008, 3080–3092 RSC; R. Carballo, B. Covelo, E. M. Vázquez-López, E. García-Martínez, A. Castiñeiras and C. Janiak, Z. Anorg. Allg. Chem., 2005, 631, 2006–2010 CrossRef CAS.
- J. Yang, J.-F. Ma, Y.-Y. Liu and S. R. Batten, CrystEngComm, 2009, 11, 151–159 RSC; Y.-Q. Lan, S.-L. Li, Y.-M. Fu, Y.-H. Xu, L. Li, Z.-M. Su and Q. Fu, Dalton Trans., 2008, 6796–6807 RSC.
- X. Zhu, Y.-M. Zhang, B.-L. Li and Y. Zhang, J. Coord. Chem., 2006, 59, 513–522 CrossRef CAS; B. Li, X. Zhu, J. Zhou, Y. Peng and Y. Zhang, Polyhedron, 2004, 23, 3133 CrossRef CAS; B. Li, B. Li, X. Zhu, X. Lu and Y. Zhang, J. Coord. Chem., 2004, 57, 1361 CrossRef CAS; J. Zhou, X. Zhu, Y. Zhang, Y. Zhang and B. Li, Inorg. Chem. Commun., 2004, 7, 949 CrossRef CAS; X. Zhu, B.-Z. Li, J.-H. Zhou, B.-L. Li and Y. Zhang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m191 CrossRef; B. Li, B. Li, X. Zhu, L. Zhu and Y. Zhang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m350 CrossRef; B. Li, Z. Xu, Z. Cao, L. Zhu and K. Yu, Transition Met. Chem., 1999, 24, 622 CrossRef CAS; B. Li, J. Zou, C. Duan, Y. Liu, X. Wei and Z. Xu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 165 CrossRef.
- A.-X. Tian, J. Ying, J. Peng, J.-Q. Sha, Z.-G. Han, J.-F. Ma, Z.-M. Su, N.-H. Hu and H.-Q. Jia, Inorg. Chem., 2008, 47, 3274–3283 CrossRef CAS.
- X.-L. Wang, C. Qin, E.-B. Wang and Z.-M. Su, Chem.–Eur. J., 2006, 12, 2680–2691 CrossRef CAS; X. Liu, H. Ge, Y. Zhang, L. Hu, B. Li and Y. Zhang, J. Mol. Struct., 2006, 796, 129–138 CrossRef CAS.
- J.-L. Du, T.-L. Hu, S.-M. Zhang, Y.-F. Zeng and X.-H. Bu, CrystEngComm, 2008, 10, 1866–1874 RSC.
- H. Ge, L. Wang, Y. Yang, B. Li and Y. Zhang, J. Mol. Struct., 2008, 876, 288–293 CrossRef CAS.
- W.-B. Wang, L.-Y. Wang, B.-L. Lia and Y. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2416–m2417 CrossRef; Y.-F. Peng, H.-Y. Ge, B.-Z. Li, B.-L. Li and Y. Zhang, Cryst. Growth Des., 2006, 6, 994–998 CrossRef CAS.
- Y.-Q. Lan, S.-L. Li, K.-Z. Shao, X.-L. Wang and Z.-M. Su, Dalton Trans., 2008, 3824–3835 RSC; S.-L. Li, Y.-Q. Lan, J.-F. Ma, J. Yang, J. Liu, Y.-M. Fu and Z.-M. Su, Dalton Trans., 2008, 2015–2025 RSC; S.-L. Li, Y.-Q. Lan, J.-F. Ma, J. Yang, X.-H. Wang and Z.-M. Su, Inorg. Chem., 2007, 46, 8283–8290 CrossRef CAS; H. Han, S. Zhang, H. Hou, Y. Fan and Y. Zhu, Eur. J. Inorg. Chem., 2006, 1594–1600 CrossRef CAS; H. Han, Y. Song, H. Hou, Y. Fan and Y. Zhu, Dalton Trans., 2006, 1972–1980 RSC.
- C. M. Fitchett and P. J. Steel, Aust. J. Chem., 2006, 59, 19 CrossRef CAS; T. Dorn, K. M. Fromm and C. Janiak, Aust. J. Chem., 2006, 59, 22 CrossRef CAS; M. B. Duriska, S. R. Batten and D. J. Price, Aust. J. Chem., 2006, 59, 26 CrossRef CAS; Y.-B. Xie, J.-R. Li and X.-H. Bu, Aust. J. Chem., 2006, 59, 34 CrossRef CAS; L. Cunha-Silva, R. Ahmad and M. J. Hardie, Aust. J. Chem., 2006, 59, 40 CrossRef CAS; S.-L. Zheng, M.-L. Tong and X.-M. Chen, Coord. Chem. Rev., 2003, 246, 185–202 CrossRef CAS; N. S. Oxtoby, A. J. Blake, N. R. Champness and C. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4905 CrossRef CAS; M. Munakata, L. P. Wu and T. Kuroda-Sowa, Adv. Inorg. Chem., 1998, 46, 173–303 CAS.
- Recent examples on magnetism in coordination polymers/metal–organic frameworks: D. G. Branzea, L. Sorace, C. Maxim, M. Andruh and A. Caneschi, Inorg. Chem., 2008, 47, 6590–6592 Search PubMed; Y.-Z. Zheng, W. Xue, M.-L. Tong, X.-M. Chen, F. Grandjean and G. J. Long, Inorg. Chem., 2008, 47, 4077–4087 CrossRef CAS; G. Agustí, M. C. Muñoz, A. B. Gaspar and J. A. Real, Inorg. Chem., 2008, 47, 2552–2561 CrossRef CAS; X.-Y. Wang and S. C. Sevov, Inorg. Chem., 2008, 47, 1037–1043 CrossRef CAS; A. M. Kirillov, Y. Y. Karabach, M. Haukka, M. F. C. G. da Silva, J. Sanchiz, M. N. Kopylovich and A. J. L. Pombeiro, Inorg. Chem., 2008, 47, 162–175 CrossRef CAS; R. Sessoli, Inorg. Chim. Acta, 2008, 361, 3356–3364 CrossRef CAS; E. Rentschler and C. von Malotki, Inorg. Chim. Acta, 2008, 361, 3646–3653 CrossRef CAS; R. A. Mole, S. P. Cottrell, J. A. Stride and P. T. Wood, Inorg. Chim. Acta, 2008, 361, 3718–3722 CrossRef CAS; M. A. M. Abu-Youssef, F. A. Mautner and R. Vicente, Inorg. Chim. Acta, 2008, 361, 2895–2900 CrossRef CAS; H.-R. Wen, C.-F. Wang, Z. Y. Du and J.-L. Zuo, Inorg. Chim. Acta, 2008, 361, 2901–2908 CrossRef CAS; A. Ray, J. Chakraborty, B. Samanta, S. Thakurta, C. Marschner, M. S. El Fallah and S. Mitra, Inorg. Chim. Acta, 2008, 361, 1850–1860 CrossRef CAS; G. S. Matouzenko, M. Perrin, B. Le Guennic, C. Genre, G. Molnar, A. Bousseksou and S. A. Borshch, Dalton Trans., 2007, 934–942 CrossRef CAS; J. Chakraborty, M. Nandi, H. Mayer-Figge, W. S. Sheldrick, L. Sorace, A. Bhaumik and P. Banerjee, Eur. J. Inorg. Chem., 2007, 5033–5044 RSC; M. Seredyuk, A. B. Gaspar, M. C. Munoz, M. Verdaguer, F. Villain and P. Gütlich, Eur. J. Inorg. Chem., 2007, 4481–4491 CrossRef CAS; K. Abu-Shandi, H. Winkler and C. Janiak, Z. Anorg. Allg. Chem., 2006, 632, 629–633 CrossRef CAS; K. Abu-Shandi and C. Janiak, Z. Naturforsch., B: Chem. Sci., 2005, 60, 1250–1254 CrossRef CAS; H.-L. Sun, Z.-M. Wang and S. Gao, Inorg. Chem., 2005, 44, 2169–2176 CAS.
- W.-X. Zhang, W. Xue, J.-B. Lin, Y.-Z. Zheng and X.-M. Chen, CrystEngComm, 2008, 10, 1770–1776 RSC.
- O. Waldmann, Coord. Chem. Rev., 2005, 249, 2550–2566 CrossRef CAS.
- J. C. Bailar, Jr., Preparative Inorganic Reactions (W. L. Jolly, ed.), Vol. 1, Interscience, New York, 1964, p. 1–25 Search PubMed.
- S. Natarajan and S. Mandal, Angew. Chem., Int. Ed., 2008, 47, 4798–4828 CrossRef CAS; P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638 CrossRef.
- Recent examples: Y. Guo, Z.-Q. Liu, B. Zhao, Y.-H. Feng, G.-F. Xu, S.-P. Yan, P. Cheng, Q.-L. Wang and D.-Z. Liao, CrystEngComm, 2009, 11, 61–66 Search PubMed; S. Pal, D. Jagadeesan, K. L. Gurunatha, M. Eswaramoorthy and T. K. Maji, J. Mater. Chem., 2008, 18, 5448–5451 RSC.
- N. Louvain, N. Mercier and M. Kurmoo, Eur. J. Inorg. Chem., 2008, 1654 CrossRef CAS; M. A. Tershansy, A. M. Goforth, M. D. Smith and H.-C. zur Loye, J. Chem. Crystallogr., 2008, 38, 453 CrossRef CAS; C.-L. Chen and A. M. Beatty, Chem. Commun., 2007, 76 RSC; N. Mercier, CrystEngComm, 2005, 7, 429 RSC; M. C. Aragoni, M. Arca, C. Caltagirone, F. A. Devillanova, F. Demartin, A. Garau, F. Isaia and V. Lippolis, CrystEngComm, 2005, 7, 544 RSC; A. M. Goforth, L. Peterson, Jr., M. D. Smith and H.-C. zur Loye, J. Solid State Chem., 2005, 178, 3529–3540 CrossRef CAS; N. Mercier and A. Riou, Chem. Commun., 2004, 844 RSC; U. Bentrup, M. Feist and E. Kemnitz, Prog. Solid State Chem., 1999, 27, 75 CrossRef CAS.
- E. Redel, C. Röhr and C. Janiak, Chem. Commun., 2009, 2103–2105 RSC; E. Redel, M. Fiederle and C. Janiak, Z. Anorg. Allg. Chem., 2009, 635, 1139–1147 CrossRef CAS; C. H. Arnby, S. Jagner and I. Dance, CrystEngComm, 2004, 6, 257 RSC; S. Jagner and G. Helgesson, Adv. Inorg. Chem., 1991, 37, 1 CAS; L. Subramanian and R. Hoffmann, Inorg. Chem., 1992, 31, 1021 CrossRef CAS.
- H.-H. Li, L.-G. Sun, Z.-R. Chen, Y.-J. Wang and J.-Q. Li, Aust. J. Chem., 2008, 61, 391–396 CrossRef CAS; M. Okubo, M. Enomoto and N. Kojima, Synth. Met., 2005, 152, 461 CrossRef CAS; D. B. Mitzi, J. Mater. Chem., 2004, 14, 2355 RSC.
- P. Rabu and M. Drillon, Adv. Eng. Mater., 2003, 5, 189 CrossRef CAS; S. Flan-drois, N. B. Chanh, R. Duplessix, T. Maris and P. Négrier, Phys. Status Solidi A, 1995, 149, 697 CrossRef CAS.
- N. Mercier, S. Poiroux, A. Riou and P. Batail, Inorg. Chem., 2004, 43, 8361 CrossRef CAS; T. Gebauer and G. Schmid, Z. Anorg. Allg. Chem., 1999, 625, 1124 CrossRef CAS; D. B. Mitzi and K. Liang, Chem. Mater., 1997, 9, 2990 CrossRef CAS; T. Ishihara, J. Takahashi and T. Goto, Solid State Commun., 1989, 69, 933 CrossRef CAS.
- C. E. Costin-Hogan, C.-L. Chen, E. Hughes, A. Pickett, R. Valencia, N. P. Rath and A. M. Beatty, CrystEngComm, 2008, 10, 1910–1915 RSC.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CAS.
- L. R. MacGillivray, CrystEngComm, 2002, 4, 37 RSC; L. R. Macgillivray, G. S. Papaefstathiou, T. Friscic, T. D. Hamilton, D. K. Bucar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280–291 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS; M. C. Etter, J. Phys. Chem., 1991, 95, 4601–4610 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546 CrossRef CAS; R. Robson, B. F. Abrahams, S. R. Batten, R. W. Gable, B. F. Hoskins and J. P. Liu, ACS Symp. Ser., 1992, 499, 256 CAS.
| This journal is © The Royal Society of Chemistry 2010 |