Mechanistic considerations on styrene–maleic anhydride copolymerization reactions
Bert
Klumperman
*
Stellenbosch University, Department of Chemistry and Polymer Science, Private Bag X1, Matieland, 7602, South Africa. E-mail: bklump@sun.ac.za; Fax: :+27 21 808 4967; Tel: +27 21 808 3988
First published on 11th January 2010
Abstract
The copolymerization of styrene and maleic anhydride has received significant attention in academia as well as in industry. Nevertheless, the underlying mechanism of the copolymerization is still a point of debate. In this paper, an overview is given that provides compelling evidence in favor of the penultimate unit model as the correct choice to describe the process of chain growth. In addition to that new developments in terms of living radical copolymerization of styrene and maleic anhydride are discussed from a mechanistic point of view.
 Bert Klumperman | Bert Klumperman obtained his PhD degree at the Eindhoven University of Technology in 1994 under the supervision of Prof. AL German and Prof. KF O’Driscoll (Univ. Waterloo, Canada). He worked for 9 years at DSM Research, mainly on the chemistry of styrene–maleic anhydride copolymers. After that he joined Eindhoven University, and from 2007 he has held a South African Research Chair on Advanced Macromolecular Architectures at Stellenbosch University in South Africa. His main research interests are the controlled synthesis of polymer materials, mainly via living radical polymerization, and the assembly of polymer materials on the nanometer length scale. |
Introduction
Copolymers of styrene and maleic anhydride have a long history. They come in a large variety of compositions and molar masses. The reactive maleic anhydride (MAnh) moiety provides the copolymers with a wide variety of options for chemical modification. The copolymerization between styrene (STY) and MAnh has a strongly alternating character. The underlying reasons for this alternating tendency have been the topic of numerous studies. The initial sections of this review will be devoted to an overview around the alternating copolymerization of STY–MAnh. In recent years, STY–MAnh copolymerization was often reported in conjunction with living radical polymerization techniques (nitroxide mediated polymerization (NMP), and reversible addition fragmentation chain transfer (RAFT) mediated polymerization). The special features arising from the use of these techniques will be summarized. Finally, an outlook will be provided into the use of STY–MAnh copolymers for a potentially wide variety of applications. The controlled synthesis and inherent post-polymerization reactivity of STY–MAnh make it an ideal starting material for complex architectures.STY–MAnh copolymers have been produced on a commercial scale for many years. Typically there were producers of low molar mass versions and producers of high molar mass versions. The low molar mass STY–MAnh copolymers are used as polymeric surfactant, as an ingredient in the papermaking industry, as the synthetic polymer component in a polymer–protein conjugate, etc. The high molar mass versions are typically used as engineering plastics. In the latter applications the polymers are often rubber-modified and sometimes glass-fiber reinforced. The main producer of low molar mass STY–MAnh copolymers is currently Sartomer. Polyscope is now the leading company to produce high molar mass copolymers, and they recently expanded their production into the low molar mass region as well. In the low as well as in the high molar mass region, copolymers are synthesized with MAnh contents varying from only a few percent to 50%.
Copolymerization
The copolymerization of STY and MAnh is interesting from a mechanistic point of view. MAnh hardly homopropagates, which means that MAnh–MAnh diads are virtually absent in the polymer chain.1 Furthermore, it is generally accepted that the copolymerization shows a strong tendency towards alternation.2 The explanation for this tendency has been debated a lot in literature. There are two schools of thought in this respect. STY is an electron-rich monomer, while MAnh is an electron-poor monomer. It has been shown via NMR and FTIR techniques that the two comonomers form charge-transfer complexes (CTCs).2 The one school of thought sees the presence of CTCs as evidence for a mechanism in which these CTCs participate in the copolymerization.3,4Copolymerization kinetics as well as copolymer composition and monomer sequence distribution can be adequately described by the so-called complex participation model. The other school of thought uses the more widely applied penultimate unit model (PUM) to describe the copolymerization.5 In the next section, the two models will be presented in more detail. Rather than performing a conventional model discrimination, compelling evidence will be reviewed in favor of the PUM.
Complex participation model
It has been shown numerous times that electron acceptor and electron donor monomer pairs are able to form charge-transfer complexes (CTCs).6 There is quite a body of older literature that explains deviation from the conventional Mayo–Lewis or terminal model (TM) via the so-called complex participation model (CPM).3,4,7 In this model, the propagation reaction is believed to involve the addition of single monomers as well as CTCs. Scheme 1 shows the reactions and their individual rate constants. Note that Scheme 1 only provides the reactions of monomer 1 chain-end radicals. An equivalent set of reactions obviously exists for monomer 2 chain-end radicals. Copolymer composition and rate of copolymerization can be expressed in terms of the individual rate constants and concentration of reactants. In the case of the CPM, this includes the concentration of the CTC, which is temperature dependent.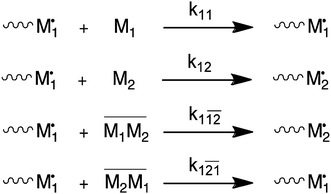 | ||
| Scheme 1 Complex participation model (CPM). Only reactions for monomer 1 chain-end radicals are shown. | ||
Penultimate unit model
The most common way to account for deviation from the TM is via the penultimate unit model (PUM).8 In this model, not only the terminal monomer in a growing chain radical, but also the penultimate unit determines the rate constants of monomer addition for the two comonomers. The model is depicted in Scheme 2, where kijk is the rate constant for the addition of monomer k to a chain-end with terminal unit j and penultimate unit i. Note that Scheme 2 only provides the reactions of monomer 1 chain-end radicals. Also here, an equivalent set of reactions exists for monomer 2 chain-end radicals. The two common versions of the model are the implicit PUM and the explicit PUM.8 The implicit PUM is quite common in that it is necessary in the majority of copolymerization reactions to describe the average propagation rate constant (〈kp〉) as a function of comonomer ratio. Briefly, the majority of copolymerizations appear to obey the TM when it comes to describing copolymer composition versus monomer feed composition (F–f). However, when 〈kp〉 versus monomer feed composition of the same copolymerization is measured by the rotating sector method, large deviations from the TM are observed.9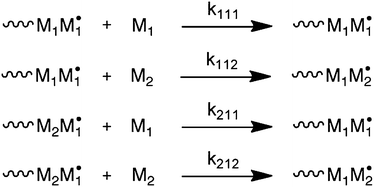 | ||
| Scheme 2 Penultimate unit model (PUM). Only reactions for monomer 1 chain-end radicals are shown. | ||
These deviations are very well captured by the implicit PUM, which means that the penultimate unit effect on the ratio of homopropagation and cross-propagation rate constants is negligible. In other words, the reactivity ratio that describes the ratio of rate constants between homopropagation and cross-propagation is independent of the penultimate unit. This in essence means that the copolymerization can be described with the TM when it comes to copolymer composition versus monomer feed ratio. The implicit PUM further says that the rate constant of homopropagation is affected by the penultimate unit. Written in terms of the rate constants in Scheme 2, this means that kiii ≠ kjii. The explicit PUM on the other hand requires all eight rate constants. In practice, four reactivity ratios are used to describe copolymer composition. The copolymerization of STY and acrylonitrile is a typical example that does not obey the implicit PUM.
STY–MAnh copolymerization kinetics
The large alternating tendency of the STY–MAnh copolymerization makes it difficult to determine reactivity ratios via conventional low conversion copolymerizations. The best way to approach this copolymerization is via a continuous polymerization process in a continuous ideal stirred tank reactor (CSTR).10This system relies on a steady state, which is established after approximately three times the mean residence time. At steady state a feed consisting of solvent, monomers and initiator is continuously fed into the reactor and instantaneously mixed with the reactor contents. Simultaneously, reactor content, i.e. solvent, residual monomers, some residual initiator and polymer are taken out of the reactor. The system works according to the principle that the exiting polymer solution has the exact same composition as the reactor content. Due to the steady state situation, copolymers with a narrow chemical composition can be readily synthesized, even at compositions that would result in major composition drift during batch polymerization.
Fig. 1 shows the results of copolymerization experiments at four different temperatures. The copolymer is isolated from the reaction mixture after dilution with butanon (MEK) and precipitation in isopropanol. This procedure is important since isopropanol is able to dissolve all the residual monomers and precipitate the copolymer. At the same time, isopropanol, unlike e.g. methanol does not lead to esterification of the MAnh residue in the copolymer under ambient conditions. The monomer feed composition is determined from a mass balance, i.e. the amount of STY and MAnh fed to the reactor per unit of time is accurately known, just as the amount of copolymer and its composition. Hence, the steady state concentrations of STY and MAnh in the reactor can be back-calculated with quite good precision.
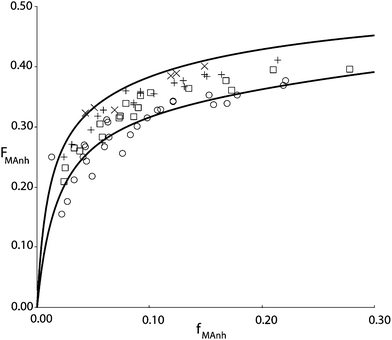 | ||
| Fig. 1 STY–MAnh copolymer composition (FMAnh) versus monomer feed composition (fMAnh) data from experiments in a CSTR at 60 °C (×), 90 °C (+), 110 °C (□) and 140 °C (○). Drawn curves are based on the penultimate unit model according to parameters shown in Table 1, at 60 °C (top curve) and 140 °C (bottom curve).10 | ||
It is interesting to note that the alternating tendency of the copolymerization decreases with increasing reaction temperature. In earlier days this observation was often linked to the decreasing concentration of the CTC with increasing temperature. The variation in concentration as a function of temperature can be described, and subsequently, the reactivity of the complex in the copolymerization can be fitted to match the temperature dependence of the copolymerization reaction. The number of parameters in the CPM is large, and easily accommodates the variations in rate, composition, and monomer sequence distribution as a function of temperature. However, it is also well documented that copolymerization reactions have a tendency to become “more random” with increasing temperature. O'Driscoll explained this behavior on the basis of Arrhenius expressions of the individual propagation rate constants.11 In actual fact, it turns out that the temperature dependence of copolymer composition versus monomer feed composition can equally well be described via CPM as via PUM. In this case, a restricted version of the explicit PUM can be employed that takes into account the absence of MAnh homopropagation. In terms of the PUM rate parameters this means that kSMM and kMMM both equal zero and therefore also the reactivity ratios rMM = rSM = 0. On the basis of this restricted PUM, copolymer composition versus comonomer feed composition curves can be fitted as shown in Fig. 1. It needs to be stressed though that on the basis of this type of experimental data, no discrimination can be made between PUM and CPM. The same is true for the monomer sequence distribution versus comonomer feed composition. Parameters estimated from monomer sequence data can be used to adequately describe the copolymer composition versus comonomer feed composition and vice versa. It is clearly necessary to adopt a different type of experimental data to perform adequate model discrimination. This was found in average propagation rate coefficient as a function of comonomer feed composition.
Fig. 2 shows data from an earlier publication5 in which pulsed laser polymerization (PLP) was used to measure average propagation rate constant (〈kp〉) versus fraction of MAnh in the comonomer feed. It can clearly be seen that the rate coefficient increases strongly towards high MAnh fraction. Parameter fitting on the basis of the PUM results in an adequate description of the experimental data as can be seen in Fig. 2. Conversely, the CPM fails to describe the experimental data. Without going into great detail it can easily be envisaged that the introduction of a fast propagation reaction of the CTC leads to a maximum in 〈kp〉 versus the fraction of MAnh which lies close to fMAnh = 0.5, which clearly differs from the experimental observation. If the combination of copolymer composition, monomer sequence distribution and average propagation rate coefficient versus comonomer feed is fitted at a variety of reaction temperatures, Arrhenius coefficients for the individual rate parameters from the restricted PUM can be determined. Table 1 shows the parameters that result from this exercise. The various experiments were carried out at temperatures in the range of 25 to 140 °C. It needs to be stressed that for practical reasons, PLP experiments were carried out at temperatures from 25–50 °C whereas the polymerization reactions in the CSTR were carried out from 90–140 °C. The rate parameters as depicted in Table 1 show significant cross-dependence, which would clearly show up in joint confidence intervals. Due to the multi-dimensionality of the system, these intervals cannot be shown graphically. Part of the cross-dependence may be due to the two different temperature regimes, although the occurrence of cross-dependence is not uncommon in copolymerization rate parameters.
| Parameter | A/L mol−1 s−1 | E a/kJ mol−1 |
|---|---|---|
| k SSS | 1.10 × 107 | 29.5 |
| k MSS | 3.2 × 106 | 26.1 |
| r SS | 0.79 | 9.3 |
| r MS | 126.5 | 19.6 |
| k SMS | >105 | — |
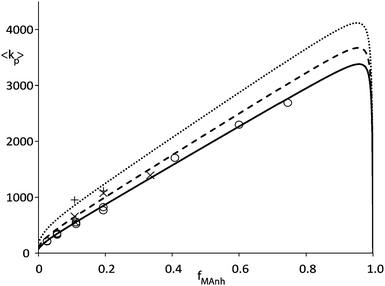 | ||
| Fig. 2 Average propagation rate coefficient (〈kp〉) as a function of fraction MAnh in STY–MAnh copolymerization (fMAnh) at 25 °C (○), 35 °C (×) and 50 °C (+). Curves are calculated on the basis of the PUM and parameters from Table 1 (solid curve: 25 °C, dashed curve: 35 °C, dotted curve: 50 °C).5,10 | ||
There are some interesting implications that arise from the rate parameters in STY–MAnh copolymerization. The one that will be highlighted here is the fraction of MAnh chain-end radicals in a polymerizing system. For any copolymerization, a steady state assumption can be written. In the present case, the rate at which MAnh chain-end radicals are formed is equated to the rate at which they disappear. Mathematically this can be written as shown in eqn (1), where pij is the fraction of chain-end radicals carrying monomer i, the penultimate unit, and monomer j, the terminal unit.
| kSSMpSSfM + kMSMpMSfM − kSMSpSMfS = 0 | (1) |
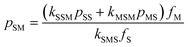 | (2) |
Based on the alternating character of the STY–MAnh copolymerization, it will be obvious that pMS > pSS. Hence, for the overriding majority of comonomer feed compositions, the fraction of MAnh chain-end radicals will be very small.
STY–MAnh living radical polymerization
As indicated above, the STY–MAnh copolymerization has been carried out via nitroxide mediated polymerization (NMP) as well as via reversible addition–fragmentation chain transfer (RAFT) mediated polymerization. Atom transfer radical polymerization (ATRP) seems incompatible with STY–MAnh copolymerization. This is most likely due to interactions of MAnh with the transition metal complex used to mediate such polymerizations.NMP of STY–MAnh has been reported in a few studies.12–14 One of the interesting features of the living radical polymerization (LRP) of STY–MAnh is a direct consequence of its strongly alternating character. If one starts with a comonomer ratio STY–MAnh larger than unity, the polymerization will start as an alternating copolymerization. However, at some point, MAnh is depleted, and the polymerization continues via the homopolymerization of STY. In a conventional radical polymerization, this would have resulted in the synthesis of a heterogeneous mixture of copolymer and homopolymer. In LRP, the result is the in situ formation of a block copolymer. The first block is composed of poly(STY-alt-MAnh), whereas the second block is composed of polySTY. This phenomenon was discovered and employed in early studies. In a very recent study, a kinetic description of the process was provided. In the latter publication, the dependence of the alternating character on polymerization temperature was discussed. This temperature dependence was used to explain that for the synthesis of pure block copolymers, a relatively low polymerization temperature is beneficial.15
In terms of polymerization temperature, reversible addition–fragmentation chain transfer (RAFT) mediated polymerization is superior over NMP. The generation of propagating radicals is accomplished by a conventional initiation process, e.g.via the thermal decomposition of an azo-initiator. As a consequence, polymerization can easily be carried out at temperatures as low as 60–80 °C.16 Due to the temperature dependence of the reactivity ratios of the STY–MAnh copolymerization, these low polymerization temperatures will lead to almost perfectly alternating copolymers. Several aspects of the RAFT-mediated copolymerization of STY–MAnh have been investigated in terms of underlying kinetics and mechanisms.
The initialization process, i.e. the conversion of the original RAFT agent into a single monomer adduct in the STY–MAnh case was studied and compared to STY homopolymerization.17–19 Initialization studies have been carried out in which in situ1H NMR was used to track the concentration profiles of important species as a function of reaction time. Two typical STY homopolymerizations were conducted under identical conditions. It was found that the initialization time was around 45 min when cyanoisopropyl dithiobenzoate (CiPDB) was used.18 When cumyl dithiobenzoate (CDB) was used, the initialization time was around 240 min.19 Under very similar conditions, completely different results were found when the STY–MAnh copolymerization was investigated.17 In the case of CiPDB, the initialization time was virtually identical to that of STY homopolymerization. In this case, addition of the leaving group radical was almost exclusively to the STY monomer. Only after complete initialization, MAnh was added to the cyanopropyl-styryl radical. Also this second reaction occurred with quite high selectivity. In the case of CDB, the initialization of STY–MAnh was extremely fast. Under the conditions where STY initialization took 240 min, the STY–MAnh initialization took less than 5 min. The addition of the cumyl leaving group radical is exclusively to MAnh. This observation was not unexpected, since the cumyl radical is electron-rich, and MAnh is electron-poor. It is known from copolymerization kinetics that this leads to high addition rates.
The other interesting mechanistic study on the RAFT-mediated STY–MAnh copolymerization used ESR spectroscopy to investigate the nature of the intermediate radical.20 In dithiobenzoate-mediated polymerizations, the concentration of intermediate radicals is reasonably high. This means that measurement of concentration and identification of the nature of these radicals are possible with ESR spectroscopy. The study by Du et al. shows experimental spectra of the STY–MAnh copolymerization and a comparison with predictions. Based on the predictions it is clear that there are large differences among intermediate radicals with a STY moiety at each side of the intermediate radical, a MAnh moiety at each side, or a STY moiety at one side and a MAnh at the other side. Comparison of the predictions with the experimental spectra reveals that the dominant structure of the intermediate radical is the one with a MAnh moiety at both sides. Du et al. conclude on the basis of their findings that propagating STY–MAnh chains carry predominantly MAnh moieties at the propagating chain end. This conclusion is in direct contradiction with the calculations shown above, based on the reactivity ratios. Du et al. overlooked the effect of addition rate coefficient to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond of the RAFT agent. Clearly, the rate coefficient of the MAnh chain-end radical addition to the CS double bond is much higher than that of the STY chain-end radical. This effect is apparently large enough to end up with MAnh moieties as the neighboring groups in the intermediate radical, despite their low fraction in the propagating chain-end radicals.
S double bond of the RAFT agent. Clearly, the rate coefficient of the MAnh chain-end radical addition to the CS double bond is much higher than that of the STY chain-end radical. This effect is apparently large enough to end up with MAnh moieties as the neighboring groups in the intermediate radical, despite their low fraction in the propagating chain-end radicals.
Outlook and conclusions
Styrene–maleic anhydride copolymers are highly interesting functional polymers. Commercially available polymers over a wide range of molar mass and chemical composition are used for a wide variety of applications. Among those applications are polymer–protein conjugates as drugs (poly(styrene-co-maleic acid)–neocarzinostatin conjugate (SMANCS)),21 an ingredient for paper-sizing, and a component in glass-fiber reinforced dashboard supports in cars. In recent years, the electrospinning of STY–MAnh copolymers has been reported.22 On the basis of the MAnh reactivity, these electrospun materials can be further modified. One recent example is the immobilization of an enzyme on electrospun STY–MAnh membranes.23 The majority of applications and developments at present are based on STY–MAnh copolymers synthesized through conventional radical copolymerization. The use of living radical polymerization techniques as highlighted in this overview will further expand the possibilities for STY–MAnh. Star-shaped polymers and graft polymers will allow the construction of highly functional shape-controlled materials.Acknowledgements
The author gratefully acknowledges support by the South African Research Chair Initiative of the Department of Science and Technology and NRF.Notes and references
- D. J. T. Hill, J. H. O'Donnell and P. W. O'Sullivan, Macromolecules, 1985, 18, 9–17 CrossRef CAS.
- E. Tsuchida and T. Tomono, Makromol. Chem., 1971, 141, 265–298 CrossRef CAS.
- P. C. Deb and G. Meyerhoff, Polymer, 1985, 26, 629–635 CrossRef CAS.
- K. Dodgson and J. R. Ebdon, Eur. Polym. J., 1977, 13, 791–797 CrossRef CAS.
- R. A. Sanayei, K. F. O'Driscoll and B. Klumperman, Macromolecules, 1994, 27, 5577–5582 CrossRef CAS.
- B. Sandner, Acta Polym., 1984, 35, 359–363 CrossRef CAS.
- M. Rätzsch and V. Steinert, Makromol. Chem., 1984, 185, 2411–2420 CrossRef CAS.
- T. Fukuda, K. Kubo and Y.-D. Ma, Prog. Polym. Sci., 1992, 17, 875–916 CrossRef CAS.
- T. Fukuda, Y. D. Ma, H. Inagaki and K. Kubo, Macromolecules, 1991, 24, 370–375 CrossRef CAS.
- B. Klumperman, PhD Thesis, Free radical copolymerization of styrene and maleic anhydride - Kinetic studies at low and intermediate conversion, Eindhoven University of Technology, Eindhoven, The Netherlands, 1994, p. 120 Search PubMed.
- K. F. O'Driscoll, J. Macromol. Sci.: Chem., 1969, 3, 335–337 CAS.
- J. Bonilla-Cruz, L. Caballero, M. Albores-Velasco, E. SaldÌvar-Guerra, J. Percino and V. Chapela, Macromol. Symp., 2007, 248, 132–140 CrossRef CAS.
- Y. Wang, Y. Shen, X. Pei, S. Zhang, H. Liu and J. Ren, React. Funct. Polym., 2008, 68, 1225–1230 CrossRef CAS.
- D. Benoit, C. J. Hawker, E. E. Huang, Z. Lin and T. P. Russell, Macromolecules, 2000, 33, 1505–1507 CrossRef CAS.
- B. Lessard and M. Milan, Macromolecules DOI:10.1021/ma902234t.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- E. T. A. van den Dungen, J. Rinquest, N. O. Pretorius, J. M. McKenzie, J. B. McLeary, R. D. Sanderson and B. Klumperman, Aust. J. Chem., 2006, 59, 742–748 CrossRef CAS.
- J. B. McLeary, F. M. Calitz, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Macromolecules, 2004, 37, 2383–2394 CrossRef CAS.
- J. B. McLeary, F. M. Calitz, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Macromolecules, 2005, 38, 3151–3161 CrossRef CAS.
- F.-S. Du, M.-Q. Zhu, H.-Q. Guo, Z.-C. Li, F.-M. Li, M. Kamachi and A. Kajiwara, Macromolecules, 2002, 35, 6739–6741 CrossRef CAS.
- H. Maeda, Adv. Drug Delivery Rev., 2001, 46, 169–185 CrossRef CAS.
- C. Tang, S. Ye and H. Liu, Polymer, 2007, 48, 4482–4491 CrossRef CAS.
- M. Ignatova, O. Stoilova, N. Manolova, D. G. Mita, N. Diano, C. Nicolucci and I. Rashkov, Eur. Polym. J., 2009, 45, 2494–2504 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |