Biocatalytic carboxylation†
Silvia M. Glueckab, Selcuc Gümüs‡b, Walter M. F. Fabianb and Kurt Faber*b
aResearch Centre Applied Biocatalysis, University of Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: Kurt.Faber@Uni-graz.at; Fax: +43-316-380-9840; Tel: +43-316-380-5332
bDepartment of Chemistry, Organic and Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, A-8010 Graz, Austria
First published on 8th September 2009
Abstract
Dwindling petroleum feedstocks and increased CO2-concentrations in the atmosphere currently open the concept of using CO2 as raw material for the synthesis of well-defined organic compounds. In parallel to recent advances in the chemical CO2-fixation, enzymatic (biocatalytic) carboxylation is currently being investigated at an increased pace. On the one hand, this critical review provides a concise overview on highly specific biosynthetic pathways for CO2-fixation and, on the other hand, a summary of biodegradation (detoxification) processes involving enzymes which possess relaxed substrate specificities, which allow their application for the regioselective carboxylation of organic substrates to furnish the corresponding carboxylic acids (145 references).
 Silvia M. Glueck | Silvia M. Glueck, born 1973 in Bruck/Mur (Austria), studied Chemistry at the University of Graz, where she received her PhD under the supervision of Prof. K. Faber in 2004. From 2005 to 2006 she worked as a post-doctoral fellow with Prof. N. J. Turner at the University of Edinburgh and the University of Manchester (MIB). In 2007 she returned to the University of Graz to join Prof. W. Kroutil, and in 2008 she became Research Scientist at the Research Centre Applied Biocatalysis in the group of Prof. K. Faber. Her research interests are focusing on biocatalysis, molecular biology and organic synthesis. |
 Selcuk Gümüs | Selcuk Gümüs was born in Bursa, Turkey. He received his BSc degree in chemistry from the Middle East Technical University in 2000. He is currently a PhD candidate working on theoretical and computational organic chemistry. His research mainly focuses on the investigation of novel topological indices and their applications to organic chemistry. |
 Walter M. F. Fabian | Walter M. F. Fabian was born in 1950. He received his PhD in 1976 from Karl-Franzens University Graz in the NMR group of Prof. H. Sterk. After post-doctoral positions at the Max-Planck Institute of Radiation Chemistry in Mülheim/Ruhr with Prof. O. E. Polanski and at the University of Florida at Gainesville with Prof. M. Zerner, he became associate professor at the Institute of Chemistry, Karl-Franzens University Graz, as a member of the computational chemistry group and the division of Organic and Bioorganic Chemistry. His principal research interests are the use of computational chemistry methods in mechanistic organic chemistry. |
 Kurt Faber | Kurt Faber, born 1953 in Klagenfurt (Carinthia/Austria), studied chemistry at the University of Graz, where he received his PhD in 1982. After a post-doctoral fellowship from 1982 to 1983 in St. John’s (Canada) he continued his career at the University of Technology in Graz, where he became associate professor in 1997. The following year he was appointed full professor at the University of Graz, where he since heads a research group devoted to the use of biocatalysts for the transformation of non-natural compounds. He was a visiting scientist at University of Tokyo (1987/1988), Exeter University (1990), University of Trondheim (1994), Stockholm University (2001) and University of Minnesota (2005). |
1. Introduction—carbon, the Janus-faced element
Located at the centre of the main group of the periodic system of elements, carbon has the ability to form stable compounds within a wide range of all oxidation states ranging from −4 (methane) to +4 (carbon dioxide) (Scheme 1). This richness in its chemistry, which is unique among all elements, has allowed Nature to use carbon as the universal atomic ‘Lego-block’ for the synthesis of all essential molecules of life—proteins, DNA/RNA, lipids, hormones, natural products and biopolymers,1 such as cellulose, lignin and chitin.2 Along the same lines, this extreme flexibility in redox states has made carbon the main element for synthetic organic chemistry to build a vast variety of compounds and materials which have considerably contributed to the improvement of the quality of life of modern societies by providing pharmaceuticals, polymers, dyes, agrochemicals, and vitamins, etc.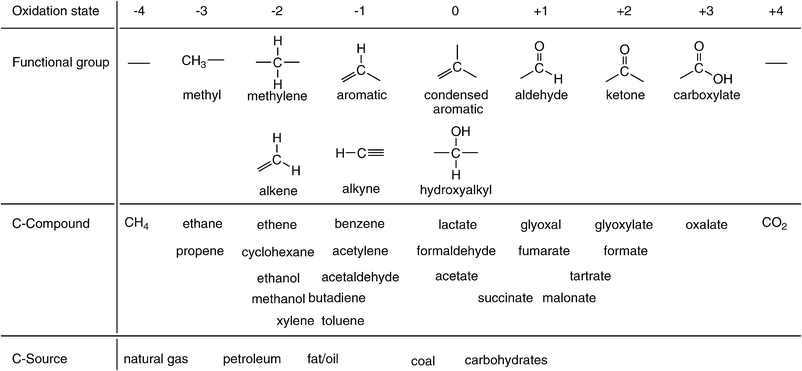 | ||
| Scheme 1 Schematic representation of oxidation states of carbon with corresponding functional groups and representative carbon compounds or materials. | ||
Whereas the synthetic methodology of (industrial-scale) organic synthesis underwent rather gradual changes over the past century—stoichiometric reagents were gradually replaced by catalysts, environmentally hazardous metals are currently being replaced by bio- and organo-catalysts, materials are increasingly being designed to be biodegradable—the ultimate origin of the carbon atoms used as raw materials for organic synthesis has switched rather sharply: whereas coal provided approx. 90% of the carbon atoms required for synthesis until the 1950s—the remainder being petroleum—this ratio flipped only within a remarkably short period of ca. 20 years between 1950 and 1970. To date, approx. 90% of all carbon atoms for chemical industrial synthesis are derived from natural gas and petroleum, 9% from coal and only ∼1% originate from renewable resources.3
However, the unique flexibility of carbon regarding its redox states had undesirable consequences. Since the beginning of industrialisation, carbon was (and still is) the main element of energy-management for the following reasons: the predominant primary carbon sources of the past—coal and petroleum—are stable and contain a high energy-density, which allows easy storage and transport. They provide a high number of electrons per carbon atom upon combustion—4 el from coal, ≥6 from petroleum—which can be transformed through more or less efficient physical principles into heat and/or mechanical energy (Scheme 1).4 Finally, the terminal oxidation product of all primary carbon sources is carbon dioxide, which—considered as a harmless by-product—was released into the atmosphere in massive amounts over the past century.5 To date, the global net CO2-emissions predominantly derived from the combustion of fossil fuel amount to 3.2 Gt of carbon per annum.6
The consequences of the increasing CO2-concentration in the atmosphere have been understood rather slowly.7 It is a commonly accepted fact that the atmospheric CO2-concentration has risen from a pre-industrial level of 280 ppm by 37% since the beginning of large-scale industrialisation in the mid 18th century to an alarming level of 383 ppm today.8 Given the projected maximal tolerance of 450 to 550 ppm for atmospheric CO2, it has been estimated that the ecological buffer capacity of the biosphere has been reached already in the 1980s and that we are currently in the process of over-stretching.9 The climatic consequences of increased CO2-levels are manifested in the rise of the mean temperature in the biosphere,10 elevated sea levels11 and ocean acidification.12
Besides the considerable political and social noise generated around the global warming due to the greenhouse-effect caused by CO2-generation originating from energy-production,13 an equally serious side effect of energy-production by combustion of non-renewable primary carbon sources is largely overlooked. From the 87 million barrels of crude oil produced daily (2008), >90% are converted directly to CO2 for energy-production and only the remainder of <10% is meaningfully used as raw material—carbon source—for comparatively long-lasting products of the chemical industry (Scheme 2).14 Without drastic action taken to decouple energy-generation from the combustion of primary carbon sources,15,16 the resources of crude oil and natural gas are currently estimated to last for approximately 40 and 60 years, respectively (Scheme 2). In this context it becomes clear that the large-scale carbon dioxide capture and storage (CCS) only provides a short-term pressure relief, but certainly no long-term solution.17
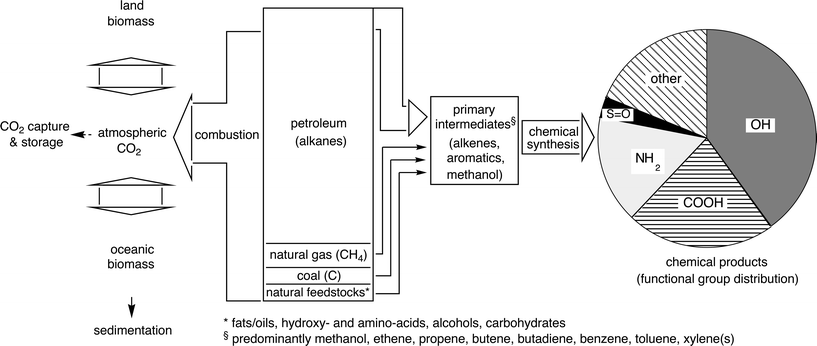 | ||
| Scheme 2 Schematic representation of the interconnection of man-made and natural carbon flux, the arrow-width roughly indicates relative proportions.18 | ||
Considering the fate of carbon derived from crude oil used as raw material for the chemical industry, it is an alarming fact that the main flux of carbon is passing through a remarkably small number of high-volume intermediates, such as methanol, alkenes (ethene, propene, butene, butadiene), and aromatics [benzene, toluene, xylene(s)].19 Although the use of renewable resources and fermentation products is picking up rapidly, they contribute only a few percent of the carbon for chemical synthesis.20 Overall, we are currently burning the non-renewable carbon source(s) for the chemical industry at an alarming rate due to our inability to provide carbon-independent energy-sources. Solutions to this dilemma are not yet visible: First-generation biofuels, such as ethanol and fatty acid methyl esters (biodiesel), are in direct competition with food production and are thus not considered to provide a long-term solution for political and ethical reasons21 and, in addition, simple overall calculations have shown that they are far from being able to meet the energy demands of today.22 The production of second-generation biofuels, which are ultimately derived from non-edible biopolymers,1 such as cellulose and lignin is technically still in its infancy.23 The same holds for scenarios depicted for the so-called hydrogen-24 and methanol-based economies.25,26
In order to widen the unilateral dependence of the chemical industry on petroleum-derived platform intermediates, bulk chemicals from biomass were intensely scouted over the past years.27 The portfolio of proposed candidates ranges from relatively inexpensive carbohydrates (sucrose, xylose, fructose), over alcohols (ethanol, sorbitol, glycerol, propane-1,2- and 1,3-diol, ethylene glycol), to more expensive functionalised (amino- and hydroxy-)carboxylic acids.27,28 It is interesting to note that the average oxidation state of these renewable ‘platform-chemicals’ is closer to that of the final products (Scheme 1), which is certainly beneficial in view of their use in chemical synthesis, but their elevated oxidation state makes them less efficient as biofuels due to their limited energy-content in comparison to hydrocarbons.29 In contrast, the use of the most inexpensive, non-toxic and readily available carbon source as platform material for the synthesis of chemicals, carbon dioxide, is often overlooked.
2. Chemical transformation of carbon dioxide
The development of CO2-fixation reactions for the production of organic molecules is one of the major challenges in synthetic organic chemistry, which would allow to convert a (nowadays problematic) waste gas into an economic profit.30–32 Since CO2 has the highest oxidised state of carbon, the biggest obstacle to using it as the starting material in a chemical reaction is its low energy level. Consequently, a large energy input is required for its transformation. There are four principles to convert CO2 into useful chemicals (Table 1):33| Starting material | Product |
|---|---|
| H2 | Alcohol, carboxylic acid, ester, carboxamides |
| Alkane | Aliphatic carboxylic acid |
| Aromatics | Aromatic carboxylic acid |
| Alkene | Alcohol, aldehyde, carboxylic acid, ester, lactone |
| Diene | Carboxylic acid, ester, lactone, pyrone |
| Alkyne | Carboxylic acid, polyester, (poly)pyrone |
| Amine | Carbamate, urea |
| Epoxide | Cyclic or polymeric carbonate |
| C–H acidic compound | β-Ketocarboxylic acid, α-nitrocarboxylic acid |
(i) The use of high-energy starting materials, such as H2, organometallics, unsaturated compounds (e.g. alkenes, alkynes, allenes34) or compounds possessing high ring-strain (e.g. epoxides35).
(ii) The choice of low-energy products containing carbon at high oxidation state (e.g. organic carbonates, carbamates, carboxylic acids, esters36 or lactones37).
(iii) Equilibrium-controlled reactions can be shifted towards the product-side by e.g. (co)product removal.
(iv) Transformations may be driven by external energy input, such as light (photoirradiation) or electricity (electrolysis).38
The widespread notion that CO2 is thermodynamically and kinetically too stable to react has discouraged its use as carbon source. However, due to its electron-deficiency as ‘carbonic acid anhydride’, CO2 has a high reactivity towards nucleophilic attack. Many nucleophiles, such as organometallics, alkoxides, amines, electron-rich (hetero)aromatics and even water, readily react with CO2 forming the corresponding carboxyl or carboxylate addition products, which further react with carbonic and carbamic acids. The latter are converted to organic carbonates and carbamates with electrophiles.
Despite impressive recent advances in ‘chemical carbon fixation’ (cf.Table 1), only a few processes are performed on an industrial scale (Scheme 3).30 Historically, the synthesis of urea (dating back to 1870) and the Kolbe–Schmitt reaction for the production of salicylic acid (1890)39 are the pioneering examples, whereas methanol production (using H2 from syngas) and the carboxylation of epoxides to yield cyclic or polymeric carbonate esters are a more recent invention. Nevertheless, an impressive amount of 56.1 Mt of CO2 is ‘chemically recycled’ by these processes to date.40 Despite these isolated success stories, the use of CO2 as a raw material for organic synthesis is still heavily underdeveloped.
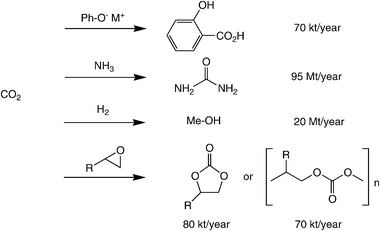 | ||
| Scheme 3 State-of-the-art industrial processes utilising CO2 as carbon source. | ||
3. Principles of biological CO2-fixation in biosynthesis
As a counterpart to the formation of CO2 from geological (volcanic) activity and the decay of biomass, several natural processes for its fixation proceed on a large scale.41 Autotrophic (‘self-nourishing’) organisms42 synthesise complex organic molecules (e.g. carbohydrates, proteins, fatty acids, etc.) by assimilation of CO2via reduction. Depending on the type of redox equivalents, two different main pathways exist, which are largely divided into organisms living in the dark or in light:(i) Organisms called chemoautotrophs derive electrons required for CO2-reduction generally from inorganic (mineral) sources, such as hydrogen, H2S, elemental sulfur, metal ions (Fe2+, Mn2+), ammonia or nitrite, often (but not always) involving oxidation with O2. Most of these organisms are bacteria or Archea living in dark and hostile environments, such as the deep sea. These pathways are summarised under the umbrella of chemosynthesis.43,44
(ii) In contrast, photoautotrophic organisms derive the energy of CO2-fixation from sunlight and use water as an electron-source, thereby producing O2. In an ecological context, they provide nutrients for a plethora of other organisms—the heterotrophs. In terrestrial environments, plants are the predominant variety, while in aquatic systems algae, protists and (cyano)bacteria depend on this pathway.
To date, four major pathways of biological CO2-fixation are known:45
(1) Calvin–Benson–Bassham-cycle,46
(2) reductive TCA (Arnon–Buchanan) cycle,47
(3) reductive Acetyl-CoA (Wood–Ljungdahl) pathway,48
(4) acyl-CoA carboxylase pathways: 3-hydroxypropionate/malonyl-CoA cycle,49 3-hydroxypropionate/4-hydroxybutyrate cycle,50 dicarboxylate/4-hydroxybutyrate pathway,51 and the ethylmalonyl-CoA pathway.52
3.1 Calvin–Benson–Bassham-cycle
The vast majority of atmospheric CO2 is assimilated through the Calvin–Benson–Bassham-cycle, the ‘chemical gearbox’ of photosynthesis.53 The key carboxylating enzyme—D-ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO, [EC 4.1.1.39]54)—binds CO2 onto a pentose derivative55—ribulose-1,5-bisphosphate. The corresponding activated enediolate species launches a nucleophilic attack onto CO2 assisted by an essential Mg2+, to produce a labile C6-β-ketoacid intermediate, which is hydrolytically cleaved into two identical triose-units—3-phosphoglycerate (Scheme 4). Overall, the acid–base catalytic mechanism of RubisCO is a distant relative of Zn2+-dependent type-II aldolases.56 The importance of this enzyme is difficult to exaggerate, since it constitutes the only significant link between the huge pools of inorganic (atmospheric) and organic carbon in the biosphere and its universal distribution is presumably due to the oxygen-insensitivity of its key enzyme and its easy regulation. As catalyst for the net biosynthesis of carbohydrates and fats from atmospheric CO2, RubisCO enables plants and photosynthetic organisms to meet their life-sustaining requirements for organic carbon, while providing food, oxygen and energy for the heterotrophic animal kingdom.57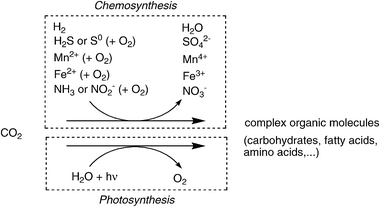 | ||
| Scheme 4 Principles of biological CO2-fixation. | ||
Despite its key position in the ecosphere, the evolution of RubisCO seems to be less than perfect as it shows two major flaws:58
(i) For biocatalytic standards, it is a sluggish catalyst with an average turnover number of 5 s−1, which requires enormous amounts of proteins to overcome the low catalytic performance.59 As a matter of fact, RubisCO is the most abundant single protein in the world.60 In order to circumvent this limitation, intense biotechnological efforts are being undertaken to improve the catalytic properties of RubisCO by enzyme engineering.61
(ii) The enzyme is promiscuous, as it also catalyses the oxygenation of the activated enediolate intermediate of the substrate, leading to the oxidative C–C bond cleavage of the pentose to yield the triose 3-phospho-D-glycerate and the C2-unit 2-phosphoglycolate (Scheme 5). This ‘side-reaction’ constitutes a considerable burden for all photosynthetic organisms with energy-wasting photorespiration.62 The competition between carboxylation and oxygenation arises from the confusion of RubisCO between the substrate of photosynthesis (CO2) with its product O2. These difficulties stem from the inevitable O2-sensitivity of the enolate intermediate, which is exacerbated by the need to discriminate between two apparently featureless and thus only weakly bound gaseous molecules—CO2 and O2.
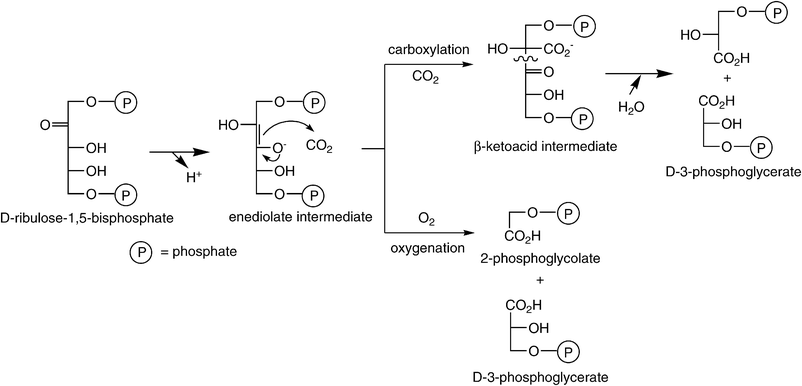 | ||
| Scheme 5 Key carboxylation step in the Calvin–Benson–Bassham-cycle catalysed by RubisCO. | ||
Overall, for each molecule of CO2 to be assimilated through the Calvin-cycle, 3ATP and 2NAD(P)H are required, an energy-equivalent of approx. 114 kcal, which is ultimately derived from sunlight in photosynthesis. Due to the high complexity of the catalytic mechanism and the unique position of RubisCO in biosynthesis, it is very unlikely that this enzyme might be able to accept non-natural substrate analogues, thereby producing carboxylic acids for organic synthesis. To date, non-selective fermentative CO2-sequestration using algae and plants is the most important impact of RubisCO for the production of biomaterials, such as fats/oils and carbohydrates.
3.2 Reductive TCA (Arnon–Buchanan) cycle
In combination with the respiratory chain, aerobic organisms use the oxidative degradation of acetyl-CoA into two CO2 molecules through the tricarboxylic acid (TCA)-cycle to provide energy.63 Consequently, the same catalytic machinery can be applied to CO2-fixation in the reverse direction to produce acetyl-CoA—the ‘reductive TCA cycle’.64 Subsequently, acetyl-CoA can be converted to pyruvate and phosphoenol pyruvate, which is required to regenerate the intermediates of the cycle in an anaplerotic manner. Overall, the energy input required to transform two CO2 units per cycle into acetyl-CoA amounts to approximately one ATP and four NAD(P)H units. Although the basic steps of the TCA-cycle are identical for both directions, several enzymes have been adapted to match the energetically unfavourable uphill direction. This cycle uses a set of enzymes that are central to all organisms, however, with some modifications that are characteristic for anaerobic bacteria.The reductive TCA-cycle is believed to have evolved in hyperthermophilic chemoautotrophs, which emerged in seawater when the earth’s atmosphere was devoid of oxygen and is thus the evolutionary origin of the oxidative TCA-cycle;65 it was even speculated that the reductive TCA cycle might have had non-enzymatic surface-bound autocatalytic ancestors.66 The full cycle was first reported from a green sulfur photosynthetic bacterium (Chlorobium limicola)43 and was later found to operate also in Aquificales, archeal Crenarcheota and various types of proteobacteria.
In the reductive TCA-cycle, both carboxylation steps occur in a successive fashion (Scheme 6). First, succinyl-CoA is reductively carboxylated by 2-oxoglutarate synthase [EC 1.2.7.3] to form 2-oxoglutarate at the expense of two equivalents of reduced ferredoxin, which is ultimately recycled via NAD(P)H. In the successive step, the α-ketoglutarate thus formed is again reductively carboxylated by isocitrate dehydrogenase [EC 1.1.1.41] and [EC 1.1.1.42] at the expense of NAD(P)H. In the latter reaction, the (de)carboxylation step is assumed to proceed via the enolate intermediate of α-oxoglutarate, which is formed through assistance of an essential divalent Mg2+ or Mn2+ ion (cf. RubisCO), leading to a highly unstable keto-tricarboxylic acid intermediate, which is immediately reduced to yield stable isocitrate. As in the Calvin-cycle, the enzymes for both carboxylation steps in the reductive TCA-cycle are highly specialised and it is extremely unlikely that they are able to accept (man-made) surrogate substrates.
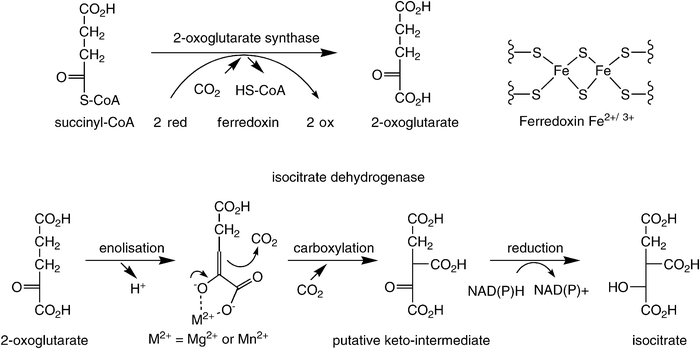 | ||
| Scheme 6 Carboxylation steps in the reductive TCA cycle. | ||
3.3 Reductive acetyl-CoA (Wood–Ljungdahl) pathway
In contrast to the cyclic Calvin- and the reductive TCA-pathways, the reductive acetyl-CoA process is exceptional in two ways: (i) in contrast to other CO2-fixation processes, which are cyclic, it is a linear unidirectional pathway, through which two molecules of CO2 are reductively coupled to furnish acetyl-CoA, and (ii) it proceeds through some rarely occurring organometallic enzyme–substrate intermediates, such as methyl-Co- and Ni-carbonyl-species.67 This pathway contains some of the most sophisticated enzyme catalysis known to date (Scheme 7) and it is not surprising that some of its components are extremely oxygen-sensitive. Overall, it represents the most important method by which anaerobic organisms, such as acetogenic bacteria and methanogenic Archaea, sequester carbon for biomass and energy for life.68,69 In a certain way, it resembles the Monsanto process of acetate synthesis from CO and methanol.70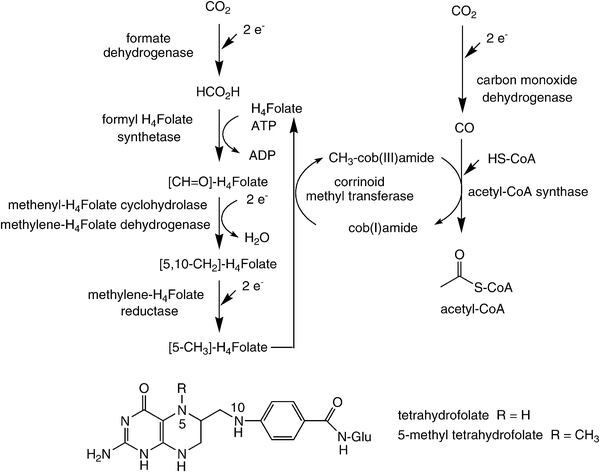 | ||
| Scheme 7 Carbon dioxide fixation via the reductive acetyl-CoA pathway. | ||
The reductive acetyl-CoA pathway consists of two parallel branches. The so-called ‘eastern branch’ starts with the reduction of CO2 to formate. The latter is coupled at the expense of ATP onto N5 of the cofactor tetrahydrofolate, where it is stepwise reduced to finally become an activated methyl group on 5-methyl-tetrahydrofolate involving a series of enzymes. The methyl group is then transferred from folate onto the cobalt centre of a corrinoid iron–sulfur-protein by a corrinoid methyltransferase.71 This step goes in hand with the liberation of tetrahydrofolate, which re-enters the pathway.
The so-called ‘western branch’ begins with the reduction of CO2 to CO catalysed by carbon monoxide dehydrogenase, which is an integral component of the central biocatalyst in the pathway: acetyl-CoA synthase. Despite the availability of high-resolution crystal structures of this enzyme72 and approximately two decades of biochemical studies, many questions remain concerning the mechanism of this intriguing enzyme.73 Both carbon monoxide dehydrogenase and acetyl-CoA synthase are integrally linked by a gas channel connecting the two active sites thereby ensuring that the CO produced is efficiently incorporated into acetyl-CoA by combination with the activated methyl group derived from the ‘eastern branch’.
3.4 Acyl-CoA carboxylation pathways
In contrast to the above-mentioned pathways for CO2-fixation, which have been known for decades, several autotrophic CO2-assimilation pathways occurring in anaerobic autotrophs have been elucidated only recently. Although the 3-hydroxypropionate/4-hydroxybutyrate pathway has been termed the ‘fifth’ pathway of CO2-assimilation,74 for the sake of clarity all of these pathways are treated together under a single paragraph, since all the carboxylation steps show a recurring feature: they all take place on a (CoA-activated) carboxylic acid substrate.In the 3-hydroxypropionate/malyl-CoA cycle and the 3-hydroxypropionate/4-hydroxybutyrate pathway, one molecule of CO2 is bound onto acetyl-CoA via an ATP-dependent step catalysed by acetyl-CoA-carboxylase. After sequential reduction of the terminal carboxylate group, propionyl-CoA is carboxylated in the same fashion yielding methylmalonyl-CoA (Scheme 8, top). Due to the similarity of these transformations, it was assumed that both enzymes are identical.75 The terminal net assimilation product is a C2-unit: acetyl-CoA in the 3-hydroxypropionate/malyl-CoA cycle and glyoxylate 3-hydroxypropionate/4-hydroxybutyrate pathway.
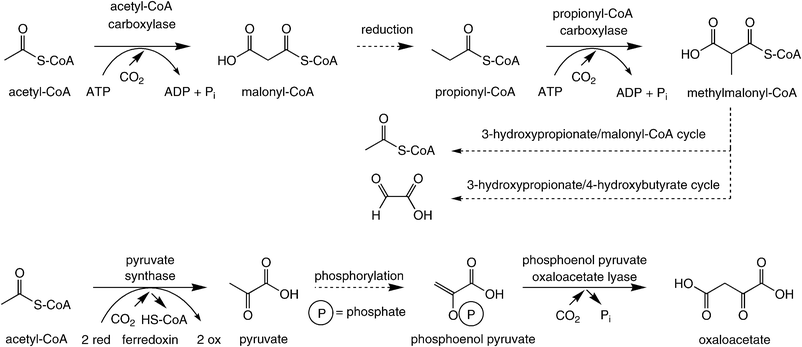 | ||
| Scheme 8 Carboxylation of acetyl/propionyl-CoA and phosphoenol pyruvate. | ||
A variant of the above-mentioned carboxylation was found in the dicarboxylate/4-hydroxybutyrate pathway76 (Scheme 8, bottom). Here, acetyl-CoA is reductively carboxylated by pyruvate synthase at the expense of two equivalents of ferredoxin yielding pyruvate. After phosphorylation, phosphoenol pyruvate is carboxylated by phosphoenol pyruvate oxaloacetate lyase to furnish oxaloacetate. The latter is transformed into two equivalents of acetyl-CoA, one of which re-enters the cycle.76
An intriguing carboxylation of an enoyl-CoA derivative was recently observed in the ethylmalonyl-CoA pathway (Scheme 9). In this case, an α,β-unsaturated acyl-CoA derivative is reductively carboxylated by crotonyl-CoA carboxylase/reductase at the expense of a hydride from nicotinamide. The mechanism of this unprecedented biotransformation was assumed to proceed via nucleophilic hydride attack at Cβ onto the activated enoyl-CoA ester via a Michael-type addition, while the enolate is trapped by CO2 as the electrophile forming ethylmalonyl-CoA. The fact that crotonyl-CoA reductase/carboxylase showed weak enoate reductase activity in the absence of CO2 (forming butyryl-CoA) suggests a certain resemblance to the conjugate reduction of activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds by the OYE-type family, where the enolate is trapped by a proton.77
C bonds by the OYE-type family, where the enolate is trapped by a proton.77
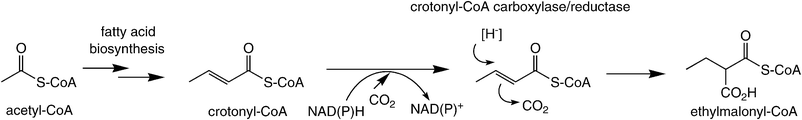 | ||
| Scheme 9 Reductive carboxylation of an enoyl-CoA. | ||
According to a generally accepted hypothesis, the first organisms were strict autotrophic anaerobes, which predominantly used the reductive acetyl-CoA pathway still found today in strictly anaerobic Archea and bacteria78 for the synthesis of biomass from CO2.79 After the gradual emergence of molecular oxygen in the atmosphere during the ‘great oxidation event’,80 this pathway could no longer operate due to the extreme oxygen-sensitivity of some of its key enzymes. As a consequence, autotrophic CO2-fixation had to be ‘re-invented’, leading to various solutions depending on environmental conditions, such as the availability of light, metal ions, etc.81 This led to drastic adaption of the carbon-metabolism in biology going in hand with the largest extinction of species which took place during evolution so far. Whereas the reductive acetyl-CoA-cycle with its O2-sensitive enzymes was forced into oxygen-free ecological niches, the TCA-cycle switched its direction and started to operate in the reverse direction. The Calvin-cycle with its O2- and light-stable enzymes was presumably evolved late in evolution.66
Due to the high complexity of these specialised biosynthetic pathways, their application for the production of well-defined organic materials requires highly sophisticated metabolic engineering. Since these are important biosynthetic processes, the enzymes involved are usually highly substrate-specific and consequently it is rather unlikely that non-natural (man-made) substrates will be accepted at reasonable rates. On the contrary, carboxylases involved in biodegradation are more likely to possess a desired broad substrate spectrum.
Overall, all biological carboxylation reactions discussed above require a significant amount of energy.82,83 This may be provided either via ATP-dependent covalent linkage of CO2 onto the cofactor biotin (vitamin B7 or H),84 or from redox equivalents, such as NAD(P)H or ferredoxin in reductive carboxylations. Likewise, the net energy of photosynthesis is derived from light. Consequently, such carboxylations were classified as high-energy (class-B) reactions.85
4. Biological carboxylation in detoxification
In contrast to highly directed biosynthetic carboxylation pathways, several unspecific CO2-fixation reactions occur in catabolic (biodegradation) pathways. The prime goal of biodegradation is making toxic compounds more hydrophilic, thereby enhancing their solubility in water in order to reduce their affinity for sensitive lipophilic biological components, such as membranes, proteins, etc. In order to make these processes more efficient, catabolic enzymes usually possess relaxed substrate specificities and are able to act on a large variety of substrates.Depending on the organism and its environment, several pathways are available. (i) Oxidation at the expense of O2 (usually catalysed by cytochrome P450 enzymes86) renders the product bearing hydroxy- and/or carboxy-groups. (ii) Glycosylation of hydroxy- and/or amino-functionalities lead to water-soluble glycosides. (iii) Under anoxic conditions, where oxidation is impossible, redox-neutral carboxylation leads to polar carboxylic acids.
Carboxylases involved in biodegradation have been exploited to an astonishingly limited extent for preparative biotransformations.87–89 Although the exact role of these enzymes and their ‘true’ substrates in Nature are currently unknown, it is assumed that they serve as detoxification mechanism under oxygen-limited conditions for lipophilic electron-rich (hetero)aromatics, such as phenols and pyrroles, to yield the corresponding water-soluble carboxylic acids.90 Overall, these reactions require little (or no) energy and thus have been classified as low-energy (class-A) carboxylation reactions.85 Due to the lack of a strong thermodynamic driving force, equilibria close to unity are common. In view of preparative-scale applications, equilibria may be driven towards the synthesis/carboxylation direction by the use of excess CO2—applied either under atmospheric pressure, as concentrated carbonate/bicarbonate solution or in supercritical form.91
In this article, a summary of enzymatic carboxylation reactions mainly derived from catabolic pathways with potential for biocatalytic applications is given. The reactions are classified into four groups according to the type of substrate:
(i) carboxylation of epoxides,
(ii) carboxylation of aromatics,
(iii) carboxylation of hetero-aromatic systems, and
(iv) carboxylation of aliphatic substrates.
4.1 Biocatalytic carboxylation of epoxides
Alkenes are usually degraded via epoxidation (catalysed by cytochrome P450 monooxygenases) to yield the corresponding epoxides, which are subsequently hydrolysed by epoxide hydrolases. Some bacteria have developed an alternative epoxide-degradation system based on enzyme-catalysed carboxylation, which leads to the corresponding β-keto acids. In Xanthobacter Py2, this pathway serves for the generation of poly-β-hydroxyalkanoates, which are central building blocks for carbon-storage (Scheme 10).92–94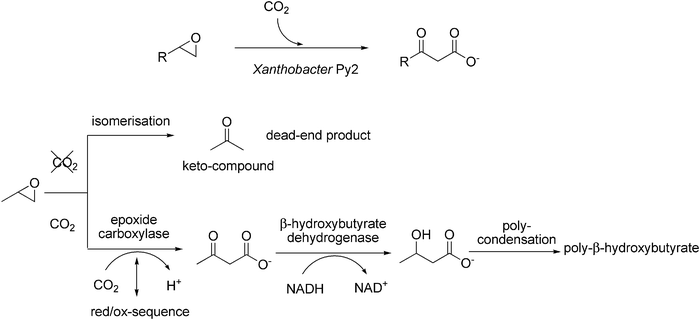 | ||
| Scheme 10 Biocatalytic carboxylation of epoxides by the aerobic bacterium Xanthobacter Py2 (top). Proposed mechanism of biodegradation of epoxypropane (bottom). | ||
In this organism, epoxide-degradation pathways appear to be regulated by the presence or absence of CO2 (Scheme 10, bottom). In the absence of CO2, whole cell suspensions95 or cell extracts96 of propene-grown Xanthobacter Py2 catalyse the isomerisation of aliphatic epoxides (e.g. epoxypropane) to the corresponding keto-compound (e.g. acetone) as dead-end products, which are not further metabolised. On the other hand, in the presence of CO2, the carboxylation reaction prevails.93,94 Both pathways were reported to be dependent on NAD+ and an unidentified (low molecular weight) factor which could be replaced by a range of artificial dithiol compounds.96 Since the net redox balance of both the isomerisation and the carboxylation reactions are zero, it was concluded that the degradation pathways presumably proceed via an oxidation–reduction sequence. The key (ORF3) protein essential for epoxide degradation represents a novel type of NADPH-dependent pyridine nucleotide-disulfide oxidoreductase. Since NADPH could replace the low molecular weight factor (or dithiol derivative, resp.) it was suggested that NADPH is the true physiological cofactor for the epoxide degradation.97
Based on (limited) biochemical evidence, the following mechanism for the carboxylation of epoxypropane was proposed (Scheme 10, bottom).93,94 In a first step, the epoxide is carboxylated by an epoxide carboxylase forming acetoacetate as intermediate. This reaction might be coupled to a (NAD+/NADPH- or DTT-dependent) reduction–oxidation sequence.96,97 Acetoacetate then undergoes NADH-dependent reduction catalysed by β-hydroxybutyrate dehydrogenase (which was detected at high levels in the cell extract) leading to β-hydroxybutyrate, which is condensed into poly-β-hydroxyalkanoates for storage.
The fact that the responsible enzymes could not be purified makes the in vitro application of this system hardly possible and is thus of limited use.98 Although this transformation shows certain academic appeal, its potential for practical applicability is low.
4.2 Biocatalytic carboxylation of aromatic (phenolic) compounds
The metabolism of aromatic compounds in aerobic bacteria commonly proceeds via oxidation catalysed by dioxygenases using molecular oxygen as co-substrate. In anaerobic bacteria, however, oxygen-independent metabolic pathways are required,99 one of which is the enzymatic carboxylation of phenols to more polar benzoic acid derivatives.85,99 The carboxylation of phenol in the strict anaerobe Thauera aromatica proceeds via a two-enzyme step involving the (i) ATP-dependent activation of phenol to phenylphosphate mediated by phenylphosphate synthase, followed by (ii) regioselective (para-)carboxylation of the activated intermediate to p-hydroxybenzoic acid catalysed by the metal (Mg2+ or Mn2+ and K+) dependent phenylphosphate carboxylase (Scheme 11). The divalent metal ion is supposed to act as a Lewis acid by increasing the electrophilic character of CO2, whereas K+ is assumed to support the regioselectivity in close analogy to the chemical process.100,101 The actual substrate for oxygen-sensitive phenylphosphate synthase is CO2 rather than bicarbonate as in the case of biotin-dependent carboxylases. In vivo, the carboxylation product is further metabolised to acetyl-CoA as the final product of phenol degradation.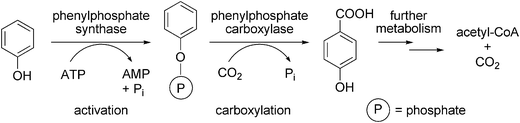 | ||
| Scheme 11 Carboxylation of phenol in Thauera aromatica. | ||
The first example of a biotechnological application of a carboxylase was reported by the group of Aresta and co-workers102 in 1998 using a crude cell-free extract or a partially purified enzyme of T. aromatica for the carboxylation of phenol to p-hydroxybenzoic acid at ambient temperature and pressure. Selective carboxylation of phenylphosphate to p-hydroxybenzoic acid proceeded in 70% yield, with moderate efficiency (estimated TON 16.000). Attempts to enhance the low stability by supporting the enzyme on low melting agar has been successful.85
Although (semi-purified) phenylphosphate carboxylase of T. aromatica showed comparable activities in supercritical CO2 as solvent/reagent (at 25 MPa and 35 °C) to that under CO2 pressure in aqueous media, the yields of carboxylation product were low (5%).91 However, the use of scCO2 offers a technological advantage in product recovery in comparison to aqueous media.
Besides phenol, phenylphosphate carboxylase is also able to carboxylate catechol and it has been proposed that the enzymatic carboxylation proceeds via the same pathway as for phenol.103 Thus, catechol is activated to catechylphosphate by ATP-dependent phenylphosphate synthase followed by regioselective carboxylation in the para-position to the phosphorylated hydroxyl group catalysed by phenylphosphate carboxylase to yield 3,4-dihydroxybenzoate. In a similar fashion, o-cresol was converted to 4-hydroxy-3-methylbenzoate.103
The obligate anaerobic bacterium Sedimentibacter hydroxybenzoicus104 catalyses the decarboxylation of a narrow range of benzoic acid derivatives (Scheme 12). The responsible enzymes (4-hydroxy- and 3,4-dihydroxybenzoate decarboxylase, respectively) were also able to catalyse the corresponding reverse (carboxylation) reaction of phenol and catechol in the presence of bicarbonate as CO2-source to yield 4-hydroxy- or 3,4-dihydroxybenzoate, respectively.105–107 Both oxygen-sensitive enzymes originating from S. hydroxybenzoicus strain JW/Z-1T were purified and characterised and (in case of 4-hydroxybenzoate decarboxylase) also cloned and overexpressed in Escherichia coli.108 Biochemical characterisation revealed that the reaction can be run in both directions and does not require ATP, biotin, TPP, PLP or a metal ion for the (de)carboxylation reaction. N-Terminal sequence analysis showed a certain similarity to uroporphyrinogen decarboxylase from Synechococcus and Saccharomyces sp. Reversible (de)carboxylation of 4-hydroxybenzoate–phenol was shown for purified 4-hydroxybenzoate decarboxylases from Enterobacter cloacae P240109 and Chlamydophila pneumoniae AR39.110 However, in all cases, the equilibrium heavily favours the decarboxylation.
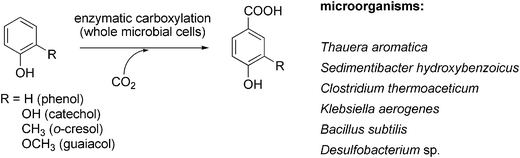 | ||
| Scheme 12 Regioselective para-carboxylation of non-activated phenolic compounds catalysed by whole (viable) microbial cells. | ||
Bacillus subtilis which is known to decarboxylate various hydroxy-aryl carboxylic acids111 is able to catalyse the carboxylation of guaiacol and phenol to yield vanillate (3-methoxy-4-hydroxybenzoate) and 4-hydroxybenzoate, respectively.112 The reversible decarboxylation–carboxylation reaction proceeds under both aerobic and anoxic conditions. However, the carboxylation rate for both substrates was low in relation to decarboxylation (3–6%). Anaerobic degradation of phenol by a microbial consortium113 and of catechol by Desulfobacterium sp. strain Cat2114 has been shown to involve carboxylation.
Besides the regioselective carboxylation in para-position to the (directing) phenolic hydroxyl group, several 2,6-dihydroxybenzoate decarboxylases (or γ-resorcylic acid decarboxylases, respectively) catalysing the regioselective ortho-carboxylation of γ-resorcinol (1,3-dihydroxybenzene) to yield resorcylic acid (2,6-dihydroxybenzoic acid) without the formation of regio-isomeric α- or β-resorcylic acid derived from meta-, or para-carboxylation have been discovered (Scheme 13). For instance, 2,6-dihydroxybenzoate decarboxylase from Agrobacterium tumefaciens IAM12048 catalysing the non-oxidative decarboxylation of 2,6- and 2,3-dihydroxybenzoate has been purified and characterised.115,116 In the presence of bicarbonate, the reverse carboxylation reaction of 1,3-dihydroxybenzene took place. Although the reaction was rather slow (30% conversion after 30 h), the potential of these cofactor-independent and oxygen-stable enzymes for biocatalytic applications is high.
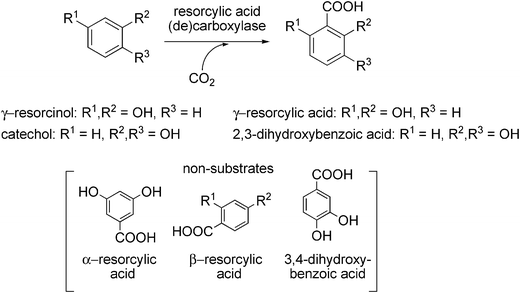 | ||
| Scheme 13 Regioselective (ortho) carboxylation of non-activated phenolic compounds catalysed by resorcylic acid (de)carboxylases. | ||
γ-Resorcylic acid decarboxylases derived from Rhizobium radiobacter WU-0108117 and Rhizobium sp. MTP-1005118 also act in a reversible fashion using bicarbonate as CO2-source; like the decarboxylase from A. tumefaciens, these enzymes are oxygen-stable and do not depend on any cofactors. Besides γ-resorcinol, the enzyme from R. radiobacter also transforms catechol to 2,3-dihydroxybenzoic acid in an ortho-selective fashion (Scheme 13). γ-Resorcylic acid is employed as an intermediate for the synthesis of a range of compounds and is industrially produced in analogy to the Kolbe–Schmitt reaction requiring high pressure and temperature. In an enzymatic process (employing a cell-free extract of recombinant E. coli overexpressing γ-resorcylic acid decarboxylase), resorcinol was carboxylated to γ-resorcylic acid in 44% yield after 7 h at 20 mM substrate concentration.119
Examples for the purification and characterisation of enzymes catalysing the decarboxylation of electron-rich (hydroxy)aryl carboxylic acids have been reported: 2,3-dihydroxybenzoate decarboxylase from fungal (Aspergillus)120 and yeast origin,121 4,5-dihydroxyphthalate decarboxylase from Pseudomonads,122 hydroxycinnamate decarboxylase from Klebsiella oxytoca123 and Aerobacter,124 gallic acid decarboxylase from Pantoea agglomerans,125 ferulate decarboxylase from Pseudomonas fluorescens126 and Bacillus pumilus,127p-coumarate decarboxylase from B. pumilus127 and Lactobacillus plantarum.128 Decarboxylase activity towards phenol and derivatives has also been reported for the obligate anaerobe Clostridium thermoaceticum129 and the facultative anaerobe Klebsiella pneumoniae (syn. aerogenes).130
However, the corresponding reverse carboxylation reaction has not been reported for the latter enzymes, but might be possible in view of the reversibility of (de)carboxylase reactions discussed above. The distribution of genes encoding microbial non-oxidative reversible hydroxyarylic acid decarboxylases/phenol carboxylases was shown to be distributed in all three microbial domains.112,131
4.3 Biocatalytic carboxylation of heteroaromatics
Besides phenols, also electron-rich heteroaromatics, such as pyrroles or indoles are good substrates for enzymatic (de)carboxylation. For instance, regioselective ortho-carboxylation of pyrrole was accomplished by pyrrole-2-carboxylate decarboxylase132–135 from Bacillus megaterium and Serratia sp. The homodimeric enzyme isolated from B. megaterium PYR2910 is unique as it requires the presence of an organic acid (such as acetate, propionate, butyrate or pimelate) for catalytic activity. A possible mechanism of action of pyrrole-2-carboxylate decarboxylase has been proposed,133,136 which suggests that the organic acid acting as ‘cofactor’ deprotonates the NH-moiety, causing a nucleophilic attack of Cα at the electrophile CO2. After tautomerisation, the intermediate regains aromaticity (Scheme 14, top). However, in view of the low acidity of the NH-moiety of pyrrole (pKa 16.5), this mechanism would need further support.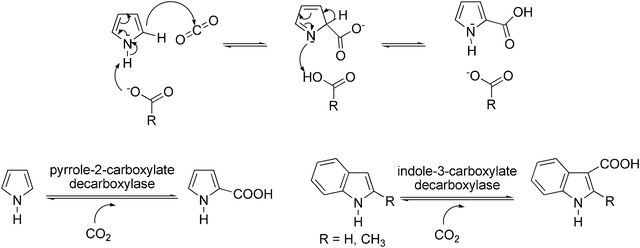 | ||
| Scheme 14 Proposed mechanism of carboxylation of pyrrole-2-carboxylate decarboxylase; R-CO2− represents the organic acid required as ‘cofactor’ (top); enzymatic carboxylation of pyrrole and indole (bottom). | ||
Since the carboxylation of pyrrole possesses an equilibrium constant not far from unity (0.3–0.4 M), the equilibrium could be shifted towards carboxylation by using saturated aqueous bicarbonate or the use of supercritical CO2,91,137,138 leading to impressive substrate concentrations (300 mM) and high conversions (81%) (Scheme 14, bottom). However, pyrrole-2-carboxylate decarboxylase from B. megaterium exerts a rather narrow substrate specificity, and related (electron-rich) substrates, such as indole and 2-methylpyrrole, were not accepted. Furthermore, electron-deficient heteroaromatics, such as pyrazine, pyridine, imidazole and various triazoles, were unreactive altogether.
A related non-oxidative reversible decarboxylase—indole-3-carboxylate decarboxylase—isolated from Arthrobacter nicotianae and several molds has been explored for the (reversible) carboxylation of indole and 2-methylindole to furnish the corresponding carboxylic acids139 in the presence of KHCO3 (Scheme 14, bottom). In whole resting cells grown under aerobic conditions, enzyme activity was fairly stable, but oxygen-sensitivity was detected in cell-free extracts. In contrast to the high conversion in the carboxylation of pyrrole, only modest conversion of indole to indole-3-carboxylate was reported (34%). The activity towards 2-methylindole was approximately one third of that of indole.
Orotidine 5′-monophosphate decarboxylase involved in the de novo biosynthesis of pyrimidine nucleotides catalyses the decarboxylation of orotidine 5′-monophosphate;140 the reverse carboxylation reaction was not reported to date.
4.4 Biocatalytic carboxylation of aliphatic compounds
Pyruvate decarboxylase (EC 4.1.1.1) requires thiamine pyrophosphate (TPP) for catalytic activity and catalyses the decarboxylation of pyruvic acid into acetaldehyde and CO2 (Scheme 15). This enzyme has been successfully applied for the reverse carboxylation reaction to yield pyruvic acid.141 The biotransformation was performed using pyruvate decarboxylase from brewer’s yeast and addition of TPP as cofactor. In order to favour the carboxylation, aqueous sodium bicarbonate buffer at high pH serving both as solvent and CO2-source was employed, the highest conversion being obtained at pH 11. An impressive yield of 81% was achieved at 500 mM bicarbonate concentration, however, the substrate concentration was in the sub-millimolar range (100 μM). It was speculated that supercritical CO2 might be effective to improve the yield of pyruvic acid synthesis.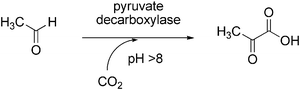 | ||
| Scheme 15 Carboxylation of acetaldehyde by pyruvate decarboxylase. | ||
Arylmalonate decarboxylases from Alcaligenes bronchisepticus (EC 4.1.1.76)142 and Achromobacter sp.,143 which catalyse the asymmetric decarboxylation of arylmalonate yielding non-racemic α-substituted propionic acids, have been purified and characterised, and the A. bronchisepticus enzyme was even engineered144 with respect to reverse its stereoselectivity. However the reverse carboxylation reaction has not been reported so far.
As depicted in Table 2, enzymatic carboxylation activities are distributed widely among the microbial world, both in aerobic and anaerobic habitats.
| Entry | Microorganism | Responsible enzyme | Substrates | Conditions |
|---|---|---|---|---|
| 1 | Xanthobacter | Epoxide carboxylase | Epoxides | Aerobe |
| 2 | Thauera aromatica | Phenylphosphate carboxylase | Phenol, catechol, o-cresol | Obligate anaerobe |
| 3 | Enterobacter cloacae | 4-Hydroxybenzoate decarboxylase | Phenol | Anaerobe |
| 4 | Chlamydophila pneumoniae | 4-Hydroxybenzoate decarboxylase | Phenol | Anaerobe |
| 5 | Sedimentibacter hydroxybenzoicus104 | 4-Hydroxy- and 3,4-dihydroxybenozate decarboxylase | Phenol, catechol | Obligate anaerobe |
| 6 | Bacillus subtilis | Vanillate/4-hydroxybenzoate decarboxylase | Guaiacol, phenol | Aerobe, anaerobe |
| 7 | Desulfobacterium sp. | Unknown | Catechol | Anaerobe |
| 8 | Agrobacterium tumefaciens | 2,6-Dihydroxybenzoate decarboxylases | γ-Resorcinol | Aerobe |
| 9 | Pandoraea sp. | 2,6-Dihydroxybenzoate decarboxylases | Phenol, 1,2- and 1,3-dihydroxybenzene | Aerobe |
| 10 | Rhizobium radiobacter, Rhizobium sp. | γ-Resorcylic acid decarboxylase | γ-Resorcinol, catechol | Aerobe |
| 11 | Bacillus megaterium, Serratia sp. | Pyrrole-2-carboxylate decarboxylase | Pyrrole | Aerobe |
| 12 | Arthrobacter nicotianae | Indole-3-carboxylate decarboxylase | Indole, 2-methylindole | Aerobe |
| 13 | Brewer’s yeast | Pyruvate decarboxylase | Acetaldehyde | Aerobe |
5. Energy-requirement of carboxylation reactions
Depending on their energy-requirement, carboxylation reactions were classified into ‘low-energy’ (class A) and ‘high-energy’ (class B) carboxylation reactions.85,145 However, this classification was never defined in a quantitative fashion. In order to provide deeper insight into this classification, we determined the Gibbs free energies (ΔGreact) of a representative set of (de)carboxylation reactions by quantum chemical calculations. For the sake of clarity, the reactions were grouped according to the type of CO2-acceptor (for details see ESI†).In general, all carboxylation reactions were calculated to be endergonic (ΔGreact >0) thus corroborating the notion of an energetically unfavourable uphill direction, which includes all major types of CO2 acceptors: aldehydes, amines and aliphatic acidic C–H and (hetero)aryl C–H acceptors within a range of +5 to +18 kcal mol−1 (Table S1, ESI†). This trend was only inverted in the case of activated acceptor substrates. Whereas carboxylation of phenylphosphate (going in hand with phosphate ester hydrolysis) turned out to be approximately thermoneutral, the carboxylation of a highly strained oxirane and an activated alkene were found to be strongly exergonic (−19 and −8 kcal mol−1, resp.).
6. Summary
By surveying the data from biochemical and metabolic studies on (de)carboxylation reactions available to date, it appears that the enzymatic fixation of CO2 to produce well-defined low molecular weight organic compounds—carboxylic acids in a broadest sense—appears feasible through two different strategies:(i) Metabolic engineering enables the redirecting of well-known metabolic pathways for the production of a limited set of carboxylic acids occurring as intermediates (or end products) in these pathways. Based on the high substrate specificity of biosynthetic pathways, it is unlikely that these pathways might be exploited to convert non-natural substrate surrogates also. Few recent examples also suggest that decarboxylases may be forced to run the reverse carboxylation reaction under appropriate reaction conditions, such as high-concentration carbonate buffer or supercritical CO2.
(ii) In contrast, the data on carboxylases occurring in biodegradation pathways suggest that these enzymes possess relaxed substrate specificities, which enable the regioselective carboxylation of various types of substrates, in particular epoxides or electron-rich (hetero)aromatics. Based on lead-sequence data, genome mining will certainly provide more oxygen-stable (de)carboxylases possessing a broad substrate tolerance in the near future.
Acknowledgements
Hans-Peter Meyer (Visp, Switzerland) and Martin Mittelbach (Graz) are cordially thanked for sharing their data, ideas and concepts with us. The Middle East Technical University of Ankara is cordially thanked for supporting the leave of S. Gümüs to Graz.References and notes
- The annual worldwide production of biopolymers has been estimated to amount 170–200 Gt, including cellulose (90 Gt), wood (1.75 Gt) and lignin (0.39–0.58 Gt).
- C. Jeuniaux and M. F. Voss-Foucart, Biochem. Syst. Ecol., 1991, 19, 347–356 CrossRef CAS.
- H. A. Wittcoff and B. G. Reuben, Industrial organic chemicals, Wiley, New York, 1996 Search PubMed.
- The current efficiency limits are very limited: petroleum-powered combustion engine 33%, fuel cell 40–50%, fuel cell including hydrogen-production via electrolysis 25%, electric motor via battery 86%.
- CO2 is sequestered weakly by land-biomass (−0.5 Gt carbon per annum) and through aquatic systems (−2 to −4 Gt carbon per annum); however, deforestation accounts for additional +1.6 Gt carbon per annum globally.
- About half of the CO2 released since 1750 remains in the atmosphere, with the majority of the rest sequestered in the oceans and the remainder stored in terrestrial ecosystems, see: R. M. Gifford, Aust. J. Plant Physiol., 2004, 21, 1–15 Search PubMed.
- It must be kept in mind that due to the complexity of the ecosphere and the immense number of variables implied, accurate analytical data cannot be obtained. Thus, depending on the source, values generally vary considerably, in extreme cases up to one order of magnitude.
- According to the World Meterological Organisation, the atmospheric CO2-concentration increased by 0.5% in the year 2007 alone.
- M. Wackernagel, N. B. Schulz, D. Deumling, A. C. Linares, M. Jenkins, V. Kapos, C. Monfreda, J. Loh, N. Myers, R. Norgaard and J. Randers, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9266–9271 CrossRef CAS.
- The mean air/earth surface temperature rose +0.3 to +0.6 °C since 1900, tropic ocean surface temperature rose +0.5 °C since 1500.
- Average rise worldwide are +10 to +25 cm during the past century.
- J. C. Orr, V. J. Fabry, O. Aumont, L. Bopp, S. C. Doney, R. A. Feely, A. Gnanadesikan, N. Gruber, A. Ishida, F. Joos, R. M. Key, K. Lindsay, E. Maier-Reimer, R. Matear, P. Monfrey, A. Mouchet, R. G. Najjar, G.-K. Plattner, K. B. Rodgers, C. L. Sabine, J. L. Sarmiento, R. Schlitzer, R. D. Slater, I. J. Totterdell, M.-F. Weirig, Y. Yamanaka and A. Yool, Nature, 2005, 437, 681–686 CrossRef CAS.
- About 2/3 of the greenhouse gas emission account for CO2 derived from combustion, see: M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981–987 Search PubMed.
- Depending on the reference, worldwide estimates for the chemical industry range from 10 to 6.2%; the chemical industry in Germany uses 80% petroleum, 8% natural gas, 2% coal, and 10–12% renewable resources as primary carbon source, see: P. Welters and A. Müller, GIT Labor-Fachz., 2008, 3, 246–248 Search PubMed ; Evonik Magazine, 2008, 4, 18–23.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS.
- Shell energy scenarios to 2050, pdf file http://www.shell.com/home/content/aboutshell/our_strategy/shell_global_scenarios/dir_global_scenarios_07112006.html available at http://www.shell.com.
- W. G. Ormerod, P. Freund and A. Smith, IEA Greenhouse Gas R&D Programme, 2002; W. G. Ormerod, P. Freund and A. Smith, Ocean storage of CO2, 2002; J. Davison, P. Freund and A. Smith, Putting carbon back into the ground, 2001; IEA Greenhouse Gas R&D Programme, all pdf-files available at: http://www.ieagreen.org.uk/publications.html. Large-scale CO2 capture and storage is estimated to start by 2020 and will be effective at an estimated level of ∼6 GT per annum by 2050 Search PubMed.
- The approximate size of carbon-deposits and carbon-fluxes have been estimated, data were taken from ref. 25 and J. A. Raven and A. J. Karley, Curr. Biol., 2006, 16, R165–R167 Search PubMed; P. Friedlingstein, I. Fung, E. Holland, J. John, G. Brasseur, D. Erickson and D. Schimel, Global Biogeochem. Cycles, 1995, 9, 541–556 CrossRef CAS; R. A. Houghton and G. M. Woodwell, Sci. Am., 1989, 260, 36–44 CrossRef CAS (i) Deposits (in Gt per carbon): terrestrial vegetation (500–600), soil (1500–1600), atmosphere (600–760), surface waters (770–1020), dissolved organic carbon (700), surface sediments (150), intermediate and deep ocean (38.000–40.000), marine sediments (20.000), fossil fuel (oil and coal, 7500). (ii) Fluxes (in Gt per carbon per annum): fossil fuel into CO2 (5.5–6.3), ocean uptake and release (90–105), plant growth and decay (60–100). (iii) CO2 Sequestration (in Gt per carbon per annum): plants (1), oceans (2–4).
- Whereas the crude material of industrial (organic) synthesis has a very low degree of functionalisation, i.e. mainly alkenes and aromatics, the average functional groups in the final products are as follows: OH (∼40%), CO2H (∼22%), NH2 (∼16%), SO (∼3%), miscellaneous (19%); in this overall context, industrial-scale (organic) synthesis constitutes oxidation at carbon.
- J. van Haveren, E. L. Scott and J. Sanders, Biofuels, Bioprod. Biorefin., 2008, 2, 41–57 Search PubMed.
- K. Kleiner, Nat. Rep. Clim. Change, 2008, 2, 9–11 Search PubMed.
- B. E. Rittmann, Biotechnol. Bioeng., 2008, 100, 203–212 CrossRef CAS.
- L. D. Schmidt and P. J. Dauenhauer, Nature, 2007, 447, 914–915 CrossRef CAS; G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS; J. N. Chedha, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS; B. E. Rittmann, Trends Biotechnol., 2006, 24, 261–266 CrossRef CAS.
- A. Melis and M. R. Melnicki, Int. J. Hydrogen Energy, 2006, 31, 1563–1573 CrossRef CAS; J. W. Tester, E. M. Drake, M. J. Driscoll, M. W. Golay and W. A. Peters, Sustainable Energy, MIT Press, Cambridge, MA, USA, 2005 Search PubMed; J. Rifkin, The Hydrogen Economy, Penguin Putnam Inc., New York, 2002 Search PubMed; F. Kreith, J. Energy Resour. Technol., 2004, 126, 249–257 Search PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond oil and gas: the methanol economy, Wiley-VCH, Weinheim, 2006 Search PubMed.
- The ‘holy grail’ of catalysis is the direct selective air-oxidation of methane to methanol, see: A. Behr, Angewandte homogene Katalyse, Wiley-VCH, Weinheim, 2008, pp. 656–657 Search PubMed.
- T. A. Werpy, J. E. Holladay and J. F. White, Top Value Added Chemicals From Biomass, 2004, pdf-file available from http://www.pnl.gov/publications Search PubMed.
- Prime candidates are: acetic, succinic, fumaric, itaconic, levulinic, furan-2,5-dicarboxylic, aspartic, glutamic, malic, glucaric, lactic, citric, and 3-hydroxypropionic acid, various fatty acids.
- C. H. Christensen, J. Rass-Hansen, C. C. Marsden, E. Taarning and K. Egeblad, ChemSusChem, 2008, 1, 283–289 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Carbon dioxide fixation into organic compounds, in Carbon dioxide recovery and utilisation, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2003, pp. 211–260 Search PubMed; S. Fujita, B. M. Bhanage and M. Arai, Chemical fixation of carbon dioxide: green processes to valuable chemicals, in Progress in Catalysis Research, ed. L. P. Bevy, Nova Science Publishers, Hauppauge, NY, USA, 2005, pp. 57–79 Search PubMed; M. Shi and Y.-M. Shen, Curr. Org. Chem., 2003, 7, 737–745 Search PubMed.
- See ref. 26, pp. 441–464.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1999, 99, 475–493 CrossRef CAS; M. Yoshida, N. Hara and S. Okuyama, Chem. Commun., 2000, 151–2 RSC; T. Sakakura, J.-C. Choi, Y. Saito, T. Masuda, T. Sako and T. Oriyama, J. Org. Chem., 1999, 64, 4506–4508 CrossRef CAS; H. Kawanami and Y. Ikushima, Chem. Commun., 2000, 2089–2090 RSC; R. J. Sowden, M. F. Sellin, N. D. Blasio and D. J. Cole-Hamilton, Chem. Commun., 1999, 2511–2512 RSC; T. Mizuno, N. Okamoto, T. Ito and T. Miyata, Tetrahedron Lett., 2000, 41, 1051–1053 CrossRef CAS; M. Kunert, M. Brauer, O. Klobes, H. Gorls and E. Dinjus, Eur. J. Inorg. Chem., 2000, 1803–1809 CrossRef CAS; Y. Wada, T. Kitamura and S. Yanagida, Res. Chem. Intermed., 2000, 26, 153–159 CAS; M. Tokuda, T. Kabuki, Y. Kato and H. Suginome, Tetrahedron Lett., 1995, 36, 3345–3348 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- M. Mori, Eur. J. Org. Chem., 2007, 4981–4993 CrossRef CAS.
- R. Zevenhofen, S. Eloneva and S. Teir, Catal. Today, 2006, 115, 73–79 CrossRef CAS; A. Behr, Angew. Chem., Int. Ed. Engl., 1988, 27, 661–678 CrossRef; D. H. Gibson, Chem. Rev., 1996, 96, 2063–2095 CrossRef CAS; W. Leitner, Coord. Chem. Rev., 1996, 153, 257–284 CrossRef CAS; M. Yoshida and M. Ihara, Chem.–Eur. J., 2004, 10, 2886–2893 CrossRef CAS.
- R. J. De Pasquale, J. Chem. Soc., Chem. Commun., 1973, 157–158 RSC; S. Inoue, in Carbon Dioxide as a Source of Carbon, ed. M. Aresta and G. Forti, Elsevier, Amsterdam, 1987, pp. 206–331 Search PubMed; D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 Search PubMed.
- M. Aresta, E. Quaranta and I. Tommasi, New J. Chem., 1994, 18, 133–142 Search PubMed.
- M. Tokuda, J. Nat. Gas Chem., 2006, 15, 275–281 Search PubMed.
- A. S. Lindsey and H. Jeskey, Chem. Rev., 1957, 57, 583–620 CrossRef CAS.
- Estimates taken from ref. 30, including inorganic products, the total amount of chemical CO2-sequestration equals to ca. 100 Mt per annum.
- I. Karube, T. Takeuchi and D. J. Barnes, Adv. Biochem. Eng. Biotechnol., 1992, 46, 63–79 CAS; B. Wang, Y. Li, N. Wu and C. Q. Lan, Appl. Microbiol. Biotechnol., 2008, 79, 707–718 CrossRef CAS; K. Asada, Biological Carboxylations, in Organic and Bio-Organic Chemistry of Carbon Dioxide, ed. S. Inoue and N. Yamazaki, Kodansha, Tokyo, 1982, ch. 5, pp. 185–251 Search PubMed.
- In contrast, heterotrophic organisms obtain their carbon through uptake of organic compounds (other than CO2) from the environment.
- CO2-fixation occurs in autotrophic procaryotes at a scale of 2 × 1011 tons per annum globally via four major pathways: (i) Calvin-cycle, (ii) acetyl-CoA pathway, (iii) 3-hydroxypropionate pathway, (iv) reductive tricarboxylic acid cycle; see: (a) J. M. Shively, G. van Keulen and W. G. Meijer, Annu. Rev. Microbiol., 1998, 52, 191–230 CrossRef CAS; (b) H. G. Wood, S. W. Ragsdale and E. Pezacka, Trends Biochem. Sci., 1986, 11, 14–18 CrossRef CAS; S. W. Ragsdale, Crit. Rev. Biochem. Mol. Biol., 1991, 26, 261–300 CrossRef CAS; (c) S. Herter, G. Fuchs, A. Bacher and W. Eisenreich, J. Biol. Chem., 2002, 277, 20277–20283 CrossRef CAS; (d) M. C. Evans, B. B. Buchanan and D. I. Arnon, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 928–934 CrossRef CAS; B. B. Buchanan and D. I. Arnon, Photosynth. Res., 1990, 24, 47–53 CrossRef CAS; G. Fuchs, Prepr. Pap.-Am. Chem. Soc., Div. Fuel Chem., 2008, 53, 381–382 Search PubMed.
- Although the term ‘chemosynthesis’ was coined by S. N. Vinogradskii already in 1890, the existence of light-independent CO2-assimilation was proven only in the late 1970s through the discovery of strange life forms in the vicinity of deep-sea hydrothermal vents.
- M. Hügler, H. Huber, K. O. Stetter and G. Fuchs, Arch. Microbiol., 2003, 179, 160–173; R. K. Thauer, Science, 2007, 318, 1732–1733 CrossRef CAS.
- M. Calvin, Nature, 1961, 192, 799–234; F. C. Hartman and M. R. Harpel, Annu. Rev. Biochem., 1994, 63, 197–234 CrossRef CAS.
- M. C. Evans, B. B. Buchanan and D. I. Arnon, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 928–934 CrossRef CAS.
- S. W. Ragsdale, Ann. N. Y. Acad. Sci., 2008, 1125, 129–136 CAS; H. L. Drake, A. S. Gößner and S. L. Daniel, Ann. N. Y. Acad. Sci., 2008, 1125, 100–128 CAS; S. W. Ragsdale and E. Pierce, Biochim. Biophys. Acta, 2008, 1784, 1873–1898 CAS.
- S. Herter, G. Fuchs, A. Bacher and W. Eisenreich, J. Biol. Chem., 2002, 277, 20277–20283 CrossRef CAS; B. Alber, M. Olinger, A. Rieder, D. Kockelkorn, B. Jobst, M. Hügler and G. Fuchs, J. Bacteriol., 2006, 188, 8551–8559 CrossRef CAS.
- I. A. Berg, D. Kockelkorn, W. Buckel and G. Fuchs, Science, 2007, 318, 1782–1786 CrossRef CAS.
- H. Huber, M. Gallenberger, U. Jahn, E. Eylert, I. A. Berg, D. Kockelkorn, W. Eisenreich and G. Fuchs, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7851–7856 CrossRef CAS.
- T. J. Erb, I. A. Berg, V. Brecht, M. Müller, G. Fuchs and B. E. Alber, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10631–10636 CrossRef CAS.
- F. C. Hartman and M. R. Harpel, Annu. Rev. Biochem., 1994, 63, 197–234 CrossRef CAS; I. Andersson and A. Backlund, Plant Physiol. Biochem., 2008, 46, 275–291 CAS; J. R. Bowyer and R. C. Leegood, Photosynthesis, in Plant Biochemistry, ed. P. Dey and J. Harborne, Academic Press, New York, 1997, pp. 49–110 Search PubMed; R. J. Ellis, Trends Biochem. Sci., 1979, 4, 241–244 Search PubMed.
- G. Schneider, Y. Lindqvist and C.-I. Bränden, Annu. Rev. Biophys. Biomol. Struct., 1992, 21, 119–143 CrossRef CAS.
- J. Pierce, N. E. Tolbert and R. Barker, J. Biol. Chem., 1980, 255, 509–511 CAS.
- W.-D. Fessner, A. Schneider, H. Held, G. Sinerius, C. Walter, M. Hixon and J. V. Schloss, Angew. Chem., Int. Ed. Engl., 1996, 35, 2219–2221 CrossRef CAS.
- Besides the well-known photosynthetic organisms (algae and plants), the Calvin-cycle is also utilised by purple nonsulfur bacteria (Rhodobacter, Rhodospirillium, Rhodopseudomonas), and purple sulfur bacteria (Chromatium), cyanobacteria (Synechococcus, Anacystis, Anabaena), hydrogen bacteria (Rastonia, Hydrogenovibrio) and chemoautotrophs (Thiobacillus).
- G. G. B. Tcherkez, G. D. Farquhar and T. J. Andrews, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7246–7251 CrossRef CAS.
- S. Gutteridge and J. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7203–7204 CrossRef CAS.
- The total amount of RubisCO has been estimated to be 4 × 1013 g, which accounts for approx. 0.2% of the total protein mass on earth; depending on the species, it counts up to 65% of the total soluble protein in leaf extracts, see ref. 53.
- R. J. Spreitzer and M. E. Salvucci, Annu. Rev. Plant Biol., 2002, 53, 449–475 CrossRef CAS; T. J. Andrews and S. M. Whitney, Arch. Biochem. Biophys., 2003, 414, 159–169; C. C. Mann, Science, 1999, 283, 314–316 CrossRef CAS.
- T. J. Andrews and G. H. Lorimer, in The Biochemistry of Plants, ed. M. D. Hatch and N. K. Boardman, Academic Press, NY, 1987, vol. 10, pp. 131–218 Search PubMed.
- Also denoted as citric acid cycle, Krebs-cycle or (more rarely) Szent-Györgyi–Krebs cycle.
- B. B. Buchanan and D. I. Arnon, Photosynth. Res., 1990, 24, 47–53 CrossRef CAS; M. Aoshima, Appl. Microbiol. Biotechnol., 2007, 75, 249–255 CrossRef CAS.
- D. E. Canfield and A. Teske, Nature, 1996, 382, 127–132 CrossRef CAS; C. H. House and S. T. Fitz-Gibbon, J. Mol. Evol., 2002, 54, 539–547 CrossRef CAS; N. R. Pace, Cell (Cambridge, Mass.), 1991, 65, 531–533 CrossRef CAS.
- G. Wächtershäuser, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 200–204 CAS; D. S. Ross, Origins Life Evol. Biosphere, 2007, 37, 61–65 CrossRef CAS.
- S. W. Ragsdale, J. Inorg. Biochem., 2007, 101, 1657–1666 CrossRef.
- S. W. Ragsdale, BioFactors, 1997, 6, 3–11 Search PubMed; S. W. Ragsdale, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 165–195 CrossRef CAS; P. A. Lindahl and B. Chang, Origins Life Evol. Biosphere, 2001, 31, 403–434 CrossRef CAS.
- It has been estimated that acetogens produce over 1013 kg of acetic acid annually, see ref. 48.
- P. M. Maitlis, A. Haynes, G. J. Sunley and M. J. Howard, J. Chem. Soc., Dalton Trans., 1996, 2187–2196 RSC.
- J. Seravalli, S. Zhao and S. W. Ragsdale, Biochemistry, 1999, 38, 5728–5735 CrossRef CAS.
- T. I. Doukov, T. M. Iverson, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Science, 2002, 298, 567–572 CrossRef CAS; C. Darnault, A. Volbeda, E. J. Kim, P. Legrqand, X. Vernede and P. A. Lindahl, Nat. Struct. Biol., 2003, 10, 271–279 CrossRef CAS; V. Svetlitchnyi, H. Dobbek, W. Meyer-Klaucke, T. Mains, B. Thiele, P. Römer, R. Huber and O. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 446–451 CrossRef CAS.
- E. L. Hegg, Acc. Chem. Res., 2004, 37, 775–783 CrossRef CAS.
- R. K. Thauer, Science, 2007, 318, 1732–1733 CrossRef CAS.
- M. Hügler, R. S. Krieger, M. Jahn and G. Fuchs, Eur. J. Biochem., 2003, 270, 736–744 CrossRef CAS.
- U. Jahn, H. Huber, W. Eisenreich, M. Hügler and G. Fuchs, J. Bacteriol., 2007, 189, 4108–4119 CrossRef CAS.
- R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 CrossRef CAS; R. M. Kohli and V. Massey, J. Biol. Chem., 1998, 273, 32763–32770 CrossRef CAS.
- G. Wächtershäuser, Chem. Biodiversity, 2007, 4, 584–602 CrossRef; W. Martin and M. J. Russel, Philos. Trans. R. Soc. London, Ser. B, 2007, 362, 1887–1926 CrossRef CAS.
- It is quite likely that the major pathways for energy-production in aerobic organisms—β-oxidation of fatty acids and the TCA-(Krebs)-cycle—were presumably ‘invented’ during evolution in the reverse direction.
- R. Buick, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 2731–2743 CrossRef CAS; T. Cavalier-Smith, M. Brasier and T. M. Embley, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 845–850 CrossRef CAS; A. D. Anbar, Y. Duan, T. W. Lyons, G. L. Arnold, B. Kendall, R. A. Creaser, A. J. Kaufman, G. W. Gordon, C. Scott, J. Garvin and R. Buick, Science, 2007, 317, 1903–1906 CrossRef CAS.
- H. Atomi, J. Biosci. Bioeng., 2002, 94, 497–505 CAS.
- K. Sugimura, S. Kuwabata and H. Yoneyama, J. Am. Chem. Soc., 1989, 111, 2361–2362 CrossRef CAS.
- Y. F. Cheung, C. H. Fung and C. Walsh, Biochemistry, 1975, 14, 2981–2986 CrossRef CAS.
- T. C. Bruice and A. F. Hegarty, Proc. Natl. Acad. Sci. U. S. A., 1970, 65, 805–809 CrossRef CAS; I. Climent and V. Rubio, Arch. Biochem. Biophys., 1986, 251, 465–470 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Rev. Mol. Biotechnol., 2002, 90, 113–128 CrossRef CAS.
- R. L. Slaughter and D. J. Edwards, Ann. Pharmacother., 1995, 29, 619–624 CAS.
- T. Yoshida and T. Nagasawa, J. Biosci. Bioeng., 2000, 89, 111–118 CrossRef CAS.
- T. Matsuda, T. Harada and K. Nakamura, Green Chem., 2004, 6, 440–444 RSC.
- A. Liese and M. V. Filho, Curr. Opin. Biotechnol., 1999, 10, 595–603 CrossRef CAS.
- This activity seems to be widespread among the microbial community and was reported from Aspergillus niger, Aspergillus oryzae, Pseudomonas testosteroni, K. pneumoniae, B. megaterium, Serratia spp., and even sulfur-metabolisers: see ref. 112, 119, 121 and: J. C. Patel and D. J. W. Grant, Antonie van Leeuwenhoek, 1969, 35, 53–64 Search PubMed.
- A. Dibenedetto, R. Lo Noce, C. Pastore, M. Aresta and C. Fragale, Environ. Chem. Lett., 2006, 3, 145–148 Search PubMed.
- J. Swaving and J. A. M. de Bont, Enzyme Microb. Technol., 1998, 22, 19–26 CrossRef CAS.
- J. R. Allen and S. A. Ensign, J. Bacteriol., 1996, 178, 1469–1471 CAS.
- F. J. Small and S. A. Ensign, J. Bacteriol., 1995, 177, 6170–6175 CAS.
- F. J. Small, J. K. Tilley and S. A. Ensign, Appl. Environ. Microbiol., 1995, 61, 1507–1513 CAS.
- C. A. G. M. Weijers, H. Jongejan, M. C. R. Franssen, A. de Groot and J. A. M. de Bont, Appl. Microbiol. Biotechnol., 1995, 42, 775–781 CrossRef CAS.
- J. Swaving, J. A. M. de Bont, A. Westphal and A. de Kok, J. Bacteriol., 1996, 178, 6644–6646 CAS.
- At about the same time of these studies, we investigated the closely related enzymatic rearrangement of epoxides furnishing aldehydes/ketones in Actinomyces spp. Overall, the enzymes involved turned out to be extremely sensitive, and could not even survive lyophilisation, W. Kroutil and K. Faber, unpublished.
- M. Boll and G. Fuchs, Biol. Chem., 2005, 386, 989–997 CrossRef CAS.
- K. Schühle and G. Fuchs, J. Bacteriol., 2004, 186, 4556–4567 CrossRef CAS.
- The regioselectivity in the chemical carboxylation of phenol via the Kolbe–Schmitt-reaction depends on the type of countercation: whereas Na-phenolates predominantly furnish salicylic acid, K-salts predominantly form p-hydroxybenzoic acid.
- M. Aresta, E. Quaranta, R. Liberio, C. Dileo and I. Tommasi, Tetrahedron, 1998, 54, 8841–8846 CrossRef CAS.
- B. Ding, S. Schmeling and G. Fuchs, J. Bacteriol., 2008, 190, 1620–1630 CrossRef CAS.
- For the reclassification of Clostridium hydroybenzoicum as S. hydroxybenzoicus, see: A. Breitenstein, J. Wiegel, C. Haertig, N. Weiss, J. R. Andreesen and U. Lechner, Int. J. Syst. Evol. Microbiol., 2002, 52, 801–807 Search PubMed.
- X. Zhang and J. Wiegel, Appl. Environ. Microbiol., 1994, 60, 4182–4185 CAS.
- Z. He and J. Wiegel, Eur. J. Biochem., 1995, 229, 77–82 CAS.
- Z. He and J. Wiegel, J. Bacteriol., 1996, 178, 3539–4353 CAS.
- J. Huang, Z. He and J. Wiegel, J. Bacteriol., 1999, 181, 5119–5122 CAS.
- T. Matsui, T. Yoshida, T. Hayashi and T. Nagasawa, Arch. Microbiol., 2006, 186, 21–29 CrossRef CAS.
- J. Liu, X. Zhang, S. Zhou, P. Tao and J. Liu, Curr. Microbiol., 2007, 54, 102–107 CrossRef CAS.
- J. F. Cavin, V. Dartois and C. Divies, Appl. Environ. Microbiol., 1998, 64, 1466–1471 CAS.
- B. Lupa, D. Lyon, L. N. Shaw, M. Sieprawska-Lupa and J. Wiegel, Can. J. Microbiol., 2008, 54, 75–81 CrossRef CAS.
- C. Gallert and J. Winter, Appl. Microbiol. Biotechnol., 1992, 37, 119–124 CAS.
- N. Gorny and B. Schink, Appl. Environ. Microbiol., 1994, 60, 3396–3400 CAS.
- T. Yoshida, Y. Hayakawa, T. Matsui and T. Nagasawa, Arch. Microbiol., 2004, 181, 391–397 CrossRef CAS.
- Carboxylation of phenol, 1,2- and 1,3-dihydroxbenzene by 2,6-dihydroxybenzoate decarboxylase from Pandoraea sp. 12B-2 see: T. Matsui, T. Yoshida, T. Yoshimura and T. Nagasawa, Appl. Microbiol. Biotechnol., 2006, 73, 95–102 Search PubMed.
- Y. Ishii, Y. Narimatsu, Y. Iwasaki, N. Arai, K. Kino and K. Kirimura, Biochem. Biophys. Res. Commun., 2004, 324, 611–620 CrossRef CAS.
- M. Yoshida, N. Fukuhara and T. Oikawa, J. Bacteriol., 2004, 186, 6855–6863 CrossRef CAS.
- Y. Iwasaki, K. Kino, H. Nishide and K. Kirimura, Biotechnol. Lett., 2007, 29, 819–822 CrossRef CAS.
- A. V. Kamath and C. S. Vaidyanathan, Biochem. Biophys. Res. Commun., 1987, 145, 586–595 CrossRef CAS; R. Santha, N. Appaji Rao and C. S. Vaidyanathan, Biochem. Biophys. Acta, 1996, 1293, 191–200 CrossRef.
- J. J. Anderson and S. Dagley, J. Bacteriol., 1981, 146, 291–297 CAS.
- T. Nakazawa and E. Hayashi, Appl. Environ. Microbiol., 1978, 36, 264–269 CAS; B. G. Pujar and D. W. Ribbons, Appl. Environ. Microbiol., 1985, 49, 374–376 CAS.
- Y. Hashidoko and S. Tahara, Arch. Biochem. Biophys., 1998, 359, 225–230 CrossRef CAS.
- B. J. Finkle, J. C. Lewis, J. W. Corse and R. E. Lundin, J. Biol. Chem., 1962, 37, 2926–2931.
- M. Zeida, M. Wieser, T. Yoshida, T. Sugio and T. Nagasawa, Appl. Environ. Microbiol., 1998, 64, 4743–4747 CAS.
- Z. Huang, L. Dosta and J. P. N. Rosazza, J. Bacteriol., 1994, 176, 5912–5918 CAS.
- G. Degrassi, P. P. de Laureto and C. V. Bruschi, Appl. Environ. Microbiol., 1995, 61, 326–332 CAS.
- J.-F. Cavin, L. Barthelmebs and C. Diviès, Appl. Environ. Microbiol., 1997, 63, 1939–1944 CAS.
- T. Hsu, M. F. Lux and H. L. Drake, J. Bacteriol., 1990, 172, 5901–5907 CAS.
- D. J. W. Grant and J. C. Patel, Antonie van Leeuwenhoek, 1969, 35, 325–341 CrossRef CAS.
- B. Lupa, D. Lyon, M. D. Gibbs, R. A. Reeves and J. Wiegel, Genomics, 2005, 86, 342–351 CrossRef CAS.
- T. Yoshida and T. Nagasawa, J. Biosci. Bioeng., 2000, 89, 111–118 CrossRef CAS.
- M. Wieser, T. Yoshida and T. Nagasawa, J. Mol. Catal. B: Enzym., 2001, 11, 179–184 CrossRef CAS.
- M. Wieser, T. Yoshida and T. Nagasawa, Tetrahedron Lett., 1998, 39, 4309–4310 CrossRef CAS.
- M. Wieser, N. Fujii, T. Yoshida and T. Nagasawa, Eur. J. Biochem., 1998, 257, 495–499 CrossRef CAS.
- H. Omura, M. Wieser and T. Nagasawa, Eur. J. Biochem., 1998, 253, 480–484 CrossRef CAS.
- T. Matsuda, Y. Ohashi, T. Harada, R. Yanagihara, T. Nagasawa and K. Nakamura, Chem. Commun., 2001, 2194–2195 RSC.
- T. Matsuda, T. Harada and K. Nakamura, Green Chem., 2004, 6, 440–444 RSC.
- T. Yoshida, K. Fujita and T. Nagasawa, Biosci. Biotechnol. Biochem., 2002, 66, 2388–2394 CrossRef CAS.
- (a) T. C. Appleby, C. Kinsland, T. P. Begley and S. E. Ealick, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2005–2010 CrossRef CAS; (b) B. G. Miller, A. M. Hassell, R. Wolfenden, M. V. Milburn and S. A. Short, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2011–2016 CrossRef CAS; (c) W. Cui, J. G. de Witt, S. M. Miller and W. Wu, Biochem. Biophys. Res. Commun., 1999, 259, 133–135 CrossRef CAS.
- M. Miyazaki, M. Shibue, K. Ogino, H. Nakamura and H. Maeda, Chem. Commun., 2001, 1800–1801 RSC.
- K. Miyamoto and H. Ohta, J. Am. Chem. Soc., 1990, 112, 4077–4078 CrossRef CAS; K. Miyamoto and H. Ohta, Eur. J. Biochem., 1992, 210, 475–481 CAS; K. Matoishi, M. Ueda, K. Miyamoto and H. Ohta, J. Mol. Catal. B: Enzym., 2004, 27, 161–168 CrossRef CAS.
- K. Miyamoto, Y. Yoshito, K. Tamura, Y. Terao and H. Ohta, J. Biosci. Bioeng., 2007, 104, 263–267 CrossRef CAS.
- Y. Terao, Y. Ijima, K. Miyamoto and H. Ohta, J. Mol. Catal. B: Enzym., 2007, 45, 15–20 CrossRef CAS.
- M. Aresta, E. Quaranta, I. Tommasi, P. Giannoccaro and A. Ciccarese, Gazz. Chim. Ital., 1995, 125, 509–538 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Quantum chemical calculation of Gibbs free energy values (ΔGreact) for a representative set of 28 carboxylation reactions. See DOI: 10.1039/b807875k |
| ‡ On leave from Middle East Technical University, Ankara, Turkey. |
| This journal is © The Royal Society of Chemistry 2010 |