Reactive photoinduced species in estuarine waters. Characterization of hydroxyl radical, singlet oxygen and dissolved organic matter triplet state in natural oxidation processes
Received 17th June 2009, Accepted 13th October 2009
First published on 2nd December 2009
Abstract
This paper describes the reactive photo-induced species (RPS) hydroxyl radical (HO˙), singlet oxygen (1O2) and chromophoric dissolved organic matter triplet state (3CDOM*) in fresh water (Canal Fumemorte) and estuarine water (Vaccarès), sampled in the Camargue region, southern France. Experiments were conducted with a medium-pressure Hg lamp in a glass photoreactor (λ > 290 nm, 220 W m−2 irradiance between 290 and 400 nm). Steady-state concentration and initial production rate of RPS were determined for HO˙ and for 1O2. HO˙ and 1O2 were indirectly identified in the presence of benzene and furfuryl alcohol, respectively, as specific probes. The steady-state measured concentration of HO˙was (1.72 ± 0.01) × 10−16 M and (9.41 ± 0.12)×10−17 M for Vaccarès and Canal waters samples, respectively, and the respective concentrations of 1O2 was (2.06 ± 0.22) × 10−13 M and (5.44 ± 0.04) × 10−14 M. The interference of 3CDOM* or other species in the determination of 1O2 with furfuryl alcohol, and of 1O2 in the quantification of 3CDOM* with 2,4,6-trimethylphenol was also quantitatively assessed. We developed a kinetic model describing the solar photo-transformation of xenobiotic organic compounds induced by the three different photooxidants HO˙, 1O2 and 3CDOM*.
Introduction
Aqueous photochemistry is an important process for the transformation of endogenous and exogenous organic components in natural waters. Under solar irradiation, photochemical transformations in aquifers, defined here as natural oxidation processes (NOP), involve direct and indirect photochemical reactions. In direct photolysis, organic compounds absorb radiation themselves and undergo chemical changes. In indirect photochemical pathways, chromophoric dissolved organic matter (CDOM), nitrate, nitrite and Fe(III) act as sensitizers by absorbing radiation, then inducing indirect degradation of surrounding organic compounds. Several important intermediates play a key role in the photodegradation pathways, including the reactive photoinduced species (RPS) HO˙, 1O2, ROO˙, HOO˙, O2˙−, CO3˙−, NO2˙, eaq−, and 3CDOM*.1,2 Particularly, HO˙, 1O2 and 3CDOM* are of prime importance for their contribution to organic fate in sunlit aquifers.3 Hydroxyl radical is one of the most reactive free radicals, reacting at nearly diffusion-controlled rate,4 that is involved in the mineralization of organic compounds. HO˙ has a central role in advanced oxidation processes (AOPs), following the definition by Glaze et al.:5 AOPs are near-ambient temperature and pressure water treatment processes that involve the generation of HO˙ in sufficient quantity to affect water purification. In sunlit natural water, HO˙ is mainly produced by photolytic reactions involving NO2−, NO3−,6 and irradiated CDOM.7–9 Additionally, Fe(III) has also been reported to enhance HO˙ production via the photo-Fenton reaction,10,11 or from the photoreduction of Fe(III) hydroxocomplexes.12,13 The photoproduction rate of HO˙ in many river waters and seawaters has been determined to be in the order 6–150 × 10−12 M s−1 and 4–50 × 10−12 M s−1, respectively.14 It is noteworthy that HO˙ is also efficiently consumed by natural dissolved organic matter, which limits its steady-state concentration in waters illuminated by sunlight.3 CDOM is known to promote organic pollutant degradation in sunlight8via its excited triplet states, which induce electron transfer with organic compounds, or via singlet oxygen (1Δg) produced from energy transfer of 3CDOM* to molecular oxygen.8,15 Photochemical processes are strongly related to the chemical composition of water, because of the possible reaction between RPS and intrinsic components of natural water.3,16 A major parameter in natural water is the concentration of chloride that distinguishes marine water from fresh water. The oceans, covering 71% of the Earth's surface, form a significant reservoir and reaction medium for exogenous contaminants, particularly in coastal water. In estuarine areas, the ionic composition of river water is strongly modified by chloride enrichment due to the proximity of seawater. Chloride may affect the photochemistry of exogenous organics and lead to the production of chloro derivatives.17 Additionally, it has been reported that estuarine waters contain higher DOM compared to river water.18 To our best knowledge the RPS HO˙, 1O2 and 3CDOM* have never been studied and identified together in freshwater samples and nor in estuarine waters.The aim of this study was then to quantify the primary RPS in estuarine waters and to determine their production kinetics in real samples in order to bring a comprehensive method to model photochemical reactions in natural waters characterized by high salinity and high DOM. The specific probes benzene, furfuryl alcohol and 2,4,6-trimethylphenol (TMP) were used to characterize the reactive species HO˙, 1O2 and the triplet states of CDOM, respectively.
Experimental
Chemicals
All reagents and chemicals were purchased commercially from the following suppliers: 2,4,6-trimethylphenol (TMP) and furfuryl alcohol (FFA) (98%) from Alfa Aesar; NaN3 (99%), methanol and acetonitrile from Sigma-Aldrich; isopropanol from Carlo Erba. All chemicals and reagents were used without further purification. All glassware and plastic were acid washed with HCl (5%) and rinsed with distilled water followed by Milli-Q water prior to use. HPLC eluents were prepared with Milli-Q water.Two sampling sites from Camargue were adopted, taking water from about 15 cm below the surface. Estuarine water was taken from Vaccarès, and fresh water samples were collected in Canal Fumemorte (04° 38′ 11″ E, 43° 31′ 25″ N; hereafter referred to as “Canal”). Water was then filtered through 0.45 μm pore-size membranes, and stored at 4 °C in the dark until analysis. Storage and all measurements were performed at the natural water pH. Sample analysis is presented in Table 1. Spectra were recorded in a cell with 1 cm optical path length. UV-Vis spectra of the samples are presented in Fig. 1. It is noticeable that the Vaccarès sample shows a much higher absorption than the Canal one. This can be attributed to the higher organic matter content of the Vaccarès sample (see Table 1) for wavelength values down to 230–240 nm where CDOM dominates the absorption.7,9 Interestingly, the absorbance ratio of the two samples is much higher than the non-purgeable organic carbon (NPOC) ratio. This implies that the specific absorbance of CDOM is higher in the case of Vaccarès. Below 230 nm other water components would also play a very important role (e.g. nitrate, nitrite and Fe species).6,7,9 Nitrate and nitrite cannot account for the difference as they are more concentrated in the case of Canal. In contrast, the higher Fe concentration of the Vaccarès sample could play a role below 230 nm.
Table 1 Physical and chemical parameters of water samples. Error intervals represent μ±σ
| Sample | Canal | Vaccarès |
|---|
| Sampling date | 28 April 2008 | 28 April 2008 |
| Cl−/M | (1.18 ± 0.03) × 10−3 | (1.90 ± 0.05) × 10−1 |
| NO3−/M | (4.36 ± 0.03) × 10−5 | (1.64 ± 0.01) × 10−5 |
| NO2−/M | (9.57 ± 0.05) × 10−7 | (8.26 ± 0.05) × 10−7 |
| Fe (ppb) | 3.81 ± 0.90 | 22.81 ± 3.06 |
| NPOC/mg(C) L−1 | 19.83 ± 0.30 | 32.11 ± 0.60 |
| pH | 7.81 | 8.01 |
| Conductivity/mS | 0.62 | 36.50 |
| Salinity/g L−1 | 0.10 | 26.20 |
| rHO˙/M s−1 | (1.52 ± 0.01) × 10−11 | (1.93 ± 0.01) × 10−11 |
| [HO˙]ss/M | (9.41 ± 0.12) × 10−17 | (1.72 ± 0.01) × 10−16 |
| ∑iki[Si]/s−1 | (1.62 ± 0.03) × 105 | (1.12 ± 0.01) × 105 |
| r1O2/M s−1 | (1.36 ± 0.01) × 10−8 | (5.14 ± 0.54) × 10−8 |
| [1O2]ss/M | (5.44 ± 0.04) × 10−14 | (2.06 ± 0.22) × 10−13 |
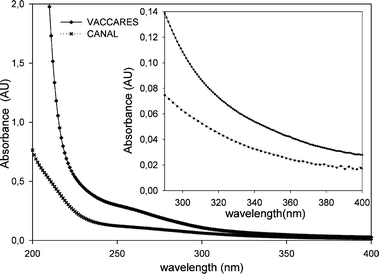 |
| | Fig. 1 UV-Vis spectra for Canal and Vaccarès water samples collected on 28 April 2008. | |
Irradiation experiments
Stationary photolysis experiments were carried out in a cylindrical immersion-type glass photoreactor (0.5 L; Heraeus TQ 150 Model) equipped with a water-cooled medium-pressure Hg lamp (glass cut off (λ < 290 nm), maximum emission wavelengths at 313, 366, 406, 436, 546 and 578 nm). The incident, volumetric photon flux in solution was 1.0 × 10−4 einstein L−1 s−1. The irradiance between 290 and 400 nm was around 220 W m−2. The radiation path length inside the reactor was 2 cm. The whole assembly was wrapped with aluminium foil. Upon irradiation, aliquots were removed at various time intervals and analyzed by HPLC-UV. Each experiment was carried out in triplicate.Analytical determinations
pH was measured by a pH meter ORION model 420A. The measures of iron were performed by ICP-AES with a JY2000 ULTRACE apparatus (Jobin Yvon Horiba). ICP-AES conditions: Plasma argon flow 12 L min−1, plasma power 1000 W, nebulisation pressure 3.2 bars. The standard solutions were supplied by Aldrich (concentration of metal 1000 mg L−1 in acidified solution containing 1% HNO3). The identification of Fe was performed at 260 nm. UV-Vis absorption spectra of water samples were recorded using a spectrophotometer model Agilent 8453 and ChemStation software (quartz cells, 1 cm path length). Anions (NO3−, NO2−, Cl−) were analyzed with a DIONEX ICS 3000 Ion chromatograph, equipped with an Autosampler AS 40, a gradient pump, a Dionex Ion Pac AS11-HC 4 × 250 mm anion exchange column, a Dionex IonPac AG 11-HC 4 × 5 mm guard column, a ASRS-ULTRA 300–4 mm suppressor, and two detectors: Ultimate 3000 Photodiode Array Detector UV/VIS and a conductimetric one at 35 °C. The eluents were H2O and 100 mM NaOH. Flow rate was 1.5 mL min−1. Elution gradient: 0–16 min (NaOH 1.3%/H2O 98.7%); 16–29 min (from NaOH 1.3% to NaOH 60%). The retention times were (min): Cl− (13.64), NO3− (21.8). NO2− concentration was measured by the sulfanilamide method.19Organic carbon was quantified as NPOC with 5050A/SSM 5000 Shimadzu carbon analyzer, after acidification of the sample with concentrated HClO4 and a 20 min air purge to eliminate CO2. Three measures were systematically acquired for each experiment, and averaged to give the final value. HPLC analysis was carried out by a Hitachi HPLC chromatograph equipped with a L-2400 UV detector and using a RP-C18 LiChrospher column (Merck; length 250 mm, diameter 4.6 mm, particle size 5 μm). Samples were eluted with a mixture of acetonitrile (ACN) and water acidified with orthophosphoric acid (0.1%) at a flow rate of 0.8 mL min−1. The percentage volume of ACN/acidified water was 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 and 60:40 for FFA and TMP, respectively. Retention times (min)/detection wavelength (nm) were: FFA (7.06/218), 6-hydroxy(2H)pyran-3(6H)-one (pyranone) (4.30/218), TMP (9.97/205). Benzene consumption and phenol formation were monitored with the same HPLC apparatus. Samples were eluted with a 40:60 mixture of ACN/water. Retention times (min)/detection wavelength (nm) were: phenol (4.62/210), benzene (9.50/210).
:80 and 60:40 for FFA and TMP, respectively. Retention times (min)/detection wavelength (nm) were: FFA (7.06/218), 6-hydroxy(2H)pyran-3(6H)-one (pyranone) (4.30/218), TMP (9.97/205). Benzene consumption and phenol formation were monitored with the same HPLC apparatus. Samples were eluted with a 40:60 mixture of ACN/water. Retention times (min)/detection wavelength (nm) were: phenol (4.62/210), benzene (9.50/210).
Methodology
The adopted methology was to measure HO˙ with the formation of phenol from benzene as a probe reaction (95% yield),9,141O2 upon transformation of FFA plus control experiments (quenching with azide),15 and 3DOM* with the phototransformation of TMP.8 The errors associated to the relevant quantities (±σ) were derived from the goodness of the fit of the theoretical kinetic functions to the experimental data or, where relevant, from the rules of error propagation.Results and discussion
Measurement of hydroxyl radical
Hydroxyl radicals were identified and quantified in natural water samples with the benzene probe as described by Vione et al.9 Benzene was chosen as a model compound for examining indirect photolysis due to its fast reaction with HO˙ (second-order rate constant 7.8 × 109 M−1 s−1)4 and to its lack of reactivity by direct photolysis. The transformation reaction of benzene to phenol was monitored as a function of time assuming that phenol was produced only upon reaction between benzene and HO˙ (95% yield). This hypothesis is justified by the fact that natural waters afford limited interference on this reaction by other reactive transients.20 Applying the steady-state approximation to [HO˙] the rate of benzene reaction is given by:| | 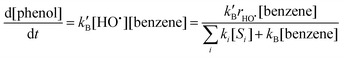 | (1) |
where kB is the second-order rate constant for the reaction between benzene and HO˙ (7.8 × 109 M−1 s−1), and k′B = 0.95kB. Si is a generic HO˙ scavenger present in the natural water sample, having rate constant ki for reaction with HO˙. rHO˙ is the rate of HO˙ photoformation. The reaction of phenol with HO˙ was not considered because only the initial rates were taken into account. Eqn (1) was resolved for low concentration of benzene to determine [HO˙] (denominator becoming equal to ∑ikSi[Si] by neglecting the reaction of benzene with HO˙ compared to the reaction of Si with HO˙) and at high concentration of benzene to determine rHO˙ (denominator equal to kB[benzene] by neglecting the reaction of Si with HO˙ compared to the reaction of benzene with HO˙). The time evolution of phenol upon irradiation of the two natural water samples in the presence of benzene at different initial concentrations is shown in Fig. 2. The initial formation rate of phenol was calculated as the slope of the tangent at t = 0 to the curve fitting the experimental concentration values of phenol. Following the procedure of Vione et al.,9 [HO˙] was determined from the tangent at t = 0 to the curve representing the initial formation rate of phenol as a function of [benzene] (Fig. 3). The values of rHO˙, [HO˙] and ∑ikSi[Si] are reported in Table 1. The associated errors are referred to the goodness of the fit of the theoretical curves (eqn (1)) to the experimental data shown in Fig. 3.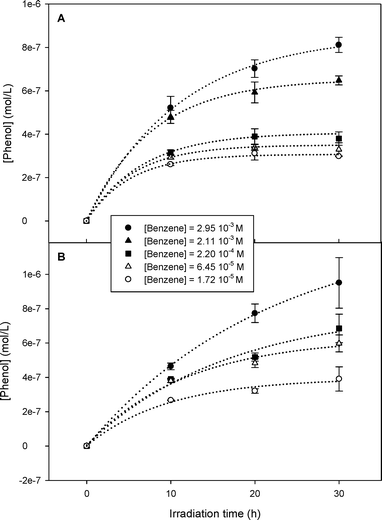 |
| | Fig. 2 Time evolution of phenol upon irradiation of water from (A) Vaccarès and (B) Canal for different concentration values of benzene. SD is given for three replicates. | |
![Comparative initial formation rate of phenol upon irradiation of water from Vaccarès and Canal, as a function of the initial benzene concentration. The tangents representing the lim[benzene]→ 0{d[phenol]/dt× [benzene]−1} give access to [HO˙].](/image/article/2010/PP/b9pp00030e/b9pp00030e-f3.gif) |
| | Fig. 3 Comparative initial formation rate of phenol upon irradiation of water from Vaccarès and Canal, as a function of the initial benzene concentration. The tangents representing the lim[benzene]→ 0{d[phenol]/dt× [benzene]−1} give access to [HO˙]. | |
The adopted procedure is based on the hypothesis of a selective reaction of HO˙ with benzene. Phenol could also be produced from alternative sources like 1O2 and humic acids via the triplet state 3CDOM*. However, 1O2 is weakly reactive toward benzene,21 and the addition of HO˙ scavengers in excess is able to completely quench the formation of phenol from benzene upon irradiation of humic acids or lake water.20 The deduced steady-state [HO˙] in the Vaccarès water sample ((1.72 ± 0.01) × 10−16 M) was about 1.8 times higher than that of Canal ((9.41 ± 0.12) × 10−17 M). The overall scavenging rate constant (∑ikSi[Si]) values were (1.12 ± 0.01) × 105 and (1.62 ± 0.03) × 105 s−1 for the Vaccarès and Canal water samples, respectively (Table 1).
Dissolved organic matter is most likely the main scavenger of HO˙ in the two samples. In contrast, chloride even at elevated concentration is extremely unlikely to be a significant HO˙ scavenger at neutral to basic pH. While significant consumption of hydroxyl radicals by Cl− is operational under acidic conditions, no scavenging could be detected at circumneutral pH in the presence of chloride at concentration up to at least 1 M.9,17 Interestingly, the lower scavenging rate constant of the Vaccarès sample in the presence of higher NPOC suggests that the Vaccarès organic matter is considerably less reactive toward HO˙ than the Canal one.
Also interesting is the higher rHO˙ in the case of Vaccarès. Among the possible HO˙ sources, nitrate is much more concentrated in Canal, and the concentration of nitrite is similar in the two samples but slightly higher for Canal. Moreover, note that [NO3−][NO2−]−1 is <100 in both cases, which suggests a more important role of nitrite as HO˙ source compared to nitrate.6 Of the other possible species that could be involved in the photoinduced generation of HO˙, CDOM and Fe are more concentrated in the Vaccarès sample. The two compounds together could be involved into photo-Fenton reactions, which would therefore be presumably more important in the estuarine sample. However, it should be pointed out that the photo-Fenton process is for instance the prevailing HO˙ source in the Satilla river (USA), characterized by high Fe levels (5–14 μM) and relatively acidic pH (<7).22 In the present case, even in the Vaccarès sample the concentration of Fe is sub-μM and the pH is quite high. Under basic conditions the generation of HO˙ by the Fenton reaction is inhibited,23 for which reason the Vaccarès sample could hardly be considered as an ideal system for the photo-Fenton process to be an important source of HO˙. Additionally, the generation rate of HO˙ upon direct irradiation of Fe(III) species under neutral to basic conditions is quite low.24 Another possibility is that the high generation rate of HO˙ in the Vaccarès sample is due to the presence of more concentrated and more active CDOM (see Fig. 1 and Table 1), which could produce hydroxyl radicals directly upon irradiation.9
Involvement of singlet oxygen (1O2)
Under solar irradiation, natural waters are known to produce hydroxyl radical and singlet oxygen. Consequently, these reactive species should be involved in the transformation pathways of organic compounds (R) that are present in water. The relevant processes are described in eqn (2)–(3), where P1 and P2 are the photoproducts of the reaction of R with HO˙ and 1O2, respectively. The reactivity of the organic compound R with RPS may be affected by various scavengers present in the natural water matrix (Si, Sj), which can compete with R for reaction with the photogenerated transients (eqn (4)–(5)). The equations representing the oxidation rate of R (rR) and the reaction rates of HO˙ (rHO˙) and 1O2 (r1O2) are given by eqn (7)–(9), where k1 is the second-order rate constant of the reaction of R with HO˙, k2 that with 1O2, ki and kj the second-order rate constants of the reaction of the scavenger Si and Sj with HO˙ and 1O2, respectively. Finally, kd is the first-order rate constant for the deactivation of 1O2 in water (2.5 × 105 s−1).25| | 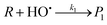 | (2) |
| | 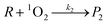 | (3) |
| | 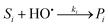 | (4) |
| | 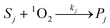 | (5) |
| |  | (6) |
| |  | (7) |
| | | rHO˙ = (k1[R] + ∑iki[Si])[HO˙] | (8) |
| | | r1O2 = (k2[R] + kd + ∑jkj[Sj])[1O2] | (9) |
Note that under the steady-state conditions that are operational for HO˙ and 1O2 in surface waters, rHO˙ and r1O2 would also represent the formation rates of the relevant species under irradiation.
The characterisation of 1O2 was carried out with furfuryl alcohol (FFA), which is known to react with singlet oxygen (k2 = 1.2 × 108 M−1 s−1) to give the corresponding 6-hydroxy(2H)pyran-3(6H)-one (pyranone) in 85% yield.15 The initial rate of FFA photooxydation (rFFA) was measured for several initial FFA concentrations. Assuming that FFA reacts with both hydroxyl radical and singlet oxygen, rFFA can be expressed as follows (note that R = FFA in eqn (2)–(3)):
| | 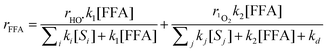 | (10) |
In this equation, the fitting variables are r1O2 and ∑jkj[Sj]. The variables rHO˙ and ∑iki[Si] were deduced from the measurement of HO˙ in the presence of benzene (see above). The preliminary fit of the experimental data with eqn (10) (data not shown) yielded ∑jkj[Sj] << k2[FFA] + kd, which suggests that the term ∑jkj[Sj] can be neglected.
The irradiation of FFA (0.25 × 10−4 M) in natural waters indicated that FFA depletion reached 44.5% and 21.1% after 240 min for the Vaccarès and Canal water samples, respectively (Fig. 4). Only a small percentage of FFA reacted under irradiation, compared to Richard and co-workers who obtained 80% FFA removal in similar conditions after 200 min of irradiation in laboratory-grade water in the presence of fulvic acid.15
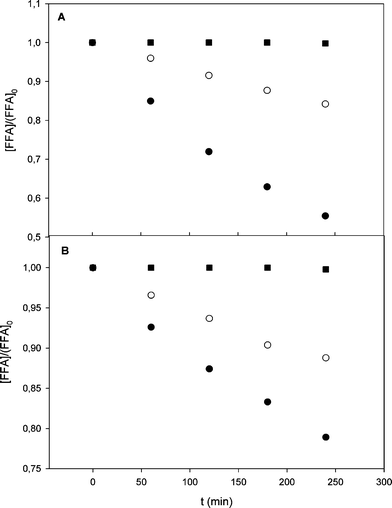 |
| | Fig. 4 Normalized evolution of FFA vs. time under polychromatic light in (A) Vaccarès water and (B) Canal water. (■) FFA (0.25 × 10−4 M) in milliQ water, (●) FFA (0.25 × 10−4 M) in real water; (○) FFA (0.25 × 10−4 M) in real water + sodium azide (3 × 10−3 M). | |
The low degradation yields observed here could be explained by the lower photochemical reactivity of whole CDOM samples compared to fulvic acids, or by the matrix effect of the natural waters because of the presence of dissolved antioxidants.26 The control of the reaction in milliQ water indicated that FFA did not undergo direct phototransformation under irradiation. The effect of the azide anion (3 × 10−3 M) is also reported in Fig. 4. Azide is known to react with HO˙ and 1O2 with second-order rate constants k3 = 1.4 × 1010 M−1 s−127 and k4 = 7.8 × 108 M−1 s−1,28 respectively. In the presence of the azide, eqn (10) would be modified as follows:
| | 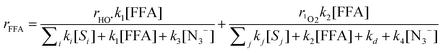 | (11) |
Eqn (11) foresees a 90% inhibition of FFA degradation in the presence of 3 mM N3−, but the experimental data (Fig. 4) show that the decrease of FFA was only partially inhibited for both the Vaccarès sample (65%) and the Canal sample (47%). These results indicate that [FFA] continued decreasing, albeit to a reduced extent, when 1O2 and HO˙ were quenched by azide. The oxidation of N3− in the studied system is expected to produce the azide radical N3˙, which shows significant reactivity toward many substituted phenols. Interestingly, no reactivity data of N3˙ with FFA or other alcohols are reported,29 which suggests that N3˙ would not be involved to a significant extent in the transformation of FFA. It is therefore hypothesized that additional reactive species, possibly 3CDOM* or organic matter-derived radicals (e.g. RO˙ and/or ROO˙),3 could be involved in FFA depletion upon irradiation of natural water.
The next section discusses in greater detail the hypothesis that the residual transformation of FFA in the presence of the azide is due to the reaction with 3CDOM*, or other species generated upon irradiation and transformation of organic matter.
Involvement of species derived from dissolved organic matter in the transformation of FFA
If the degradation of FFA is carried out by 3CDOM*, the process would take place according to the following eqn (12)–(15):30,31| | | CDOM + hν→3CDOM* [r3CDOM*] | (12) |
| | | 3CDOM* + FFA → Products [k5] | (14) |
| | | 3CDOM* + O2→ CDOM + 1O2 [k6] | (15) |
The chemistry of species such as RO˙ and/or ROO˙ is not known in sufficient detail to enable the setting up of a similar reaction scheme, but equivalent reactions could possibly be operational.
The role of the interfering species has been approximately assessed from the difference (rdiff) between the experimental (Table 2) and the calculated (eqn (11)) FFA rates in the presence of the azide. It was possible to obtain rdiff = 7 × 10−6[FFA] for Canal and rdiff = 3 × 10−6[FFA] for Vaccarès.
Table 2 Experimental rate of FFA phototransformation in the absence (rFFA) and in the presence (rFFAq) of sodium azide (3 × 10−3 M) in the Canal and Vaccarès water samples
| [FFA]/106 M | Canal | Vaccarès |
|---|
| rFFA/10−10 M s−1 | rFFAq/10−10 M s−1 | rFFA/10−10 M s−1 | rFFAq/10−10 M s−1 |
|---|
| 25 | 4.0 | 2.1 | 10.3 | 2.9 |
| 50 | 8.2 | 4.5 | 19.0 | 3.9 |
| 100 | 15.8 | 8.1 | 36.5 | 6.4 |
| 200 | 24.3 | 4.8 | 46.9 | 8.7 |
Under the hypothesis that the difference between the experimental and the calculated values is due to 3CDOM*, and applying the steady-state approximation to [3CDOM*], an exact expression for rdiff would be:
| | 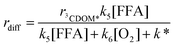 | (16) |
where
r3CDOM* and
k* are the formation rate of
3CDOM* and the deactivation rate constant of
3CDOM*, respectively. It would also be
k5[FFA] <<
k6[O
2] + k*.
30 Considering that the involvement of
3CDOM* is not demonstrated,
eqn (16) can be more generically written as
rdiff =
α[FFA] (
α has units of s
−1). The rate data of FFA without
sodium azide would then be given by the sum of the contributions of HO˙,
1O
2, and
3CDOM* or other species. The following, simplified fitting function was obtained:
| | 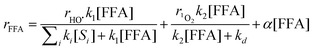 | (17) |
considering that ∑
jksj[
Sj] <<
kd.
31,32 From the
experimental data of
rdiff one gets
α = 7 × 10
−6 s
−1 for Canal and 3 × 10
−6 s
−1 for Vaccarès.
Fig. 5 reports the experimental rates of
rFFAvs. [FFA] for the Canal and Vaccarès waters (solid squares). The dashed lines represent the fit with
eqn (17), where
r1O2 was the only fit variable. It was found that
r1O2 = (1.36 ± 0.01) × 10
−8 M s
−1 and (5.14 ± 0.54) × 10
−8 M s
−1 for Canal and Vaccarès, respectively. The errors represent the goodness of the fit of
eqn (17) to the
experimental data. The dotted line indicates what would be the model without taking into account the contribution of
3CDOM* or other interfering species (
α[FFA] term in
eqn (17)). It is apparent that the contribution of the interfering agents is much more important in the case of Canal, for which the value of
α was more than double compared to Vaccarès. The fact that
r1O2 is higher for Vaccarès compared to Canal is consistent with the higher NPOC (see
Table 1) and even more with the higher content of CDOM (see
Fig. 1) of Vaccarès. Indeed,
1O
2 is produced by the excited triplet states
3CDOM* (
reaction 15). It is also possible to derive the steady-state [
1O
2] in the absence of FFA or the
azide. Consider
eqn (9) with [
R] = 0 and ∑
jksj[
Sj] <<
kd,
31,32 from which one gets [
1O
2] =
r1O2kd−1. The steady-state [
1O
2] values thus derived are reported in
Table 1.
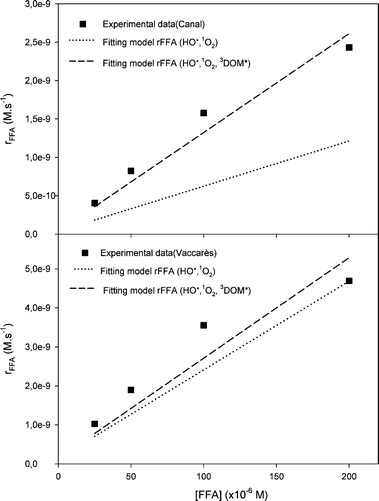 |
| | Fig. 5 Fitting model of rFFA taking in account the three RPS in (A) Canal and (B) Vaccarès water samples. FFA concentrations: 0.25, 5.0, 1.0 and 2.0 × 10−4 M. | |
Quantitation of 3CDOM*
2,4,6-Trimethylphenol (TMP) has successfully been used to probe the photosensitising properties of natural waters. This electron-rich phenol is readily transformed upon irradiation in the presence of chromophoric dissolved organic matter or humic substances.30,33 TMP can react with singlet oxygen (k2 = 6.3 × 107 M−1 s−1),34 triplet states or other radicals (e.g. HO˙). However, it has been reported that the rate of TMP phototransformation in the presence of humic or fulvic acids was not affected by the presence of the azide, confirming the negligible involvement of 1O2 under such conditions.35 The formation of 3CDOM* was specifically determined from the irradiation of TMP in Vaccarès and Canal water samples. Fig. 6 reports the transformation rate of TMP at different initial concentrations upon irradiation of the natural water samples. The transformation rate rTMP can be written as follows:| | 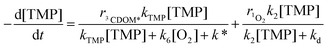 | (18) |
![Plot of rTMPvs. [TMP] for Canal and Vaccarès water samples. Experimental data and fitting curves.](/image/article/2010/PP/b9pp00030e/b9pp00030e-f6.gif) |
| | Fig. 6 Plot of rTMPvs. [TMP] for Canal and Vaccarès water samples. Experimental data and fitting curves. | |
The reaction rate constant of TMP with hydroxyl radicals was neglected, based on previous findings that k1[HO˙] << kTMP[3CDOM*] in natural waters,30 where kTMP is the second-order reaction rate constant between TMP and 3CDOM*.
The fit of the experimental data was carried out with eqn (18), with k2 = 6.3 × 107 M−1 s−1, kd = 2.5 × 105 s−1, k6[O2] + k* = 5 × 105 s−1,25r1O2 = (1.36 ± 0.01) × 10−8 M s−1 and (5.14 ± 0.54) × 10−8 M s−1 for Canal and Vaccarès, respectively, and r3CDOM* and kTMP as fit variables. The fit yielded r3CDOM* = (1.8 ± 0.3) × 10−8 M s−1 and kTMP = (4.8 ± 4.4) × 109 M−1 s−1 for Vaccarès, r3CDOM* = (1.4 ± 0.1) × 10−8 M s−1 and kTMP = (3.0 ± 1.1) × 109 M−1 s−1 for Canal. Fig. 6 also reports the calculated contribution of 1O2 to the transformation of TMP in the two samples, which is definitely not negligible in the case of Vaccarès. It is noticeable the elevated error on kTMP in the case of the Vaccarès sample. However, a further fit carried out on the same data by taking fixed kTMP = 3.0 × 109 M−1 s−1 as for Canal yielded a very similar r3CDOM* = 1.9 × 10−8 M s−1. The determination of r3CDOM* from the Vaccarès data can therefore be considered as sufficiently robust.
Interestingly, in the case of Canal it is r3CDOM*≈r1O2, which is reasonable because 3CDOM* is the main source of 1O2 in surface waters.15,30 However, for the Vaccarès sample one finds r3CDOM* < r1O2. Possible explanations are: (i) the addition of the azide to the Vaccarès sample was able to quench both 1O2 and some of the interfering species. The role of these species would be underestimated from the azide data and, as a consequence, one would overestimate r1O2. (ii) TMP was not able to react with the whole of 3CDOM* in the Vaccarès sample, possibly because some transients would preferentially react with O2 to yield 1O2, even with TMP in large excess. One should assume that such peculiar transients are particularly common in the estuarine sample. (iii) The adopted literature value of kd is too elevated. Note that the model fit to the data of Fig. 5 would be largely improved by adopting a lower value of kd. In that case r1O2 would be considerably lower. Interestingly, in cases (i) and (ii) one has to assume a rather peculiar behavior of the estuarine sample.
Expected transformation kinetics of model substrates
The data obtained or available for HO˙ (rHO˙ and ∑iki[Si]), 1O2 (r1O2 and kd) and 3CDOM* (r3CDOM* and k6[O2] + k*) can be used for the assessment of the transformation kinetics of model substrates in the two water samples under the adopted irradiation conditions. It would therefore be possible to compare the relative importance of the different transformation pathways. Unfortunately, while a wide data set is available for the reactivity of the dissolved molecules with HO˙, that with 3CDOM* has been studied for relatively few compounds (mainly phenylurea herbicides and sulfonamides),8 for which additionally there is no data concerning the (presumably low, however) reaction rate constant with 1O2. Table 3 reports the reaction rate constants with HO˙ of two amino acids (tyrosine and histidine), 4-chlorophenol (4CP) and the corresponding phenolate (4CP−), and the phenylurea herbicides diuron and fenuron.4,36,37 For phenylureas the reaction rate constants with 3CDOM* are also reported,38 while for the other compounds the rate constants with 1O2 only are available.36,39
Table 3 Rate constants for the reaction of some model substrates with HO˙, 1O2, and 3CDOM*.36–39 The interval in the case of 3CDOM* shows the different reactivity of 3BP* and 3MAP* with diuron and fenuron. The corresponding values of t½ for the Vaccarès (V) and Canal (C) samples are based on the reported literature rate constants, the results of the irradiation experiments carried out in this work, and eqn (19)–(21). The values of t½ are expressed in hours of continuous irradiation under the adopted device. n/a = not available
| | Tyrosine | Histidine | 4-CP | 4-CP− | Diuron | Fenuron |
|---|
| k˙OH/M−1 s−1 | 1.3 × 1010 | 5.0 × 109 | 7.6 × 109 | 4.1 × 109 | 5 × 109 | 7 × 109 |
| k1O2/M−1 s−1 | 9.0 × 106 | 6.6 × 107 | 6.0 × 106 | 1.9 × 108 | n/a | n/a |
| k3CDOM*/M−1 s−1 | n/a | n/a | n/a | n/a | (9–520) × 106 | (8.1–200) × 107 |
| t½˙OH(C)/h | 160 ± 5 | 410 ± 10 | 270 ± 10 | 500 ± 15 | 410 ± 10 | 290 ± 10 |
| t½1O2(C)/h | 390 ± 5 | 54 ± 1 | 590 ± 10 | 19 ± 1 | n/a | n/a |
| t½3CDOM*(C)/h | n/a | n/a | n/a | n/a | 13–760 | 3.4–85 |
| t½˙OH(V), h | 90 ± 1 | 220 ± 2 | 150 ± 1 | 270 ± 3 | 220 ± 2 | 160 ± 2 |
| t½1O2(V)/h | 100 ± 10 | 14 ± 1 | 160 ± 20 | 4.9 ± 0.5 | n/a | n/a |
| t½3CDOM*(V)/h | n/a | n/a | n/a | n/a | 10–590 | 2.7–66 |
Let R be a generic compound dissolved in water. Under reasonable pseudo-first order conditions, the half life time of R for reaction with HO˙, 1O2 and 3CDOM*, respectively, is expressed by:
| | 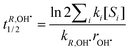 | (19) |
| | 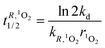 | (20) |
| | 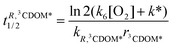 | (21) |
where
kd = 2.5 × 10
5 s
−1,
21 and
k6 [O
2] +
k* = 5 × 10
5 s
−1.
25 The values of
rHO˙, ∑
iki[
Si],
r1O2 and
r3CDOM* for the Vaccarès and Canal samples are reported in
Table 1, together with the associated errors. The values of the reaction rate constants of the different model compounds (substrates) with the transient species HO˙,
1O
2 and
3CDOM*, and the corresponding half-life times of the substrates are reported in
Table 3. They are expressed in hours of continuous irradiation under the adopted device. The UV irradiance was 220 W m
−2, namely around 7 times more intense than the irradiance of sunlight at noon during the summer solstice at mid-latitude.
40 Also note that the errors associated to the half-life times have been derived by error propagation, from the relevant quantities reported in
Table 1, and rounded to the precision of the
t½ values.
In the case of kR,3CDOM*, the main problem is that CDOM is not a species of definite chemical composition. Accordingly, the relevant reaction rate constants can only be measured relative to model molecules that are representative of the nature of CDOM. It has been reported that 3′-methoxyacetophenone (MAP) and benzophenone (BP) can be used to this purpose.38 The reactivity of the relevant excited triplet states toward fenuron and diuron is quite different, with MAP being considerably less reactive than BP. For this reason Table 3 reports an interval for the values of k3CDOM*, with the upper end corresponding to k3BP* and the lower end to k3MAP*.
From the t½ data reported in Table 3 it is apparent that, first of all, the photochemical reactivity of the Vaccarès (estuarine, brackish) sample is higher compared to Canal (freshwater). The difference is highest for the reaction with 1O2, and lowest for 3CDOM*. It is remarkable that the estuarine sample is photochemically more reactive than the riverine/freshwater one. The most plausible explanation is the higher occurrence and reactivity of CDOM in the studied estuarine sample, which in the case of the Rhône river would be combined with a delta where the water is quite shallow. Higher intrinsic photoreactivity and shallow water would combine together in producing a delta environment where photochemical processes could play a very important role, also considering that such an environment is located in the Mediterranean region where sunlight is abundant. Such a result strengthens the previous findings that the Rhône delta is a very useful field photochemical reactor, where for instance the photoinduced nitration processes can be easily detected.41
As far as the relative reactivity with HO˙ and 1O2 is concerned, in both samples tyrosine and 4CP are expected to react faster with the hydroxyl radical, histidine and the 4-chlorophenolate with singlet oxygen. In the case of the compounds that react faster with HO˙, the reactivity with 1O2 is of secondary importance but it is not negligible. In contrast, for the molecules that react faster with 1O2, the reaction with HO˙ is unimportant. Note that 4CP−, in analogy with many chlorophenolates, would also undergo direct photolysis to a significant extent.36,42–44
In the case of the two phenylurea pesticides chosen as model substrates, the comparison of the reactivity with HO˙ and with 3CDOM* should take into account the relatively large interval of t½3CDOM*. Such an interval depends on the choice of BP and MAP as model molecules for CDOM. By comparison with t½HO˙ one gets that, in the case of diuron, the reaction with the hydroxyl radical is more important if the reactivity of 3CDOM* is more similar to that of 3MAP*, while 3CDOM* is a more significant sink if its reactivity resembles that of 3BP*. In case the reaction with HO˙ is more important, it would be around twice as fast as that with 3CDOM*. In the opposite case, the reaction with 3CDOM* could out-compete that of HO˙ by an order of magnitude or more.
In the case of fenuron the reaction with 3CDOM* is expected to be more important compared to HO˙, irrespective of the hypothesized reactivity of 3CDOM* (3BP*-like or 3MAP*-like). Accordingly, the present results confirm the findings of Canonica and coworkers that 3CDOM* is a very important sink of phenylurea herbicides in surface water.8 However, under definite circumstances of CDOM photoreactivity and with certain substrates (e.g. diuron), the reaction with HO˙ cannot be neglected as a sink.
Conclusions
In this study, for the first time to our knowledge, the photochemical formation of HO˙, 1O2 and 3CDOM* was determined in both a freshwater and an estuarine environment (Canal and Vaccarés samples, respectively). The estuarine sample contained more organic matter as measured by NPOC, as expected, but interestingly the organic matter of the estuarine sample also showed a higher intrinsic absorption of radiation. As a consequence, the absorbance ratio between the estuarine and the freshwater sample is higher than the ratio of NPOC, which could make the CDOM in the estuarine sample considerably more photoreactive.The reported CDOM features in the two samples are reflected in the respective photochemical activity as measured by the formation of HO˙, 1O2, and 3CDOM*. The formation rates of all these species are considerably higher in the estuarine sample, where the indirect photolysis of organic contaminants could therefore be faster. Such a finding is somewhat expected in the case of 1O2 and 3CDOM* that are produced by irradiated colored organic matter. The radical HO˙ is also produced by nitrate and nitrite, which have equal to higher concentration in the freshwater sample. It can therefore be inferred that the higher photoproduction of HO˙ in the estuarine sample can be accounted for by irradiated CDOM. Overall, the transition from the Rhône freshwater to the delta results in more favorable conditions for photochemistry, because of the combination of higher intrinsic reactivity, shallower water columns and longer residence time of water.41
In this work it was also shown that the quantification of 1O2 through the degradation of FFA suffers from some interference by 3CDOM* or other species such as ROO˙ and/or RO˙. At the same time, the determination of 3CDOM* using TMP is also sensitive to the interference by 1O2. However, it was possible to overcome the problem upon addition of NaN3 to the FFA system, which allowed for the quantification of the contribution of the interfering species to the degradation of FFA. In a similar way, 1O2 is able to react with TMP and therefore to interfere with the determination of 3CDOM*. The knowledge of the formation rate of 1O2 allows its inclusion into the kinetic model that describes the degradation of TMP, and the measurement of the formation rate of 3CDOM* is therefore possible despite the interference. Interestingly, Fig. 6 shows that the reaction rate between TMP and 1O2 increases linearly with [TMP], while the total transformation rate of TMP (rTMP) has a less-than-linear increase with [TMP]. The consequence is that the interference by the reaction between TMP and 1O2 becomes more important at elevated [TMP].
The procedure depicted in the present paper (measurement of 1O2 with FFA, of other interfering species with FFA + NaN3, and inclusion of 1O2 into the kinetic model of TMP degradation to exactly quantify 3CDOM*) could therefore be useful for future studies into the quantification of 1O2 and 3CDOM* upon irradiation of surface waters.
Acknowledgements
DV acknowledges financial support by the Italian Inter-University Consortium of Chemistry and the Environment (INCA) and Università di Torino–Ricerca locale.Notes and references
- S. S. Walse, S. L. Morgan, L. Kong and J. L. Ferry, Role of dissolved organic matter, nitrate, and bicarbonate in the photolysis of aqueous fipronil, Environ. Sci. Technol., 2004, 38, 3908–3915 CrossRef CAS.
- R. Huie and P. Neta, Kinetics of one-electron transfer-reactions involving ClO2 and NO2, J. Phys. Chem., 1986, 90, 1193–1198 CrossRef CAS.
- C. Richard and S. Canonica, Aquatic phototransformation of organic contaminants induced by coloured dissolved natural organic matter, Handb. Environ. Chem., 2005, 2m, 299–323 Search PubMed.
- G. Buxton, C. Greenstock, W. Helman and A. Ross, Critical-review of rate constants for reactions of hydrated electrons, hydrogen-atoms and hydroxyl radicals (˙OH/˙O−) in aqueous-solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CAS.
- W. Glaze, J. Kang and D. Chapin, The chemistry of water-treatment processes involving ozone, hydrogen-peroxide and ultraviolet-radiation, Ozone Sci. Eng., 1987, 9, 335–352 CAS.
- C. Minero, S. Chiron, G. Falletti, V. Maurino, E. Pelizzetti, R. Ajassa, M. E. Carlotti and D. Vione, Photochemical processes involving nitrite in surface water samples, Aquat. Sci., 2007, 69, 71–85 CrossRef CAS.
- P. Vaughan and N. Blough, Photochemical formation of hydroxyl radical by constituents of natural waters, Environ. Sci. Technol., 1998, 32, 2947–2953 CrossRef.
- S. Canonica, Oxidation of aquatic organic contaminants induced by excited triplet states, Chimia, 2007, 61, 641–644 CrossRef CAS.
- D. Vione, G. Falletti, V. Maurino, C. Minero, E. Pelizzetti, M. Malandrino, R. Ajassa, R. Olariu and C. Arsene, Sources and sinks of hydroxyl radicals upon irradiation of natural water samples, Environ. Sci. Technol., 2006, 40, 3775–3781 CrossRef CAS.
- B. Voelker and B. Sulzberger, Effects of fulvic acid on Fe(II) oxidation by hydrogen peroxide, Environ. Sci. Technol., 1996, 30, 1106–1114 CrossRef CAS.
- B. Voelker, F. Morel and B. Sulzberger, Iron redox cycling in surface waters: effects of humic substances and light, Environ. Sci. Technol., 1997, 31, 1004–1011 CrossRef CAS.
- I. Pozdnyakov, Y. Sosedova, V. Plyusnin, V. Grivin, D. Vorob'ev and N. Bazhin, Optical spectra and kinetic characteristics of radicals formed upon the photolysis of aqueous solutions of a FeOHaq2+ complex and phenol, Russ. Chem. Bull., 2004, 53, 2715–2722 Search PubMed.
- I. Pozdnyakov, Y. Sosedova, V. Plyusnin, E. Glebov, V. Grivin, D. Vorobyev and N. Bazhin, Photodegradation of organic pollutants in aqueous solutions caused by Fe(OH)aq2+ photolysis: Evidence of OH radical formation, Int. J. Photoenergy, 2004, 6, 89–93 CrossRef CAS.
- K. Takeda, H. Takedoi, S. Yamaji, K. Ohta and H. Sakugawa, Determination of hydroxyl radical photoproduction rates in natural waters, Anal. Sci., 2004, 20, 153–158 CAS.
- S. Halladja, A. Ter Halle, J. P. Aguer, A. Boulkamh and C. Richard, Inhihition of humic substances mediated photooxygenation of furfuryl alcohol by 2,4,6-trimethylphenol. Evidence for reactivity of the phenol with humic triplet excited states, Environ. Sci. Technol., 2007, 41, 6066–6073 CrossRef CAS.
- S. Canonica and P. G. Tratnyek, Quantitative structure-activity relationships for oxidation reactions of organic chemicals in water, Environ. Toxicol. Chem., 2003, 22, 1743–1754 CrossRef CAS.
- S. Chiron, C. Minero and D. Vione, Photodegradation processes of the Antiepileptic drug carbamazepine, relevant to estuarine waters, Environ. Sci. Technol., 2006, 40, 5977–5983 CrossRef CAS.
- S. Mylon, K. Chen and M. Elimelech, Influence of natural organic matter and ionic composition on the kinetics and structure of hematite colloid aggregation: Implications to iron depletion in estuaries, Langmuir, 2004, 20, 9000–9006 CrossRef CAS.
- J. Rodier, in L'analyse de l'eau, eaux naturelles, eaux résiduaires, eau de mer, Dunod, Paris, 7th edition, 1984, p. 1365 Search PubMed.
- D. Vione, V. Lauri, C. Minero, V. Maurino, M. Malandrino, M. E. Carlotti, R. I. Olariu and C. Arsene, Photostability and photolability of dissolved organic matter upon irradiation of natural water samples under simulated sunlight, Aquat. Sci., 2009, 71, 34–45 CrossRef CAS.
- S. H. Chien, M. F. Cheng, K. C. Lau and W. K. Li, Theoretical study of the Diels-Alder reactions between singlet (1Dg) oxygen and acenes, J. Phys. Chem. A, 2005, 109, 7509–7518 CrossRef CAS.
- E. M. White, P. P. Vaughan and R. G. Zepp, Role of the photo-Fenton reaction in the production of hydroxyl radicals and photobleaching of colored dissolved organic matter in a coastal river of the southeastern United States, Aquat. Sci., 2003, 65, 402–414 CrossRef CAS.
- S. J. Hug and O. Leupin, Iron-catalyzed oxidation of arsenic(III) by oxygen and by hydrogen peroxide: pH-dependent formation of oxidants in the Fenton reaction, Environ. Sci. Technol., 2003, 37, 2734–2742 CrossRef CAS.
- S. Chiron, S. Barbati, S. Khanra, B. K. Dutta, M. Minella, C. Minero, V. Maurino, E. Pelizzetti and D. Vione, Bicarbonate-enhanced transformatino of phenol upon irradiation of hematite, nitrate and nitrite, Photochem. Photobiol. Sci., 2009, 8, 91–100 RSC.
- M. A. J. Rodgers and P. T. Snowden, Lifetime of O21Dg) in liquid water as dermined by time-resolved infrared luminescence measurements, J. Am. Chem. Soc., 1982, 104, 5541–5543 CrossRef CAS.
- S. Canonica and H. U. Laubscher, Inhibitory effect of dissolved organic matter on triplet-induced oxidation of aquatic contaminants, Photochem. Photobiol. Sci., 2008, 7, 547–551 RSC.
- N. Motohashi and Y. Saito, competitive measurement of rate constants for hydroxyl radical reactions using radiolytic hydroxylation of benzoate, Chem. Pharm. Bull., 1993, 41, 1842–1845 CAS.
- F. Wilkinson and J. Brummer, Rate constants for the decay and reactions of the lowest electronically excited singlet-state of molecular-oxygen in solution, J. Phys. Chem. Ref. Data, 1981, 10, 809–1000 CAS.
- P. Neta, R. E. Huie and A. B. Ross, Rate constants for reactions of inorganic radicals in aqueous solution, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1230 CAS.
- S. Canonica and M. Freiburghaus, Electron-rich phenols for probing the photochemical reactivity of freshwaters, Environ. Sci. Technol., 2001, 35, 690–695 CrossRef CAS.
- J. Hoigné, Formulation and calibration of environmental reaction kinetics: Oxidations by aqueous photooxidants as an example, in Aquatic Chemical Kinetics, ed. W. Stumm, Wiley, New York, 1990, pp. 43–70 Search PubMed.
- D. Vione, D. Bagnus, V. Maurino and C. Minero, Quantification of singlet oxygen and hydroxyl radicals upon UV irradiation of surface water, Environ. Chem. Lett., 2009 DOI:10.1007/s10311-009-0208-z.
- B. C. Faust and J. Hoigné, Sensitized photooxidation of phenols by fulvic acid and in natural waters, Environ. Sci. Technol., 1987, 21, 957–964 CrossRef CAS.
- P. G. Tratnyek and J. Hoigné, Photo-oxidation of 2,4,6-trimethylphenol in aqueous laboratory solutions and natural waters: kinetics of reaction with singlet oxygen, J. Photochem. Photobiol., A, 1994, 84, 153–160 CrossRef CAS.
- J.-P. Aguer, D. Tétégan and C. Richard, Humic substances mediated phototrensformation of 2,4,6-trimethylphenol: a catalytic reaction, Photochem. Photobiol. Sci., 2005, 4, 451–453 RSC.
- M. Czaplicka, Photo-degradation of chlorophenols in the aqueous solution, J. Hazard. Mater., 2006, 134, 45–59 CrossRef CAS.
- F. Javier Benitez, F. J. Real, J. L. Acero and C. Garcia, Kinetics of the transformation of phenyl-urea herbicides during ozonation of natural waters: Rate constants and model predictions, Water Res., 2007, 41, 4073–4084 CrossRef.
- S. Canonica, B. Hellrung, P. Müller and J. Wirz, Aqueous oxidation of phenylurea herbicides by triplet aromatic ketones, Environ. Sci. Technol., 2006, 40, 6636–6641 CrossRef CAS.
- A. L. Boreen, B. L. Edhlund, J. B. Cotner and K. McNeill, Indirect photodegradation of dissolved free amino acids: the contribution of singlet oxygen nd the differential reactivity of DOM from various sources, Environ. Sci. Technol., 2008, 42, 5492–5498 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, M. E. Carlotti, S. Chiron and S. Barbati, Modelling the occurrence and reactivity of the carbonate radical in surface freshwater, C. R. Chim., 2009, 12, 865–871 CrossRef CAS.
- S. Chiron, C. Minero and D. Vione, Occurrence of 2,3-dichlorophenol and of 2,4-dichloro-6-nitrophenol in the Rhône River Delta (Southern France), Environ. Sci. Technol., 2007, 41, 3127–3133 CrossRef CAS.
- D. Vione, C. Minero, F. Housari and S. Chiron, Photoinduced transformation processes of 2,4-dichlorophenol and 2,6-dichlorophenol on nitrate irradiation, Chemosphere, 2007, 69, 1548–1554 CrossRef CAS.
- S. Chiron, L. Comoretto, E. Rinaldi, V. Maurino, C. Minero and D. Vione, Pesticide by-products in the Rhône delta (Southern France). The case of 4-chloro-2-methylphenol and of its nitroderivative, Chemosphere, 2009, 74, 599–604 CrossRef CAS.
- G. Grabner, C. Richard and G. Kohler, Formation and reactivity of 4-oxocyclohexa-2,5-dienyldiene in the photolysis of 4-chlorophenol in aqueous solution at ambient temperature, J. Am. Chem. Soc., 1994, 116, 11470–11480 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2010 |