An organic matrix-mediated processing methodology to fabricate hydroxyapatite based nanostructured biocomposites†
Prakash Hariram
Kithva
a,
Lisbeth
Grøndahl
b,
Rajendra
Kumar
c,
Darren
Martin
a and
Matt
Trau
*a
aAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, 4072, Australia. E-mail: m.trau@uq.edu.au; Fax: +61 7 33463973; Tel: +61 7 33464173
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, 4072, Australia
cDivision of Bioengineering, MontanaTech of the University of Montana, Butte, MT 59701, USA
First published on 13th August 2009
Abstract
An amorphous calcium phosphate precursor phase, which forms by adding orthophosphoric acid to a calcium hydroxide suspension, is transformed into crystalline hydroxyapatite by introducing polymer solutions. The nanostructured composite films formed by a solvent casting technique from the concentrated hybrid suspension are characterised for structure and mechanical properties.
Many biological structural materials, composed of a brittle inorganic phase and a soft organic phase, exhibit an unusual combination of high stiffness and strength coupled with high toughness.1–4 This has inspired materials scientists to develop synthetic biocomposite materials having similar properties. In particular, hydroxyapatite (HA, Ca5(PO4)3OH) based resorbable biocomposites with mechanical properties close to bone are the object of much research aiming at producing load-bearing implants.5,6 Human cortical bone exhibits an elastic modulus (E) and maximum strength (UTS) in the long axis of bone under tension in the range 12–18 GPa and 140–180 MPa, respectively.7 As the superior mechanical properties of bone are greatly attributed to the hierarchically organized building of the organic/inorganic nano-structured hybrid,3,4 several scientists have attempted to create nano-structured biocomposites of HA and collagen similar to those found in bone.8–11 However, the mechanical properties of thus produced composites either displayed a low strength (a bending strength of 50 MPa for an 80 wt% HA and collagen composite)9 or they were not reported. Therefore, much attention has been focussed on HA composites of biopolymers other than collagen. In particular, water soluble polysaccharides such as alginate and chitosan have been investigated.12–20
Chitosan, a deacetylated derivative of chitin,12 which is found in shells of crustaceans, is a linear polymer composed of glucosamine (2-amino-2-deoxy-D-gluco-pyranose) and it is soluble in weak acidic solution. Alginate, derived from seaweed, possesses a polysaccharide backbone comprised of two repeating carboxylated monosaccharide units (mannuronic acid, M, and guluronic acid, G), the ratio of which influences the physical properties of the biopolymer.21 HA biocomposites of these polymers have been produced by physical blending15–17 and co-precipitation.14,18–20 Although these processing techniques allow the production of dispersed HA particles within the polymer matrix, the HA particles are often on the micron scale15–17,20 (due to using micron-sized HA or to agglomeration during processing). A few studies reporting on chitosan/HA composites do display nano-sized HA particles;14,18,19 however, the resulting biocomposite materials exhibit poor mechanical properties. In the current work we present an advanced methodology to produce high mineral fraction HA/biopolymer composite films with nano-sized HA particles well dispersed in the polymer matrix that show tensile properties close to those of cortical bone. The method involves in situcrystallization of an amorphous calcium phosphate (ACP, Ca3(PO4)2·xH2O)22 precursor in the presence of a biopolymer. The two biopolymers tested in the current study were cationic chitosan (pre-treated with formaldehyde) and anionic alginate. The thus formed hybrid suspensions were cast into films with thicknesses of about 25–30 µm. These films were analyzed by XRD, TGA, SEM and tensile testing revealing the formation of phase-pure HA, and this was independent of the polymer used.
In the wet chemical synthesis of HA using aqueous solutions of Ca(OH)2 and H3PO4, two events take place concurrently (Fig. 1). They are the onset of decrease in pH of the reaction mixture (event 1) and the transformation of ACP into crystalline HA (event 2).22 With the addition of H3PO4 to a Ca(OH)2 suspension, the pH of the reaction mixture is initially maintained until the complete dissolution of dispersed Ca(OH)2, after which it starts decreasing. At the same time, formation of ACP begins and progressively increases in concentration up to a critical level beyond which it starts transforming into crystalline HA. The times at which event 1 (t1) and event 2 (t2) start depend on the temperature and the concentration of the Ca(OH)2 suspension as determined from pH and conductivity measurements.22 The critical factor in the present process of biocomposite fabrication is that the addition of polymer solutions to the reaction mixture should be performed before the ACP starts transforming into crystalline HA and after the pH starts decreasing (i.e.t1 < t2, Fig. 1). This can be achieved by using a high temperature (at 98 °C) and a reasonably diluted Ca(OH)2 suspension.
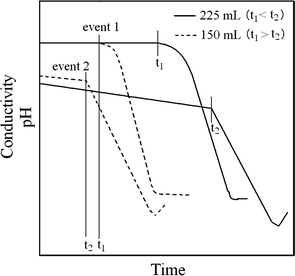 | ||
| Fig. 1 Schematic showing the effect of initial Ca(OH)2 suspension concentration on the start of the pH decrease and ACP → HA transformation reaction in the reaction mixture at 98 °C. This diagram is in part based on previously published work.22 | ||
In the present study, a formaldehyde treated chitosan (HCS) solution and a sodium alginate (SA) solution were used to fabricate HCS–HA and SA–HA composite films, respectively. Both composites contained about 55 wt% HA (ESI,† Fig. 1S and Table 1S). The polymer solution was added to the reaction mixture when the pH began to decrease (that is, after the event 1) and the initial concentration of Ca(OH)2 suspension was adjusted so that event 1 takes place before event 2 (ACP → HA). This is a key factor in this process. The suspensions were heated whilst stirring to increase the concentration of polymer and the concentrated suspensions were used to cast films. Attempts to produce a high HA content (>50%) biocomposite of untreated chitosan (CS) (without formaldehyde addition) following the same method used to prepare the HCS–HA films failed as the composite precipitated out in solution during solvent evaporation and films with adequate thickness could not be produced. However, the as-synthesized suspension produced films of thickness less than 10 µm. Although the adsorption of chitosan onto HA has been shown to lead to colloidal stability as well as the long term chemical stability of HA in solution,23 the level of binding interaction between HA particles and chitosan molecules without formaldehyde treatment appears to be insufficient. This emphasizes the importance of formaldehyde treatment to chitosan in improving the binding interaction between HA and chitosan.
The nanostructure of the composite films at high magnification (Fig. 2a and b) shows an array of HA crystals which appear to be surrounded by the polymer. The array of HA crystals are randomly orientated and appear very similar in the two biocomposites. It can be seen in Fig. 2 that the biocomposites contain nano-sized and nearly spherical shaped HA particles. In the low magnification images (Fig. 2c and d) it is evident that the spatial distribution of the HA particles is uniform in the matrix with no major agglomeration occurring in these hybrid films. A similar nanostructure (nearly spherical shaped HA particles) has been reported in chitosan–HA hybrid films where chitosan was used in trace quantities without formaldehyde treatment (maximum 3 wt% with respect to nHA) and the hybrid suspension was not concentrated prior to solvent casting.24 It is well documented that the reaction between Ca(OH)2 and H3PO4 produces HA crystals whose shape changes from needle-like at 40 °C to nearly spherical at 95 °C (decrease in aspect ratio).25,26 Thus, in the present composites, a combination of high temperature and interaction of polymer chains with ACP particles resulted in the formation of near spherical HA nano-sized particles.
 | ||
| Fig. 2 FESEM micrographs (secondary electron images) showing the nanostructures of (a) HCS–HA and (b) SA–HA composite films. Images (c) and (d) are lower magnifications of (a) and (b) respectively, showing the spatial distribution of HA particles in the polymer matrix. | ||
The X-ray diffraction (XRD) patterns of both composite films showed diffractions which can all be assigned to crystalline HA using JCPDS card no. 09-432 (Fig. 3) thus indicating that highly phase pure HA has formed. The relative diffraction intensities for different crystallographic planes of both biocomposites were similar, however they did not match with the JCPDS card. The relative intensity of the (002) plane, in particular, is weaker and that of the (300) plane is stronger compared to the JCPDS card. Kumar et al.24 have also shown similar weaker (002) diffraction and stronger (300) diffraction intensities in untreated chitosan–97 wt% HA hybrids.
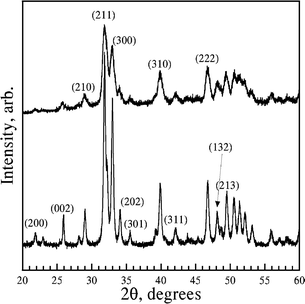 | ||
| Fig. 3 X-Ray diffraction pattern of HCS–HA (bottom) and SA–HA (top). The diffractions were indexed using JCPDS card no. 09-432 for synthetic HA. | ||
The Rietveld analysis of the XRD patterns (Table 1) revealed the March–Dollase (MD) number for the plane (300) to be 0.83 for both the biocomposites indicating the HA crystallites are preferentially oriented in the [100] direction and the degree of preferred orientation is comparable in the biocomposites. The calculated average crystallite size indicated that they are larger in HCS–HA films compared to those in SA–HA films (Table 1). As these crystallite sizes are in close agreement with the particle size shown in Fig. 2, it can be said that each particle is a single crystal of HA.
The tensile properties of the biocomposites formed in the current study show pronounced increases in both strength and modulus compared to the pure biopolymer films (p < 0.05) (Table 2 and ESI† Fig. 2S). The strain at failure was significantly different from that of the pure polymer only in the SA–HA but not in the HCS–HA film. The HCS films showed a large standard deviation in strain at failure; however, the reason for this is not known. The significant improvements in both elastic modulus and UTS of the biocomposite films are likely to be due to the strong interfacial bonding between the nano-sized HA particles and the biopolymer as well as the uniform dispersion of the nano-sized HA particles within the polymer matrix. In the SA–HA composite, the strong Coulombic interactions between the carboxylate groups in alginate and calcium ions in nHA particles appear to prevent the sedimentation of the HA particles even in the concentrated suspension. As such, alginate has a strong tendency for binding towards calcium ions, which are also used as a cross-linking agent for SA.27,28 In HCS–HA composites, at present it is clear that formaldehyde aids chitosan to interact strongly with the ACP particles (only formaldehyde chitosan could be used to produce composites of high HA content). It has been reported previously that formaldehyde can shield the amine groups through Schiff's base formation.29 However it is not clear if this is its role in the current work and further studies are currently under way to elucidate this.
The tensile properties obtained for the biocomposites in the current study (Table 2) are comparable to those of cortical bone (tensile modulus 12–18 GPa and strength 140–180 MPa7) and thus represent a significant advance compared to those previously reported for composite materials of similar composition. Composite films of chitosan–60 wt% HA prepared by a physical blending process where a slurry of HA was mixed with a chitosan solution (dissolved in acetic acid) showed a coarse microstructure with agglomerated HA particles and displayed a UTS of 29 MPa.15 Alternatively, in the co-precipitation method where a mixture of a chitosan solution and phosphoric acid was added to a suspension of Ca(OH)2 in water18 or in ethanol,19 a mixture of chitosan and HA precipitates yielding a material with poor mechanical properties (maximum bending modulus 260 MPa18 and compressive strength 109 MPa19). Mechanical properties of dense alginate–HA composites are not available in the literature, however Turco et al.17 have reported the mechanical properties of an 88% porous alginate–HA composite prepared by physical blending. They observed an 80% increase in compressive modulus and a 26% increase in ultimate compressive strength with respect to pure porous alginate.17 In the present study, the elastic modulus of the SA–HA composite is increased about 277% and the UTS is increased by about 65% with respect to pure SA (Table 2).
Experimental
Chitosan (∼85% deacetylated, low molecular weight), alginate sodium salt (extracted from Macrocystis pyrifera (kelp), high viscosity), calcium hydroxide, (Ca(OH)2, 96% pure and max. 3% CaCO3) were procured from Sigma-Aldrich. Acetic acid (CH3COOH, glacial, reagent grade), orthophosphoric acid (H3PO4, 85%) and formaldehyde (HCHO, 35%) were purchased from Univar. All the chemicals were used as-received and the solutions and suspensions were prepared using deionized (DI) water (MilliQ, Millipore, USA).Chitosan solution with a concentration of 10 g L−1 was prepared by dissolving chitosan powder in acetic acid solution (1 vol%) heated to 70 °C whilst stirring. After complete dissolution of chitosan, 35% formaldehyde (1 mL per g of chitosan) was added and stirred for 6 h at 70 °C. Alginate solution with a concentration of 10 g L−1 was prepared by dissolving sodium alginate powder in DI water heated at 70 °C and stirred. To a suspension of Ca(OH)2 (15 mmol or 1.11 g in 225 mL water) heated to 98 °C and stirred, H3PO4 solution (1.04 g 85% H3PO4 in 225 mL water) was added at a rate approximately 4 mL min−1 using a peristaltic pump (Cole-Palmer). After adding about 200 mL of acid, where the pH of the reaction mixture starts decreasing and the ACP concentration in the mixture is about 9.4 mM, 100 mL of HCS or SA solution was added, whilst acid addition continued, this mixture was heated at 98 °C for 2 h and then allowed to cool to room temperature (further details are given in ESI† ). This yields suspensions containing approximately 60 wt% HA.
The concentration of polymer in the as-synthesized hybrid suspension is about 1.8 g L−1. To cast films with adequate thickness (about 20 µm) to handle and test, at least 75 mL of the as-synthesized suspension should be dried on a 80 mm diameter Petri dish, which is practically not possible. Therefore, the suspension was concentrated by heating at 80 °C until the polymer concentration reached about 10 g L−1. The suspension was cooled whilst stirring and then stored at room temperature for two days to monitor any sedimentation. Sedimentation of nHA was not observed. Hybrid films were formed by transferring about 20 mL suspension onto glass Petri dishes and drying them at 37 °C in an oven overnight. Similarly, HCS and SApolymer films were also formed. The HCS and HCS–HA films were removed by soaking the dishes in 1 M NaOH solution for at least 30 min, the films were then washed with water and ethanol. Alginate and SA–nHA films were removed by soaking in acetone for 5 min and dried. The films had thicknesses in the range 25–30 µm and were stored in between glass plates under ambient conditions.
A Mettler-Toledo TGA LF1600 was used for thermo-gravimetric analysis (TGA) of films. The films were heated to 650 °C at a heating rate of 10 °C min−1 under reactive gas (air) atmosphere and held at 650 °C for 15 min before cooling to room temperature. The weight fraction of nHA particles in the composites was estimated using eqn (1) (more details and thermograms are provided in ESI† ).
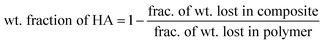 | (1) |
A Bruker-AXS D8 Advance X-ray diffractometer utilizing Cu Kα radiation and a graphite monochromator was used to identify the calcium phosphate phase in the composite films. The accelerating voltage and current used were 40 kV and 30 mA, respectively. The scanning was done over the 2θ range of 10–60° with step size 0.01° and step time 8 s. Quantitative analysis of the XRD data was carried out by Rietveld full profile fitting using MAUD (v. 2.04) crystallographic refinement software. The details of the refinement are reported in detail elsewhere.24,30–32 The HA crystal model was built using the information from the International Crystal Structure Database (ICSD) for hydroxyapatite. Peak shapes were modeled using the pseudo-Voigt function and two asymmetry parameters were refined. In each case, four background parameters, a scale factor, five peak shape parameters, 2θ offset (zero point correction), sample displacement, cell parameters and atomic positions were refined. After refinement of the parameters, the atomic position occupancies and thermal vibration factors for the various atomic species were refined till convergence was reached. The occupancy of the oxygen and hydrogen atoms associated with the –OH group was refined as a group (i.e. OH− occupancy) using the same reasoning given by Knowles et al.33 Lastly, the XRD data were also refined to construct the crystallite shape and size based on the Popa model,34 which refines crystallite size using the broadening of all diffraction peaks and calculates the individual coherent lengths for the hkl planes under consideration.
A JEOL 6300F field emission scanning electron microscope (FESEM) was used for structural characterization and all the images were taken in secondary electron imaging mode. Specimens were placed onto aluminium stubs using carbon adhesive tape and coated with platinum (thickness ∼10 nm) using an EIKO IB-5 Sputter coater.
Specimens for tensile testing were cut from the films using a dumbbell cutter (gauge length 14 mm, width 4 mm) and an Instron 5543 universal testing machine was used at a constant cross-head speed of 1 mm min−1 (strain rate about 10−3 s−1). For each sample, at least 4 specimens were tested and the average of the results with standard deviation is reported. Statistical significance was determined by Student's t-test using Instat Software (GraphPad Software Inc., USA). A value of p < 0.05 was considered to be statistically significant.
Conclusions
The fabrication method presented here produces strong composites with nano-sized, well dispersed HA particles in a polymer matrix. It is very versatile allowing strong composites to be formed from both anionic and cationic polysaccharides. The novelty of the fabrication method is that the polymers are added to an ACP particle suspension, which is more reactive than crystalline HA. The ACP particles then transform into crystalline HA with the matrix leading to a strong polymer–HA interface in the composites and hence much improved mechanical properties compared to composites prepared by physical blending or co-precipitation methods.Acknowledgements
This work was supported by the Australian Research Council (ARC) Centre of Excellence for Functional Nanomaterials, Australia. Prof. Matt Trau acknowledges the support of the ARC Federation Fellowship (FF0455861). The electron micrographs were taken in the Australian Microscopy and Microanalysis Research Facility (AMMRF), University of Queensland, Australia and the XRD was performed by run by Ms Anya Yago and Dr Kevin Jack of Brisbane Surface Analysis Facility, AMMRF, Australia.References
- J. D. Currey, Proc. R. Soc. London, Ser. B, 1977, 196, 443 CrossRef.
- G. Mayer, Science, 2005, 310, 1144 CrossRef CAS.
- S. Weiner and H. D. Wagner, Annu. Rev. Mater. Sci., 1998, 28, 271 CrossRef.
- T. Hassenkam, G. E. Fantner, J. A. Cutroni, J. C. Weaver, D. E. Morse and P. K. Hansma, Bone, 2004, 35, 4 CrossRef.
- W. Bonfield, M. D. Grynpas, A. E. Tully, J. Bowman and J. Abram, Biomaterials, 1981, 2, 185 CrossRef CAS.
- W. Suchanek and M. Yoshimura, J. Mater. Res., 1998, 13, 94 CrossRef CAS.
- P. Zioupos and J. D. Currey, Bone, 1998, 22, 57 CrossRef CAS.
- M. Kikuchi, S. Itoh, S. Ichinose, K. Shinomiya and J. Tanaka, Biomaterials, 2001, 22, 1705 CrossRef CAS.
- S. H. Rhee and J. Tanaka, J. Am. Ceram. Soc., 2001, 84, 459 CAS.
- N. Roveri, G. Falini, M. C. Sidoti, A. Tampieri, E. Landi, M. Sandri and B. Parma, Mater. Sci. Eng., C, 2003, 23, 441 CrossRef.
- M. J. Olszta, E. P. Douglas and L. B. Gower, Calcif. Tissue Int., 2003, 72, 583 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603 CrossRef CAS.
- A. D. Martino, M. Sittinger and M. V. Risbud, Biomaterials, 2005, 26, 5983 CrossRef.
- V. M. Rusu, C.-H. Ng, M. Wilke, B. Tiersch, P. Fratzl and M. G. Peter, Biomaterials, 2005, 26, 5414 CrossRef CAS.
- C. Xiammiao, L. Yuabo, Z. Yi, Z. Li, L. Jidong and W. Huanan, Mater. Sci. Eng., C, 2009, 29, 29 CrossRef.
- P. Parhi, A. Ramanan and A. R. Ray, J. Appl. Polym. Sci., 2006, 102, 5162 CrossRef CAS.
- G. Turco, E. Marsich, F. Bellomo, S. Semeraro, I. Donati, F. Brun, M. Grandolfo, A. Accardo and S. Paoletti, Biomacromolecules, 2009, 10, 1575 CrossRef CAS.
- I. Yamaguchi, K. Tokuchi, H. Fukuzaki, Y. Koyama, K. Takakuda, H. Monma and J. Tanaka, J. Biomed. Mater. Res., 2001, 55, 20 CrossRef CAS.
- Z. Li, L. Yubao, Y. Aiping, P. Xuelin, W. Xuejiang and Z. Xiang, J. Mater. Sci.: Mater. Med., 2005, 16, 213 CrossRef CAS.
- L. Wang, L. Yue and C. Li, J. Nanopart. Res., 2009, 11, 691 CrossRef CAS.
- D. A. Rees and E. J. Walsh, Angew. Chem., Int. Ed. Engl., 1977, 16, 214 CrossRef.
- K. H. Prakash, R. Kumar, C. P. Ooi, P. Cheang and K. A. Khor, J. Phys. Chem. B, 2006, 110, 24457 CrossRef CAS.
- O. C. Wilson Jr. and J. R. Hull, Mater. Sci. Eng., C, 2008, 28, 434 CrossRef.
- R. Kumar, K. H. Prakash, P. Cheang, L. Gower and K. A. Khor, J. R. Soc. Interface, 2008, 5, 427 CrossRef CAS.
- R. Kumar, K. H. Prakash, P. Cheang and K. A. Khor, Langmuir, 2004, 20, 5196 CrossRef CAS.
- K. H. Prakash, R. Kumar, C. P. Ooi, P. Cheang and K. A. Khor, Langmuir, 2006, 22, 11002 CrossRef CAS.
- I. Braccini and S. Pérez, Biomacromolecules, 2001, 2, 1089 CrossRef CAS.
- L. Li, Y. Fang, R. Vreeker and I. Mendes, Biomacromolecules, 2007, 8, 464 CrossRef CAS.
- N. Li and R. Bai, Ind. Eng. Chem. Res., 2005, 44, 6692 CrossRef CAS.
- R. Kumar, P. Cheang and K. A. Khor, Acta Mater., 2004, 52, 1171 CrossRef CAS.
- R. Kumar, K. H. Prakash, P. Cheang and K. A. Khor, Acta Mater., 2005, 53, 2327 CrossRef CAS.
- L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Louer and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36 CrossRef CAS.
- J. C. Knowles, K. A. Gross, C. C. Berndt and W. Bonfield, Biomaterials, 1996, 17, 639 CrossRef CAS.
- N. C. Popa, J. Appl. Crystallogr., 1998, 31, 176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of timing of polymer solution addition to the reaction mixture; TGA details; stress–strain curves. See DOI: 10.1039/b9nr00062c |
| This journal is © The Royal Society of Chemistry 2009 |